

Abstract
Modified peptide nucleic acids (PNA) containing one or two thymine PNA monomers derived from phenylalanine were synthesized. Triple helix formation by these modified PNAs with RNA and DNA hairpins having a variable base pair in the middle of the helix were studied using isothermal titration calorimetry and compared with triple helix formation by non-modified PNAs. While unmodified PNA had low sequence selectivity against mismatched hairpins, introduction of one or two phenylalanine-derived monomers significantly increased the mismatch discrimination and sequence selectivity of the modified PNA. Consistent with our previous observations, PNA formed more stable triple helices with RNA than with DNA. Interestingly, the phenylalanine modification further improved the preference of PNA for RNA over DNA hairpin.
Keywords: PNA backbone modification, PNA-RNA triple helix, isothermal titration calorimetry, peptide nucleic acid, recognition of double-stranded RNA
Introduction
Over the last few decades we have realized that RNA is not just a passive messenger molecule that translates genetic information from DNA to proteins. Just as proteins, RNA can catalyze chemical reactions and regulate gene expression via complex mechanisms. In particular, various non-coding RNAs play a central role in temporal and spatial gene regulation ensuring proper development and function of various cell types and tissues.1,2 Sequence selective molecular recognition of such non-coding RNA would be of high importance for fundamental science and practical applications.
Most non-coding RNAs exist either as double helical structures (e.g., tRNA and rRNA) or feature double helical intermediates during their biogenesis. For example, microRNAs (miRNAs) are transcribed as long hairpin structures, pri-miRNAs, which are processed to shorter pre-miRNA hairpins of up to 70 bases.3-5 Chemical probes capable of detecting and inhibiting the function of such intermediates would be highly useful. However, sequence selective recognition of double helical RNA is a formidable challenge.6,7 The standard paradigm for targeting biological receptors relies largely on shape selectivity in recognition of a relatively rigid binding pocket. Shape selective RNA recognition has had limited success because of conformational flexibility of RNA. Strong RNA binders typically are cationic compounds that rely on electrostatic interaction with the negatively charged phosphate backbone of RNA. Although cationic compounds, such as aminoglycosides, exhibit high affinity for RNA, the sequence selectivity is typically low.6
Recently, we proposed that triple helix formation using peptide nucleic acids might be a new approach for sequence selective recognition of double helical RNA.8 We showed that PNAs as short as six nucleobases were surprisingly strong RNA binders and exhibited high sequence selectivity. The introduction of just one mismatch in a PNA hexamer led to little or no binding.8 Isolated pyrimidines interrupting longer purine tracts could also be recognized using nucleobase-modified PNA nonamers.9 However, the sequence selectivity for the PNA nonamers9 was somewhat lower than that observed for the hexamers in our initial study.8 As the length of PNA increased, the effect of a mismatch was diminished, eroding the excellent sequence selectivity. In the present study we demonstrate that the sequence selectivity of PNA-RNA triple helix formation can be enhanced by using backbone-modified PNA derived from phenylalanine instead of glycine (Fig. 1).
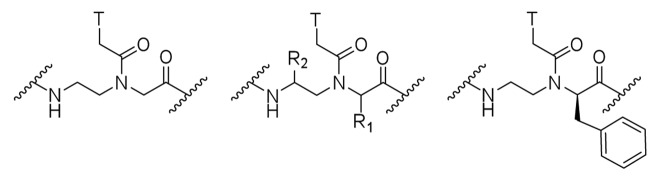
Figure 1. Structures of unmodified PNA, α (R1) and γ (R2) modified PNA, and phenylalanine-derived PNA (modified at the α position).
