

Abstract
Aims
Genistein, an isoflavone derivative found in soy, is known as a promising treatment for rheumatoid arthritis (RA). However, the detailed molecular mechanism of genistein in suppression of proinflammatory cytokine production remains ambiguous. The aim of this work was to evaluate the signal pathway by which genistein modulates inflammatory cytokine expression.
Materials and methods
MH7A cells were stimulated with tumor necrosis factor (TNF)-α and incubated with genistein, and interleukin (IL)-1β, IL-6, and IL-8 production was measured by enzyme-linked immunosorbent assay. Nuclear translocation of nuclear factor (NF)-κB was measured by a confocal fluorescence microscopy. The intracellular accumulation of reactive oxygen species (ROS) was monitored using the fluorescent probe 5-6-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate. Signal-transduction protein expression was measured by Western blot.
Results
Genistein decreased the secretion of IL-1β, IL-6, and IL-8 from TNF-α-stimulated MH7A cells in a dose-dependent manner. Genistein prevented TNF-α-induced NF-κB translocation as well as phosphorylation of IκB kinase-α/β and IκBα, and also suppressed TNF-α-induced AMPK inhibition. The production of IL-1β, IL-6, and IL-8 induced by TNF-α was decreased by the phosphatidylinositol-3 kinase inhibitor LY294002, suggesting that inhibition of Akt activation might inhibit IL-1β, IL-6, and IL-8 production induced by TNF-α. In addition, we also found that pretreatment with the adenosine monophosphate-activated protein kinase (AMPK) agonist 5-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside obviously inhibited TNF-α-induced proinflammatory cytokine production. These observations suggest that the inhibitory effect of genistein on TNF-α-induced proinflammatory cytokine production is dependent on AMPK activation.
Conclusion
These findings indicate that genistein suppressed TNF-α-induced inflammation by inhibiting the ROS/Akt/NF-κB pathway and promoting AMPK activation in MH7A cells.
Keywords: genistein, rheumatoid arthritis, cytokine, signal transduction, inflammation
Introduction
Rheumatoid arthritis (RA) is a chronic autoimmune disease that causes inflammation and joint destruction with a prevalence of about 1% of the general population. Without treatment, inflammation leads to cartilage damage, bone erosions, joint destruction, and impaired movement. Despite aggressive immunosuppression with biologics and traditional disease-modifying antirheumatic drugs, 30%–40% of RA patients are still not adequately controlled.1 In this context, there is a demand for exploration of new antirheumatic drugs with high efficacy and less toxicity.
Genistein (4′,5,7-trihydroxyisoflavone) is the principal isoflavone found predominantly in soy beans, and has attracted considerable attention due to its potential effects on some of the degenerative diseases, such as cardiovascular disease, osteoporosis, and hormone-related cancers. Interest in genistein as a potential therapeutic agent for RA has recently risen, as studies have shown that genistein exerts evident anti-inflammatory properties in a collagen-induced RA (CIA) model.2–5 However, detailed molecular mechanisms of the anti-inflammatory effects of genistein are still elusive. Here, we found that genistein suppressed tumor necrosis factor (TNF)-α-induced inflammation by inhibiting the reactive oxygen species (ROS)/Akt/nuclear factor (NF)-κB pathway and promoting adenosine monophosphate-activated protein kinase (AMPK) activation in MH7A cells.
What is already known about this subject
Genistein exerts evident anti-inflammatory properties in a CIA model. However, detailed molecular mechanisms of the anti-inflammatory effects of genistein are still elusive.
What this study adds
Our studies provided new insights regarding the anti-inflammation activities of genistein against RA (modulating ROS/Akt/NF-κB and AMPK signal pathway in human synoviocyte MH7A cells), and may contribute to the rational utility and pharmacological study of genistein in future anti-RA research.
Materials and methods
Reagents
Genistein was purchased from Longpu Technology (Shenyang, People’s Republic of China). Enzyme-linked immunosorbent assay (ELISA) kits for interleukin (IL)-1β, IL-6,and IL-8 quantification were purchased from NeoBioscience Technology (Shenzhen, People’s Republic of China). Recombinant human TNF-α was obtained from PeproTech (Rocky Hill, NJ, USA). N-acetyl-L-cysteine (NAC), phosphoinositide-3 kinase (PI3K) inhibitor LY294002, 5-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside (AICAR), and 5-(6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate (CM-H2DCFDA) were purchased from Sigma-Aldrich (St Louis, MO, USA). Antibodies for glyceraldehyde 3- phosphate dehydrogenase, vascular endothelial growth factor, and matrix metalloproteinases 3 and 9 were purchased from Abcam (Cambridge, UK). Antibodies against phosphorylated (p)-Akt (Ser473), p-IκB kinase (IKK)-α/β (Ser176/180), p-IκBα (Ser32), NF-κB p65, p-NF-κB p65 (Ser536), and p-AMPK (Thr172) were purchased from Cell Signaling Technology (Danvers, MA, USA).
Cell culture
MH7A cells were obtained from the Riken cell bank (Tsukuba, Japan). Cells were maintained in Roswell Park Memorial Institute 1640 medium (Sigma-Aldrich), supplemented with 10% fetal bovine serum (Sigma-Aldrich) and penicillin/streptomycin (1:100; Sigma-Aldrich), in a CO2 incubator at 37°C.
Cell-viability assay (MTT dye assay)
Cell viability was measured by the 3-(4,5-dimethylthylthiazol-2-yl)-2,5 diphenyltetrazolium bromide (MTT) method, as reported previously.6
Enzyme-linked immunosorbent assay
MH7A cells were seeded in 24-well plates. The cells were pretreated with various concentrations of genistein for 2 hours, and then incubated for another 24 hours with or without 10 ng/mL of TNF-α. The culture medium was collected, and the concentrations of the cytokines were determined by ELISA using a commercial kit (NeoBioscience Technology), according to the manufacturer’s instructions.
