

Abstract
The role of Mer kinase in regulating the second phase of platelet activation generates an opportunity to use Mer inhibitors for preventing thrombosis with diminished likelihood for bleeding as compared to current therapies. Toward this end, we have discovered a novel, Mer kinase specific substituted-pyrimidine scaffold using a structure-based drug design and a pseudo-ring replacement strategy. The co-crystal structure of Mer with two compounds (7 & 22) possessing distinct activity have been determined. Subsequent SAR studies identified compound 23 (UNC2881) as a lead compound for in vivo evaluation. When applied to live cells, 23 inhibits steady-state Mer kinase phosphorylation with an IC50 value of 22 nM. Treatment with 23 is also sufficient to block EGF-mediated stimulation of a chimeric receptor containing the intracellular domain of Mer fused to the extracellular domain of EGFR. In addition, 23 potently inhibits collagen-induced platelet aggregation, suggesting that this class of inhibitors may have utility for prevention and/or treatment of pathologic thrombosis.
Keywords: Mer inhibitors, TAM RTK, platelet aggregation, thrombosis, pyrimidines, pseudo-ring replacement
INTRODUCTION
Platelets are small cells derived from precursor megakaryocytes. The physiologic procoagulant activity of platelets helps prevent excessive bleeding, while increased platelet activation and overactive coagulation can lead to pathologic thrombus formation which may result in stroke or heart attack. Anti-platelet compounds are therefore an important family of drugs for cardiovascular diseases and for certain surgical procedures where a risk of stroke or thrombosis is prevalent. However, current anti-platelet therapies are often complicated by significant bleeding and other side effects. For example, aspirin, an antiplatelet drug that functions through inhibiting the production of thromboxane, has a baseline major bleeding risk (gastrointestinal or intracranial) of 1–4%1 and is associated with other toxicities, including gastric side effects. The addition of clopidogrel (Plavix), another widely used antiplatelet drug, as a combination therapy further increases bleeding risk by 1%.2 Moreover, not all patients respond to aspirin and clopidogrel: the non-response rates are 5.5–60% in patients treated with aspirin3 and 4–30% in those treated with clopidogrel4 based on meta-analyses. Consequently, there is still a significant need to develop novel therapies for treatment of thrombosis, especially ones that do not increase the risk for bleeding and other side effects associated with current anti-thrombotic agents.
Mer is a member of the TAM (Tyro3, Axl and Mer) receptor tyrosine kinase (RTK) subfamily with growth-arrest-specific-6 (Gas6) as one of the endogenous ligands.5 Elevated Mer activation has been strongly associated with the oncogenesis of a number of human cancers.6 Recently, Mer has also been shown to play important roles in regulating macrophage activity and platelet aggregation.7 Mer knock-out mice have decreased platelet aggregation while maintaining normal bleeding times and coagulation parameters. Consequently, these mice are protected from thrombosis without increased spontaneous bleeding.7a, 8 These observations indicate that small molecule Mer kinase inhibitors have potential as new anti-platelet drugs with decreased bleeding complications, a profile that confers a major advantage over currently available anti-platelet therapies.
In our efforts to develop novel therapeutic agents for cancer treatment, we have discovered several potent small molecule Mer inhibitors. However, the first generation of these molecules possess either low selectivity for Mer relative to other TAM family members (1(UNC569))9 or low solubility and poor pharmacokinetic (PK) properties, such that they are not suitable for in vivo study (UNC1062)10. While efforts to optimize these molecules to yield a Mer selective, in vivo active pyrazolopyrimdine are ongoing, here we report the discovery of a new substituted-pyrimidine scaffold with significantly improved selectivity to further validate Mer as a potential target for thrombosis prevention.
In the co-crystal structure of Mer in complex with compound 1 (Figure 1a),9 the inhibitor is fully confined to the relatively small adenine pocket, forming three hydrogen bonds: two with the hinge region of Mer using one nitrogen of the pyrimidine ring (with residue Met674) and the NH from the butyl amino side chain (with residue Pro672) and another one with the carbonyl of Arg727 via the methylcyclohexylamino group. Since the pyrazole ring does not appear to interact with the Mer active site directly, its major role may be to rigidify the molecule. Therefore, replacement of the pyrazole ring with a pseudo-ring11 constrained by an intramolecular hydrogen bond while maintaining functionality to create the three hydrogen bonds observed with 1 may mimic the binding conformation in Figure 1a and retain the potency observed with compound 1. One such design is shown in Figure 1b where the intramolecular hydrogen bond in 2 will be formed between the carbonyl oxygen of the amide group and the hydrogen on the adjacent amino side chain. The other substituents are not modified and will likely occupy the same regions as in 1. However, because the pseudo ring is less rigid and larger in size than the pyrazole ring, this new scaffold may have a distinct kinase specificity profile or pharmacokinetic (PK) properties due to subtle conformational and physical property changes. Furthermore, the synthesis of 2 is straightforward making efficient structure-activity relationship (SAR) studies feasible.
Figure 1.

Structure-based design of a scaffold that features pseudo-ring formation through an intramolecular hydrogen bond. A). X-ray structure of 1 complexed with Mer protein (kinase domain) (PDB ID code 3TCP); B). Docking model (based on X-ray structure PDB ID code 3TCP) of the designed molecule 2.