To improve binding affinity, solubility, and cellular uptake of PNA several laboratories have introduced modifications at α or γ positions of the PNA backbone (Fig. 1). Nielsen and coworkers showed that even sterically demanding substituents like benzyl (derived from tyrosine or phenylalanine) could be incorporated at the α-position in PNA leading to moderate loss in stability of PNA-DNA and PNA-RNA duplexes.10,11 These authors reported that positively charged side chains such as aminobutyl (derived from lysine) at the α-position led to higher thermal stability of PNA-DNA duplexes than neutral side chains, which, in turn, were less destabilizing than negatively charged side chains, such as derivatives of glutamic and aspartic acid.11 Ly and coworkers reported that PNA modification with positively charged guanidine groups (GPNA derived from arginine) at both the α or γ position improved binding affinity, sequence selectivity, and cellular uptake.12-15 Inspired by these results, our group explored the potential of Guanidine-PNA (GPNA) in triple helical recognition of RNA.16 Contrary to our expectations, we discovered that in the case of triple helices, the guanidine modification reduced the affinity and sequence selectivity of PNA for double helical RNA.16 However, strong and selective binding was observed to the transactivation response element RNA of HIV-1 presumably through a strand invasion and Watson-Crick duplex formation.16
Ly, Armitage and coworkers incorporated backbone modifications derived from l-and d-alanine into G-rich PNAs to improve selectivity for quadruplex vs. duplex formations.17 Appella and coworkers studied PNA in which the ethylenediamine portion of the PNA backbone was replaced by (S,S)-trans-cyclopentane diamine.18 This modification significantly increased binding affinity and sequence specificity to cDNA.17 Later, Englund and Appella reported that PNA derived from l-lysine showed better single-base-mismatch discrimination than unmodified PNA.19
Nielsen and coworkers found that α-modified PNA derived from d- and l-lysine, d- glutamate, l-isoleucine and d-serine were better at discriminating mismatches in PNA-DNA and PNA-RNA duplexes than unmodified PNA.20 These authors suggested that the better discrimination was due to the bulky substituent at the α-position making it difficult for the backbone to adopt a conformation required to accommodate the mismatched base pairs in the helix.20 In the present study, we hypothesized that the benzyl group in PNA derived from phenylalanine may have a similar selectivity enhancing effect on PNA-RNA triple helices. We found that the benzyl substituent at the α-position somewhat decreased the stability of PNA-RNA triple helices. However, confirming our hypothesis, the benzyl modification significantly increased the sequence selectivity of the PNA-RNA and PNA-DNA triple helix formation.
Results and Discussion
To test whether the benzyl substituent may increase the selectivity of triple helical binding of PNA to double-stranded RNA, we synthesized PNAs containing one or two phenylalanine-derived α-modified monomers. Since Nielsen and coworkers reported that the backbone modifications derived from either d- or l-amino acids both improved sequence selectivity,20 we chose l-phenylalanine as the least expensive starting material for our studies. To simplify synthetic effort, we chose thymine as the modified PNA monomer to study the effect of benzyl group at the α-position.
The synthesis of the benzyl-modified PNA monomer 3 (Scheme 1) was achieved following the procedures by Kleiner et. al.21 Parikh-Doering oxidation of (9H-fluoren-9-yl)methyl 2-hydroxyethylcarbamate gave Fmoc-glycinal 1, which was coupled with l-phenylalanine allyl ester to give the α-modified PNA backbone 2. Acylation of the PNA backbone 2 with 1-thymine acetic acid, followed by ester deprotection gave the target PNA monomer 3.
Scheme 1.
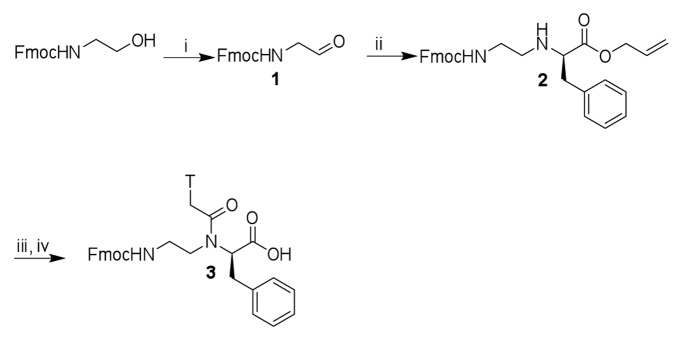
Synthesis of benzyl-modified PNA monomer T. Reagents and conditions: (i) dichloromethane:dimethyl sulfoxide (2:1 v/v), Et3N, SO3Py, rt, 1.5 h, 82%; (ii) methanol, l-phenylalanine allyl ester, acetic acid, 2h 15min, then NaBH3CN, 30 min, 74%; (iii) dimethylformamide, 1-thymine acetic acid, 3,4-dihydro-3-hydroxy-4-oxo-1,2,3-benzotriazine (DhbtOH), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC), 40 °C, overnight, 82%;(iv) THF, N-ethylaniline, Pd(PPh3)4, rt, 1 h, 87%.
