

Abstract
Background
Brief isoflurane anesthesia induces neuroapoptosis in the developing rodent brain, but susceptibility of nonhuman primates to the apoptogenic action of isoflurane has not been studied. Therefore, we exposed postnatal day 6 (P6) rhesus macaques to a surgical plane of isoflurane anesthesia for 5 h, and studied the brains 3 h later for histopathological changes.
Method
With the same intensity of physiological monitoring typical for human neonatal anesthesia, five P6 rhesus macaques were exposed for 5 h to isoflurane maintained between 0.7 and 1.5 end tidal Vol% (endotracheally intubated, mechanically ventilated), and five controls were exposed for 5 h to room air without further intervention. Three hours later, the brains were harvested and serially sectioned across the entire forebrain and midbrain, and stained immunohistochemically with antibodies to activated caspase-3 for detection and quantification of apoptotic neurons.
Results
Quantitative evaluation of brain sections revealed a median of 32.5 (range, 18.0 to 48.2) apoptotic cells per mm3 of brain tissue in the isoflurane group and only 2.5 (range, 1.9 to 3.8) in the control group (difference significant at p = 0.008). Apoptotic neuronal profiles were largely confined to the cerebral cortex. In the control brains, they were sparse and randomly distributed, whereas in the isoflurane brains they were abundant and preferentially concentrated in specific cortical layers and regions.
Conclusion
The developing nonhuman primate brain is sensitive to the apoptogenic action of isoflurane, and displays a 13-fold increase in neuroapoptosis after 5 h exposure to a surgical plane of isoflurane anesthesia.
INTRODUCTION
Acute exposure of infant rats or mice to several classes of drugs, including those that block N-methyl-D-aspartate glutamate receptors, those that activate γ-aminobutyric acid-A receptors, and ethanol (which has both N-methyl-D-aspartate antagonist and γ-aminobutyric acid-mimetic properties), triggers widespread apoptotic death of neurons in the developing brain1-5. The window of peak vulnerability to the apoptogenic mechanism coincides with the developmental period of rapid synaptogenesis,1, 2 also known as the brain growth spurt period, which in mice and rats occurs primarily during the first 2 weeks after birth, but in humans extends from about mid-gestation to several years after birth.6
The cell death process triggered by these drugs has been demonstrated at both light and electron microscopic levels to have classical morphological characteristics of apoptosis1,3,5,7,8, a form of cell death in which the cell actively mediates its own demise. Studies undertaken to examine gene-regulated biochemical pathways have revealed that the cell death process induced by these drugs involves translocation of Bax protein to mitochondrial membranes where it disrupts membrane permeability, allowing extramitochondrial leakage of cytochrome c, followed by a sequence of changes culminating in activation of caspase-3.9-11 Commitment to cell death occurs prior to the caspase-3 activation step11; therefore, immunohistochemical detection of neurons positive for activated caspase-3 (AC3) provides a reliable means of mapping and quantifying dying cells that have already progressed beyond the point of cell death commitment. Accordingly, AC3 immunohistochemistry has been used extensively for marking dying neurons in recent studies focusing on drug-induced developmental neuroapoptosis.3,8-25.
The observation that N-methyl-D-aspartate antagonist and γ-aminobutyric acid-mimetic drugs induce widespread neuronal degeneration in the developing brain has relevance in a public health context, because there are many agents in the human environment that have N-methyl-D-aspartate antagonist or γ-aminobutyric acid-mimetic properties, including drugs that are sometimes abused by pregnant mothers (ethanol, phencyclidine, ketamine, nitrous oxide, barbiturates, benzodiazepines), and many drugs used worldwide in obstetric and pediatric medicine as anticonvulsants, sedatives or anesthetics. Particularly concerning is mounting evidence that even single or brief exposure to clinically relevant doses of commonly used anesthetics (ketamine, midazolam, propofol, isoflurane, sevoflurane, chloral hydrate*) may trigger a significant neuroapoptosis response in the developing rodent brain.5,13-17,22,26-28 It has also been reported that exposure of the developing rodent brain to these agents can result in long-term neurobehavioral impairments.5,25-29 In addition, there is preliminary evidence potentially linking anesthesia exposure in infancy with long-term neurobehavioral deficits in both humans30,31,+ and nonhuman primates#.
Research aimed at clarifying whether anesthetic drugs can trigger neuroapoptosis in the developing non-human primate brain is limited to studies19,20 in which it was reported that intravenous infusion of ketamine triggered neuroapoptosis in the postnatal day 5 (P5) rhesus macaque brain if the infusion was continued for 9 or 24 h, but not if it was stopped at 3 h. Potent volatile anesthetic agents, such as isoflurane, are frequently used to administer general anesthesia to pregnant women, and to human neonates and infants requiring surgery. Therefore, we undertook the present study in which P6 rhesus macaque neonates were exposed to a surgical plane of isoflurane anesthesia for 5 h, and the brains were processed 3 h later for subsequent histopathological evaluation.