Western blot
As reported before,7 40 μg of protein from each treatment was separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred onto a polyvinylidene fluoride membrane. After blocking with 10% instant nonfat dry milk, membranes were incubated with specific antibodies overnight at 4°C, followed by incubation with the secondary antibody. Antibody binding was detected with the enhanced ECL detection system. (Beyotime Institute of Biotechnology, Haimen, People’s Republic of China) Western blot results were quantified using ImageJ software (downloaded from the NIH website http://rsb.info.nih.gov/ij/download.html) after normalizing to corresponding loading controls.
Analysis of p-Akt and p-AMPK activity
In all the experiments, MH7A cells were serum-starved overnight. To evaluate the effects on Akt and AMPK activity, cells were preexposed to drugs (genistein, LY294002, or AICAR) for 2 hours, and then incubated with TNF-α for the indicated times. After treatment, the cells were scraped from the dishes and lysed in ice-cold lysis buffer. p-Akt and p-AMPK activity were assessed by the levels of p-Akt (Ser473) and p-AMPK (Thr172).
Detection of reactive oxygen species
The intracellular accumulation of ROS was monitored using the fluorescent probe CM-H2DCFDA. At the end of the treatment, cells were loaded with 10 μM CM-H2DCFDA and incubated at 37°C for 30 minutes in the dark. Cells were then cleaned and resuspended in phosphate-buffered saline (PBS). Samples were observed under a fluorescence microscope (DMI 3000 B; Leica Microsystems, Wetzlar, Germany) at 488 nm excitation and 530 nm emission.
Immunocytochemistry
MH7A cells cultured on glass coverslips were fixed with 4% paraformaldehyde in PBS and stained with anti-NF-κB p65 antibody (diluted 1:100) for 1 hour at room temperature, and then incubated with fluorescein isothiocyanate-conjugated second antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA). After washing in PBS, nuclei were stained for 3 minutes with the fluorescent dye 4-,6-diamidino-2- phenylindole. The stained cells were analyzed with confocal fluorescence microscopy (TCS SP5-II; Leica Microsystems).
Statistical analysis
All quantitative data were expressed as means ± standard deviation from at least three independent experiments. Statistical analysis was carried out by a one-way analysis of variance followed by a Dunnett’s t-test, in which all groups were tested against a control group as a reference using SPSS 13.0.1 (SPSS Inc., Chicago, IL, USA) software. A difference was considered significant when P<0.05.
Results
Cytotoxicity of genistein on MH7A cells
To avoid any cytotoxic effects caused by genistein, we investigated the cytotoxicity of genistein, which was assessed using the MTT assay. No cytotoxicity was observed when the cells were exposed to up to 20 μM genistein for 24 hours (Figure 1). We then decided to set the concentration of genistein at 20 μM for the following experiment.
Figure 1.
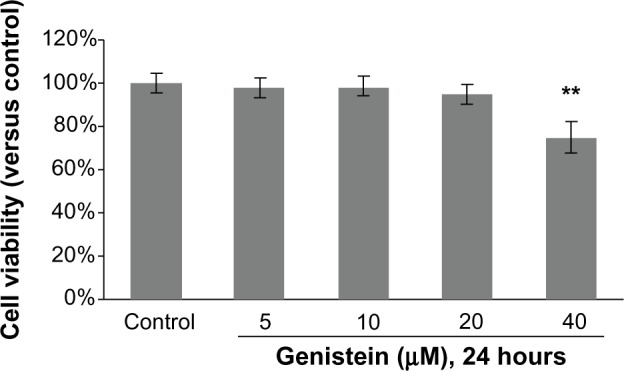
Cytotoxicity of genistein on MH7A cells.
Notes: Cells were treated with different concentrations of genistein for 24 hours, and their viability was determined using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay. **P<0.01 versus group without genistein treatment. The data are presented as the means ± standard deviation from three independent experiments.
Effects of genistein on TNF-α-induced proinflammatory cytokine production
It has been well documented that overproduction of inflammatory cytokines, such as TNF-α and IL-1β, is implicated in the pathogenesis of RA. Therefore, we explored whether genistein could suppress TNF-α-induced proinflammatory cytokine production. To investigate the anti-inflammatory activity of genistein, we examined the production of IL-1β, IL-6, and IL-8 in TNF-α-activated MH7A cells. MH7A cells were pretreated with genistein (5, 10, 20 μM) for 2 hours prior to 24-hour stimulation using 10 ng/mL TNF-α. As shown in Figure 2, the stimulation of MH7A cells with TNF-α significantly increased the expression of IL-1β, IL-6, and IL-8 in the culture supernatant. However, pretreatment of MH7A cells with genistein reduced the release of IL-1β, IL-6, and IL-8 in a concentration-dependent manner in TNF-α-stimulated MH7A cells.
Figure 2.