A series of modified PNAs were synthesized (Fig. 2) using the standard PNA synthesis protocol on an Expedite 8909 DNA Synthesizer. These PNAs were similar to unmodified PNA1 and PNA3 from our previous study8 except that one or two of the standard T monomers were substituted with the phenylalanine derived T. The modifications were placed near the middle of the PNA since it has been reported in literature that modification in the center of the PNA has the most significant effect on affinity and selectivity.11 As in our previous studies,8,9 we used RNA hairpins (HRP1–4) having a variable base pair in the middle of the stem as the targets for PNA binding and model system to test the binding affinity and sequence selectivity.
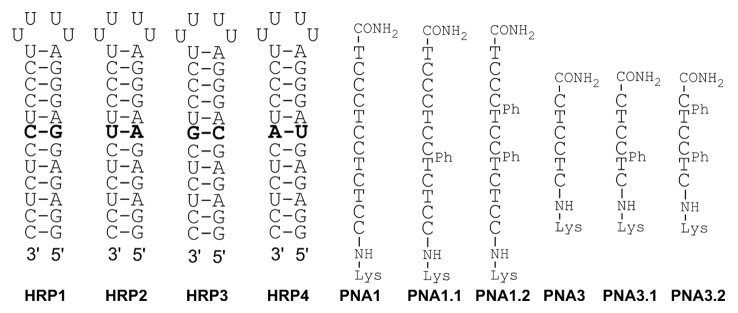
Figure 2. Sequences of RNA hairpins and PNAs. The numbering of RNA and PNA is retained from reference 8; TPh is the phenylalanine-derived monomer T.
To study the affinity and selectivity of the modified PNAs for RNA hairpins HRP1–4, we used isothermal titration calorimetry (ITC). ITC directly measures the enthalpy of binding and, through fitting of the binding curve, provides the association constant (Ka in M−1) and binding stoichiometry.22,23 We have previously used ITC to study triple helical binding of PNA to double stranded RNA.8,9 Typical ITC curves are shown in Figure 3.

Figure 3. ITC titration curves of PNA1.2 binding to HRP1–4 (2.6 µM).
In 100 mM sodium acetate buffer at pH 5.5, PNA1 had high binding affinity for its target hairpin HRP1.8 However, PNA1 had poor sequence selectivity and was binding to HRP3 (G-C mismatch, Table 1) with higher affinity than to the matched HRP1. Introduction of one phenylalanine-derived monomer in PNA1.1 slightly reduced the binding affinity but improved the mismatch discrimination. PNA1.1 had the highest affinity for matched HRP1, and the affinities for the mismatched hairpins were two to eight times lower.
Table 1. Sequence selectivity of PNA binding to RNA hairpinsa.
| PNA | HRP1 (C-G) |
HRP2 (U-A) |
HRP3 (G-C) |
HRP4 (A-U) |
|---|---|---|---|---|
| PNA1 | 35b (1.2) | 16 (0.9) | 39 (1.2) | 12 (1.5) |
| PNA1.1c | 23 (1.2) | 5.8 (1.1) | 10 (1.5) | 2.8 (0.9) |
| PNA1.2c | 16.7 (1.1) | 3.4 (1.2) | 3.7 (1.2) | 0.7 (1.4) |
| PNA3 | 8.4b (1.1) | 0.04b (2.1) | 0.05b (1.1) | 0.02b (2.5) |
| PNA3.1c | 2.1 (1.3) | 0.0005 (1.2) | 0.007 (0.0) | NBd |
| PNA3.2c | 0.3 (0.7) | NBd | NBd | NBd |
a Association constants Ka × 107 M−1, binding stoichiometry is given in parentheses, 100 mM sodium acetate, 1.0 mM EDTA, pH 5.5; bData from our previous study, ref. 8; c.1 or .2 designates the number of modified monomers in PNA1 and PNA3; dNB, no binding, Ka < 103.