MATERIALS AND METHODS
Animals and Experimental Procedures
All animal procedures were approved by the Oregon National Primate Research Center (Beaverton, Oregon,) and Washington University Medical School (St. Louis, Missouri) Institutional Animal Care and Use Committees and were conducted in full accordance with the Public Health Service Policy on Humane Care and Use of Laboratory Animals. The subjects for these experiments were infant rhesus macaques that were exposed for 5 h either to room air (n = 5) or to isoflurane (end tidal 0.7-1.5 Vol%; n = 5). The sex distribution in both the control and isoflurane groups was three females and two males. The mean age of the control and isoflurane animals was 5.6 and 6.0 days old, respectively. Criteria for maintaining a surgical plane of anesthesia were: no movement and not more than 10% increase in heart rate or blood pressure in response to a profound moskito-clamp pinch at hand and foot (checked every 30 min). After mask induction (isoflurane), animals were tracheally intubated (semi-rigid fiberoptic endoscope; Karl Storz America, El Segundo, CA; 2.0 ID endotracheal tube, Mallinckrodt, Hazelwood, MO), mechanically ventilated (small animal ventilator; Harvard Apparatus, Holliston, MA), and maintained using full physiologic monitoring (continuous end tidal-carbon dioxide, end tidal-Isoflurane, FiO2 [Capnomac, Datex Ohmeda, Madison, WI], electrocardiogram, peripheral oxygen saturation, noninvasive blood pressure [every 15 min], rectal temperature [Surgivet Advisor V9200, Smith Medical, Wankesha, WI], blood gases, metabolic profile including pH, base excess, blood urea nitrogen, hematocrit, hemoglobin, Na, Cl, K, and serum glucose and lactate levels [every 2 hrs; i-STAT, Princeton, NJ]). After extubation of the trachea, animals were kept for 3 h in an infant monkey incubator (Snyder ICU cage, Snyder MFG Co., Centennial, CO), and were formula-fed as tolerated. After the 3-h observation period, the animals received high-dose phenobarbital and were transcardially perfusion-fixed to prepare the brain for histopathological analysis.
Histopathology Methods
For histopathological analysis, brains were cut serially into 70 μM coronal sections across the entire rostro-caudal extent of the forebrain and midbrain (approximately 800 sections per brain). Sections were selected at 2.24 mm intervals (approximately 25 sections per brain) and stained for detection of apoptotic profiles by AC3 immunohistochemistry, as described previously.3,8-25 Before applying the AC3 stain, sections were processed for antigen retrieval to reduce background nonspecific staining and maximize the AC3-specific signal. For antigen retrieval, sections were immersed in a citrate buffer, pH 6.0 and subjected to heat in a pressure cooker for 10 min.
We chose the AC3 staining procedure for several reasons: 1) It has been shown in numerous prior studies3,8-25 to faithfully mark neurons that are undergoing apoptotic degeneration following exposure to various apoptogenic drugs, including isoflurane;15,16,22 2) It has been demonstrated that cellular profiles marked by the AC3 stain are also marked by the cupric silver stain which selectively marks neurons that are dead or dying;1-5,12 3) It has been confirmed by electron microscopy that that these cells display all of the classical ultrastructural characteristics of neurons undergoing apoptotic cell death.1-3,7,8,12 In addition, it has been found9 that the AC3 stain is valuable, not only for identifying neurons that are undergoing apoptosis, but for revealing whether the cell is in an early or advanced stage of degeneration, and for revealing what type of cell (based on location and morphological appearance) is undergoing degeneration. The AC3 stain can provide this information because in the early stages of apoptotic degeneration neurons generate copious amounts of AC3 protein, which fills the cell body and its dendritic processes, so that the full microanatomy of the neuron can be visualized by immunohistochemical staining with AC3 antibody in this early stage. In the ensuing several hours, the dendritic processes shrivel and become fragmented, the cell body becomes condensed or rounded up into a spherical shape, and these changes signify that the cell is progressing from early to advanced stages of cell death.
Quantification of Neuroapoptosis
AC3-positive neuronal profiles were counted by an investigator who was blinded to the treatment condition. Each stained section was comprehensively scanned by light microscopy using a 10x objective lens and a computer-assisted StereoInvestigator system (Microbrightfield Inc., Williston, VT) with an electronically driven automatic stage to plot the number and location of each AC3 stained profile in each section. The total area scanned and the total number of stained neurons per section and per brain were computer-recorded. The total area was converted to volume by multiplying by the thickness of the section (70 μM), and the total number of neurons was divided by the total volume of tissue within which counts were made to yield a density count (number of stained profiles per mm3) for each brain.