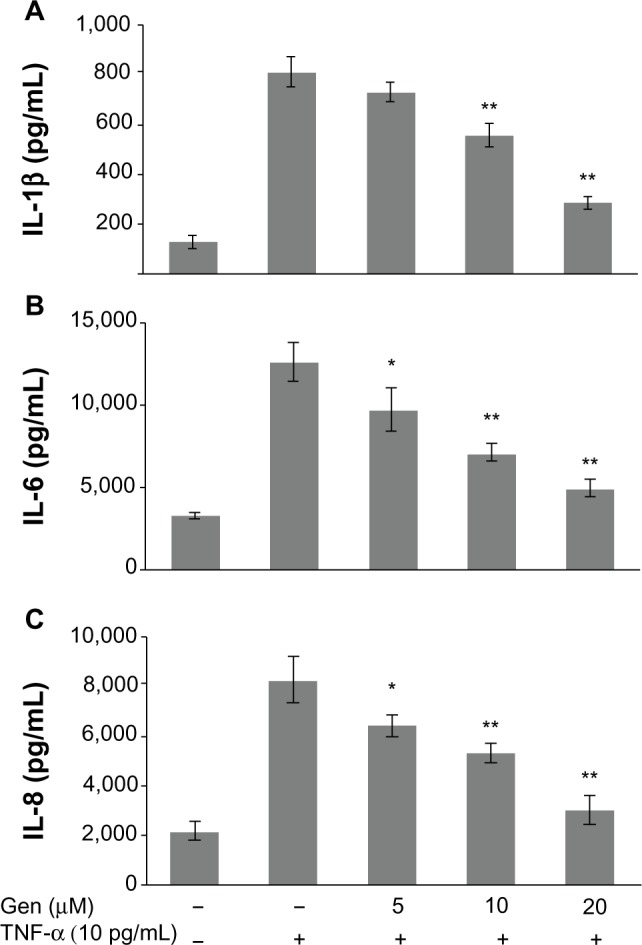
(A–C) Effects of genistein (Gen) on tumor necrosis factor (TNF)-α-induced proinflammatory cytokine production.
Notes: MH7A cells were pretreated with Gen or not for 2 hours, and then stimulated with TNF-α. After 24 hours, the levels of interleukin (IL)-1β, IL-6, and IL-8 were measured in the culture medium by enzyme-linked immunosorbent assay. *P<0.05 and **P<0.01 versus group with TNF-α treatment alone. The data are presented as the means ± standard deviation from three independent experiments.
Effects of genistein on NF-κB activation in MH7A cells
NF-κB is a transcriptional regulator that plays a central role in responses to inflammatory signaling in RA. Phosphorylation of NF-κB p65 is an important step for its transcriptional activity.8,9 Thus, we examined whether genistein could suppress TNF-α-induced p65 phosphorylation. Cells were pretreated with or without genistein (20 μM) for 2 hours, and then stimulated with TNF-α (10 ng/mL) at the indicated times. As shown in Figure 3A, treatment of MH7A cells with TNF-α enhanced phosphorylation of Ser536 of NF-κB p65 (which closely correlates with NF-κB transcriptional activation), which was inhibited by pretreatment with genistein. To further investigate the effect of genistein on NF-κB signaling, we used immunocytochemistry to examine the nuclear translocation of NF-κB p65. We found that treatment with genistein (20 μM) significantly inhibited TNF-α-induced NF-κB p65 nuclear translocation (Figure 3B). NF-κB is associated with IκB protein in the cytoplasm, which inhibits NF-κB activity. IKK is an important upstream kinase for phosphorylation of IκB. Thus, we examined the phosphorylation of IKKα/β and IκBα by Western blot analysis. As expected, TNF-α (10 ng/mL) induced IKKα/β and IκBα phosphorylation. This effect was dramatically blocked by treatment with genistein (Figure 3A). Based on these results, we conclude that genistein suppresses TNF-α-induced proinflammatory cytokine production at least partly by inhibiting the IKK/IκB/NF-κB pathway in MH7A cells.
Figure 3.
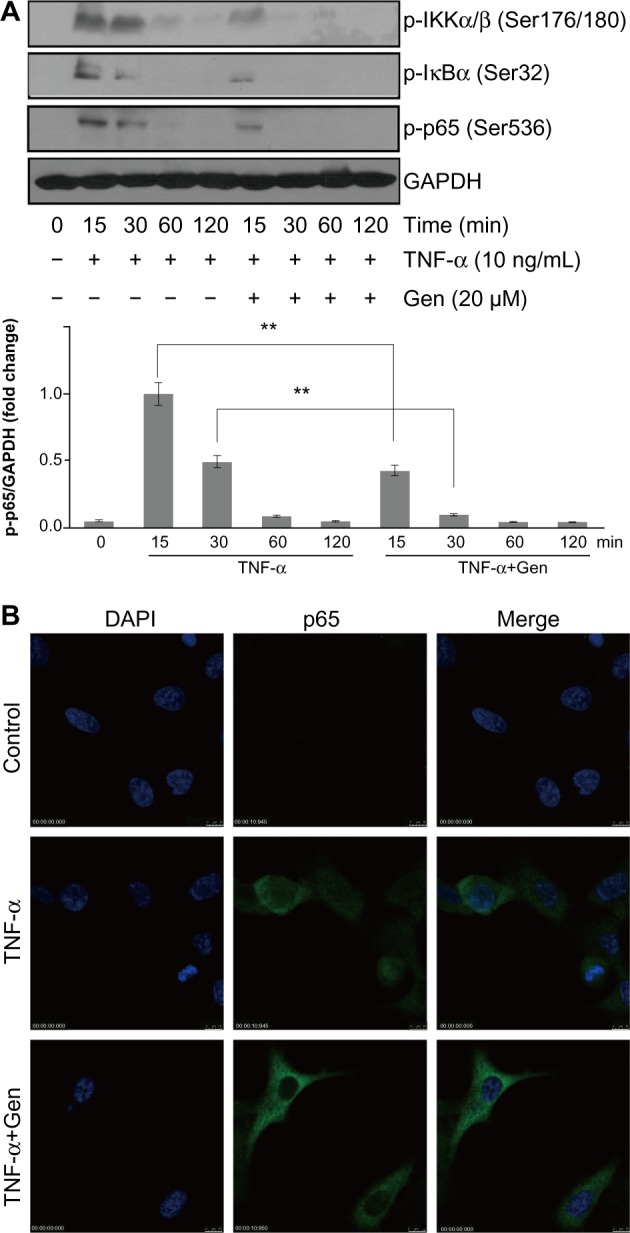
(A and B) Effects of genistein (Gen) on nuclear factor (NF)-κB activation in MH7A cells.