PNA1.2 having two phenylalanine-derived monomers, had somewhat lower but comparable binding affinity for the matched HRP1 than unmodified PNA1. However, selectivity significantly improved with affinity for the mismatched hairpins being up to 23 times lower. In the shorter hexamers, the effect was much greater. Even though PNA3 had good sequence selectivity, introduction of a benzyl group in the backbone (PNA3.1, Table 1) strongly improved the sequence selectivity. Increasing the number of benzyl modifications lowered binding affinity. The binding affinity of PNA3.1, having one benzyl group, to HRP1 was 4 times lower than that of unmodified PNA3. PNA3.2 that had two benzyl groups in its backbone, had Ka = 3.3 × 106, which was 25 times lower than that of PNA3. The binding order of all PNAs to the matched HRP1 was approximately one (Table 1, HRP1 column), which was consistent with the proposed triple helix formation.
Our results on PNA-RNA triple helices are consistent with observations by Nielsen and coworkers12 on PNA-DNA and PNA-RNA double helices and suggest that the destabilizing effect of the α-benzyl modification is greater for the mismatched complexes than for the matched ones. Thus, a slight decrease in affinity is accompanied by a significant increase in sequence selectivity, which was our desired objective.
UV thermal melting experiments were consistent with the observations made by ITC. In general, triple helix melting is expected to show a biphasic UV thermal melting curve where the lower temperature transition is attributed to the triplex melting and the higher temperature transition is due to melting of the more stable duplex (hairpin in our case). However, we have previously observed that for PNA-RNA triplexes the low temperature transitions were fairly weak and might not give reliable melting temperatures.8,9 Moreover, the matched triplexes typically had similar or higher thermal stability than the target hairpins. UV thermal melting experiments for the triple helices of PNA1.2 with HRP1–4 (Fig. 4) were consistent with our previous observations.8,9 The matched triplex, PNA 1.2 with HRP1 showed only one high temperature (>90 °C) transition. The mismatched triplexes showed weak transitions at lower temperatures that were not well defined to give quantitative melting temperatures. As in our previous studies, we found that UV thermal melting data, while generally consistent with the ITC results, was less informative for quantitative evaluation of stability of the PNA-RNA triple helices.

Figure 4. UV thermal melting curves of PNA1.2‒HRP1–4 complexes.
To explore further the effect of the benzyl group on PNA binding to double-stranded RNA, we performed CD thermal melting experiments. The introduction of additional chirality by one (PNA1.1 or PNA3.1) or two (PNA1.2 or PNA3.2) benzyl modifications did not result in observable changes in the CD spectra of modified PNA compared with unmodified PNA (data not shown). CD thermal melting of PNA1.2-HRP1–4 triple helices yielded results that were consistent with the ITC and UV thermal melting data (Figs. 5 and 6). The CD thermal melting of PNA1.2-HRP1 triplex showed only one transition at >90°C (Fig. 6). CD thermal melting of complexes between PNA1.2 and the mismatched hairpins HRP2‒4 showed more complex transitions at lower temperatures, which could be attributed to lower thermal stability of the mismatched triplexes (Fig. 6).

Figure 5. Experimental CD thermal melting curves of PNA1.2‒HRP1–4 complexes.

Figure 6. Color-coded 3D representation of the CD melting surface of PNA1.2‒HRP1–4 complexes.
In our previous studies,8,24 we observed that PNA exhibited unique selectivity for triple helical recognition of RNA over DNA. For example, PNA1 had an order of magnitude higher affinity for HRP1 than for HRP5 (Fig. 2, the DNA version of HRP1)8 and the nucleobase-modified PNAs exhibited at least two orders of magnitude higher affinity for the double stranded RNAs than for the same DNA sequences.24 We decided to further explore this phenomenon and test if the phenylalanine modification would also enhance selectivity of triple helical binding of PNA to DNA.
Consistent with previous results,8 PNA1 had about an order of magnitude higher affinity for the RNA hairpin HRP1 (Table 1) than for the DNA hairpin HRP5 (Table 2). Binding of PNA1.2, having two phenylalanine modifications, to the RNA hairpin HRP1 was almost two orders of magnitude higher than its binding to DNA hairpin HRP5 (Ka = 1.7 × 108 in Table 1 vs. 6 × 106 in Table 2). Taken together these data show that the phenylalanine modification improved the sequence selectivity of PNA binding to DNA hairpins (Table 2) and further enhanced the preference of PNA to form stronger triple helices with RNA than with DNA (c.f., Table 1 and 2).