Statistical Evaluation
There are important ethical and logistical considerations regarding non-human primate experimentation. It is important to include as few animals as are needed to answer research questions with as much precision as possible. As there are no published data indicating the amount of potential neuroapoptosis following exposure of the neonatal macaque to isoflurane, it was difficult to estimate how many animals might be needed for the present study. However, previous experiments in non-human primates and rodents have shown that, when neonatal animals are exposed to drugs that consistently and potently induce neuroapoptosis, the apoptosis is extensive and can be demonstrated compellingly with a small number of animals, typically between three and seven per group.13,14,16,19-21,22-24 Therefore, we decided a-priori to use five experimental and five control animals, and to present the raw data for each group graphically, with medians and full ranges for density counts of apoptotic neurons per mm3 of brain tissue, and to compare median difference in density counts between the isoflurane and control groups using the Mann-Whittney-U statistical test. A two sided p value < 0.05 would be judged significant, and the 95% confidence interval for the median difference would provide a measure of precision. Statistical analysis was performed with Analyse-It® Statistical Software (Leeds, United Kingdom) for Microsoft Excel® (Redmond, WA).
RESULTS
All animals randomized to the experimental protocol survived induction and maintenance of anesthesia. Endotracheal intubation and mechanical ventilation allowed the controlled administration of the volatile anesthetic at the desired level, and maintenance of vital signs, blood gases and metabolic values within normal limits (see figs. 1-3, Supplemental Digital Content 1, which contains graphs showing mean arterial pressure, serum glucose, and hemoglobin levels; also see table 1, Supplemental Digital Content 2, which is a table listing physiological data). Continuous fluid and glucose application as well as active warming ensured homeostasis throughout the entire experimental period. Recovery was fast after cessation of isoflurane delivery and animals were extubated without complications and maintained in an animal incubator at physiologic conditions under direct visual observation. All infants tolerated formula milk within the first 2 h after the end of the anesthetic.
Figure 1.

Density of activated caspase 3-stained neurons (profiles/mm3) in isoflurane-exposed versus control brains. Transverse sections (approximately 800) were cut serially across the entire forebrain and midbrain and counts were performed on sections selected at 2.24 mm intervals (every 32nd section). An outline of each section was entered into the computer and all stained neuronal profiles within the outlined space were counted and marked for location. The total number of stained neurons divided by the total tissue space (determined by the applied software) yielded a neuronal density count (number of activated caspase 3-stained profiles per mm3) for each brain. The median density (range) of activated caspase 3-positive neurons per mm3 for the isoflurane-exposed brains was 32.5 (18.0 to 48.2), and for the control brains was 2.5 (1.1 to 5.2). This amounts to a 13-fold increase in density of apoptotic neurons in the isoflurane-exposed brains. The difference in median density of apoptotic neurons between the isoflurane and control groups was 30 (95% CI, 15.5 to 45.7, p = 0.008).
Figure 3.

The appearance of activated caspase 3-stained neurons in layer II (panel A & B) and layer V (panel C) of the primary visual cortex of an isoflurane-exposed brain. The layer II profiles have the shape and arborization pattern characteristic of γ-aminobutyric acid-ergic inhibitory interneurons. The layer V profiles are predominantly small pyramidal neurons (presumably glutamatergic) of a type that are thought to project to visual neurons in the contralateral hemisphere33. The large activated caspase 3-positive multipolar neuron in panel C was seen occasionally in layer V and more frequently in the superficial portion of layer VI. This cell has the appearance of a Martinotti cell, which has been described as γ-aminobutyric acid-ergic34 and is thought to mediate inhibition.
Quantitative evaluation of AC3-stained brain sections across the entire extent of the forebrain and midbrain revealed that the median density (range) of AC3-positive neuronal cells per mm3 of brain tissue in the five isoflurane-exposed brains was 32.5 (18.0 to 48.2), and in the five control brains was only 2.5 (1.1 to 5.2) (fig. 1). This translates to a 13-fold increase in density of apoptotic cells in the brains of the animals that were exposed to isoflurane. The difference in median density of apoptotic cells per mm3 of brain tissue between the isoflurane and control groups was 30 (95% CI, 15.5 to 45.7, p = 0.008).
