

Summary
Mutations or duplications in MECP2 cause Rett and Rett-like syndromes, neurodevelopmental disorders characterized by mental retardation, motor dysfunction, and autistic behaviors. MeCP2 is expressed in many mammalian tissues and functions as a global repressor of transcription; however, the molecular mechanisms by which MeCP2 dysfunction leads to the neural-specific phenotypes of RTT remain poorly understood. Here, we show that neuronal activity and subsequent calcium influx trigger the de novo phosphorylation of MeCP2 at serine 421 (S421) by a CaMKII- dependent mechanism. MeCP2 S421 phosphorylation is induced selectively in the brain in response to physiological stimuli. Significantly, we find that S421 phosphorylation controls the ability of MeCP2 to regulate dendritic patterning, spine morphogenesis, and the activity-dependent induction of Bdnf transcription. These findings suggest that, by triggering MeCP2 phosphorylation, neuronal activity regulates a program of gene expression that mediates nervous system maturation and that disruption of this process in individuals with mutations in MeCP2 may underlie the neural-specific pathology of RTT.
Introduction
Rett Syndrome (RTT) is an X-linked neurological disorder and a leading cause of mental retardation in females. Classical RTT is characterized by relatively normal development through the first 6–18 months of life, followed by an abrupt neurodevelopmental regression and stagnation that may result in loss of acquired skills such as purposeful movement and speech, deceleration of head growth, irregular breathing, stereotypical hand wringing, and the emergence of autistic behaviors (Hagberg et al., 1983; Moretti and Zoghbi, 2006; Rett, 1966). The symptoms of RTT appear during early childhood when sensory experience is driving the synaptic reorganization required for the emergence of appropriately functioning circuits in the mature brain. This observation raised the hypothesis that the underlying cause of RTT is inappropriate synaptic connectivity or plasticity, possibly resulting from abnormal experience-dependent synaptic maturation, refinement, and/or maintenance (Zoghbi, 2003).
The majority of RTT cases are caused by mutations in the methyl-CpG-binding protein 2 (MECP2) gene (Amir et al., 1999). MeCP2 is a member of the family of methyl-CpG-binding domain (MBD) proteins that function as long-range transcriptional repressors that mediate developmental silencing through binding to methylated DNA (Klose and Bird, 2006; Lewis et al., 1992). In addition to its amino-terminal MBD, MeCP2 contains a transcriptional-repressor domain (TRD) and a C terminus that facilitates binding to methylated DNA (Chandler et al., 1999; Kriaucionis and Bird, 2003). The mutations found in RTT span the entire MeCP2 protein and include missense, nonsense, insertion, deletion, and splice-site variations (Bienvenu and Chelly, 2006; Kriaucionis and Bird, 2003). Recently, reports of duplications of the entire MECP2 locus in some individuals with a RTT-like phenotype suggest that overexpression of MeCP2 also leads to Rett Syndrome (Ariani et al., 2004; Lugtenberg et al., 2006; Meins et al., 2005; Van Esch et al., 2005).
Like other MBD family proteins,MeCP2 is expressed in many somatic tissues. How disruption of this ubiquitously expressed protein leads to a predominantly neurological phenotype remains a central unresolved issue in the RTT field. One hint may lie in the fact that MeCP2 protein levels increase as neurons mature (Zoghbi, 2003). The high level of MeCP2 protein in mature neurons is consistent with a possible role for MeCP2 in synaptic processes.
Knockout mouse models with disrupted MeCP2 function mimic many key clinical features of RTT, including normal early postnatal life followed by developmental regression that results in motor impairment, irregular breathing, hindlimb clasping, and early mortality (Chen et al., 2001; Guy et al., 2001; Shahbazian et al., 2002). In addition, transgenic mice overexpressing wild-type MeCP2 protein also develop a RTT-like phenotype, suggesting that a precise level of MeCP2 expression is critical for proper brain development (Collins et al., 2004; Luikenhuis et al., 2004). Importantly, the conditional deletion of Mecp2 in postmitotic neurons recapitulates many of the phenotypes seen in the Mecp2 total null mouse (Chen et al., 2001; Guy et al., 2001), indicating that RTT is due to a specific defect in MeCP2 function in mature neurons.
Further supporting a role for MeCP2 in mature synaptic function and plasticity, Mecp2 null mice exhibit abnormalities in dendritic arborization (Chen et al., 2001; Kishi and Macklis, 2004), basal synaptic transmission (Moretti et al., 2006), presynaptic function (Asaka et al., 2006; Moretti et al., 2006; Nelson et al., 2006), excitatory synaptic plasticity (Asaka et al., 2006; Moretti et al., 2006), and hippocampal and amygdalar learning (Moretti et al., 2006; Pelka et al., 2006). In addition, Mecp2 mutant mice exhibit reduced spontaneous cortical activity due to an imbalance between excitatory and inhibitory circuitry in the cortex (Dani et al., 2005). Notably, many of the phenotypes observed in RTT individuals and Mecp2 mutant mice—defects in dendritic branching, spine density, synaptic plasticity, learning and memory, and the maturation of inhibitory circuits— involve processes known to be influenced by experience- dependent neuronal activity (Katz and Shatz, 1996). However, the molecular mechanisms by which MeCP2 dysfunction leads to these physiological and cellular defects and the relationship between MeCP2 and experience-dependent synaptic development remain undefined.
We recently found that membrane depolarization of neurons leads to phosphorylation of MeCP2, which correlates with the transcriptional induction of an activity- regulated gene, Bdnf (Chen et al., 2003). These findings, together with those of other groups (Ballas et al., 2005; Martinowich et al., 2003), suggested that dynamic regulation of MeCP2 by calcium influx may play a pivotal role in regulating specific programs of activity-dependent gene transcription that are important for nervous system function. In the present study, we investigated the significance of the calcium-dependent phosphorylation of MeCP2 for nervous system maturation and RTT. We find that phosphorylation of MeCP2 at a specific amino acid residue, S421, occurs selectively in the nervous system in response to neuronal activity. Mutation of MeCP2 at S421 blocks the ability of MeCP2 to restrict dendritic growth, spine maturation, and activate calcium- dependent Bdnf transcription in an in vitro overexpression model of RTT. These findings suggest that, by triggering MeCP2 phosphorylation, neuronal activity regulates a program of gene expression that mediates neuronal connectivity in the nervous system. The disruption of this process in individuals with mutations in MeCP2 may underlie the neural-specific pathology of RTT.
Results
Identification of S421 as a Site Required for Membrane Depolarization-Induced Phosphorylation of MeCP2
We have previously shown that membrane depolarization of neurons by exposure to elevated levels of extracellular potassium leads to the production of a slow-migrating species of MeCP2 as detected on a denaturing SDS-polyacrylamide gel (SDS-PAGE; Figure 1A, left). This slow-migrating form of MeCP2 disappears when extracts from membrane-depolarized neurons are treated with alkaline phosphatase (Figure 1A, right), suggesting that phosphorylation of MeCP2 may slow its migration on an SDS-PAGE gel. The slow-migrating form of MeCP2 exhibits reduced binding to methylated DNA, and its presence correlates with the induction of Bdnf promoter IV transcription in cortical cultures (see Figure 5 legend for Bdnf promoter nomenclature), raising the possibility that the phosphorylation of MeCP2 may inactivate the repressor function of MeCP2 (Chen et al., 2003). However, it remained to be determined whether phosphorylation, or another modification, regulates MeCP2 function in membrane-depolarized neurons and whether these modifications are relevant to the etiology of RTT.
Figure 1. Identification of MeCP2 S421 as a Site of Membrane Depolarization-Induced Phosphorylation.

(A) Antitotal MeCP2 western blots of whole-cell lysates (lanes 1–3) or nuclear extracts treated with alkaline phosphatase (lanes 5– 7) prepared from E18 + 5 DIV dissociated rat cortical neurons harvested at the indicated times following 55 mM KCl stimulation. The arrow indicates the slow-migrating form of MeCP2 induced in response to membrane depolarization. AP: Nuclear extracts were treated with 2, 4, or 8 U of calf intestinal alkaline phosphatase for 30 min at 37°C prior to SDS-PAGE separation. Note: MeCP2 runs at approximately 75 kDa on an 8% SDS-PAGE gel. All protein samples in this study were separated on an 8% gel unless otherwise noted.
(B) Coomassie blue staining of MeCP2 protein purified from rat brain nuclear extract. Both bands were excised and subjected to tandem MS/MS sequencing for identification of phosphorylation sites.
(C) A schematic of MeCP2 illustrating the positions of phosphorylation sites relative to the methyl-CpG-binding domain (MBD), nuclear localization signal (NLS), and the transcriptional repression domain (TRD).
(D) Identification of S421 as a residue required for membrane depolarization-induced phosphorylation of MeCP2. FLAG-tagged wild-type or S421A MeCP2 was transfected into E18 + 2 DIV cortical neurons. Two days later, extracts were prepared from untreated neurons or neurons membrane depolarized for 60 min and subjected to western blotting with the anti-FLAG antibody.
(E) A phospho specific antibody detects phosphorylation of MeCP2 at S421. FLAG immunoprecipitates from E18 + 5 DIV cortical neurons transfected with FLAG-tagged wild-type or S421A MeCP2 were immunoblotted with antitotal MeCP2 or anti-MeCP2 pS421 antibodies. The anti-MeCP2 pS421 antibody recognizes a band at a position corresponding to that of the slow-migrating species of MeCP2.