Table 2. Sequence selectivity of PNA binding to DNA hairpinsa.
| PNA | HRP5 (G-C) |
HRP6 (A-T) |
HRP7 (C-G) |
HRP8 (T-A) |
|---|---|---|---|---|
| PNA1 | 4.1 (1.1) | NDb | 1.5 (0.9) | 0.8 (0.5) |
| PNA1.2 | 0.6 (0.9) | NBc | NBc | NBc |
a Association constants Ka x 107 M−1, binding stoichiometry is given in parentheses, 100 mM sodium acetate, 1.0 mM EDTA, pH 5.5. bND, not determined; cNB, no binding, Ka < 103. HRP5-8 are the DNA versions of HRP1-4, respectively.
In summary, we have shown that the phenylalanine modification improved sequence selectivity of triple helical binding of PNA to double helical RNA at a small cost of slightly decreased binding affinity. Introduction of one or two phenylalanine-derived monomers in PNA1.1 and PNA1.2 significantly increased the mismatch discrimination in RNA hairpins and also enhanced the preference of PNA to form stronger triple helices with RNA than with DNA. The latter observation is important for future applications to insure that the triple helix forming PNA will not have undesired binding to genomic DNA in cells. Taken together with our previous studies,8,9,24 our results suggest that modified PNAs have the potential to be used as chemical probes for interrogating the function of non-coding double helical RNA, which will be important for fundamental science and practical applications.
Materials and Methods
Synthesis of PNA
Phenylalanine derived PNA monomer 3 was synthesized as described by Kleiner et. al.21 PNA synthesis was done on an Expedite 8909 synthesizer following the standard manufacturer’s protocol (2 µmol scale) and using NovaSyn TG Sieber resin (Novabiochem) as a support, HATU (Oakwood Chemicals) as an activator and PNA monomer 3, Fmoc-PNA-C(Bhoc)-OH and Fmoc-PNA-T-OH as monomers (purchased from Link Technologies Ltd). l-lysine was coupled to the N-terminus of PNA on Expedite 8909 (using standard PNA coupling protocol) using Fmoc-l-lys (Boc)-OH (Novabiochem). Chain extension followed a three-step cycle: (1) removal of the Fmoc-protecting group from the terminal amine with 20% piperidine (Sigma-Aldrich) in dimethylformamide, (2) coupling of the next monomer onto the N-terminus of the growing chain, and (3) capping of the unreacted amines with acetic anhydride. The PNA oligomers were cleaved off the Sieber resin using 80% trifluoroacetic acid; 20% m-cresol mixture (0.9 mL) for 2.5 h at room temperature. The crude PNA samples were precipitated by adding anhydrous diethyl ether (4 mL) to the deprotection mixture. The solid was collected, lyophilized, dissolved in HPLC grade water and purified by RP-HPLC on Xbridge Prep C-18 column (5 µm, 10 × 150 mm, Waters) at 60 °C eluting with a linear gradient of 10–45% of acetonitrile in water containing 0.1% of TFA over 40 min, flow rate of 3 mL/min. Absorbency was monitored at 254 nm and 280 nm. The fraction containing the major peak was collected, lyophilized to dryness to afford pure PNA samples. The PNA was quantified following the procedure described for DNA and RNA.25 The molecular weight of the synthesized PNAs was confirmed by ESI or MALDI TOF mass spectrometry:
PNA1.1
ESI found m/z 3312.1 [M+H]+, calculated for C137H181N59O41: 3310.3.
PNA1.2
MALDI found m/z 3401.2 [M+H]+, calculated for C144H187N59O41: 3400.4.
PNA3.1
ESI found m/z 1774.1 [M+H]+, calculated for C75H101N31O21: 1772.8.
PNA3.2
ESI found m/z 1864.4 [M+H]+, calculated for C82H107N31O21: 1862.9.
RNA
RNA hairpins were purchased from Thermo Fisher Genomics, deprotected according to manufacturer’s recommendations and purified by HPLC. After deprotection RNA samples were purified using RP-HPLC on Xbridge Prep C-18 column (5 µm, 10 × 150 mm, Waters) at 60 °C eluting with a linear gradient (5–20%) of mobile phase B in mobile phase A over 40 min, flow rate 5 ml/min. Mobile phase A was 0.1 M of triethylammonium acetate (pH = 7.0) in HPLC water and mobile phase B was a mixture of 0.1 M of triethylammonium acetate (pH = 7.0) in HPLC water and HPLC grade acetonitrile (60/40, v/v). Absorbency was monitored at 254 nm and 280 nm. The fraction containing the major peak was collected, lyophilized to dryness, redissolved in HPLC water and lyophilized twice (2 × 2 mL) to afford pure RNA samples. RNAs were quantified using the extinction coefficients provided by Thermo Fisher Genomics.