CHEMISTRY
The syntheses of pyrimidine analogs are shown in Scheme 1. An amide coupling reaction is used to introduce the R1 group while an SNAr reaction is used to introduce the R2 and R3 groups. Path A is designed for SAR exploration of the R2 and R3 positions while path B is designed for diversifying the R1 position.
Scheme 1.
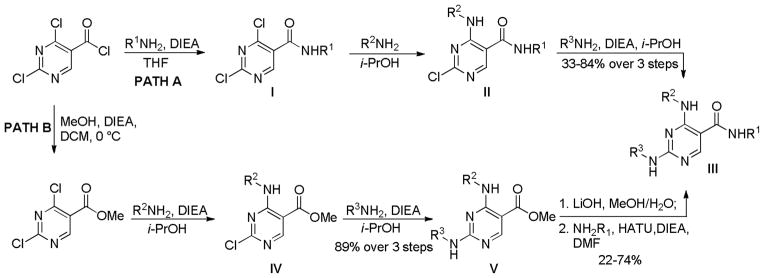
The synthetic routes for pyrimidine analogs.
RESULTS AND DISCUSSION
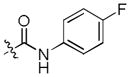
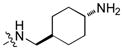
To test our pseudo-ring replacement hypothesis, a small set of compounds were synthesized using the route depicted in Scheme 1 (compound 3 and 5 started with 2,4-dichloropyrimidin-5-amine and 4-fluorobenzoyl chloride) (Table 1). Inhibition of Mer kinase activity by these compounds was tested using a microfluidic capillary electrophoresis (MCE) assay.12 Indeed, compound 2 was very potent against Mer while its close analog 3 exhibited only weak activity. The only structural difference between 2 & 3 was the regiochemistry of the key hydrogen bond enabling amide functionality. The reverse amide bond in 3 is unable to form the pseudo-ring forming intramolecular hydrogen bond with the hydrogen on the amino side chain at the R2 position resulting in greatly diminished Mer activity. Comparison of the activity of 4 and 5 further confirmed the important role of the intramolecular hydrogen bond and validated our design of the pseudo-ring replacement. To monitor selectivity within the TAM family, the ability of these analogs to inhibit Axl and Tyro3 was also tested; and they were significantly more active against Mer than Axl and Tyro3 (Table 1). In addition, the trans-4-aminocyclohexylmethylamino group at the R2 position in 2 can be replaced by a trans-4-hydroxycyclohexylamino group in 4 and the activity against Mer is retained. In our previous studies, the primary amino group at the R2 position could result in undesired hERG activity.10a Therefore, the trans-4-hydroxycyclohexylamino group was used at the R2 position for further SAR exploration at the R1 site.
Table 1.
Key SAR observations
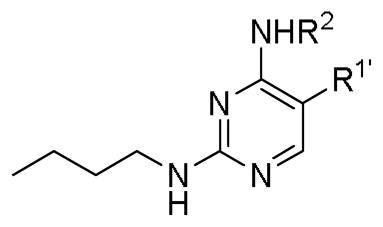
| |||||
|---|---|---|---|---|---|
| Compound | R1′ | NHR2 | IC50 (μM)a
|
||
| Mer | Axl | Tyro3 | |||
| 2 |
|
|
0.040 | 2.0 | 0.90 |
| 3 |
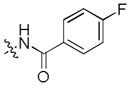
|
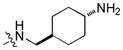
|
12.6 | >30 | >30 |
| 4 |
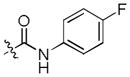
|
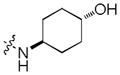
|
0.023 | 1.7 | 2.2 |
| 5 |
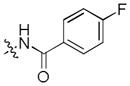
|
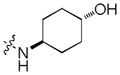
|
>30 | >30 | >30 |
Values are the mean of two or more independent assays.
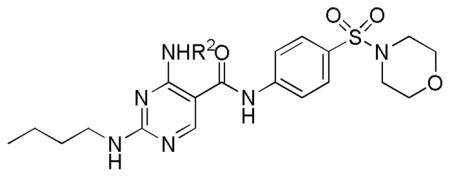
Next, a focused library of compounds with different R1 groups was synthesized while keeping R3 fixed as a butylamino group (Table 2). Interestingly, the amide nitrogen at the R1 site could not be further substituted. Even simple methylation of the amide bond almost abolished the Mer activity (6 vs. 7 and 8 vs. 9). This may be due to weaker binding of the ligand to the hinge motif (residues 672 to 677). Not only does amide methylation of the ligand preclude a hydrogen bond to the Met674-backbone carbonyl, it also contributes to a less favorable orientation of the aminopyrimidine group with respect to Pro672 and Met674. Since 8 was more active than the open chain analog 6, our focus was initially on further modification of 8. However, the addition of extra groups such as iPr (10) and pyrimidine (11) to the nitrogen of the piperidine ring did not boost the activity. Exchange of the piperidine nitrogen with oxygen (12) or carbon (13) also did not significantly affect the activity. Interestingly, when the piperidine ring was replaced with an aromatic ring, the activity of the analogs was modulated by the substituents on the aromatic ring: an electron-withdrawing group such as CF3 on the aromatic ring yielded a much weaker Mer inhibitor (14) compared to 8 while an electron-donating group like MeO on the aromatic ring produced an analog with similar activity (15). Inhibition of Mer was not sensitive to the substitution pattern on the aromatic ring (15–17). In addition, larger groups such as morpholine (18), morpholinomethyl (19) and morpholinosulfonyl (which was the R1 group of UNC106210a) (20) could be tolerated on the aromatic ring. This is consistent with the prediction that the R1 group interacting with the solvent front based on the docking model. Furthermore, a methylene group could be inserted between the amide bond and the aromatic ring without a change in activity (4 vs. 21, Table 1). However, the activity of the analogs was increased by introducing a larger group on the aromatic ring as in 22 & 23 (UNC2881). In general, these analogs are significantly more Mer-selective than 2, especially compound 23 (84-fold over Axl & 58-fold over Tyro3).