(F) Membrane depolarization triggers the phosphorylation of MeCP2 at S421. Antitotal MeCP2 and anti-MeCP2 pS421 western blots of nuclear extracts prepared from unstimulated or membrane-depolarized E18 + 5 DIV cortical neurons, either untreated (control) or incubated with alkaline phosphatase for 30 min at 37°C (AP). Unlike other experiments, cultures were not treated with TTX prior to stimulation in this experiment.
(G) The anti-MeCP2 pS421 antibody specifically recognizes MeCP2. Brain lysates from C57BL/6 wild-type Mecp2+/+, heterozygous Mecp2−/+, or Mecp2−/y null mice were probed with anti-MeCP2 pS421 or anti-actin antibodies.
Figure 5. Phosphorylation of MeCP2 at S421 Is Required for Activity-Induced Induction of Bdnf Transcription.

(A) The lentivirus-mediated protein-replacement assay (LEMPRA) construct contains two expression cassettes: (1) the pU6 pol III promoter directing the expression of an shRNA directed against endogenous MeCP2 and (2) the human Ubiquitin-C promoter driving the expression of a bicistronic cassette comprised of FLAG-tagged shRNA-resistant wild-type or S421A mutant MeCP2 followed by an internal ribosomal entry site (IRES) directing GFP expression. When packaged into and delivered by a lentivirus, the construct is effective in replacing MeCP2 in greater than 90% of neurons in a culture.
(B and C) LEMPRA can effectively reduce endogenous MeCP2 and express functional FLAG-tagged MeCP2 variants of choice. E18 rat hippocampal neurons were infected at 1 DIV with indicated lentiviruses. Extracts were prepared at 4 DIV (B) or 7 DIV (C) and incubated with antibodies specific to FLAG, MeCP2 pS421, or total MeCP2. shRNA, small hairpin RNA against MeCP2; sr-FgMP2, FLAG-tagged shRNA-resistant mouse MeCP2, e2 form; scr shRNA, scrambled shRNA; rMeCP2, endogenous rat MeCP2. Note that in (C), 5-fold more control lysates were loaded than wild-type or S421A-MeCP2 lysates for comparison of expression levels.
(D and E) E18 + 1 DIV hippocampal neurons were infected with control, wild-type, or S421A mutant MeCP2 LEMPRA lentiviruses. Total RNA was collected at 7 DIV from infected neurons that were either left untreated (−) or membrane depolarized with 55 mM KCl for 3 hr (+). Bdnf exon IV (D) and c-fos (E) mRNA levels were measured by quantitative RT-PCR and normalized to a Gapdh control (data are mean±SEM; *p < 0.01, Bonferroni-corrected multiple comparison after ANOVA). Note that the nomenclature for the exons of the Bdnf gene has changed due to the identification of additional Bdnf transcripts in rodent; Bdnf exon IV was previously known as Bdnf exon III, for example, in Chen et al. [2003]).
To address these issues, we sought to identify the amino acid residues on MeCP2 that become newly phosphorylated upon membrane depolarization of cortical neurons. We used tandem mass spectrometry to identify phosphorylation sites present on endogenous MeCP2 protein purified from P15 rat brain nuclear extracts (Figure 1B), as well as MeCP2 expressed in HEK293T cells (data not shown). Taken together, the mass spectrometry analyses revealed the presence of three major sites of phosphorylation—serine 80 (S80), serine 229 (S229), and serine 421 (S421) (Figure 1C). We next generated nonphosphorylatable mutant forms of MeCP2 to determine if the phosphorylation events identified by mass spectrometry were required for the generation of the slowly migrating species of MeCP2 in response to membrane depolarization. FLAG-tagged wild-type MeCP2 or MeCP2 constructs bearing various serine to alanine mutations were transfected into cortical neurons, and the tagged MeCP2 was detected by western blot with an antibody directed against the FLAG epitope. Whereas mutation of S80 and S229 of FLAG-MeCP2 to alanine did not affect the production of the slow-migrating species of MeCP2 in response to membrane depolarization, mutation of S421 to alanine (S421A) resulted in the loss of the activity-dependent MeCP2 mobility shift (Figure 1D). Moreover, mutation of 17 additional MeCP2 serine or threonine residues predicted by the Scansite program (http://scansite.mit.edu) to be phosphorylated (S13, S86, S113, S164, S178, S216, S229, S341, S350, S357, S360, S385, S399, T308, T311, T443, T477), either individually or in combinations, did not affect the production of the slow-migrating form of MeCP2 (data not shown). Thus, S421 is required for the generation of the slow-migrating form of MeCP2 in membrane-depolarized neurons, suggesting that membrane depolarization triggers the phosphorylation of MeCP2 at S421. Although we cannot exclude the possibility that membrane depolarization may concomitantly trigger the phosphorylation or dephosphorylation of MeCP2 at other sites, the phosphorylation status of these sites does not appear to affect the mobility of MeCP2 on an SDS-PAGE gel.
MeCP2 Is Phosphorylated at S421 in Response to Neuronal Activity
To directly assess whether membrane depolarization induces the phosphorylation of MeCP2 at S421, we raised an anti-MeCP2 S421 phospho-site-specific antibody (anti-MeCP2 pS421) and used the antibody to assess the phosphorylation status of MeCP2 under various conditions of neuronal stimulation in culture and in vivo. We first tested affinity-purified fractions of sera from rabbits immunized with a synthetic phosphopeptide containing phosphorylated MeCP2 S421 for their ability to recognize the nonphosphorylated and S421-phosphorylated forms of MeCP2. Western blotting of FLAG immunoprecipitates from cortical neurons expressing FLAG-tagged MeCP2 indicated that the anti-MeCP2 pS421 antibody specifically recognizes a band corresponding to the slow-migrating species of MeCP2 in membrane-depolarized neurons (Figure 1E). However, the anti-MeCP2 pS421 antibody failed to recognize a mutant version of FLAG-MeCP2 in which S421 was replaced with an alanine (S421A) in either untreated or membrane-depolarized neurons (Figure 1E). Furthermore, the anti-MeCP2 pS421 antibody effectively recognizes endogenous MeCP2 in extracts from membrane- depolarized neurons (Figure 1F), but not in extracts from unstimulated neurons (Figure 1F, left), membrane-depolarized neurons treated with alkaline phosphatase (Figure 1F, right) or brain extracts from Mecp2−/y null mice (Figure 1G). Together, these findings indicate that the anti-MeCP2 pS421 antibody specifically recognizes the S421-phosphorylated form of MeCP2. In addition, these results demonstrate that phosphorylation of MeCP2 at S421 is an inducible event that is triggered by membrane depolarization in cultured neurons.
Neurotransmitter Release at Synapses and Neurotrophin Stimulation Both Trigger MeCP2 S421 Phosphorylation
Although MeCP2 has been thought to function primarily as a stable, long-range transcriptional silencer, the finding that MeCP2 undergoes dynamic de novo phosphorylation at S421 in response to membrane depolarization suggests that MeCP2 may also function in a novel manner in the nervous system. To gain further insight into how MeCP2 function might be affected by neuronal activity, we used the anti-MeCP2 pS421 antibody to investigate the regulation of MeCP2 S421 phosphorylation in cultured neurons.
When cortical neurons were membrane depolarized for different periods of time using elevated levels of extracellular potassium to activate L-type voltage-sensitive calcium channels (L-VSCCs), MeCP2 S421 phosphorylation could be detected as early as 5 min after depolarization and reached its maximal level 60 min after stimulation (Figures 2A–2C). The relatively slow kinetics of MeCP2 S421 phosphorylation contrast with that of other phosphorylation events induced by membrane depolarization, such as the phosphorylation of CREB at S133, which is maximally activated just minutes after membrane depolarization (Figure 2A; Kornhauser et al., 2002). The slow kinetics of activity-dependent MeCP2 S421 phosphorylation correlates with the relatively slow induction of a subset of activity-regulated genes such as Bdnf, Narp, Ves1/Homer1a, and Crem/Icer (Lanahan and Worley, 1998), suggesting that the phosphorylation of MeCP2 at S421 may be a critical step in the activation of this class of genes.
Figure 2. Characterization of S421 as a Site of Activity-Dependent Phosphorylation on MeCP2.

(A–C) Kinetics of MeCP2 S421 phosphorylation and dephosphorylation. Lysates were prepared from rat E18 + 5 DIV cortical neurons membrane depolarized for the indicated times (A and B) or membrane depolarized for 30 min and repolarized with culture medium for the indicated times (C).
(D–F) MeCP2 is phosphorylated at S421 in response to glutamate (D), NMDA (E), and bicuculline (F) in a calcium-dependent manner. Antitotal MeCP2 and anti-MeCP2 pS421 western blots of extracts from E18 + 12 DIV hippocampal neurons (D and E) or P1 + 12 DIV hippocampal neurons (F) treated for 1 hr with the indicated stimuli after a 1 hr pretreatment with the indicated blockers. Nimo, nimodipine.
(G) MeCP2 S421 phosphorylation is triggered by neurotrophins. Western blot analysis of whole-cell extracts prepared from E18 + 12 DIV hippocampal neurons stimulated for 1 hr with BDNF, CNTF, EGF, IGF, NGF, NT3, NT4, or PDGF.