DNA
DNA hairpins were purchased from Eurofins MWG Operon. The DNA hairpins were purified by RP-HPLC following the same procedure described for the purification of RNA hairpins. DNAs were quantified using the extinction coefficient provided by Eurofins MWG Operon.
Isothermal titration calorimetry
ITC Experiments were done on a Nano ITC G2 (TA Instruments). RNA/DNA stock solution (17.5 µL, 0.24 mM) was evaporated to dryness and the solid was dissolved in 1.6 mL of acetate buffer (100 mM of sodium acetate, 1.0 mM of EDTA, pH = 5.5). After degassing, the RNA solution (0.95 mL, 2.63 μM) was loaded into the ITC reaction cell and the reference cell was loaded with degassed HPLC water. PNA stock solution (70 µL, 0.24 mM) was evaporated to dryness and the solid was dissolved in 350 µL of acetate buffer. After degassing, the PNA solution (250 µL, 48 µM) was loaded in the titration syringe. The syringe was inserted into the reaction cell, the stirring was set on 250 rpm, and the instrument was equilibrated at 25 °C until the baseline was flat and stable (~1 h). The titration experiment was done by performing 50 injections of 5 µl with 4–13 min intervals. The titration data were analyzed using NanoAnalyze software (TA Instruments) and independent model to obtain the fitting graph and binding constants (Ka).
Acknowledgments
We thank Binghamton University and NIH (R01 GM071461) for support of this research. The Regional NMR Facility (600 MHz instrument) at Binghamton University is supported by NSF (CHE-0922815).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/artificialdna/article/26599
References
- 1.Meister G, Tuschl T. Mechanisms of gene silencing by double-stranded RNA. Nature. 2004;431:343–9. doi: 10.1038/nature02873. [DOI] [PubMed] [Google Scholar]
- 2.Plasterk RHA. Micro RNAs in animal development. Cell. 2006;124:877–81. doi: 10.1016/j.cell.2006.02.030. [DOI] [PubMed] [Google Scholar]
- 3.Krol J, Loedige I, Filipowicz W. The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet. 2010;11:597–610. doi: 10.1038/nrg2843. [DOI] [PubMed] [Google Scholar]
- 4.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–97. doi: 10.1016/S0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 5.Siomi H, Siomi MC. Posttranscriptional regulation of microRNA biogenesis in animals. Mol Cell. 2010;38:323–32. doi: 10.1016/j.molcel.2010.03.013. [DOI] [PubMed] [Google Scholar]
- 6.Thomas JR, Hergenrother PJ. Targeting RNA with small molecules. Chem Rev. 2008;108:1171–224. doi: 10.1021/cr0681546. [DOI] [PubMed] [Google Scholar]
- 7.Guan L, Disney MD. Recent advances in developing small molecules targeting RNA. ACS Chem Biol. 2012;7:73–86. doi: 10.1021/cb200447r. [DOI] [PubMed] [Google Scholar]
- 8.Li M, Zengeya T, Rozners E. Short peptide nucleic acids bind strongly to homopurine tract of double helical RNA at pH 5.5. J Am Chem Soc. 2010;132:8676–81. doi: 10.1021/ja101384k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gupta P, Zengeya T, Rozners E. Triple helical recognition of pyrimidine inversions in polypurine tracts of RNA by nucleobase-modified PNA. Chem Commun (Camb) 2011;47:11125–7. doi: 10.1039/c1cc14706d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dueholm KL, Petersen KH, Jensen DK, Egholm M, Nielsen PE, Buchardt O. Bioorg. Peptide nucleic acid (PNA) with chiral backbone based on alanine. Med. Chem. Lett. 1994;4:1077–80. doi: 10.1016/S0960-894X(01)80684-3. [DOI] [Google Scholar]
- 11.Puschl A, Sforza S, Haaima G, Dahl O, Nielsen PE. Peptide nucleic acids with functional backbone. Tetrahedron Lett. 1998;39:4707–10. doi: 10.1016/S0040-4039(98)00862-4. [DOI] [Google Scholar]