Table 2.
Preliminary SAR at the R1 position
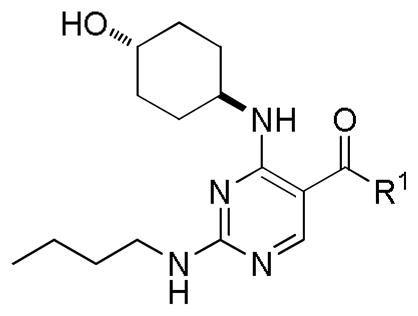
| ||||
|---|---|---|---|---|
| Compound | R1 | IC50 (μM)a
|
||
| Mer | Axl | Tyro3 | ||
| 6 |
|
0.067 | 3.0 | 6.7 |
| 7 |
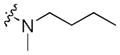
|
5.9 | >30 | >30 |
| 8 |
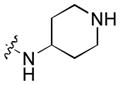
|
0.0090 | 0.48 | 0.53 |
| 9 |
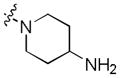
|
2.5 | >30 | >30 |
| 10 |
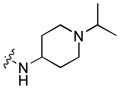
|
0.0084 | 0.43 | 0.19 |
| 11 |
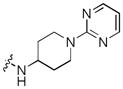
|
0.0041 | 0.56 | 0.48 |
| 12 |
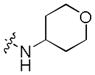
|
0.013 | 0.58 | 0.54 |
| 13 |
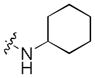
|
0.0080 | 0.37 | 0.37 |
| 14 |
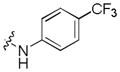
|
0.23 | 16.6 | 17.9 |
| 15 |
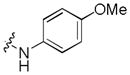
|
0.015 | 0.88 | 0.66 |
| 16 |
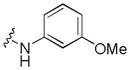
|
0.029 | 1.7 | 0.83 |
| 17 |
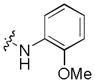
|
0.039 | 3.8 | 1.5 |
| 18 |
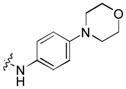
|
0.014 | 0.57 | 0.45 |
| 19 |
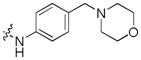
|
0.0067 | 0.37 | 0.24 |
| 20 |
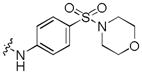
|
0.0063 | 0.53 | 0.70 |
| 21 |
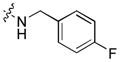
|
0.014 | 0.79 | 0.57 |
| 22 |
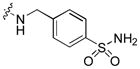
|
0.0052 | 0.26 | 0.28 |
| 23 |
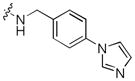
|
0.0043 | 0.36 | 0.25 |
Values are the mean of two or more independent assays.
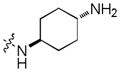
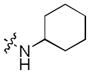
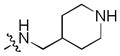
In addition, we explored SAR at the R2 site while keeping R1 fixed as morpholinosulfonylphenyl carboxamide and R3 fixed as butylamino group. A few examples are included in Table 3. When introducing the trans-4-aminocyclohexylamino group at the R2 site, the corresponding analog 24 had activity similar to 20 (Table 2). However, removal of the polar group NH2 on the cyclohexyl ring (25) reduced the activity dramatically (over 280-fold). This is consistent with the SAR observed with analogs of 1: a hydrogen bond between a polar substituent at the R2 position with the carbonyl of Arg727 of the Mer protein is important.9 Compared to 25, secondary amines such as 4-piperidinylmethylamino (26), (R)-3-piperidinylmethylamino (27) and 4-piperidinylamino (28) at the R2 position increased the activity compared to 25 but were less suitable than the primary amine group. Interestingly, introduction of an (S)-3-pyrrolodinylamino group in (30) led to a 9-fold higher activity compared to the (R)-3-pyrrolodinylamino substitution (29). The structural analysis suggests that this 9-fold activity gain is most likely due to an optimal interaction of the pyrrolidine nitrogen in 30 with the backbone carbonyl of Leu593, while the same nitrogen in 29 would not be able to optimally approach Leu593. A 4 substituted-pyridine ring at the R2 position resulted in a much less active analog 31 than the corresponding 4 substituted-piperidine ring (26). Replacement of the 4-piperidinylamino group (28) with a 4-pyranylamino group (32) decreased the activity slightly. However, a morpholine at the R2 position (33) almost abolished the Mer activity due to the absence of the pseudo-ring forming intramolecular hydrogen bond. In general, these analogs have good selectivity for Mer over Axl and Tyro3.
Table 3.