Glutamate or NMDA stimulation of cultured hippocampal neurons also induced the phosphorylation of MeCP2 at S421 (Figures 2D and 2E), indicating that stimulation of ligand-gated ion channels that mediate excitatory neurotransmission could also trigger MeCP2 phosphorylation at S421. Since both synaptic and extrasynaptic pools of glutamate receptors can potentially be activated by bath application of glutamate or NMDA (Hardingham et al., 2002), we also treated hippocampal neurons with the GABAA-receptor antagonist bicuculline to derepress tonic inhibition of endogenous excitatory neurotransmission and selectively allow glutamate release at synapses (Hardingham et al., 2002). We found that bicuculline stimulation also induces the phosphorylation of MeCP2 at S421 (Figure 2F). This event was dependent in part on activation of NMDA receptors, AMPA receptors, and L-VSCCs, as antagonists of each of these channels partially blocked the response (Figure 2F). Thus, endogenous glutamate release at synapses triggers the phosphorylation of MeCP2 at S421 directly via the gating of calcium influx through NMDA-type receptors and/or indirectly due to glutamate-mediated rises in membrane potential that activate calcium flux through L-VSCCs.
We next investigated the range of stimuli that induce the phosphorylation of MeCP2 at S421 in neurons. We found that the neurotrophins BDNF, NT3, and NT4 induced the acute phosphorylation of MeCP2 at S421, whereas CNTF, EGF, IGF, NGF, and PDGF each had no effect on MeCP2 S421 phosphorylation, even though these growth factors induced Akt and/or MAPK activation (Figure 2G). The ability of neurotrophins to induce MeCP2 S421 phosphorylation appears to be developmentally regulated and present primarily in older neurons (12 DIV), since no effect of neurotrophin treatment on MeCP2 S421 phosphorylation is observed in younger cultures (Chen et al., 2003). These results demonstrate that, among the many growth factors tested, only neurotrophins trigger the phosphorylation of MeCP2 at S421 in neurons and suggest that neurotrophin signaling may regulate MeCP2 function in addition to or in cooperation with neuronal activity.
Synaptic Activity Driven by Sensory Experience Induces Acute Phosphorylation of MeCP2 S421 in the Intact Brain
We next examined whether MeCP2 is phosphorylated at S421 in the brains of intact animals in response to synaptic activity. We first asked if synchronized synaptic activity induced by chemoconvulsants triggers MeCP2 S421 phosphorylation in the brain. Seizures were induced in rats or mice by intraperitoneal injection of metrazole (pentylenetetrazole) or kainic acid, respectively, and brain extracts or histological sections, respectively, were prepared from the cortex and hippocampus of seized and saline-injected control animals. Western blotting revealed that metrazole-based seizures induced a significant increase in the levels of S421-phosphorylated MeCP2 in the rat brain compared to saline-injected controls (Figure 3A). Furthermore, immunohistochemistry with the anti-MeCP2 pS421 antibody detected a significant increase in the number of neuronal nuclei immunopositive for S421-phosphorylated MeCP2 in cortical layers II–VI and in all regions of the hippocampus of seized brains (Figure 3B and data not shown). Parallel immunostaining with an antibody that recognizes MeCP2 regardless of its phosphorylation state (antitotal MeCP2) showed no significant change in the level of MeCP2 in response to seizures (Figure 3B), suggesting that, in the intact brain, neuronal activity triggers the phosphorylation of preexisting MeCP2. To confirm the specificity of the anti-MeCP2 pS421 and antitotal MeCP2 antibodies for immunohistochemistry, we stained brain sections from pairs of Mecp2−/y null mice left untreated or stimulated with kainic acid injection and observed no signal with either antibody under either condition (Figure 3B, top). Together, these findings indicate that pharmacologically induced increases in synaptic activity trigger the acute phosphorylation of MeCP2 at S421 within neuronal nuclei in the intact brain.
Figure 3. Phosphorylation of MeCP2 at S421 Is Triggered by Synaptic Activity in the Intact Brain.

(A) Whole-cell lysates were prepared from P19 rat brains 0, 10, or 30 min after metrazole-based seizure induction and incubated with antibodies specific to MeCP2 pS421, total MeCP2, pCREB (S133), or total CREB.
(B) Fluorescent immunohistochemistry of coronal cryosections from mouse parietal cortex collected from wild-type (WT) or Mecp2−/y (KO) mice 3 hr after kainic acid-based seizure induction using antibodies specific for MeCP2 pS421 or total MeCP2. Sections were counterstained with the nuclear dye Hoechst 33342.
(C) Light exposure during subjective night induces the phosphorylation of MeCP2 S421 in the suprachiasmatic nucleus (SCN). Wild-type mice were entrained to a 12hr:12hr light-dark cycle and then transferred to constant darkness. Two days later, entrained mice were either maintained in darkness (DD) or exposed to light for 2, 4, or 6 hr (LP2, LP4, LP6). Adjacent cryosections were alternately immunostained with antibodies specific for MeCP2 pS421 or total MeCP2.
We next asked whether neural activity driven by a physiologically relevant environmental stimulus induces the phosphorylation of MeCP2 at S421. To obtain the synchronous activation of a specific population of neurons, we examined light entrainment of circadian rhythms, a process that, in mammals, is regulated by a central pacemaker residing in the suprachiasmatic nuclei (SCN) of the hypothalamus (Reppert and Weaver, 2001). Light in the context of the day-night cycle serves as an important external sensory stimulus to synchronize the near 24 hr internal biological clock to the external 24 hr cycle. Mice that have been entrained to a 12 hr:12 hr light-dark cycle for several days will maintain a circadian rhythm for many more days, as measured by locomotor activity during the subjective night, even when the animals are kept in continuous darkness. Exposure of entrained mice to light during the subjective night triggers a behavioral shift in the phase of the circadian rhythm that is believed to be mediated by a subset of retinal ganglion cells that project directly to the SCN, releasing glutamate onto SCN neurons (Reppert and Weaver, 2001). This synaptic release of glutamate triggers NMDA receptor-mediated calcium influx, leading to the induction of CREB S133 and S142 phosphorylation and the activation of immediate early genes, such as c-fos or jun-B, within SCN neurons (Kornhauser et al., 1996).
We investigated whether phosphorylation of MeCP2 at S421 is induced in the SCN by such a light stimulus that triggers a shift in circadian rhythm. Circadian-entrained adult mice were transferred to constant darkness for 48 hr and then were either kept in darkness (DD), or exposed during subjective night to a light pulse of 2, 4, or 6 hr (LP2, LP4, LP6), a stimulus previously shown to be sufficient to trigger a behavioral shift in circadian rhythm (Reppert and Weaver, 2001). Tissue sections were then prepared from the SCN and stained with the anti-MeCP2 pS421 or antitotal MeCP2 antibody. We found that a 2, 4, or 6 hr light pulse induced a significant increase in the levels of MeCP2 S421 phosphorylation in the SCN (Figure 3C, right), whereas only background staining was observed in the SCN of DD animals that received no light pulse (Figure 3C, top). Staining of adjacent sections with the total-MeCP2 antibody showed that MeCP2 protein is well expressed in the SCN, even in DD animals (Figure 3C, left) and that the overall level of MeCP2 is not significantly affected by light stimulation. Taken together, these results demonstrate that light, a physiologically and behaviorally important cue, triggers the de novo phosphorylation of MeCP2 at S421 in the intact brain.
MeCP2 Is Phosphorylated at S421 Selectively in the Brain
MeCP2 is ubiquitously expressed in mammalian tissues. Indeed, its widespread expression has been puzzling, given the predominantly neurological phenotype of RTT (Zoghbi, 2003). To assess the presence of MeCP2 S421 phosphorylation in various mammalian tissues, we homogenized equal wet masses of different adult mouse tissues directly into SDS sample buffer and measured the level of MeCP2 S421 phosphorylation by western blotting. Among the 12 different tissue lysates tested in this experiment, S421-phosphorylated MeCP2 was detected only in brain lysate, even though, consistent with previous reports, total MeCP2 protein was present in many tissues (Figure 4). The lack of MeCP2 S421 phosphorylation in most tissues was not likely due to dephosphorylation during extract preparation since (1) the tissues were lysed rapidly in SDS sample buffer and (2) the phosphorylation of CREB S133 and p42/44 MAPK was observed in most tissues (Figure 4 and data not shown). In addition, serum or growth-factor stimulation of a fibroblast cell line (3T3) failed to induce MeCP2 phosphorylation at S421, although these stimuli are known to induce the release of calcium from internal stores and effectively induced CREB (S133) and/or Akt (S473) phosphorylation (data not shown). These findings suggest that MeCP2 is selectively phosphorylated at S421 in neural tissues and raise the possibility that MeCP2 phosphorylation at S421 may have a specialized function in the brain. Thus, further investigation into the functional significance of MeCP2 S421 phosphorylation may provide insight into the specific role of MeCP2 in the brain and how mutations in MeCP2 lead to the neurological outcome in RTT.
Figure 4. MeCP2 Is Phosphorylated at S421 Specifically in the Brain.