The apoptotic neurons were located primarily in various divisions of the cerebral cortex. In the controls they were sparse and randomly distributed, whereas in the isoflurane-exposed brains they were more abundant and tended to be densely concentrated in specific cortical regions or layers. In the isoflurane-exposed brains, the primary visual cortex, known as the striate cortex because of its conspicuous laminar organization, had an exceedingly high density of AC3 positive profiles, which were concentrated primarily in layers II and V (figs. 2 and 3). The next most severely affected regions were the temporal and somatosensory cortices (figs. 4 and 5). All cortical regions of the isoflurane-exposed brains were more severely affected than any cortical region in the control brains. The frontal cortex, which reportedly is more sensitive than other brain regions to ketamine-induced neuroapoptosis in P5 rhesus macaques19,20, was more severely affected in the isoflurane brains than in the concurrent controls, but was relatively spared compared to other cortical regions of the isoflurane-exposed brains (fig. 4).
Figure 2.

Computer plots based on the number and location of each activated caspase 3-stained neuronal profile in homologous sections from the primary visual cortex of a control and isoflurane brain. The sections shown are from the animal in each treatment group (isoflurane and control) that had the highest mean activated caspase 3 density count. Note the striking laminar pattern of distribution of stained neuronal profiles in the isoflurane brain and the randomly scattered pattern in the control brain. The layers primarily affected in the isoflurane brain are layers II and V, but there are also many stained profiles in between these two layers. The primary cell types affected are depicted in figure 3.
Figure 4.

Computer plots based on the number and location of each activated caspase 3-stained profile in homologous sections from three different divisions of the neocortex (temporal, somatosensory, frontal) of control versus isoflurane-exposed brains. In all of the control sections the activated caspase 3-stained profiles are sparse and randomly distributed in no particular relationship to the laminar organization of the cortical tissue. In the temporal cortex of the isoflurane-exposed brain a laminar pattern is evident, with layers II and IV being the most severely affected. The density count in these particular temporal cortical sections is 17-fold higher in the isoflurane compared to the control section. In the somatosensory cortex, a laminar pattern is also evident, but not quite as pronounced, and the overall count is 11.5 fold higher in the isoflurane than control section. In the frontal cortex, the laminar pattern is only faintly evident and the overall count is only 3-fold greater in the isoflurane than control section.
Figure 5.

The appearance of activated caspase 3-stained (AC3) neurons in layer II (panel A) and deeper layers (panel B) of the temporal cortex of an isoflurane-exposed brain. The layer II AC3-positive profiles are interneurons, presumably γ-aminobutyric acid-ergic, and are identical in appearance to layer II neurons that frequently undergo apoptosis in various divisions of the infant mouse neocortex following exposure to either anesthetics or ethanol. Panel B is a scene from deeper layers of the isoflurane-exposed temporal cortex depicting the appearance of several types of pyramidal neurons in an early stage of degeneration. In layer IV, many AC3-positive profiles are large pyramidal neurons having a typical triangular cell body and long apical dendrites. These are accompanied in the same layer by very small AC3-positive neurons and in layer III by occasional AC3-positive neurons that are relatively large. The latter two cell types would probably be classified as pyramidal neurons, although their cell bodies are shaped more like a tear- drop than a pyramid. These several types of pyramidal neurons are identical in appearance to those that are preferentially affected in the same cortical layers of infant rodent brain following exposure to either anesthetics or ethanol.
Morphological analysis of the stained profiles of individual cells revealed that both early and advanced stages of degeneration could be detected (figs. 3, 5, and 6), but the majority of AC3-positive profiles were in a relatively early stage. The reaction was not confined to a specific cell type, but rather affected several different cell types, including large multipolar neurons (layers V & VI), pyramidal neurons both large and small (layers IV & V), and interneurons, especially those concentrated in layer II of various divisions of the cerebral cortex (figs. 3 and 5). An important distinction between the appearance of stained cells in the control brains vs. the isoflurane-exposed brains was that the majority of stained profiles in the control brains were in a late stage (rounded up cell body with few or no intact processes), and the majority in the isoflurane brains were in an earlier stage. This reflects the fact that the latter are responding within an acute time frame to a recent drug challenge, whereas the former have begun degenerating spontaneously at various more remote intervals in the absence of any drug challenge.
Figure 6.