SAR Study of R2
| ||||
|---|---|---|---|---|
| Compound | NHR2 | IC50 (μM)a
|
||
| Mer | Axl | Tyro3 | ||
| 24 |
|
0.0021 | 0.40 | 0.40 |
| 25 |
|
0.59 | >30 | >30 |
| 26 |
|
0.037 | 4.7 | 0.62 |
| 27 |
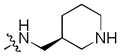
|
0.14 | 14.0 | 1.6 |
| 28 |
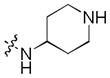
|
0.062 | 10.7 | 2.0 |
| 29 |
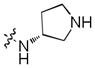
|
1.0 | >30 | 19.5 |
| 30 |
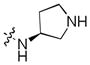
|
0.11 | 12.6 | 2.0 |
| 31 |
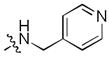
|
0.62 | 21 | 4.1 |
| 32 |
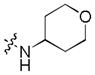
|
0.17 | >30 | >30 |
| 33 |
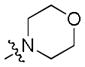
|
9.9 | >30 | >30 |
Values are the mean of two or more independent assays.
The SAR at the R3 position was further explored while fixing R1 as a morpholinosulfonylphenyl carboxamide group and R2 as a trans-4-hydroxycyclohexylamino group (Table 4). Absence of substitution on the amino group at the R3 position resulted in only a weak Mer inhibitor (34) and even a methyl group at this position significantly increased the activity (16-fold) (35 vs. 34). Analogs were progressively more active with addition of longer alkyl side chains (36 and 37) until the chain reached 4-carbons in length (20 (Table 2); the activity began to decrease when the chain was 5-carbons (38) or longer. A cyclopropyl group (39) was tolerated at the end of the propyl side chain while a polar group, such as trifluoromethyl (40) or a hydroxyl group (41), was less well tolerated. A branched alkyl side chain did not significantly affect activity (42 vs. 36). However, cyclohexyl (43), 4-tetrahydropyranyl (44), or 4-fluorobenzyl (45) at this position decreased Mer activity. Furthermore, when the amino group at this position was fully substituted, as in 46 and 47, Mer activity was completely eliminated. This result is consistent with our design hypothesis where the nitrogen of the amino group of R3 forms a hydrogen bond with residue Pro672 at Mer protein hinge area. Again, these analogs are Mer-selective as compared to Axl and Tyro3.
Table 4.
SAR Study of R3
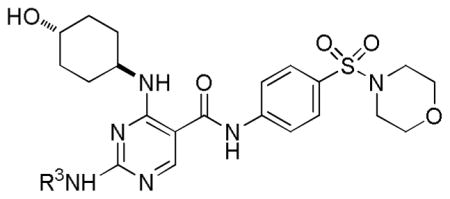
| ||||
|---|---|---|---|---|
| Compound | NHR3 | IC50 (μM)a
|
||
| Mer | Axl | Tyro3 | ||
| 34 |
|
2.4 | 16.6 | >30 |
| 35 |
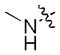
|
0.15 | 1.2 | >30 |
| 36 |
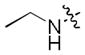
|
0.037 | 0.78 | 16.2 |
| 37 |
|
0.011 | 0.61 | 1.2 |
| 38 |
|
0.027 | 1.1 | 21.9 |
| 39 |
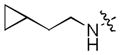
|
0.0073 | 0.30 | 0.94 |
| 40 |
|
0.086 | 5.7 | 5.1 |
| 41 |
|
0.059 | 4.3 | 6.7 |
| 42 |
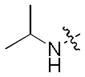
|
0.024 | 1.9 | 1.7 |
| 43 |
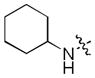
|
0.047 | 9.0 | 4.3 |
| 44 |
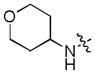
|
0.18 | 7.4 | 2.3 |
| 45 |
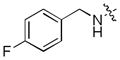
|
0.12 | >30 | >30 |
| 46 |
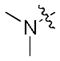
|
>30 | >30 | >30 |
| 47 |
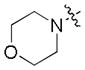
|
>30 | >30 | >30 |
Values are the mean of two or more independent assays.
To characterize the binding interactions of the reported pyrimidine inhibitors with Mer and provide a structural basis for future directions in chemical optimization, co-crystal structures of Mer in complex with compounds 7 and 22 were obtained. The structures in Figure 2 demonstrate a binding mode in which the pyrimidine ring inserts into the adenine binding site and mimics interactions of the adenine with the backbone atoms of the hinge (residues Pro672 and Met674). Similar to the previously published Mer-1 complex,9 the butyl amino side chain (R3 substituent) is folded into the relatively small adenine site instead of protruding into the lipophilic back-pocket. However, unlike the previously published structure, the hydroxyl group of the R2 substituent forms a hydrogen bond with the side chain of Asp741 instead of the nearby Arg727 (as does the R2 amino group of 1). It is noteworthy that one of the gate-forming residues, Ile650, is not conserved among other members of the TAM subfamily, Axl and Tyro3, which feature methionine and alanine in this position, respectively. This variability is quite likely the reason for the significant intra-family selectivity observed. It is remarkable that despite sharing identical core motifs, compounds 7 and 22 display very different IC50 values of 5.9 μM and 0.0052 μM, respectively. The most likely explanation for this significant difference is that the methyl group substituted on the amide of compound 7 precludes a hydrogen bond to the Met674 backbone carbonyl (present in the compound 22:Mer complex) and may also prevent the aminopyrimidine group from binding to Pro672 and Met674 in the most favorable orientation. Consistent with the SAR discussed above, the R1 substituent interacts mostly with the solvent and does not significantly impact the activity. Consequently, it can be utilized for tuning solubility and other physical or PK properties.