Notes: (A) MH7A cells were pretreated with Gen or not for 2 hours, and then incubated with tumor necrosis factor (TNF)-α for the indicated times. Phosphorylated IκB kinase (p-IKK)-α/β, p-IκBα, and p-NF-κB p65 levels were determined by Western blot analysis. (B) After being pretreated with Gen or not for 2 hours, MH7A cells were then incubated with TNF-α for 15 minutes. The nuclei translocation of NF-κB p65 was assessed by confocal fluorescence microscopy. Cells were immunostained with NF-κB p65 antibody. Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI). Results representative of three independent experiments. Bar 10 μm.
Abbreviation: GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
Involvement of Akt in NF-κB activation by TNF-α
The PI3K/Akt pathway has been demonstrated to be involved in the regulation of NF-κB activation.10 We thus examined the effects of genistein on the TNF-α-induced phosphorylation of Akt in MH7A cells. As shown in Figure 4A, genistein (20 μM) significantly suppressed TNF-α-induced phosphorylation of Akt at Ser473. To investigate the relationship between the PI3K/Akt pathway and NF-κB, we treated the cells with the PI3K inhibitor LY294002 before TNF-α (10 ng/mL) treatment. Cells were pretreated with 20 μM LY294002 for 1 hour and then exposed to TNF-α. We found that LY294002 pretreatment blocked the increased p-Akt, IKK, and p65 levels by TNF-α (Figure 4B). In addition, LY294002 significantly suppressed proinflammatory cytokine production in TNF-α-activated MH7A cells (Figure 4C–E). Based on these results, we conclude that PI3K/Akt might act as an upstream regulator on NF-κB activation, and that genistein inhibits inflammatory cytokines expression in MH7A cells by blocking the PI3K/Akt/NF-κB signaling cascades.
Figure 4.

(A–E) Involvement of Akt in nuclear factor (NF)-κB activation by tumor necrosis factor (TNF)-α.
Notes: (A) MH7A cells were pretreated with genistein (Gen) or LY294002 for 2 hours, and then incubated with TNF-α for the indicated times. Phosphorylated (p)-Akt levels were determined by Western blot analysis. (B) After being pretreated with LY294002 for 2 hours, MH7A cells were then incubated with TNF-α for 15 minutes. p-IκB kinase (p-IKK)-α/β, p-IκBα, and p-NF-κB p65 levels were determined by Western blot analysis. (C–E) Interleukin (IL)-1β, IL-6,and IL-8 production was determined in MH7A cells treated with TNF-α in the presence or absence of LY294002 for 24 hours by enzyme-linked immunosorbent assay. *P<0.05 and **P<0.01 vsersus group with TNF-α treatment alone. The data are presented as the means ± standard deviation from three independent experiments.
Abbreviation: GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
The role of ROS in TNF-α-mediated inflammatory responses
To elucidate the mechanism of genistein-induced inhibition of the PI3K/Akt/NF-κB pathway, we first performed experiments to determine the effects of genistein on intracellular ROS accumulation. Stimulation of MH7A cells with TNF-α (10 ng/mL) increased intracellular ROS levels, as determined by using the H2O2-sensitive probe CM-H2DCFDA, which was effectively attenuated by pretreatment with genistein (20 μM) or NAC (10 mM), a common ROS scavenger (Figure 5A). In addition, pretreatment with the antioxidant NAC dramatically reduced TNF-α-induced inflammatory cytokine expression (Figure 5B–D). To investigate whether genistein exhibited anti-inflammatory properties via inhibition of the ROS/Akt/NF-κB pathway, we tested the effect of NAC on TNF-α-induced Akt and downstream NF-κB activation. As shown in Figure 5E, we confirmed that TNF-α induced activation of the Akt/NF-κB pathway, while NAC (10 mM) pretreatment blocked TNF-α-induced Akt/NF-κB activation. These data indicated that genistein could attenuate TNF-α-induced inflammatory responses in MH7A cells by suppressing ROS/Akt/NF-κB activation.
Figure 5.

(A–E) The role of reactive oxygen species (ROS) in tumor necrosis factor (TNF)-α-mediated inflammatory responses.
Notes: (A) MH7A cells were stimulated with TNF-α (10 ng/mL) for 15 minutes in the presence of genistein (Gen; 20 μM) or N-acetyl-L-cysteine (NAC; 10 mM). 5-(6)-Chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate was used to determine the generation of intracellular ROS. Intracellular ROS levels were examined by fluorescence microscopy. (B–D) The levels of interleukin (IL)-1β, IL-6, and IL-8 were determined in cells treated with TNF-α alone or with NAC for 24 hours by enzymelinked immunosorbent assay. (E) MH7A cells pretreated with NAC for 2 hours before incubating with TNF-α for 15 minutes. Phosphorylated IκB kinase (p-IKK)-α/β, p-IκBα, and p-nuclear factor κB p65 levels were determined by Western blot analysis. *P<0.05 and **P<0.01 versus group with TNF-α treatment alone. The data are presented as the means ± standard deviation from three independent experiments. Bar 100 μm.