Western analysis of whole-cell lysates prepared from different mammalian tissues using antibodies specific to total MeCP2, MeCP2 pS421, and pCREB (S133). Fresh tissues were collected from a 4-week-old male mouse, directly homogenized into 1.5% SDS sample buffer at a ratio of 50 mg/ml w:v, and separated on a 12% SDS-PAGE gel. The same blot was reprobed with an anti-GAPDH antibody as an indicator of sample loading.
Phosphorylation of MeCP2 at S421 Regulates Neuronal Activity-Dependent Gene Expression
We next investigated the significance of S421 phosphorylation for MeCP2 function. Our previous studies suggested that MeCP2 may act as a transcriptional repressor that must be modified by activity-dependent phosphorylation if activity-dependent gene activation is to occur (Chen et al., 2003). To test this hypothesis, we asked whether the phosphorylation of MeCP2 at S421 affects the ability of MeCP2 to regulate neuronal activity-dependent gene expression, focusing on Bdnf promoter IV transcription, a well-characterized model for activity-dependent gene expression (West et al., 2002) and a putative target of MeCP2-mediated repression (Chen et al., 2003).
Since MeCP2 normally binds methylated DNA that is assembled into chromatin, we assessed the ability of wild-type and S421A mutant MeCP2 to regulate activity- dependent transcription at endogenous loci, rather than to assess their effect on a plasmid-based reporter gene that, when transfected into cells, might not acquire a higher-order chromatin conformation. We developed a lentivirus-mediated protein-replacement assay (LEMPRA) that allowed us to substitute endogenous MeCP2 with tagged MeCP2 variants of choice in a high percentage of neurons in a culture. A lentiviral vector was used to deliver a short-hairpin RNA (shRNA) to knock down endogenous MeCP2 and to express a FLAG-tagged, shRNA-resistant version of MeCP2 in greater than 90% of neurons in postmitotic hippocampal cultures (Figure 5A). Lentiviral infection of hippocampal neurons with this MeCP2 replacement vector resulted in a significant reduction in the level of endogenous MeCP2 (Figure 5B), along with the stable overexpression of FLAG-tagged MeCP2 at levels approximately 5-fold higher than that of normal endogenous MeCP2 (Figures 5B and 5C). Western blotting with the anti-FLAG and anti-MeCP2 pS421 antibodies revealed that membrane depolarization of lentiviral-infected neurons expressing FLAG-MeCP2, but not FLAG-S421A MeCP2, triggered phosphorylation at S421 (Figure 5C). This result demonstrates that reconstituted MeCP2 is phosphorylated in response to neuronal activity.
To determine if the phosphorylation of MeCP2 at S421 affects activity-dependent gene induction, lentiviral-infected neurons expressing either reconstituted FLAG-tagged wild-type or S421A mutant MeCP2 were left untreated or membrane depolarized, and the resulting level of Bdnf exon IV transcripts was assessed by quantitative RT-PCR. Whereas Bdnf exon IV induction in neurons that expressed wild-type MeCP2 is similar to that seen in control-infected cultures, activity-dependent Bdnf exon IV induction is significantly impaired in neurons expressing S421A mutant MeCP2 (Figure 5D). This effect did not reflect the general inhibition of calcium-dependent signaling pathways because the activity-dependent transcription of another immediate early gene, c-fos, is not inhibited in neurons expressing S421A mutant MeCP2 as compared to wild-type MeCP2 (Figure 5E). These findings are consistent with the possibility that the nonphosphorylatable S421A mutant MeCP2 remains bound to the promoters of activity-regulated genes such as Bdnf and maintains these genes in a repressed state, even under conditions of membrane depolarization. In further support of this idea, the S421A mutant MeCP2 appears to bind methylated DNA with similar affinity as non-S421-phosphorylated MeCP2 in vitro (see Figure S1 in the Supplemental Data available with this article online), and the attenuation of Bdnf exon IV transcription is not seen with an S421A mutant MeCP2 bearing an R106W point mutation that abolishes methyl-DNA binding (data not shown). Taken together, these findings support a model in which neuronal activity and subsequent calcium influx relieve MeCP2- mediated transcriptional repression via phosphorylation of MeCP2 at S421.
Phosphorylation of MeCP2 at S421 Mediates Dendritic Patterning and Dendritic Spine Development
The requirement of MeCP2 S421 phosphorylation for activity-dependent Bdnf transcription prompted us to examine whether phosphorylation of MeCP2 at S421 is important for biological processes known to be regulated by neuronal activity. We investigated the significance of MeCP2 S421 phosphorylation for dendritic patterning and dendritic spine morphogenesis, two activity-mediated processes that have been linked to RTT on the basis of neuropathological studies of RTT individuals and Mecp2−/y mutant mice. Given that overexpression of MeCP2 from duplications of MECP2 leads to RTT-like disorder(s), we reasoned that we could use MeCP2 overexpression as an experimental model to test the significance of S421 phosphorylation for activity- dependent neuronal maturation. We biolistically transfected organotypic hippocampal slice cultures with a lentiviral-based plasmid expressing both an shRNA to knockdown expression of endogenous MeCP2 and a FLAG-tagged shRNA-resistant version of MeCP2 (LEMPRA construct, Figures 5A–5C, S3, and S4C). This construct was used to reduce levels of endogenous MeCP2 protein, increasing the probability that exogenously introduced MeCP2 would efficiently replace endogenous MeCP2 that is complexed into chromatin. We also co-transfected another dual-promoter vector that expresses an eGFP reporter and the antiapoptotic protein Bcl-XL. Bcl-XL was introduced to minimize the possibility that any effects we observed on dendrites were secondary to an effect of MeCP2 knockdown or overexpression on survival. Whereas Bcl-XL is known to inhibit cell death, it does not affect dendritic arborization (Gaudilliere et al., 2004).
Dendritic branching was quantified using Sholl analysis, a method which reports the number of intersections between the dendritic arbor and a series of concentric circles of increasing radii centered at the cell body (see Supplemental Experimental Procedures). We found that replacement of endogenous MeCP2 with FLAG-tagged wild-type MeCP2 in hippocampal pyramidal neurons resulted in a significant decrease in dendritic branch complexity as compared to a vector control (p < 0.05, n = 45–51/condition; Figures 6A and 6D). This effect on dendritic branching reflects the overexpression of MeCP2 (Figures S3–S5) and may be relevant to recent reports that transgenic mouse models that overexpress MeCP2 and individuals with a duplication of the MeCP2 locus develop a RTT-like, progressive neurological disorder (see Introduction). Consistent with previous reports in Mecp2−/y null mice and RTT brains, we also observed a simplification in the dendritic arbors of hippocampal pyramidal neurons transfected only with the shRNA directed against MeCP2, as compared to the vector control (p < 0.05, n = 25/condition; Figures 6A and 6C). Efficient knockdown of MeCP2 protein by the shRNA construct was confirmed by immunostaining (Figures S3 and S4). Taken together, these results indicate that the development of appropriate dendritic morphology requires a precise set point of MeCP2 expression; either the elimination of MeCP2 or the overexpression of MeCP2 leads to a decrease in dendritic arbor complexity.
Figure 6. Phosphorylation of MeCP2 at S421 Mediates MeCP2-Dependent Regulation of Dendritic Growth and Spine Maturation.

(A) Organotypic hippocampal slices were prepared from P5–7 rat pups and biolistically transfected after 2 days in culture with LEMPRA-based plasmid DNA encoding a control vector (in which the shRNA directed against MeCP2 is replaced with a scrambled shRNA and a stop codon is introduced directly after the FLAG epitope), the anti-MeCP2 shRNA only, wild-type MeCP2, or S421A MeCP2, in combination with a dual-promoter plasmid expressing eGFP and Bcl-XL. Representative 25× images of 7 DIV transfected pyramidal neurons are shown. Scale bar, 50 μm. Pseudocoloring: green, GFP; red, anti-FLAG; blue, antitotal MeCP2.
(B) Representative 63× GFP images of dendritic spines from control-, anti-MeCP2 shRNA-, wild-type MeCP2-, or S421A MeCP2-transfected neurons. Scale bar, 2 μm.
(C and D) Quantification of dendritic branch complexity by Sholl analysis (data are presented as mean ± SEM, *p < 0.05, ANOVA).
(E and F) shRNA-mediated knockdown of MeCP2 does not affect dendritic spine morphology (p > 0.05, K-S test)
(G and H) Quantification of the effects of wild-type or S421A mutant MeCP2 overexpression on dendritic spine length (G) and width (H) (p < 0.05 for wild-type versus S421A or wild-type versus control, K-S test).
In contrast to the effect of wild-type MeCP2 expression on dendritic complexity, expression of the S421A mutant MeCP2 in hippocampal pyramidal neurons had no significant effect on dendritic branch complexity relative to vector control (p = 0.14; n = 45–51/condition; Figures 6A and 6D). This finding indicates that the mutation of MeCP2 S421 to an alanine blocks the ability of MeCP2 overexpression to restrict dendritic growth. Importantly, we confirmed that exogenously expressed FLAG-tagged wild-type and S421A mutant MeCP2 are localized to the nucleus (Figures 6A, S3, and S5) and are expressed at similar levels, demonstrated by quantification of either anti-FLAG or antitotal MeCP2 immunostaining intensity (Figures S4B and S4C and S5). Quantification of antitotal MeCP2 immunostaining also revealed that FLAG-tagged wild-type or S421A-MeCP2 are overexpressed approximately 4-fold in transfected neurons compared to endogenous MeCP2 levels in either untreated or control-transfected cells (Figure S4C). Furthermore, exogenously expressed wild-type MeCP2, but not S421A mutant MeCP2, is efficiently phosphorylated at S421 in response to conditions of membrane depolarization or glutamate stimulation (Figure S5 and data not shown). Finally, to eliminate the possibility that the observed changes in dendritic branch complexity reflect differences in neuronal viability and/or relative levels of GFP expression, we quantified the average intensity of GFP signal in dendrites lying within the Sholl radius of analysis and found no significant differences in the level of dendritic GFP among neurons transfected with wild-type, S421A mutant MeCP2, or vector control (Figure S4A). Taken together, these results suggest that the phosphorylation of MeCP2 at S421 regulates the inhibition of dendritic growth mediated by MeCP2 overexpression.