Figure 2.
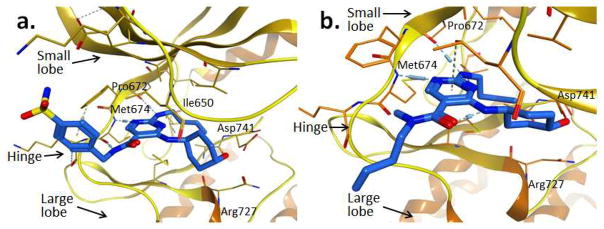
X-ray structures of compounds 22 (PDB ID code 4MHA) (a) and 7 (PDB code 4MH7) (b) in complex with the Mer kinase domain.
Based on its solubility, inhibitory activity against Mer, and remarkable intra-family selectivity, 23 was chosen as a lead compound in this series. The in vivo PK properties of 23 were assessed in mice via both intravenous (IV) and oral (PO) administration (Table 5). 23 had high systemic clearance (94.5 mL/min/kg) and 14% oral bioavailability. The terminal half-life was 0.80 hr. The volume of distribution was 2-fold greater than the normal volume of total body water (0.70 L/kg). Although the PK properties of 23 are not yet ideal and need to be further improved to enable chronic in vivo studies, this compound is sufficient for in vitro or short-term in vivo studies.
Table 5.
PK profile of 23
| Routea | T1/2 (h) | Tmax (h) | Cmax (ng/mL) | AUClast (h*ng/mL) | Cl_obs (mL/min/kg) | Vss (L/kg) | %F |
|---|---|---|---|---|---|---|---|
| IV | 0.80 | - | 2609 | 527 | 94.5 | 1.65 | - |
| PO | - | 0.30 | 90.0 | 71.7 | - | - | 14 |
a dose of 3 mg/kg for both routes.
The inhibitory activity mediated by 23 against a panel of 30 kinases was also determined at a concentration 100-fold above its Mer IC50 (Figure 3) (details in supplemental materials). This experiment provides a broad survey of kinase families emphasizing tyrosine kinases along with a selection of serine/threonine kinases. Six kinases were inhibited by greater than 50% in the presence of 430 nM 23, while none of the serine/threonine kinases were appreciably inhibited.
Figure 3.

Kinase Tree.
When 23 was tested in cell-based assays, it mediated inhibition of Mer phosphorylation in 697 B-ALL cells with an IC50 value of 22 nM (Figure 4). In addition to this effect on steady-state levels of phosphorylated Mer, 23 efficiently inhibited ligand-dependent phosphorylation of a chimeric protein consisting of the extracellular domain from the epidermal growth factor (EGF) receptor and the intracellular domain of Mer (Figure 5). Moreover, treatment with 23 inhibited platelet aggregation by greater than 25% in human platelet-rich plasma in response to stimulation with fibrillar Type I equine collagen (Figure 6). ATP release, which is a marker of platelet activation and degranulation, was decreased to a similar extent. These data are the first to demonstrate functional inhibition of platelet activity mediated by a Mer-selective small molecule tyrosine kinase inhibitor and suggest the utility of Mer inhibitors for this therapeutic application.
Figure 4. 23 inhibits endogenous Mer tyrosine kinase activation in acute lymphoblastic leukemia cells.
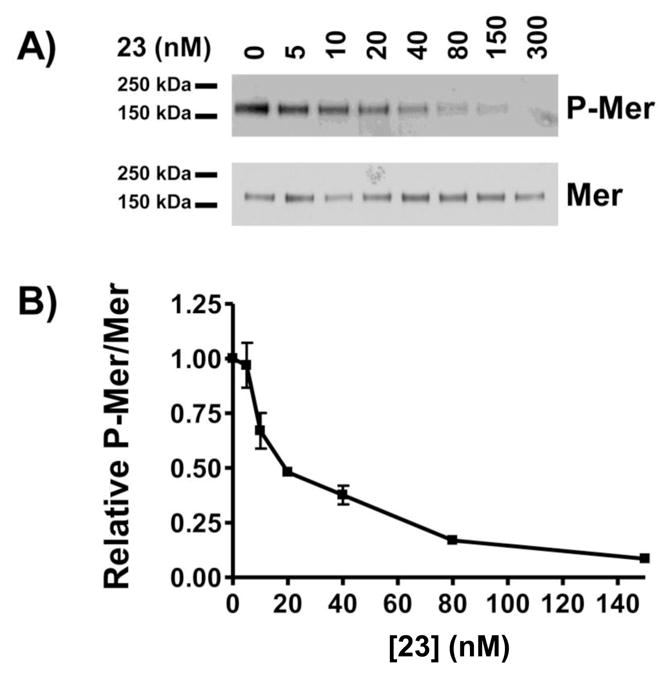
697 B-ALL cells were treated with the indicated concentrations of 23 for 1 h. Pervanadate was added to cultures for 3 min to stabilize the phosphorylated form of Mer. Mer was immunoprecipitated from cell lysates and total Mer protein and Mer phosphoprotein were detected by immunoblot. A) Representative western blots. B) Relative levels of phospho-Mer and Mer proteins were determined. Mean values +/− standard error derived from 3 independent experiments are shown. IC50 = 21.9 nM with a 95% confidence interval of 16.9 nM to 28.5 nM.