Abbreviation: GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
The role of AMPK in the anti-inflammatory effects of genistein
To characterize further the mechanism underlying the anti-inflammatory effects of genistein, we assessed the AMPK pathway, which correlated with inflammatory disease. We found that TNF-α (10 ng/mL) alone decreased AMPK phosphorylation, whereas pretreatment with genistein (20 μM) prevented this effect (Figure 6A). To determine whether AMPK is involved in TNF-α-mediated inflammatory responses, we tested the effects of AMPK agonist AICAR on TNF-α-induced inflammatory cytokine expression. As shown in Figure 6B–D, pretreatment with AICAR (1 mM) obviously inhibited TNF-α-induced proinflammatory cytokine production. These data indicate that the inhibitory effect of genistein on TNF-α-induced proinflammatory cytokine production is dependent on AMPK activation.
Figure 6.
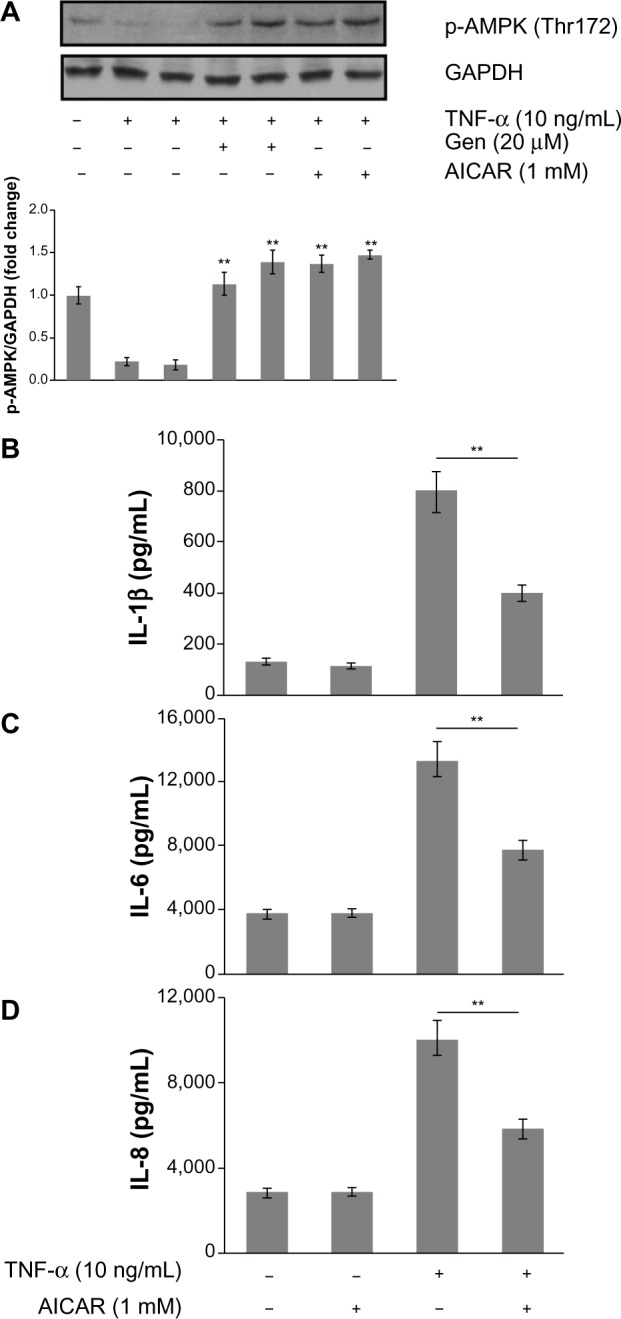
(A–D) The role of adenosine monophosphate-activated protein kinase (AMPK) in the anti-inflammatory effects of genistein (Gen).
Notes: (A) MH7A cells were either exposed to tumor necrosis factor (TNF)-α for 15 minutes, cotreated with TNF-α and Gen, treated with TNF-α and 5-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside (AICAR), or left untreated for 15 minutes. Phosphorylation (p-) of AMPK was determined by Western blotting. (B–D) The levels of interleukin (IL)-1β, IL-6, and IL-8 were determined in cells treated with TNF-α alone or with AICAR for 24 hours by enzyme-linked immunosorbent assay. *P<0.05 and **P<0.01 versus group with TNF-α treatment alone. The data are presented as the means ± standard deviation from three independent experiments.
Abbreviation: GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
Discussion
The isoflavonoid genistein represents the major active compound from soybean. Its anti-inflammatory and immune-regulation effects have been documented. Several reports have shown that genistein could reduce inflammation in a CIA rat model. For instance, Verdrengh et al suggested that subcutaneous injection of genistein exerts evident anti-inflammatory properties on CIA rats.3 Wang et al also reported that genistein treatment could reduce RA-induced inflammation and affect the immune system.2 In addition, Zhang et al suggest that genistein has a specific inhibitory effect on the anchorage-dependent and anchorage-independent growth of fibroblastlike synoviocytes in RA, arresting the synovial cells at the G1 restriction point.4 However, the mechanisms underlying these effects are not fully understood and further investigations are needed to explore the detailed molecular mechanism regulated by genistein. Here, we found that genistein significantly reduced inflammatory mediators, such as IL-1β, IL-6, and IL-8 production, in TNF-α-activated MH7A cells in vitro. Our results also showed the anti-inflammatory properties of genistein are mediated by the interruption of the ROS/Akt/NF-κB signaling pathway and promoting AMPK activation in MH7A cells. These signaling events together led to a marked decrease of TNF-α-induced proinflammatory cytokine production (Figure 7).
Figure 7.