Although RTT has been suggested to be a consequence of the disruption of experience-dependent synaptic development and refinement (Zoghbi, 2003), it has not been established how neuronal activity acting through MeCP2 might regulate synaptic maturation. We therefore tested the effect of mutating MeCP2 at its site of activity-dependent phosphorylation (S421) on the development of dendritic spines, the major sites of excitatory synaptic transmission onto neurons that are thought to be morphological correlates of synaptic contacts (Nimchinsky et al., 2002). Using the experimental paradigm described above, we observed a similar density of spines studding the dendrites of neurons in which endogenous MeCP2 was replaced with either FLAG-tagged wild-type or S421A mutant MeCP2 (Figure S2); however, we found that spines of neurons overexpressing wild-type MeCP2 were on average longer, thinner, and more filopodia-like than spines of control neurons or neurons transfected with the S421A mutant MeCP2 (p < 0.05, n = 29–35 cells and 9,561–11,553 spines; Figures 6B, 6G, and 6H), resembling immature spines. These results indicate that, although wild-type or S421A mutant MeCP2 expression does not appear to effect the formation or maintenance of spines, the overexpression of wild-type MeCP2 retards the maturation of dendritic spines in an S421 phosphorylation-dependent manner. Interestingly, transfection of only the shRNA directed against MeCP2 had no effect on either dendritic spine density or morphology (n = 25 cells and 7425–8668 spines; Figures S2, 6B, 6E, and 6F), although immunostaining for total MeCP2 indicated efficient knockdown of MeCP2 protein levels (Figures S3 and S4C). These data indicate that MeCP2 overexpression results in a distinct but overlapping set of phenotypes compared to MeCP2 loss of function—overexpression of MeCP2 affects dendritic patterning and dendritic spine morphology, whereas the reduction of MeCP2 expression by RNA interference affects dendritic patterning, but not dendritic spine morphology. These results suggest that the phosphorylation of MeCP2 at S421 by neuronal activity plays a key role in regulating the function of MeCP2 in mediating dendritic patterning and dendritic spine maturation.
CaMKII Mediates the Activity-Dependent Phosphorylation of MeCP2 at S421 in Neurons
To gain further insight into the mechanisms regulating activity-dependent MeCP2 phosphorylation and function, we investigated the signaling pathways by which activity-mediated calcium influx regulates MeCP2 S421 phosphorylation in neurons. To identify the kinase( s) that phosphorylate MeCP2 at S421, cortical neurons were incubated with a variety of kinase inhibitors, membrane depolarized with elevated levels of extracellular potassium or stimulated with NMDA, and assessed for the induction of MeCP2 S421 phosphorylation. Whereas pretreatment with pharmacological inhibitors of CaMKK, PKA, PKC, MAPK, CDK5, or PI3K each has no effect on membrane depolarization-dependent MeCP2 S421 phosphorylation (data not shown), the CaMKII inhibitor KN-93, but not its inactive analog KN-92, effectively blocks the membrane depolarization-induced phosphorylation of MeCP2 at S421 (Figures 7A and 7B). We also blocked CaMKII activity using CaMKIIN, a potent and specific endogenous protein inhibitor of CaMKII (Chang et al., 1998). Coexpression of CaMKIIN with FLAG-MeCP2 in cortical neurons blocks the ability of membrane depolarization to trigger MeCP2 S421 phosphorylation (Figure 7C). This effect was specific to CaMKII because coexpression of dominant negative versions of other CaMK family members (dn-CaMKI, dn-CaMKIV, or dn-CaMKK) with FLAG-MeCP2 had no effect (Figure 7C and data not shown).
Figure 7. CaMKII Mediates the Phosphorylation of MeCP2 at S421.

(A and B) Western blot analysis of extracts from E18 + 10 DIV hippocampal neurons that were pretreated for 60 min with the CaMKII inhibitor KN93, the inactive analog KN92, or vehicle control (DMSO), followed by stimulation with 55 mM KCl (A) or 20 μM NMDA (B) for 30 min.
(C) E18 + 5 DIV cortical neurons were cotransfected with FLAG-tagged MeCP2 and vector control, ca-CaMKK, dn-CaMKK, ca-CaMKII, or CaMKII-N. Extracts were prepared from these neurons left unstimulated (−) or membrane depolarized for 1 hr (+) 2 days after transfection and probed with antibodies specific to the FLAG epitope. ca, constitutive-active; dn, dominant-negative.
(D) Western blot analysis using anti-MeCP2 pS421 or anti-FLAG antibodies of extracts from HEK293T cells cotransfected with FLAG-MeCP2 and vector control, ca-CaMKI, ca-CaMKII, ca-CaMKIV, or ca-Akt.
(E) CaMKII phosphorylates MeCP2 in vitro. FLAG-tagged wild-type, S421A mutant MeCP2, or C5A mutant MeCP2 (S341A, S350A, S360A, S385A, S399A) were purified from HEK293T cells, coincubated with recombinant constitutively active CaMKII (left panel) and 32P-ATP, and separated on an SDS-PAGE gel. The right panel shows the results of a control reaction in the absence of any added kinase.
(F) BDNF-dependent induction of MeCP2 S421 phosphorylation is likely mediated by CaMKII. Western blot analysis of whole-cell extracts prepared from E18 + 12 DIV hippocampal neurons that were treated with indicated agents for 60 min followed by BDNF treatment for 60 min. K252a is an inhibitor of the BDNF receptor TrkB.
We also found that increasing CaMKII activity promotes the phosphorylation of MeCP2 at S421 in neurons or HEK 293T cells. Anti-FLAG western blots of extracts from neurons or HEK 293T cells that coexpress constitutively active CaMKII (ca-CaMKII) and FLAG-MeCP2 revealed the presence of the slow-migrating, phosphorylated form of MeCP2, even in the absence of membrane depolarization (Figures 7C and 7D). However, coexpression of ca-CaMKK, ca-CaMKI, or ca-CaMKIV with FLAG-MeCP2 had no effect on the phosphorylation status of MeCP2 S421 in unstimulated neurons or HEK 293T cells (Figures 7C and 7D and data not shown). Altogether, these results suggest that CaMKII mediates the phosphorylation of MeCP2 at S421 in response to neuronal activity-induced calcium influx.
We also tested whether MeCP2 is a direct substrate of CaMKII in vitro. Wild-type, S421A mutant, or C5A mutant forms of FLAG-tagged MeCP2 were purified from HEK 293T cells and analyzed in an in vitro kinase assay for their ability to be phosphorylated by recombinant, constitutively active CaMKII. The C5A mutant bears alanine mutations at S341, S350, S360, S385, and S399 in MeCP2. Recombinant CaMKII efficiently phosphorylates wild-type or C5A mutant MeCP2 in vitro, indicated by the appearance of the 32P-ATP-labeled, slow-migrating form of MeCP2 (Figure 7E, left). By contrast, significantly less phosphorylated MeCP2 was observed when S421A mutant MeCP2 was incubated with CaMKII (Figure 7E, left) or when wild-type MeCP2 was incubated with CaMKI, CaMKIV, or in the absence of recombinant kinase (data not shown, and Figure 7E, right). The residual background labeling observed was likely due to copurification of endogenous kinase(s) from the HEK 293T cell extract since this level of phosphorylation was detected in the absence of the addition of CaMKII. These results indicate that CaMKII, in contrast to many other kinases, phosphorylates MeCP2 in vitro in an S421-dependent manner. Taken together, these findings support a model in which CaMKII mediates the neural-specific phosphorylation of MeCP2 at S421 in response to synaptic activation and subsequent calcium influx. Whether MeCP2 S421 is a direct substrate of CaMKII in vivo or whether CaMKII acts through as-yet-unidentified intermediaries to mediate MeCP2 S421 phosphorylation remains an open question. Interestingly, KN93 pretreatment of hippocampal neurons also blocks the BDNF-dependent induction of MeCP2 S421 phosphorylation (Figure 7F), suggesting that CaMKII may also mediate the phosphorylation of MeCP2 at S421 in response to neurotrophin signaling.