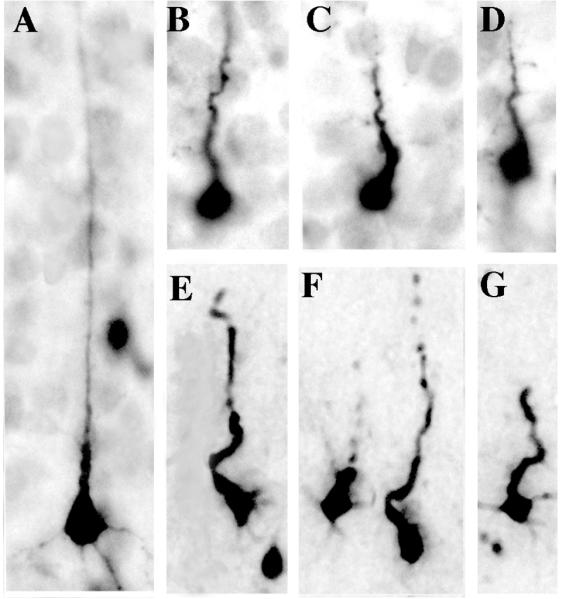
Comparison of pyramidal neurons in early versus late stages of degeneration following exposure to isoflurane. Panel A shows the typical appearance of a relatively well-preserved activated caspase 3 positive pyramidal neuron in an early stage of degeneration. Note that the shaft of the apical dendrite is straight and does not appear dysmorphic. Panels B, C, & D are from layer V of the primary visual cortex and illustrate a later stage of degeneration when the apical dendrite has begun to shrivel and assume a corkscrew-like configuration. Panels E, F, & G are from the deep layers of the temporal cortex and show neurons in a late stage of degeneration with shriveled apical dendrites that are beginning to undergo fragmentation. Compare these dysmorphic neuronal profiles with those in figure 5B that are in an earlier stage of degeneration.
DISCUSSION
We have demonstrated that exposing P6 infant rhesus macaques to a moderate surgical plane of anesthesia for 5 h, while maintaining physiologic stability, is associated with a 13-fold increase in the rate of neuroapoptosis compared to non-anesthetized controls. The increased neurodegeneration was demonstrable in widespread distribution across all divisions of the cerebral cortex. The reaction was most severe in the primary visual cortex, least severe in the frontal cortex and of intermediate severity in the remaining cortical regions. A wide variety of neuronal cell types were affected, and while the majority were in an early stage of degeneration when examined 3 h after cessation of anesthesia, some cell populations showed morphological evidence of being in various stages of advanced degeneration. This signifies, consistent with observations in rodent studies3,9,32, that some neuronal populations are more sensitive to the toxic stimulus and, therefore, begin dying earlier.
Because rhesus macaques are neurodevelopmentally precocious compared to humans, a P6 rhesus infant is approximately equivalent to a six-month-old human infant (fig. 7), or a P8 to P12 infant rodent6. In rodents during the early postnatal period (P1 to P3) subcortical neurons are vulnerable and cerebrocortical neurons are spared, then there is a period (P4 to P7) when both cortical and subcortical neurons are vulnerable, and finally a period (P8 to P12) when cerebrocortical neurons remain vulnerable while subcortical vulnerability is waning1,2. During this latter period, the cortical neurons most severely affected are in layers II, IV, and V. The pattern of neurodegeneration observed in the isoflurane-exposed P6 rhesus macaque was limited primarily to various divisions of the cerebral cortex, and preferentially affected layer II interneurons and pyramidal neurons in layers IV and V. Thus, the degenerative reaction to isoflurane in the P6 rhesus macaque is consistent with findings in P8 to P12 infant rodents, both with respect to brain regions and cell types that are preferentially involved. This suggests the type of reaction that might occur in the human infant brain if humans are susceptible to this toxic mechanism. Whether humans are susceptible is not a question that can be answered definitively by research on nonhuman species.
Figure 7.

Timing of the brain growth spurt period in human versus nonhuman primates. This graph was adapted from a publication by Dobbing and Sands6 providing comparative brain growth spurt data for numerous mammalian species. The curves represent the changing rates of brain growth for humans and rhesus monkeys in the perinatal period. In both species the rate of brain growth increases rapidly to a peak level then gradually descends to a lower level sustained into adolescence. Note that the timing of the growth spurt in relation to birth occurs earlier in monkeys than in humans, and it tapers down to a low level at an earlier age. Thus, in the monkey infant at postnatal day 6, the brain growth rate has decreased to about 25% of its peak value, whereas in the human infant at postnatal day 6 the brain growth rate is just reaching its peak and does not taper down to the 25% level until the age of 6 months (see dashed line). Therefore, in terms of comparative brain growth spurt timing, the 6-day-old monkey infant is the equivalent of a 6-month-old human infant. Another relevant comparison is that the brain weight of the infant rhesus monkey at birth is 76% of its adult brain weight, whereas the brain weight of the newborn human is only 27% of its adult brain weight6.