Figure 5. 23 inhibits ligand-stimulated activation of a chimeric EGFR-MerTK.

32D cells expressing a chimeric receptor consisting of the extracellular ligand-binding domain of the EGF receptor and the intracellular domain of Mer were treated with 23 or vehicle for 1 h prior to stimulation with 100 ng/mL EGF ligand for 15 min. Mer was immunoprecipitated from whole cell lysates and phospho-tyrosine containing and total Mer proteins were detected by western blot. Results shown are representative of 2 independent experiments.
Figure 6. 23 inhibits collagen-stimulated platelet aggregation.

Human platelet-rich plasma was normalized to a platelet count of 250,000/μL with platelet-poor plasma obtained by differential centrifugation. Samples were treated with 3 μM 23 or DMSO vehicle for 1 h at room temperature prior to stimulation with 1 μg/mL type I equine fibrillar collagen and assessment of aggregation and ATP release using an aggregometer. A) Representative aggregation from samples treated with 3 μM 23 or DMSO. B) Maximum percent aggregation and ATP release are shown for samples treated with 23 or DMSO vehicle. Mean values +/− SEM derived using plasma collected from 5 (ATP release) or 7 (aggregation) different individuals are shown. *Statistically significant results were determined using the student’s paired t test (* p = 0.05 and ** p = 0.01 relative to vehicle control).
CONCLUSIONS
We have successfully applied a pseudo-ring replacement strategy to discover a potent substituted-pyrimidine series as new Mer kinase selective inhibitors. The co-crystal structures of 7 and 22 with Mer protein have been determined and their binding modes provide a rationale for the 1000-fold difference in their IC50 values. The lead compound 23 inhibited Mer kinase activity in cell-based assays and mediated functional inhibition of human platelet activation, suggesting the utility of Mer-selective small molecule inhibitors for treatment and/or prevention of pathologic thrombosis.
EXPERIMENTAL SECTION
Details on the synthesis of all compounds are given in the Supporting Information. The purity of all tested compounds was determined by LC/MS to be >95%.
Microfluidic Capillary Electrophoresis (MCE) Assay9
Activity assays were performed in a 384 well, polypropylene microplate in a final volume of 50 μL of 50 mM Hepes, pH 7.4 containing 10 mM MgCl2, 1.0 mM DTT, 0.01% Triton X-100, 0.1% Bovine Serum Albumin (BSA), containing 1.0 μM fluorescent substrate (Table 6) and ATP at the Km for each enzyme (Table 6). All reactions were terminated by addition of 20 μL of 70 mM EDTA. After a 180 min incubation, phosphorylated and unphosphorylated substrate peptides (Table 6) were separated in buffer supplemented with 1 x CR-8 on a LabChip EZ Reader equipped with a 12-sipper chip. Data were analyzed using EZ Reader software.
Table 6.
Assay conditions for MCE assays
| Kinase | Peptide Substrate | Kinase (nM) | ATP (uM) |
|---|---|---|---|
| Mer | 5-FAM-EFPIYDFLPAKKK-CONH2 | 2.0 | 5.0 |
| Axl | 5-FAM-KKKKEEIYFFF-CONH2 | 120 | 65 |
| Tyro | 5-FAM-EFPIYDFLPAKKK-CONH2 | 10 | 21 |
Cell Based Assays for Mer Kinase Inhibition
697 B-ALL Cell Assay
697 B-ALL cells were cultured in the presence of 23 or vehicle only for 1.0 h. Pervanadate solution was prepared fresh by combining 20 mM sodium orthovanadate in 0.9x PBS in a 1:1 ratio with 0.3% (w/w) hydrogen peroxide in PBS for 15–20 min at room temperature. Cultures were treated with 120 μM pervanadate prior to collection for preparation of whole cell lysates, immunoprecipitation of Mer, and analysis by western blot.
697 B-ALL cells were treated with pervanadate for 3.0, 5.0, and 1.0 min respectively. Cell lysates were prepared in 50 mM HEPES pH 7.5, 150 mM NaCl, 10 mM EDTA, 10% glycerol, and 1% Triton X-100, supplemented with protease inhibitors (Roche Molecular Biochemicals, #11836153001). Mer protein was immunoprecipitated with a monoclonal anti-Mer antibody (R&D Systems, #MAB8912) and Protein G agarose beads (InVitrogen). Phospho-Mer was detected by western blot using a polyclonal anti-phospho-Mer antibody raised against a peptide derived from the tri-phosphorylated activation loop of Mer. Nitrocellulose membranes were stripped and total Mer protein was detected using a second anti-Mer antibody (Epitomics Inc., #1633-1). For 697 B-ALL cells, relative phosphorylated and total Mer protein levels were determined by densitometry using Image J software and IC50 values were determined by non-linear regression.