Discussion
In this study, we identify an important missing link in the synaptic hypothesis of RTT by identifying S421 as a major site of activity-dependent modification on MeCP2 that is required for the maturation of neuronal connectivity, thereby providing a potential mechanism by which experience-dependent stimuli might regulate MeCP2 function. Using a phospho-site-specific antibody, we demonstrate that neuronal activity and subsequent calcium influx induces the de novo phosphorylation of MeCP2 at S421 in neurons in vitro and in vivo. For example, physiological light stimulation that regulates circadian behavior triggers the phosphorylation of MeCP2 at S421 in the intact brain. Importantly, we show that, although MeCP2 is expressed throughout the body, it appears to be phosphorylated at S421 selectively in the brain by a CaMKII-dependent mechanism. The phosphorylation of MeCP2 at S421 is required for MeCP2-dependent regulation of dendritic patterning, spine morphogenesis, and activity-dependent gene expression (Figure 8). These findings suggest a key role for the activity-dependent regulation of MeCP2 in the maturation of neuronal connectivity and provide a new framework for understanding how mutations in MeCP2 lead to the deregulation of these processes in RTT.
Figure 8.
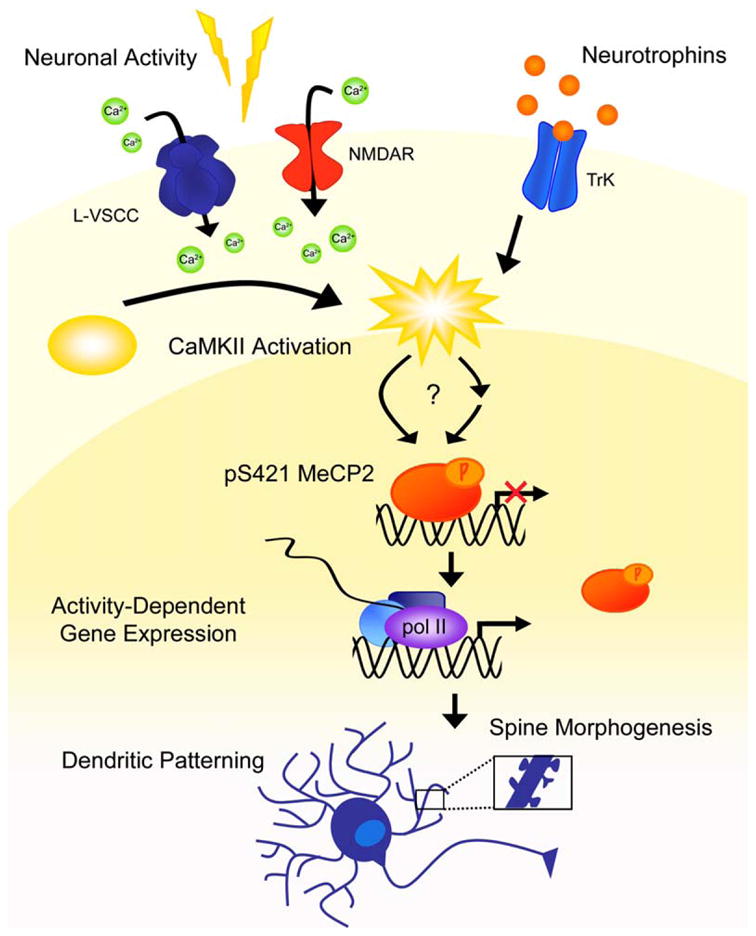
Regulation of Dendritic Morphogenesis and Spine Maturation by Activity-Dependent Phosphorylation of MeCP2
In addition to providing insight into the role of neuronal activity in regulating MeCP2 function, our study highlights the importance of exact MeCP2 expression levels in mediating neuronal development and to RTT. Although MeCP2 overexpression phenocopies a loss of function in MeCP2 in several aspects, such as a reduction in dendritic arbor complexity and reduced soma size (data not shown), MeCP2 overexpression also causes distinct phenotypes not seen in the loss of function, such as a delay in dendritic spine maturation. These findings provide clues as to how overexpression of MeCP2 leads to neurological abnormalities, a question of considerable interest given recent reports that duplications of the MECP2 locus may be the genetic basis for a subset of cases of RTT-like syndromes (Ariani et al., 2004; Lugtenberg et al., 2006; Meins et al., 2005; Van Esch et al., 2005).
Accruing evidence suggests that deregulation of the activity-responsive gene Bdnf may play a central role in RTT. Our finding that both MeCP2 S421 phosphorylation regulates the activity-dependent induction of Bdnf transcription and that BDNF treatment triggers MeCP2 S421 phosphorylation suggests that a complex set of interactions between MeCP2 and BDNF coregulates their functions. Mecp2−/y and Bdnf−/− knockout mice share many overlapping phenotypes, which include smaller brain size, reduced dendritic arbor complexity, impaired presynaptic function and synaptic plasticity, and limb clasping (Chang et al., 2006; Gorski et al., 2003; Korte et al., 1995; Patterson et al., 1996), consistent with reports that BDNF protein and transcript levels are reduced in Mecp2−/y null brains (Chang et al., 2006). Although our model posits that activity- or neurotrophin- dependent MeCP2 S421 phosphorylation relieves its transcriptional repressor function on Bdnf promoter IV and would predict an increase in BDNF levels in Mecp2−/y null mice, the decreased levels of the activity- dependent gene product BDNF that are observed in vivo may be secondary to reduced cortical activity in Mecp2−/y null brains (Chang et al., 2006; Dani et al., 2005).
Consistent with the hypothesis that MeCP2 S421 phosphorylation is required for activity-dependent activation of Bdnf promoter IV, we found that overexpression of S421A mutant MeCP2 inhibits the induction of Bdnf exon IV in response to membrane depolarization and that this inhibition is dependent on DNA binding. This result is consistent with our findings that phosphorylation of MeCP2 correlates with a reduced ability of MeCP2 to bind methylated DNA and Bdnf promoter IV (Figure S1; Chen et al., 2003). Taken together, these observations suggest that, in the absence of neuronal activity, MeCP2 binds to the promoters of activity-regulated genes such as Bdnf and represses their transcription. In the context of synaptic activity, membrane depolarization triggers the phosphorylation of MeCP2 at S421, leading to reduced binding of MeCP2 to Bdnf promoter IV and a relief of transcriptional repression, allowing for full gene activation that is mediated by the calcium-dependent modification of a host of transcriptional activators such as CREB, CaRF, and USFs (West et al., 2002).
Our finding that the S421A mutant of MeCP2 suppresses the activity-dependent transcription of Bdnf provides a possible explanation for the differential effects of wild-type and S421 mutant MeCP2 on dendrite and spine morphogenesis. When compared to overexpressed S421A mutant MeCP2, overexpressed wild-type MeCP2 may promote the induction of a variety of activity-responsive genes that regulate neuronal connectivity. Although it might seem counterintuitive that the higher levels of BDNF in neurons overexpressing wild-type MeCP2, compared to S421 mutant MeCP2, would contribute to the inhibition of dendritic growth and spine maturation, the effects of BDNF have been shown to be highly context dependent (Poo, 2001). For instance, BDNF has been shown to inhibit basal dendritic complexity of layer VI cortical pyramidal neurons in culture (McAllister et al., 1995) and to inhibit dendritic branching in Xenopus retinal ganglion cells in vivo (Lom and Cohen-Cory, 1999). In addition, MeCP2 has many other transcriptional target genes besides Bdnf, among them Dlx5, Ube3a, Sgk1, and Fkbp5 (Bienvenu and Chelly, 2006; Klose and Bird, 2006), and the effects of MeCP2 overexpression on neuronal development might also reflect the deregulation of one or several yet-to-be-discovered target genes.
An important unresolved question is how phosphorylation of MeCP2 at S421, a site near the C terminus of MeCP2 that is remote from the DNA binding domain of MeCP2, disrupts the ability of MeCP2 to repress activity- responsive promoters such as Bdnf promoter IV. One possibility is that phosphorylation of MeCP2 at S421 triggers a conformational change in MeCP2 that reduces its binding affinity for methyl-DNA sequences. Alternatively or in addition, activity-mediated S421 phosphorylation may serve as a priming event to allow additional modifications of MeCP2, perhaps on residues within the DNA binding domain itself that might alter DNA binding affinity. Interestingly, our mass spectrometry analysis of MeCP2 purified from neuronal extracts revealed two additional sites of phosphorylation on MeCP2 at S80 and S229. Whether S80 and S229 are also responsive to neuronal activity, and whether S421, S80, S229, or other still unknown sites of modification are coordinately modified to regulate the function of MeCP2 in activity-dependent gene expression warrants further investigation.
Our finding that CaMKII mediates the phosphorylation of MeCP2 at S421 may be relevant to the well-known role of CaMKII and other CaMK family members in neurite extension and dendritic patterning (Gaudilliere et al., 2004; Redmond et al., 2002; Shen et al., 1998; Wayman et al., 2006). The implication of CaMKII in the regulation of MeCP2 function is also intriguing in the context of the cognitive deficits observed in RTT because a large body of literature indicates that CaMKII plays a critical role in regulating synaptic development and plasticity (Lisman et al., 2002). Notably, many of the molecular players in the calcium-dependent signaling pathway that mediates neuronal activity-dependent gene expression have now been shown through human genetics to be important for the development of cognitive function, including CAV1.2, CBP, BDNF, and MECP2 (Hong et al., 2005). Further investigation of the mechanisms mediating neuronal activity-dependent transcription is likely to uncover additional factors important for human neural development.