- 12.Zhou P, Wang M, Du L, Fisher GW, Waggoner A, Ly DH. Novel binding and efficient cellular uptake of guanidine-based peptide nucleic acids (GPNA) J Am Chem Soc. 2003;125:6878–9. doi: 10.1021/ja029665m. [DOI] [PubMed] [Google Scholar]
- 13.Dragulescu-Andrasi A, Zhou P, He G, Ly DH. Cell-permeable GPNA with appropriate backbone stereochemistry and spacing binds sequence-specifically to RNA. Chem Commun (Camb) 2005;5:244–6. doi: 10.1039/b412522c. [DOI] [PubMed] [Google Scholar]
- 14.Zhou P, Dragulescu-Andrasi A, Bhattacharya B, O’Keefe H, Vatta P, Hyldig-Nielsen JJ, Ly DH. Synthesis of cell-permeable peptide nucleic acids and characterization of their hybridization and uptake properties. Bioorg Med Chem Lett. 2006;16:4931–5. doi: 10.1016/j.bmcl.2006.06.052. [DOI] [PubMed] [Google Scholar]
- 15.Sahu B, Chenna V, Lathrop KL, Thomas SM, Zon G, Livak KJ, Ly DH. Synthesis of conformationally preorganized and cell-permeable guanidine-based γ-peptide nucleic acids (gammaGPNAs) J Org Chem. 2009;74:1509–16. doi: 10.1021/jo802211n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gupta P, Muse O, Rozners E. Recognition of double-stranded RNA by guanidine-modified peptide nucleic acids. Biochemistry. 2012;51:63–73. doi: 10.1021/bi201570a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lusvarghi S, Murphy CT, Roy S, Tanious FA, Sacui I, Wilson WD, Ly DH, Armitage BA. Loop and backbone modifications of peptide nucleic acid improve g-quadruplex binding selectivity. J Am Chem Soc. 2009;131:18415–24. doi: 10.1021/ja907250j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pokorski JK, Witschi MA, Purnell BL, Appella DH. (S,S)-trans-cyclopentane-constrained peptide nucleic acids. a general backbone modification that improves binding affinity and sequence specificity. J Am Chem Soc. 2004;126:15067–73. doi: 10.1021/ja046280q. [DOI] [PubMed] [Google Scholar]
- 19.Englund EA, Appella DH. γ-substituted peptide nucleic acids constructed from L-lysine are a versatile scaffold for multifunctional display. Angew Chem Int Ed Engl. 2007;46:1414–8. doi: 10.1002/anie.200603483. [DOI] [PubMed] [Google Scholar]
- 20.Haaima G, Lohse A, Buchardt O, Nielsen PE. Peptide nucleic acids (PNAs) containing thymine monomers derived from chiral amino acids: hybridization and solubility properties of D-lysine PNA. Angew Chem Int Ed Engl. 1996;35:1939–42. doi: 10.1002/anie.199619391. [DOI] [Google Scholar]
- 21.Kleiner RE, Brudno Y, Birnbaum ME, Liu DR. DNA-templated polymerization of side-chain-functionalized peptide nucleic acid aldehydes. J Am Chem Soc. 2008;130:4646–59. doi: 10.1021/ja0753997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Feig AL. Applications of isothermal titration calorimetry in RNA biochemistry and biophysics. Biopolymers. 2007;87:293–301. doi: 10.1002/bip.20816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shwartz FP, Reinisch T, Hinz J, Surolia A. Recommendations on measurement and analysis of results obtained on biological substances using isothermal titration calorimetry. Pure Appl Chem. 2008;80:2025–40. doi: 10.1351/pac200880092025. [DOI] [Google Scholar]
- 24.Zengeya T, Gupta P, Rozners E. Triple-helical recognition of RNA using 2-aminopyride modified PNA at physiologically relevant conditions. Angew Chem Int Ed. 2012;51:12593–6. doi: 10.1002/anie.201207925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Puglisi JD, Tinoco I., Jr. Absorbance melting curves of RNA. Methods Enzymol. 1989;180:304–25. doi: 10.1016/0076-6879(89)80108-9. [DOI] [PubMed] [Google Scholar]