32D-EMC cell Assay
32D-EMC suspension cultures were treated with the indicated concentration of 23 or vehicle before stimulation with 100 ng/mL EGF (BD Biosciences #354010) for 15 min. Cells were centrifuged at 1000g for 5 min and washed with 1X PBS. Cell lysates were prepared in 20 mM HEPES (pH 7.5), 50 mM NaF, 500 mM NaCl, 5.0 mM EDTA, 10% glycerol, and 1% Triton X-100, supplemented with protease inhibitors (10 μg/mL leupeptin, 10 μg/mL phenylmethylsulfonyl fluoride, and 20 μg/mL aprotinin) and phosphatase inhibitors (50 mM NaF and 1.0 mM sodium orthovanadate) and Mer protein was immunoprecipitated using a custom polyclonal rabbit anti-Mer antisera raised against a GST protein derived from the C-terminus of human Mer and Protein A agarose beads (Santa Cruz Biotechnology). Phosphotyrosine-containing proteins were detected by western blot with a monoclonal HRP-conjugated anti-phosphotyrosine antibody (Santa Cruz Biotechnology, #sc-508). Antibodies were stripped from membranes and total Mer levels were determined using the custom polyclonal rabbit anti-Mer antibody raised against a peptide derived from the catalytic domain of Mer.
Platelet Aggregation Assay
Human whole blood (WB) was collected from healthy volunteers as permitted by an Institutional Review Board-approved protocol (COMIRB # 09-0816). WB was drawn by venipuncture into 3.8% sodium citrate and centrifuged at 200g for 20 min at room temperature. Platelet-rich plasma (PRP) was separated and the remaining solution was spun at 2400g for 10 minutes to create platelet poor plasma (PPP). PRP and PPP were mixed in appropriate proportions to obtain a final concentration of 2.5 × 105 platelets/μL, and then used within 3 hours of the initial blood draw.
For platelet aggregation assays, plasma was incubated for 60 minutes at RT with either 3 μM 23 or vehicle (20% DMSO in saline). Samples were analyzed using a light-transmission aggregometer (CHRONO-LOG Corporation, Havertown, PA) at 37°C in the presence of Chrono-lume reagent (CHRONO-LOG) with magnetic stirring at 1200 rpm and addition of 1 μg/mL equine type I fibrillar collagen (CHRONO-LOG) as an aggregation agonist. Optical density was determined as an indicator of aggregation. Emission of light by Chrono-lume, a luciferin-luciferase compound, occurs as a result of ATP binding and is determined relative to a 2 nM standard as a measure of the amount of ATP released from activated platelets. ATP release (nM) and the maximum percent aggregation were recorded 7 min after collagen addition.
Supplementary Material
Acknowledgments
This work was supported by the University Cancer Research Fund and Federal Funds from the National Cancer Institute, National Institute of Health, under Contract No. HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Footnotes
PDB ID Codes: The atomic coordinates for the X-ray crystal structures of 7 & 22 have been deposited with the RCSB Protein Data Bank under the accession code 4MH7 & 4MHA.
SUPPORTING INFORMATION AVAILABLE Experimental details and characterization of all compounds and biological methods. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.De Berardis G, Lucisano G, D’Ettorre A, et al. Association of aspirin use with major bleeding in patients with and without diabetes. J Am Med Association. 2012;307(21):2286–2294. doi: 10.1001/jama.2012.5034. [DOI] [PubMed] [Google Scholar]
- 2.Diener HC, Bogousslavsky J, Brass LM, Cimminiello C, Csiba L, Kaste M, Leys D, Matias-Guiu J, Rupprecht HJ. Aspirin and clopidogrel compared with clopidogrel alone after recent ischaemic stroke or transient ischaemic attack in high-risk patients (MATCH): randomised, double-blind, placebo-controlled trial. The Lancet. 2004;364(9431):331–337. doi: 10.1016/S0140-6736(04)16721-4. [DOI] [PubMed] [Google Scholar]
- 3.Sanderson S, Emery J, Baglin T, Kinmonth AL. Narrative Review: Aspirin Resistance and Its Clinical Implications. Annals of Internal Medicine. 2005;142(5):370–380. doi: 10.7326/0003-4819-142-5-200503010-00012. [DOI] [PubMed] [Google Scholar]
- 4.Nguyen TA, Diodati JG, Pharand C. Resistance to clopidogrel: A review of the evidence. Journal of the American College of Cardiology. 2005;45(8):1157–1164. doi: 10.1016/j.jacc.2005.01.034. [DOI] [PubMed] [Google Scholar]