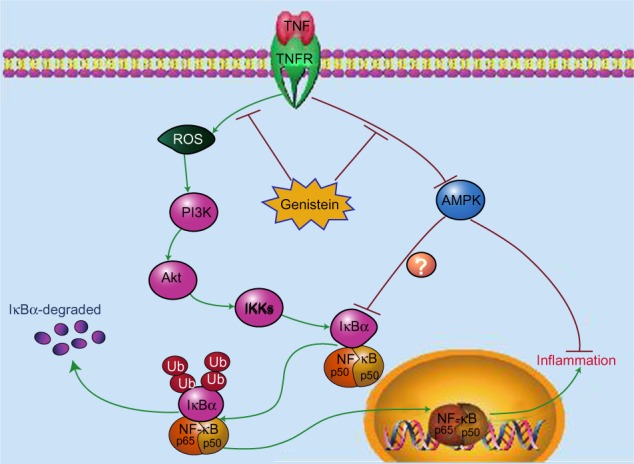
Schematic model illustrating the potential pathway associated with genistein’s inhibition of tumor necrosis factor (TNF)-α-induced inflammation.
Notes: Genistein suppresses TNF-α-induced proinflammatory cytokine production through 1) reducing intracellular reactive oxygen species (ROS) accumulation triggered by TNF-α, which represses Akt/nuclear factor (NF)-κB signaling, and 2) promoting adenosine monophosphate-activated protein kinase (AMPK) activation.
Abbreviations: IKK, IκB kinase; PI3K, phosphoinositide-3 kinase; TNFR, tumor necrosis factor receptor.
The transcription factor NF-κB has crucial roles in inflammation. In most cell types, NF-κB is represented mainly by the p65/p50 heterodimeric complex. In the resting stage, this complex is retained in the cytoplasm as an inactive form bound to an additional inhibitory subunit – IκBα.11 During the activation process, the inhibitory subunit IκBα is rapidly phosphorylated at Ser32 and Ser36 residues by IKKα/β, and subsequently ubiquitinated and degraded by the 26S proteasome complex. Once released free NF-κB translocates to the nucleus and activates transcription of various inflammatory gene products. Notably, there is accumulating evidence suggesting that NF-κB plays a pivotal role in the pathogenesis of RA.12 Thus, NF-κB may be a potential therapeutic target for RA. Moreover, genistein inhibits the activation of the NF-κB pathway in multiple cell lines, such as lymphocytes,13 PC3 cells,14 RAW 264.7 macrophages,15 breast cancer cells,16 and myeloma cells.17 In this study, we found that genistein significantly inhibited NF-κB transcriptional activity in human synoviocyte MH7A cells by attenuating TNF-α-induced IKKα/β, IκBα, and p65 phosphorylation and subsequent p65 nuclear translocation.
Genistein, a selective protein tyrosine-kinase inhibitor, could inhibit epidermal growth-factor receptor kinase, pp60v-src, and pp110 gag-fes. Moreover, recent studies have also indicated that genistein could inhibit Akt activation.14 Previous studies have reported the importance of the PI3K/Akt pathway, an important regulator of growth and inflammation, in inflammation-mediated diseases, such as RA18 and psoriasis.19 Recent studies also indicated that TNF-α could activate the PI3K/Akt pathway in synovial cells.20,21 Interestingly, the PI3K/Akt pathway has been also implicated in NF-κB transcriptional activation.22 In addition, Gong et al found that genistein might inhibit NF-κB activation via the Akt pathway in MDA-MB-231 breast cancer cells.16 However, the involvement of the PI3K/Akt pathway in NF-κB activation is a cell- and tissue-specific event. It has been confirmed that TNF-α-induced NF-κB activation is independent of the PI3K/Akt pathway in human glioma cell lines.23 Shao et al suggested that radiation-induced NF-κB activation is independent of Akt activation in parental cancer cells.24 We found that inhibition of PI3K/Akt could attenuate the phosphorylation of IKKα/β, which was accompanied by the suppression of IκBα and NF-κB phosphorylation. Furthermore, the upregulation of proinflammatory cytokine production was attenuated by LY294002. Based on these data, we argue that PI3K/Akt participates in NF-κB activation and NF-κB-dependent cytokine expression.
ROS encompass a variety of partially reduced metabolites of oxygen generated in the cell as byproducts of normal mitochondrial metabolism. Exposure to cytokines such as TNF-α induces marked transient increases in the intracellular levels of ROS, which play a key role in modulating the cellular inflammatory response.25 Furthermore, ROS are critical for the inflammatory signals through Akt activation and NF-κB activation.26,27 More importantly, it has been shown that genistein could reduce ROS by attenuating the expression of ROS-producing enzymes.28 Here, we demonstrated that the reduction of cellular ROS by genistein or NAC inhibited Akt, IκBα, and IKKα/β phosphorylation and the activation of NF-κB. Moreover, pretreatment with NAC dramatically reduced TNF-α-induced inflammatory cytokine expression. We thus hypothesize that ROS-mediated activation of the PI3K/Akt pathway plays an important role in NF-κB activation and subsequent proinflammatory cytokine production.
AMPK is a highly conserved protein kinase that participates in the cellular response to metabolic stress. Cellular stresses that inhibit adenosine triphosphate (ATP) production or increase its consumption change the AMP:ATP ratio and activate the pathway. Once switched on, AMPK activates catabolic pathways that generate ATP and inhibits biosynthetic cell-cycle progression. In addition to balancing cellular energy, it is likely that AMPK also acts to limit inflammation.29–34 Metformin, an AMPK agonist, could reduce the systemic inflammation by lowering the level of C-reactive protein and IL-6.35 Intriguingly, genistein can also activate AMPK.28,36 Here, we found that pretreatment with AICAR obviously inhibited TNF-α-induced proinflammatory cytokine production. Our results indicate that the inhibitory effect of genistein on TNF-α-induced inflammatory cytokine overproduction is dependent on AMPK activation.