Slikker and colleagues19,20 have reported that an intravenous infusion of ketamine induces a neuroapoptosis reaction in the P5 rhesus macaque brain if the infusion is continued for 9 or 24 h, but not if it is stopped at 3 h. These authors confined their quantitative evaluation to a single brain region, the frontal cortex, after making the preliminary qualitative observation that this region of the ketamine-exposed brains was rich in degenerating profiles compared to several subcortical regions (striatum, thalamus, hippocampus and amygdala). Our findings are in agreement with theirs regarding susceptibility of the neonatal macaque brain to anesthesia-induced neuroapoptosis, and with respect to cerebrocortical neurons being more sensitive than subcortical neurons at this developmental age. Regarding the potential implication of their findings that it may require at least 9 h of anesthesia exposure to induce neuroapoptosis, our findings clarify, consistent with observations in rodents13,14,16, that a 5-h exposure period is sufficient, and that the apoptotic response can be detected as early as 3 h after a 5-h exposure period. Regarding their finding that 3 h of ketamine anesthesia was not sufficient to induce neuroapoptosis in the neonatal non-human primate brain, this important issue warrants further investigation, in that their observation is based on quantitative data gathered from only a single brain region (frontal cortex), and from only a small number (n = 3) of experimental subjects. In light of our observation in P6 rhesus macaques that the frontal cortex, although susceptible to isoflurane-induced neuroapoptosis at this developmental age, was less sensitive than other regions of the cerebral cortex, in future research examining this issue it will be important to include all regions of the cerebral cortex in the quantitative evaluation.
In summary, our findings indicate that exposure of the P6 rhesus macaque brain for 5 h to isoflurane, under clinically relevant conditions, induces widespread neuroapoptosis affecting all divisions of the cerebral cortex. Our studies leave unanswered whether shorter durations of isoflurane anesthesia would also trigger acute apoptotic neurodegeneration, and whether either brief or prolonged exposure of the infant nonhuman primate brain to isoflurane can cause long-term neurocognitive deficits. Research addressing these important questions is urgently needed.
Supplementary Material
Acknowledgments
Funded by grants from the National Institutes of Health, Bethesda, Maryland, HD37100, HD052664, and RR-000163 (for operation of the Oregon National Primate Research Center).
Footnotes
Cattano D, Straiko MMW, Olney JW: Chloral hydrate induces and lithium prevents neuroapoptosis in the infant mouse brain. Presented at the Annual Meeting of the American Society of Anesthesiologists, 2008, Orlando, Florida, #A315. Accessed November 1, 2009.
DiMaggio CJ, Sun L, Kakavouli A, Guoha L: Exposure to anesthesia and the risk of developmental and behavioral disorders in young children. Presented at the Annual Meeting of the American Society of Anesthesiologists, 2008, Orlando, Florida, #A1415. Accessed November 1, 2009.
Paule MG, Li M, Zou X, Hotchkiss C, Hanig JP, Patterson TA, Slikker W, Wang C: Early postnatal ketamine anesthesia causes persistent cognitive deficits in rhesus monkeys. Presented at the Society for Neuroscience Annual Meeting, 2009, Chicago, Illinois, #413.7. Accessed November 1, 2009.
REFERENCES
- 1.Ikonomidou C, Bosch F, Miksa M, Bittigau P, Vöckler J, Dikranian K, Tenkova T, Stevoska V, Turski L, Olney JW. Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science. 1999;283:70–4. doi: 10.1126/science.283.5398.70. [DOI] [PubMed] [Google Scholar]
- 2.Ikonomidou C, Bittigau P, Ishimaru MJ, Wozniak DF, Koch C, Genz K, Price MT, Stefovska V, Hörster F, Tenkova T, Dikranian K, Olney JW. Ethanol-induced apoptotic neurodegeneration and fetal alcohol syndrome. Science. 2000;287:1056–60. doi: 10.1126/science.287.5455.1056. [DOI] [PubMed] [Google Scholar]
- 3.Olney JW, Tenkova T, Dikranian K, Qin YQ, Labruyere J, Ikonomidou C. Ethanol-induced apoptotic neurodegeneration in the developing C57BL/6 mouse brain. Dev Brain Res. 2002;133:115–26. doi: 10.1016/s0165-3806(02)00279-1. [DOI] [PubMed] [Google Scholar]