An incisive way to understand the in vivo biological importance of MeCP2 S421 phosphorylation for RTT will be to develop mice that bear a single point mutation at MeCP2 S421 that converts it to alanine, rendering endogenous MeCP2 S421 refractory to phosphorylation. This knockin mouse model will allow us to assess the hypothesis that phosphorylation of MeCP2 at S421 is an important molecular event that regulates adaptive responses to the environment and to test the idea that the inappropriate regulation of MeCP2 S421 phosphorylation in the brains of individuals with RTT could underlie some of the neurological symptoms associated with RTT. For instance, our finding that MeCP2 is rapidly phosphorylated in the SCN in response to light exposure suggests that MeCP2 may function in regulating circadian rhythms. Interestingly, sleep disturbances are frequently reported in RTT, and the Mecp2308/y C-terminal mutant mice have abnormalities in the diurnal regulation of motor activity (Moretti et al., 2005). It will be interesting to examine in more detail whether the activity-dependent phosphorylation of MeCP2 at S421 regulates sleep-wake behaviors.
Taken together, the evidence to date suggests a model in which experience-dependent stimuli trigger the dynamic modification of MeCP2, thereby modulating its function as a key transcriptional regulator that controls the normal developmental maturation of the nervous system (Figure 8). Thus, MECP2 mutations or duplications that occur in RTT might disrupt the ability of the nervous system to appropriately convert activity-dependent cues into programs of gene expression that guide the structural and functional maturation of neuronal circuits. Ultimately, the phosphorylation of MeCP2 at S421 may be only one of a number of posttranslational modifications on MeCP2; however, its selective occurrence in the brain suggests that further efforts to elucidate the importance of MeCP2 S421 phosphorylation for brain development and the mechanisms by which S421 phosphorylation regulates MeCP2 function may further our understanding of how mutations in MeCP2 lead to the neurological phenotype of RTT.
Experimental Procedures
See Supplemental Experimental Procedures for details of immunohistochemistry, imaging, image analysis, seizure induction, circadian entrainment, viral infection, quantitative real-time PCR, protein purification, kinase assay, and southwestern assay.
Plasmids and Antibodies
The antibodies that recognize either MeCP2 regardless of its phosphorylation at S421 (antitotal MeCP2) or S421-phosphorylated MeCP2 (anti-MeCP2 pS421) were both generated in rabbits using peptide antigens. MeCP2 expression vectors were constructed using the mouse e1 form of MeCP2, except where otherwise noted. See Supplemental Experimental Procedures for further details.
Cell Culture
Dissociated neuronal cultures were prepared and transfected by the calcium phosphate method as previously described (Xia et al., 1996). Three hundred fifty micrometer slices of P5–7 rat hippocampus were organotypically cultured as described in Stoppini et al. (1991) and transfected biolistically. See Supplemental Experimental Procedures for further details.
Lentiviral Production
Lentiviruses were pseudotyped with the VSV-G envelope and concentrated by centrifugation as previously described (Lois et al., 2002). See Supplemental Experimental Procedures for further details.
Supplementary Material
Acknowledgments
We are indebted to T. Soderling, T.K. Kim, J. Kornhauser, and M. Lin for valuable reagents and J. Li and S. Gygi for mass spectrometry analysis. We thank A.E. West, L. Jackson-Grusby, and members of the Greenberg laboratory for useful discussions and critical comments on the manuscript. M.E.G. acknowledges the generous support of the F.M. Kirby Foundation to the Children’s Hospital Neurobiology Program. This work was supported by the Rett Syndrome Research Foundation (M.E.G.), National Institutes of Health grants (NS048276, M.E.G.; NS43491, C.J.W.), the NIH Medical Scientist Training Program (S.C.), the Damon Runyon Cancer Research Foundation (H.-y. H.H), the Fannie and John Hertz Foundation (E.J.H.), and the Helen Hay Whitney Foundation (E.C.G., Z.Z.).
Footnotes
The Supplemental Data for this article can be found online at http://www.neuron.org/cgi/content/full/52/2/255/DC1/.
References
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- Ariani F, Mari F, Pescucci C, Longo I, Bruttini M, Meloni I, Hayek G, Rocchi R, Zappella M, Renieri A. Realtime quantitative PCR as a routine method for screening large rearrangements in Rett syndrome: report of one case of MECP2 deletion and one case of MECP2 duplication. Hum Mutat. 2004;24:172–177. doi: 10.1002/humu.20065. [DOI] [PubMed] [Google Scholar]
- Asaka Y, Jugloff DG, Zhang L, Eubanks JH, Fitzsimonds RM. Hippocampal synaptic plasticity is impaired in the Mecp2-null mouse model of Rett syndrome. Neurobiol Dis. 2006;21:217–227. doi: 10.1016/j.nbd.2005.07.005. [DOI] [PubMed] [Google Scholar]
- Ballas N, Grunseich C, Lu DD, Speh JC, Mandel G. REST and its corepressors mediate plasticity of neuronal gene chromatin throughout neurogenesis. Cell. 2005;121:645–657. doi: 10.1016/j.cell.2005.03.013. [DOI] [PubMed] [Google Scholar]
- Bienvenu T, Chelly J. Molecular genetics of Rett syndrome: when DNA methylation goes unrecognized. Nat Rev Genet. 2006;7:415–426. doi: 10.1038/nrg1878. [DOI] [PubMed] [Google Scholar]
- Chandler SP, Guschin D, Landsberger N, Wolffe AP. The methyl-CpG binding transcriptional repressor MeCP2 stably associates with nucleosomal DNA. Biochemistry. 1999;38:7008– 7018. doi: 10.1021/bi990224y. [DOI] [PubMed] [Google Scholar]
- Chang BH, Mukherji S, Soderling TR. Characterization of a calmodulin kinase II inhibitor protein in brain. Proc Natl Acad Sci USA. 1998;95:10890–10895. doi: 10.1073/pnas.95.18.10890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Q, Khare G, Dani V, Nelson S, Jaenisch R. The disease progression of Mecp2 mutant mice is affected by the level of BDNF expression. Neuron. 2006;49:341–348. doi: 10.1016/j.neuron.2005.12.027. [DOI] [PubMed] [Google Scholar]
- Chen RZ, Akbarian S, Tudor M, Jaenisch R. Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat Genet. 2001;27:327–331. doi: 10.1038/85906. [DOI] [PubMed] [Google Scholar]
- Chen WG, Chang Q, Lin Y, Meissner A, West AE, Griffith EC, Jaenisch R, Greenberg ME. Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science. 2003;302:885–889. doi: 10.1126/science.1086446. [DOI] [PubMed] [Google Scholar]
- Collins AL, Levenson JM, Vilaythong AP, Richman R, Armstrong DL, Noebels JL, David Sweatt J, Zoghbi HY. Mild overexpression of MeCP2 causes a progressive neurological disorder in mice. Hum Mol Genet. 2004;13:2679–2689. doi: 10.1093/hmg/ddh282. [DOI] [PubMed] [Google Scholar]
- Dani VS, Chang Q, Maffei A, Turrigiano GG, Jaenisch R, Nelson SB. Reduced cortical activity due to a shift in the balance between excitation and inhibition in a mouse model of Rett syndrome. Proc Natl Acad Sci USA. 2005;102:12560–12565. doi: 10.1073/pnas.0506071102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudilliere B, Konishi Y, de la Iglesia N, Yao G, Bonni A. A CaMKII-NeuroD signaling pathway specifies dendritic morphogenesis. Neuron. 2004;41:229–241. doi: 10.1016/s0896-6273(03)00841-9. [DOI] [PubMed] [Google Scholar]
- Gorski JA, Zeiler SR, Tamowski S, Jones KR. Brain-derived neurotrophic factor is required for the maintenance of cortical dendrites. J Neurosci. 2003;23:6856–6865. doi: 10.1523/JNEUROSCI.23-17-06856.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy J, Hendrich B, Holmes M, Martin JE, Bird A. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat Genet. 2001;27:322–326. doi: 10.1038/85899. [DOI] [PubMed] [Google Scholar]
- Hagberg B, Aicardi J, Dias K, Ramos O. A progressive syndrome of autism, dementia, ataxia, and loss of purposeful hand use in girls: Rett’s syndrome: report of 35 cases. Ann Neurol. 1983;14:471–479. doi: 10.1002/ana.410140412. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Fukunaga Y, Bading H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci. 2002;5:405–414. doi: 10.1038/nn835. [DOI] [PubMed] [Google Scholar]
- Hong EJ, West AE, Greenberg ME. Transcriptional control of cognitive development. Curr Opin Neurobiol. 2005;15:21–28. doi: 10.1016/j.conb.2005.01.002. [DOI] [PubMed] [Google Scholar]