In conclusion, our data provide new insight into the anti-inflammatory mechanisms of genistein, demonstrating that genistein suppresses TNF-α-induced inflammation by inhibiting the ROS/Akt/NF-κB pathway and promoting AMPK activation in MH7A cells. These mechanisms may provide scientific support for the use of genistein for RA.
Acknowledgments
This work was supported by the National Natural Foundation of China (81000786 and 81271952).
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Smolen JS, Kalden JR, Scott DL, et al. Efficacy and safety of leflunomide compared with placebo and sulphasalazine in active rheumatoid arthritis: a double-blind, randomised, multicentre trial. European Leflunomide Study Group. Lancet. 1999;353(9149):259–266. doi: 10.1016/s0140-6736(98)09403-3. [DOI] [PubMed] [Google Scholar]
- 2.Wang J, Zhang Q, Jin S, He D, Zhao S, Liu S. Genistein modulate [sic] immune responses in collagen-induced rheumatoid arthritis model. Maturitas. 2008;59(4):405–412. doi: 10.1016/j.maturitas.2008.04.003. [DOI] [PubMed] [Google Scholar]
- 3.Verdrengh M, Jonsson IM, Holmdahl R, Tarkowski A. Genistein as an anti-inflammatory agent. Inflamm Res. 2003;52(8):341–346. doi: 10.1007/s00011-003-1182-8. [DOI] [PubMed] [Google Scholar]
- 4.Zhang Y, Dong J, He P, et al. Genistein inhibit [sic] cytokines or growth factor-induced proliferation and transformation phenotype in fibroblast-like synoviocytes of rheumatoid arthritis. Inflammation. 2012;35(1):377–387. doi: 10.1007/s10753-011-9365-x. [DOI] [PubMed] [Google Scholar]
- 5.Li J, Gang D, Yu X, et al. Genistein: the potential for efficacy in rheumatoid arthritis. Clin Rheumatol. 2013;32(5):535–540. doi: 10.1007/s10067-012-2148-4. [DOI] [PubMed] [Google Scholar]
- 6.Sun H, Yu T, Li JC. Co-administration of perifosine with paclitaxel synergistically induces apoptosis in ovarian cancer cells: more than just AKT inhibition. Cancer Lett. 2011;310(1):118–128. doi: 10.1016/j.canlet.2011.06.010. [DOI] [PubMed] [Google Scholar]
- 7.Yu T, Li J, Qiu Y, Sun H. 1-Phenyl-2-decanoylamino-3-morpholino-1-propanol (PDMP) facilitates curcumin-induced melanoma cell apoptosis by enhancing ceramide accumulation, JNK activation, and inhibiting PI3K/AKT activation. Mol Cell Biochem. 2012;361(1–2):47–54. doi: 10.1007/s11010-011-1086-9. [DOI] [PubMed] [Google Scholar]
- 8.Sakurai H, Chiba H, Miyoshi H, Sugita T, Toriumi W. IkappaB kinases phosphorylate NF-kappaB p65 subunit on serine 536 in the transactivation domain. J Biol Chem. 1999;274(43):30353–30356. doi: 10.1074/jbc.274.43.30353. [DOI] [PubMed] [Google Scholar]
- 9.Madrid LV, Mayo MW, Reuther JY, Baldwin AS. Akt stimulates the transactivation potential of the RelA/p65 subunit of NF-kappa B through utilization of the Ikappa B kinase and activation of the mitogen-activated protein kinase p38. J Biol Chem. 2001;276(22):18934–18940. doi: 10.1074/jbc.M101103200. [DOI] [PubMed] [Google Scholar]
- 10.Ozes ON, Mayo LD, Gustin JA, Pfeffer SR, Pfeffer LM, Donner DB. NF-kappaB activation by tumour necrosis factor requires the Akt serine-threonine kinase. Nature. 1999;401(6748):82–85. doi: 10.1038/43466. [DOI] [PubMed] [Google Scholar]
- 11.Oeckinghaus A, Ghosh S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb Perspect Biol. 2009;1(4):a000034. doi: 10.1101/cshperspect.a000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Makarov SS. NF-kappa B in rheumatoid arthritis: a pivotal regulator of inflammation, hyperplasia, and tissue destruction. Arthritis Res. 2001;3(4):200–206. doi: 10.1186/ar300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Davis JN, Kucuk O, Djuric Z, Sarkar FH. Soy isoflavone supplementation in healthy men prevents NF-kappa B activation by TNF-alpha in blood lymphocytes. Free Radic Biol Med. 2001;30(11):1293–1302. doi: 10.1016/s0891-5849(01)00535-4. [DOI] [PubMed] [Google Scholar]
- 14.Li YW, Sarkar FH. Inhibition of nuclear factor kappaB activation in PC3 cells by genistein is mediated via Akt signaling pathway. Clin Cancer Res. 2002;8(7):2369–2377. [PubMed] [Google Scholar]
- 15.Choi C, Cho H, Park J, Cho C, Song Y. Suppressive effects of genistein on oxidative stress and NFkappaB activation in RAW 264.7 macrophages. Biosci Biotechnol Biochem. 2003;67(9):1916–1922. doi: 10.1271/bbb.67.1916. [DOI] [PubMed] [Google Scholar]
- 16.Gong LJ, Li YW, Nedeljkovic-Kurepa A, Sarkar FH. Inactivation of NF-kappa B by genistein is mediated via Akt signaling pathway in breast cancer cells. Oncogene. 2003;22(30):4702–4709. doi: 10.1038/sj.onc.1206583. [DOI] [PubMed] [Google Scholar]
- 17.He H, Chen L, Zhai M, Chen JZS. Genistein down-regulates the constitutive activation of nuclear factor-kappa B in human multiple myeloma cells, leading to suppression of proliferation and induction of apoptosis. Phytother Res. 2009;23(6):868–873. doi: 10.1002/ptr.2715. [DOI] [PubMed] [Google Scholar]