- 5.Laurance S, Lemarie CA, Blostein MD. Growth Arrest-Specific Gene 6 (gas6) and Vascular Hemostasis. Adv Nutr. 2012;3(2):196–203. doi: 10.3945/an.111.001826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Linger RMA, Keating AK, Earp HS, Graham DK. Advances in Cancer Research. Vol. 100. Academic Press; New York: 2008. TAM Receptor Tyrosine Kinases: Biologic Functions, Signaling, and Potential Therapeutic Targeting in Human Cancer; pp. 35–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Chen CL, Li Q, Darrow AL, Wang YP, Derian CK, Yang J, de Garavilla L, Andrade-Gordon P, Damiano BP. Mer receptor tyrosine kinase signaling participates in platelet function. Arterioscl Throm Vas. 2004;24(6):1118–1123. doi: 10.1161/01.ATV.0000130662.30537.08. [DOI] [PubMed] [Google Scholar]; (b) Chen J, Carey K, Godowski PJ. Identification of Gas6 as a ligand for Mer, a neural cell adhesion molecule related receptor tyrosine kinase implicated in cellular transformation. Oncogene. 1997;14(17):2033–2039. doi: 10.1038/sj.onc.1201039. [DOI] [PubMed] [Google Scholar]; (c) Sather S, Kenyon KD, Lefkowitz JB, Liang X, Varnum BC, Henson PM, Graham DK. A soluble form of the Mer receptor tyrosine kinase inhibits macrophage clearance of apoptotic cells and platelet aggregation. Blood. 2007;109(3):1026–1033. doi: 10.1182/blood-2006-05-021634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Angelillo-Scherrer A, Burnier L, Flores N, Savi P, DeMol M, Schaeffer P, Herbert JM, Lemke G, Goff SP, Matsushima GK, Earp HS, Vesin C, Hoylaerts MF, Plaisance S, Collen D, Conway EM, Wehrle-Haller B, Carmeliet P. Role of Gas6 receptors in platelet signaling during thrombus stabilization and implications for antithrombotic therapy. The Journal of clinical investigation. 2005;115(2):237–246. doi: 10.1172/JCI22079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu J, Yang C, Simpson C, DeRyckere D, Van DA, Miley MJ, Kireev D, Norris-Drouin J, Sather S, Hunter D, Korboukh VK, Patel HS, Janzen WP, Machius M, Johnson GL, Earp HS, Graham DK, Frye SV, Wang X. Discovery of Small Molecule Mer Kinase Inhibitors for the Treatment of Pediatric Acute Lymphoblastic Leukemia. ACS Med Chem Lett. 2012;3:129–134. doi: 10.1021/ml200239k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Liu J, Zhang W, Stashko MA, DeRyckere D, Cummings CT, Hunter D, Yang C, Jayakody CN, Cheng N, Simpson C, Norris-Drouin J, Sather S, Kireev D, Janzen WP, Earp HS, Graham DK, Frye SV, Wang X. UNC1062, a new and potent Mer inhibitor. European Journal of Medicinal Chemistry. 2013;65:83–93. doi: 10.1016/j.ejmech.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Schlegel J, Sambade MJ, Sather S, Moschos SJ, Tan AC, Winges A, Deryckere D, Carson CC, Trembath DG, Tentler JJ, Eckhardt SG, Kuan PF, Hamilton RL, Duncan LM, Miller CR, Nikolaishvili-Feinberg N, Midkiff BR, Liu J, Zhang W, Yang C, Wang X, Frye SV, Earp HS, Shields JM, Graham DK. MERTK receptor tyrosine kinase is a therapeutic target in melanoma. The Journal of clinical investigation. 2013;123(5):2257–2267. doi: 10.1172/JCI67816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Mathieu S, Gradl SN, Ren L, Wen Z, Aliagas I, Gunzner-Toste J, Lee W, Pulk R, Zhao G, Alicke B, Boggs JW, Buckmelter AJ, Choo EF, Dinkel V, Gloor SL, Gould SE, Hansen JD, Hastings G, Hatzivassiliou G, Laird ER, Moreno D, Ran Y, Voegtli WC, Wenglowsky S, Grina J, Rudolph J. Potent and Selective Aminopyrimidine-Based B-Raf Inhibitors with Favorable Physicochemical and Pharmacokinetic Properties. Journal of Medicinal Chemistry. 2012;55(6):2869–2881. doi: 10.1021/jm300016v. [DOI] [PubMed] [Google Scholar]; (b) Zhang G, Ren P, Gray NS, Sim T, Liu Y, Wang X, Che J, Tian S-S, Sandberg ML, Spalding TA, Romeo R, Iskandar M, Chow D, Martin Seidel H, Karanewsky DS, He Y. Discovery of pyrimidine benzimidazoles as Lck inhibitors: Part I. Bioorganic & Medicinal Chemistry Letters. 2008;18(20):5618–5621. doi: 10.1016/j.bmcl.2008.08.104. [DOI] [PubMed] [Google Scholar]; (c) Furet P, Caravatti G, Guagnano V, Lang M, Meyer T, Schoepfer J. Entry into a new class of protein kinase inhibitors by pseudo ring design. Bioorganic & Medicinal Chemistry Letters. 2008;18(3):897–900. doi: 10.1016/j.bmcl.2007.12.041. [DOI] [PubMed] [Google Scholar]
- 12.(a) Pommereau A, Pap E, Kannt A. Two simple and generic antibody-independent kinase assays: comparison of a bioluminescent and a microfluidic assay format. Journal of biomolecular screening. 2004;9(5):409–416. doi: 10.1177/1087057104264175. [DOI] [PubMed] [Google Scholar]; (b) Dunne J, Reardon H, Trinh V, Li E, Farinas J. Comparison of on-chip and off-chip microfluidic kinase assay formats. Assay Drug Dev Technol. 2004;2(2):121–129. doi: 10.1089/154065804323056468. [DOI] [PubMed] [Google Scholar]; (c) Bernasconi P, Chen M, Galasinski S, Popa-Burke I, Bobasheva A, Coudurier L, Birkos S, Hallam R, Janzen WP. A chemogenomic analysis of the human proteome: application to enzyme families. Journal of biomolecular screening. 2007;12(7):972–982. doi: 10.1177/1087057107306759. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.