- 4.Bittigau P, Sifringer M, Genz K, Reith E, Pospischil D, Govindarajalu S, Dzietko M, Pesditschek S, Mai I, Dikranian K, Olney JW, Ikonomidou C. Antiepileptic drugs and apoptotic neurodegeneration in the developing brain. Proc Natl Acad Sci USA. 2002;99:15089–94. doi: 10.1073/pnas.222550499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jevtovic-Todorovic V, Hartman RE, Izumi Y, Benshoff ND, Dikranian K, Zorumski CF, Olney JW, Wozniak DF. Early exposure to common anesthetic agents causes widespread neurodegeneration in the developing rat brain and persistent learning deficits. J Neurosci. 2003;23:876–82. doi: 10.1523/JNEUROSCI.23-03-00876.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dobbing J, Sands J. Comparative aspects of the brain growth spurt. Early Hum Dev. 1979;3:79–83. doi: 10.1016/0378-3782(79)90022-7. [DOI] [PubMed] [Google Scholar]
- 7.Dikranian K, Ishimaru MJ, Tenkova T, Labruyere J, Qin YQ, Ikonomidou C, Olney JW. Apoptosis in the in vivo mammalian forebrain. Neurobiol Dis. 2001;8:359–79. doi: 10.1006/nbdi.2001.0411. [DOI] [PubMed] [Google Scholar]
- 8.Dikranian K, Qin YQ, Labruyere J, Nemmers B, Olney JW. Ethanol-induced neuroapoptosis in the developing rodent cerebellum and related brain stem structures. Dev Brain Res. 2005;155:1–13. doi: 10.1016/j.devbrainres.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 9.Olney JW, Tenkova T, Dikranian K, Muglia LJ, Jermakowicz WJ, D'Sa C, Roth KA. Ethanol-induced caspase-3 activation in the in vivo developing mouse brain. Neurobiol Dis. 2002;9:205–19. doi: 10.1006/nbdi.2001.0475. [DOI] [PubMed] [Google Scholar]
- 10.Young C, Klocke BJ, Tenkova T, Choi J, Labruyere J, Qin YQ, Holtzman DM, Roth KA, Olney JW. Ethanol-induced neuronal apoptosis in vivo require BAX in the developing mouse brain. Cell Death Differ. 2003;10:1148–55. doi: 10.1038/sj.cdd.4401277. [DOI] [PubMed] [Google Scholar]
- 11.Young C, Roth KA, Klocke BJ, West T, Holtzman DM, Labruyere J, Qin YQ, Dikranian K, Olney JW. Role of caspase-3 in ethanol-induced developmental neurodegeneration. Neurobiol Dis. 2005;20:608–14. doi: 10.1016/j.nbd.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 12.Tenkova T, Young C, Dikranian K, Olney JW. Ethanol-induced apoptosis in the visual system during synaptogenesis. Invest Ophthalmol Vis Sci. 2003;44:2809–17. doi: 10.1167/iovs.02-0982. [DOI] [PubMed] [Google Scholar]
- 13.Young C, Jevtovic-Todorovic V, Qin YQ, Tenkova T, Wang H, Labruyere J, Olney JW. Potential of ketamine and midazolam, individually or in combination, to induce apoptotic neurodegeneration in the infant mouse brain. Brit J Pharmacol. 2005;146:189–97. doi: 10.1038/sj.bjp.0706301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cattano D, Young C, Straiko MMW, Olney JW. Subanesthetic doses of propofol induce neuroapoptosis in the infant mouse brain. Anesth Analg. 2008;106:1712–4. doi: 10.1213/ane.0b013e318172ba0a. [DOI] [PubMed] [Google Scholar]
- 15.Ma D, Williamson P, Januszewski A, Nogaro MC, Hossain M, Ong LP, Shu Y, Franks NP, Maze M. Xenon mitigates isoflurane-induced neuronal apoptosis in the developing rodent brain. Anesthesiology. 2007;106:746–53. doi: 10.1097/01.anes.0000264762.48920.80. [DOI] [PubMed] [Google Scholar]
- 16.Johnson SA, Young C, Olney JW. Isoflurane-induced neuroapoptosis in the developing brain of non-hypoglycemic mice. J Neurosurg Anesth. 2008;20:21–8. doi: 10.1097/ANA.0b013e3181271850. [DOI] [PubMed] [Google Scholar]
- 17.Zhang X, Xue Z, Sun A. Subclinical concentration of sevoflurane potentiates neuronal apoptosis in the developing C57BL/6 mouse brain. Neurosci Lett. 2008;447:109–14. doi: 10.1016/j.neulet.2008.09.083. [DOI] [PubMed] [Google Scholar]
- 18.Wozniak DF, Hartman RE, Boyle MP, Vogt SK, Brooks AR, Tenkova T, Young C, Olney JW, Muglia LJ. Apoptotic neurodegeneration induced by ethanol in neonatal mice is associated with profound learning/memory deficits in juveniles followed by progressive functional recovery in adults. Neurobiol Dis. 2004;17:403–14. doi: 10.1016/j.nbd.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 19.Slikker W, Jr, Zou X, Hotchkiss CE, Divine RL, Sadovova N, Twaddle NC, Doerge DR, Scallet AC, Patterson TA, Hanig JP, Paule MG, Wang C. Ketamine-induced neuronal cell death in the perinatal rhesus monkey. Toxicol Sci. 2007;98:145–58. doi: 10.1093/toxsci/kfm084. [DOI] [PubMed] [Google Scholar]