- Katz LC, Shatz CJ. Synaptic activity and the construction of cortical circuits. Science. 1996;274:1133–1138. doi: 10.1126/science.274.5290.1133. [DOI] [PubMed] [Google Scholar]
- Kishi N, Macklis JD. MECP2 is progressively expressed in post-migratory neurons and is involved in neuronal maturation rather than cell fate decisions. Mol Cell Neurosci. 2004;27:306–321. doi: 10.1016/j.mcn.2004.07.006. [DOI] [PubMed] [Google Scholar]
- Klose RJ, Bird AP. Genomic DNA methylation: the mark and its mediators. Trends Biochem Sci. 2006;31:89–97. doi: 10.1016/j.tibs.2005.12.008. [DOI] [PubMed] [Google Scholar]
- Kornhauser JM, Mayo KE, Takahashi JS. Light, immediate-early genes, and circadian rhythms. Behav Genet. 1996;26:221–240. doi: 10.1007/BF02359382. [DOI] [PubMed] [Google Scholar]
- Kornhauser JM, Cowan CW, Shaywitz AJ, Dolmetsch RE, Griffith EC, Hu LS, Haddad C, Xia Z, Greenberg ME. CREB transcriptional activity in neurons is regulated by multiple, calcium-specific phosphorylation events. Neuron. 2002;34:221–233. doi: 10.1016/s0896-6273(02)00655-4. [DOI] [PubMed] [Google Scholar]
- Korte M, Carroll P, Wolf E, Brem G, Thoenen H, Bonhoeffer T. Hippocampal long-term potentiation is impaired in mice lacking brain-derived neurotrophic factor. Proc Natl Acad Sci USA. 1995;92:8856–8860. doi: 10.1073/pnas.92.19.8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriaucionis S, Bird A. DNA methylation and Rett syndrome. Hum Mol Genet. 2003;12(Spec No 2):R221–R227. doi: 10.1093/hmg/ddg286. [DOI] [PubMed] [Google Scholar]
- Lanahan A, Worley P. Immediate-early genes and synaptic function. Neurobiol Learn Mem. 1998;70:37–43. doi: 10.1006/nlme.1998.3836. [DOI] [PubMed] [Google Scholar]
- Lewis JD, Meehan RR, Henzel WJ, Maurer-Fogy I, Jeppesen P, Klein F, Bird A. Purification, sequence, and cellular localization of a novel chromosomal protein that binds to methylated DNA. Cell. 1992;69:905–914. doi: 10.1016/0092-8674(92)90610-o. [DOI] [PubMed] [Google Scholar]
- Lisman J, Schulman H, Cline H. The molecular basis of CaMKII function in synaptic and behavioral memory. Nat Rev Neurosci. 2002;3:175–190. doi: 10.1038/nrn753. [DOI] [PubMed] [Google Scholar]
- Lois C, Hong EJ, Pease S, Brown EJ, Baltimore D. Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science. 2002;295:868–872. doi: 10.1126/science.1067081. [DOI] [PubMed] [Google Scholar]
- Lom B, Cohen-Cory S. Brain-derived neurotrophic factor differentially regulates retinal ganglion cell dendritic and axonal arborization in vivo. J Neurosci. 1999;19:9928–9938. doi: 10.1523/JNEUROSCI.19-22-09928.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugtenberg D, de Brouwer AP, Kleefstra T, Oudakker AR, Frints SG, Schrander-Stumpel CT, Fryns JP, Jensen LR, Chelly J, Moraine C, et al. Chromosomal copy number changes in patients with nonsyndromic X linked mental retardation detected by array CGH. J Med Genet. 2006;43:362–370. doi: 10.1136/jmg.2005.036178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luikenhuis S, Giacometti E, Beard CF, Jaenisch R. Expression ofMeCP2 in postmitotic neurons rescues Rett syndrome in mice. Proc Natl Acad Sci USA. 2004;101:6033–6038. doi: 10.1073/pnas.0401626101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinowich K, Hattori D, Wu H, Fouse S, He F, Hu Y, Fan G, Sun YE. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science. 2003;302:890–893. doi: 10.1126/science.1090842. [DOI] [PubMed] [Google Scholar]
- McAllister AK, Lo DC, Katz LC. Neurotrophins regulate dendritic growth in developing visual cortex. Neuron. 1995;15:791–803. doi: 10.1016/0896-6273(95)90171-x. [DOI] [PubMed] [Google Scholar]
- Meins M, Lehmann J, Gerresheim F, Herchenbach J, Hagedorn M, Hameister K, Epplen JT. Submicroscopic duplication in Xq28 causes increased expression of the MECP2 gene in a boy with severe mental retardation and features of Rett syndrome. J Med Genet. 2005;42:e12. doi: 10.1136/jmg.2004.023804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moretti P, Zoghbi HY. MeCP2 dysfunction in Rett syndrome and related disorders. Curr Opin Genet Dev. 2006;16:276–281. doi: 10.1016/j.gde.2006.04.009. [DOI] [PubMed] [Google Scholar]
- Moretti P, Bouwknecht JA, Teague R, Paylor R, Zoghbi HY. Abnormalities of social interactions and home-cage behavior in a mouse model of Rett syndrome. Hum Mol Genet. 2005;14:205–220. doi: 10.1093/hmg/ddi016. [DOI] [PubMed] [Google Scholar]
- Moretti P, Levenson JM, Battaglia F, Atkinson R, Teague R, Antalffy B, Armstrong D, Arancio O, Sweatt JD, Zoghbi HY. Learning and memory and synaptic plasticity are impaired in a mouse model of Rett syndrome. J Neurosci. 2006;26:319–327. doi: 10.1523/JNEUROSCI.2623-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson ED, Kavalali ET, Monteggia LM. MeCP2-dependent transcriptional repression regulates excitatory neurotransmission. Curr Biol. 2006;16:710–716. doi: 10.1016/j.cub.2006.02.062. [DOI] [PubMed] [Google Scholar]
- Nimchinsky EA, Sabatini BL, Svoboda K. Structure and function of dendritic spines. Annu Rev Physiol. 2002;64:313–353. doi: 10.1146/annurev.physiol.64.081501.160008. [DOI] [PubMed] [Google Scholar]
- Patterson SL, Abel T, Deuel TA, Martin KC, Rose JC, Kandel ER. Recombinant BDNF rescues deficits in basal synaptic transmission and hippocampal LTP in BDNF knockout mice. Neuron. 1996;16:1137–1145. doi: 10.1016/s0896-6273(00)80140-3. [DOI] [PubMed] [Google Scholar]
- Pelka GJ, Watson CM, Radziewic T, Hayward M, Lahooti H, Christodoulou J, Tam PP. Mecp2 deficiency is associated with learning and cognitive deficits and altered gene activity in the hippocampal region of mice. Brain. 2006;129:887–898. doi: 10.1093/brain/awl022. [DOI] [PubMed] [Google Scholar]
- Poo MM. Neurotrophins as synaptic modulators. Nat Rev Neurosci. 2001;2:24–32. doi: 10.1038/35049004. [DOI] [PubMed] [Google Scholar]
- Redmond L, Kashani AH, Ghosh A. Calcium regulation of dendritic growth via CaM kinase IV and CREB-mediated transcription. Neuron. 2002;34:999–1010. doi: 10.1016/s0896-6273(02)00737-7. [DOI] [PubMed] [Google Scholar]
- Reppert SM, Weaver DR. Molecular analysis of mammalian circadian rhythms. Annu Rev Physiol. 2001;63:647–676. doi: 10.1146/annurev.physiol.63.1.647. [DOI] [PubMed] [Google Scholar]
- Rett A. On a unusual brain atrophy syndrome in hyperammonemia in childhood. Wien Med Wochenschr. 1966;116:723–726. [PubMed] [Google Scholar]
- Shahbazian M, Young J, Yuva-Paylor L, Spencer C, Antalffy B, Noebels J, Armstrong D, Paylor R, Zoghbi H. Mice with truncated MeCP2 recapitulate many Rett syndrome features and display hyperacetylation of histone H3. Neuron. 2002;35:243–254. doi: 10.1016/s0896-6273(02)00768-7. [DOI] [PubMed] [Google Scholar]
- Shen K, Teruel MN, Subramanian K, Meyer T. CaMKIIβ functions as an F-actin targeting module that localizes CaMKIIα/β heterooligomers to dendritic spines. Neuron. 1998;21:593–606. doi: 10.1016/s0896-6273(00)80569-3. [DOI] [PubMed] [Google Scholar]
- Stoppini L, Buchs PA, Muller D. A simple method for organotypic cultures of nervous tissue. J Neurosci Methods. 1991;37:173–182. doi: 10.1016/0165-0270(91)90128-m. [DOI] [PubMed] [Google Scholar]
- Van Esch H, Bauters M, Ignatius J, Jansen M, Raynaud M, Hollanders K, Lugtenberg D, Bienvenu T, Jensen LR, Gecz J, et al. Duplication of the MECP2 region is a frequent cause of severe mental retardation and progressive neurological symptoms in males. Am J Hum Genet. 2005;77:442–453. doi: 10.1086/444549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayman GA, Impey S, Marks D, Saneyoshi T, Grant WF, Derkach V, Soderling TR. Activity-dependent dendritic arborization mediated by CaM-Kinase I activation and enhanced CREB-dependent transcription of Wnt-2. Neuron. 2006;50:897–909. doi: 10.1016/j.neuron.2006.05.008. [DOI] [PubMed] [Google Scholar]
- West AE, Griffith EC, Greenberg ME. Regulation of transcription factors by neuronal activity. Nat Rev Neurosci. 2002;3:921–931. doi: 10.1038/nrn987. [DOI] [PubMed] [Google Scholar]
- Xia Z, Dudek H, Miranti CK, Greenberg ME. Calcium influx via the NMDA receptor induces immediate early gene transcription by aMAP kinase/ERK-dependent mechanism. J Neurosci. 1996;16:5425–5436. doi: 10.1523/JNEUROSCI.16-17-05425.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoghbi HY. Postnatal neurodevelopmental disorders: meeting at the synapse? Science. 2003;302:826–830. doi: 10.1126/science.1089071. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.