- 18.Camps M, Ruckle T, Ji H, et al. Blockade of PI3Kgamma suppresses joint inflammation and damage in mouse models of rheumatoid arthritis. Nat Med. 2005;11(9):936–943. doi: 10.1038/nm1284. [DOI] [PubMed] [Google Scholar]
- 19.Schon MP, Boehncke WH. Medical progress – psoriasis. New Engl J Med. 2005;352(18):1899–1912. doi: 10.1056/NEJMra041320. [DOI] [PubMed] [Google Scholar]
- 20.Morel J, Audo R, Hahne M, Combe B. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) induces rheumatoid arthritis synovial fibroblast proliferation through mitogen-activated protein kinases and phosphatidylinositol 3-kinase/Akt. J Biol Chem. 2005;280(16):15709–15718. doi: 10.1074/jbc.M414469200. [DOI] [PubMed] [Google Scholar]
- 21.Chen QQ, Casali B, Pattacini L, Boiardi L, Salvarani C. Tumor necrosis factor-alpha protects synovial cells from nitric oxide induced apoptosis through phosphoinositide 3-kinase Akt signal transduction. J Rheumatol. 2006;33(6):1061–1068. [PubMed] [Google Scholar]
- 22.Dan HC, Cooper MJ, Cogswell PC, Duncan JA, Ting JP, Baldwin AS. Akt-dependent regulation of NF-kappa B is controlled by mTOR and Raptor in association with IKK. Gene Dev. 2008;22(11):1490–1500. doi: 10.1101/gad.1662308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sudheerkumar P, Shiras A, Das G, Jagtap JC, Prasad V, Shastry P. Independent activation of Akt and NF-kappaB pathways and their role in resistance to TNF-alpha mediated cytotoxicity in gliomas. Mol Carcinog. 2008;47(2):126–136. doi: 10.1002/mc.20372. [DOI] [PubMed] [Google Scholar]
- 24.Shao R, Tsai EM, Wei K, et al. E1A inhibition of radiation-induced NF-kappaB activity through suppression of IKK activity and IkappaB degradation, independent of Akt activation. Cancer Res. 2001;61(20):7413–7416. [PubMed] [Google Scholar]
- 25.Yang D, Elner SG, Bian ZM, Till GO, Petty HR, Elner VM. Pro-inflammatory cytokines increase reactive oxygen species through mitochondria and NADPH oxidase in cultured RPE cells. Exp Eye Res. 2007;85(4):462–472. doi: 10.1016/j.exer.2007.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Morgan MJ, Liu ZG. Crosstalk of reactive oxygen species and NF-kappaB signaling. Cell Res. 2011;21(1):103–115. doi: 10.1038/cr.2010.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Asehnoune K, Strassheim D, Mitra S, Kim JY, Abraham E. Involvement of reactive oxygen species in Toll-like receptor 4-dependent activation of NF-kappa B. J Immunol. 2004;172(4):2522–2529. doi: 10.4049/jimmunol.172.4.2522. [DOI] [PubMed] [Google Scholar]
- 28.Park CE, Yun H, Lee EB, et al. The antioxidant effects of genistein are associated with AMP-activated protein kinase activation and PTEN induction in prostate cancer cells. J Med Food. 2010;13(4):815–820. doi: 10.1089/jmf.2009.1359. [DOI] [PubMed] [Google Scholar]
- 29.Peairs A, Radjavi A, Davis S, et al. Activation of AMPK inhibits inflammation in MRL/lpr mouse mesangial cells. Clin Exp Immunol. 2009;156(3):542–551. doi: 10.1111/j.1365-2249.2009.03924.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jeong HW, Hsu KC, Lee JW, et al. Berberine suppresses proinflammatory responses through AMPK activation in macrophages. Am J Physiol Endocrinol Metab. 2009;296(4):E955–E964. doi: 10.1152/ajpendo.90599.2008. [DOI] [PubMed] [Google Scholar]
- 31.Zhao X, Zmijewski JW, Lorne E, et al. Activation of AMPK attenuates neutrophil proinflammatory activity and decreases the severity of acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2008;295(3):L497–L504. doi: 10.1152/ajplung.90210.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Salt IP, Palmer TM. Exploiting the anti-inflammatory effects of AMP-activated protein kinase activation. Expert Opin Investig Drugs. 2012;21(8):1155–1167. doi: 10.1517/13543784.2012.696609. [DOI] [PubMed] [Google Scholar]
- 33.Salminen A, Hyttinen JM, Kaarniranta K. AMP-activated protein kinase inhibits NF-kappaB signaling and inflammation: impact on healthspan and lifespan. J Mol Med (Berl) 2011;89(7):667–676. doi: 10.1007/s00109-011-0748-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.O’Neill LA, Hardie DG. Metabolism of inflammation limited by AMPK and pseudo-starvation. Nature. 2013;493(7432):346–355. doi: 10.1038/nature11862. [DOI] [PubMed] [Google Scholar]
- 35.Akbar DH. Effect of metformin and sulfonylurea on C-reactive protein level in well-controlled type 2 diabetics with metabolic syndrome. Endocrine. 2003;20(3):215–218. doi: 10.1385/ENDO:20:3:215. [DOI] [PubMed] [Google Scholar]
- 36.Cederroth CR, Vinciguerra M, Gjinovci A, et al. Dietary phytoestrogens activate AMP-activated protein kinase with improvement in lipid and glucose metabolism. Diabetes. 2008;57(5):1176–1185. doi: 10.2337/db07-0630. [DOI] [PubMed] [Google Scholar]