- 20.Zou X, Patterson TA, Divine RL, Sadova N, Zhang X, Hanig JP, Paule MG, Slikker W, Wang C. Prolonged exposure to ketamine increases neurodegeneration in the developing monkey brain. Int J Dev Neurosci. 2009;27:727–31. doi: 10.1016/j.ijdevneu.2009.06.010. [DOI] [PubMed] [Google Scholar]
- 21.Straiko MMW, Young C, Cattano D, Creeley CE, Wang H, Smith DJ, Johnson SA, Li ES, Olney JW. Lithium protects against anesthesia-induced developmental neuroapoptosis. Anesthesiology. 2009;110:662–8. doi: 10.1097/ALN.0b013e31819b5eab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sanders RD, Xu J, Shu Y, Fidalgo A, Ma D, Maze M. General anesthetics induce apoptotic neurodegeneration in the neonatal rat spinal cord. Anesth Analg. 2008;106:1708–11. doi: 10.1213/ane.0b013e3181733fdb. [DOI] [PubMed] [Google Scholar]
- 23.Zhong J, Yang X, Yao W, Lee W. Lithium protects ethanol-induced neuronal apoptosis. Biochem Biophys Res Commun. 2006;350:905–10. doi: 10.1016/j.bbrc.2006.09.138. [DOI] [PubMed] [Google Scholar]
- 24.Young C, Straiko MMW, Johnson SA, Creeley CE, Olney JW. Ethanol causes and lithium prevents neuroapoptosis and suppression of pERK in the infant mouse brain. Neurobiol Dis. 2008;31:355–60. doi: 10.1016/j.nbd.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Satomoto M, Satoh Y, Terui K, Miyao H, Takishima K, Ito M, Imaki J. Neonatal exposure to sevoflurane induces abnormal social behaviors and deficits in fear conditioning in mice. Anesthesiology. 2009;110:628–37. doi: 10.1097/ALN.0b013e3181974fa2. [DOI] [PubMed] [Google Scholar]
- 26.Fredriksson A, Ponten E, Gordh T, Eriksson P. Neonatal exposure to a combination of N-methyl-d-aspartate and γ-aminobutyric acid type A receptor anesthetic agents potentiates apoptotic neurodegeneration and persistent behavioral deficits. Anesthesiology. 2007;107:427–36. doi: 10.1097/01.anes.0000278892.62305.9c. [DOI] [PubMed] [Google Scholar]
- 27.Fredriksson A, Archer T, Alm H, Gordh T, Eriksson P. Neurofunctional deficits and potentiated apoptosis by neonatal NMDA antagonist administration. Behav Brain Res. 2004;153:367–76. doi: 10.1016/j.bbr.2003.12.026. [DOI] [PubMed] [Google Scholar]
- 28.Fredriksson A, Archer T. Neurobehavioural deficits associated with apoptotic neurodegeneration and vulnerability for ADHD. Neurotox Res. 2004;6:435–56. doi: 10.1007/BF03033280. [DOI] [PubMed] [Google Scholar]
- 29.Stratmann G, Sall JW, May LD, Bell JS, Magnusson KR, Rau V, Visrodia KH, Ku B, Lee MT, Dai R. Isoflurane differentially affects neurogenesis and long-term neurocognitive function in 60-day-old and 7-day-old rats. Anesthesiology. 110:834–48. doi: 10.1097/ALN.0b013e31819c463d. [DOI] [PubMed] [Google Scholar]
- 30.Wilder RT, Flick RP, Sprung J, Katusic SK, Barbaresi WJ, Mickelson C, Gleich SJ, Schroeder DR, Weaver AL, Warner DO. Early exposure to anesthesia and learning disabilities in a population-based birth cohort. Anesthesiology. 2009;110:796–804. doi: 10.1097/01.anes.0000344728.34332.5d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kalkman CJ, Peelen LM, deJong TP, Sinnema G, Moons KG. Behavior and development in children and age at time of first anesthetic exposure. Anesthesiology. 2009;110:805–12. doi: 10.1097/ALN.0b013e31819c7124. [DOI] [PubMed] [Google Scholar]
- 32.Young C, Olney JW. Neuroapoptosis in the infant mouse brain triggered by a transient small increase in blood alcohol concentration. Neurobiol Dis. 2006;22:548–54. doi: 10.1016/j.nbd.2005.12.015. [DOI] [PubMed] [Google Scholar]
- 33.Kasper EM, Larkman AU, Lubke J, Blakemore C. Pyramidal neurons in layer 5 of the rat visual cortex. I. Correlation among cell morphology, intrinsic electrophysiological properties, and axon targets. J Comp Neurol. 1994;339:459–74. doi: 10.1002/cne.903390402. [DOI] [PubMed] [Google Scholar]
- 34.Hedlich A, Luth HJ, Werner L, Bar B, Hanisch U, Winkelmann E. GABAergic NADPH-Diaphorase-positive Martinotti cells in the visual cortex in rats. Journal fur Hirnforschung. 1990;31:681–7. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.