

Abstract
Age is a key risk factor for breast cancer and epigenetic alterations may contribute to age-related increases in breast cancer risk, though the relation of age-related methylation in normal breast tissues with altered methylation in breast tumors is unclear. We investigated the relation of age with DNA methylation in normal breast tissues genome-wide using two data sets from the Gene Expression Omnibus (GEO) database (GSE32393 and GSE31979). We validated our observations in an independent set of normal breast tissues, examined age-related methylation in normal breast for enrichment of genomic features, and compared age-related methylation in normal tissue with methylation alterations in breast tumors. Between the two array-based methylation data sets, there were 204 CpG loci with significant (P < 0.05) and consistent age-related methylation, 97% of which were increases in methylation. Our validation sets confirmed the direction of age-related DNA methylation changes in all measured regions. Among the 204 age-related CpG loci, we observed a significant enrichment for CpG islands (P = 8.7E-6) and polycomb group protein target genes (P = 0.03). In addition, 24 of the 204 CpGs with age-related methylation in normal breast were significantly differentially methylated between normal and breast tumor tissues. We identified consistent age-related methylation changes in normal breast tissue that are further altered in breast tumors and may represent early events contributing to breast carcinogenesis. This work identifies age-related methylation in normal breast tissue and begins to deconstruct the contribution of aging to epigenetic alterations present in breast tumors.
Keywords: Breast, aging, methylation, normal, array, cancer
Introduction
It is well established that age is the strongest demographic risk factor for breast cancer, with breast cancer incidence rates increasing with age.1,2 Alterations to epigenetic marks such as DNA methylation are early events in breast carcinogenesis, and these modifications may contribute to age-related increases in breast cancer risk.3 These altered patterns of DNA methylation have the ability to modify the regulation of gene expression and are frequently observed in the early stages of breast tumorigenesis.4,5 However, the relationship between molecular changes in normal breast tissue and etiologic risk factors, such as age, are less well characterized.
Normal epithelial tissues exhibit age-related patterns of DNA methylation, and the link between aging and changes in DNA methylation was initially reported for the estrogen receptor gene in normal human colon.6 However, DNA methylation profiles are tissue-specific,7,8 and while others have observed age-associated methylation changes in other tissues such as blood, prostate, and colon,6,9-12 the relation of age with breast tissue methylation is less clear. In previous work, we have shown that aging is related to DNA methylation in normal human tissues from 11 distinct anatomic sites (not including breast), and that these changes are dependent upon tissue type and CpG island context.7 Evidence from candidate gene studies has shown that aberrant methylation of tumor suppressor genes in normal breast demonstrate a relationship with aging and are associated with increased risk of breast cancer.13-15 Results from these candidate gene approaches are consistent with the hypothesis that DNA methylation alterations known to contribute to invasive disease can occur with aging. However, genome-wide approaches to investigate age-related methylation in normal breast tissues are lacking. Genome-wide assessment of age-related methylation allows testing for whether certain genomic features may be enriched among regions with age-related methylation, providing insights into the underlying biology of age-related methylation. In addition, identification of age-associated methylation in the breast followed by comparison with tumor tissues may allow us to identify key methylation alterations that contribute to increased risk of breast carcinogenesis with aging.
In this work we used publicly available genome-wide methylation array data from normal breast tissue to investigate DNA methylation patterns associated with subject age, followed by validation in a third independent population and a set of normal-adjacent breast tissues from The Cancer Genome Atlas database. Among age-related loci, enrichment analyses for genomic features were implemented to discern patterns that may be common among age-related methylation alterations and important in carcinogenesis. For instance, polycomb group proteins repress genes necessary for stem cell differentiation and the target genes of these transcriptional repressors are frequently found to be hypermethylated across a variety of tumor types.12,16-19 Lastly, we compared DNA methylation levels from disease-free breast tissue to breast tumors to identify candidate age-related changes with functional relevance for neoplastic transformation.
Results
Identification of age-related methylation in breast tissue from healthy subjects
To investigate DNA methylation patterns in the normal human breast related to subject age, we analyzed two publicly available GEO Human Methylation27 BeadArray data sets that measured methylation in normal breast tissues from breast cancer-free subjects. A summary of subject age for data sets used in this study is shown in Table 1. The scheme of our analysis strategy for identifying age-related CpG loci is depicted in Figure 1.
Table 1. Summary statistics of subject age among disease-free breast tissue data sets.
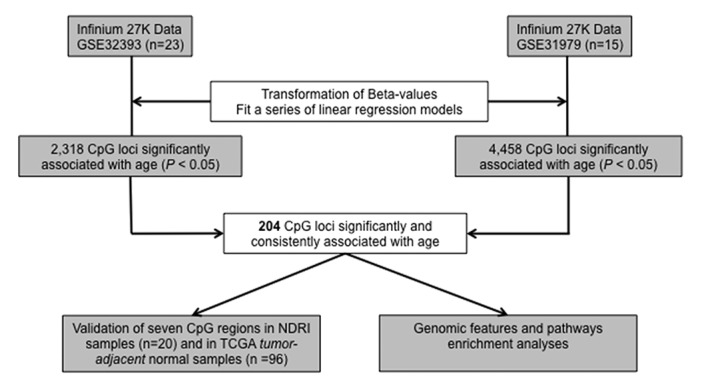
Figure 1. Diagram of the analysis strategy used in identifying CpG loci with age-related methylation in normal breast tissue.
We modeled the association of DNA methylation with age in normal breast tissue from each GEO data set in an array-wide locus-by-locus analysis. From GSE32393 (n = 23), 2318 CpG loci were significantly (P < 0.05) associated with subject age, and from GSE31979 (n = 15), 4458 CpG loci were significantly (P < 0.05) associated with subject age. Between these two sets of CpG loci with significant age-related methylation, 204 CpGs were shared and directionally consistent (Table S1). Among the 204 CpGs with significant and consistent age-related methylation 197 (97%) exhibited increased methylation as a function of age.
Genomic feature enrichment among CpGs with age-related methylation
To further investigate age-related methylation in the breast we assessed the potential enrichment of local sequence and bioinformatics features among the 204 CpG loci with age-related methylation in normal breast. Through an analysis of molecular pathways using DAVID we surveyed whether any biologic functions were enriched among loci with age-related methylation. After correcting for multiple comparisons no DAVID processes were significantly enriched among the 204 age-related loci. Next, since ERα is known to be a master transcriptional regulator in the breast,20 we tested for enrichment of ERα binding sites near the age-related CpG loci. None of the age-related CpG loci were found to be within 1 KB of ERα binding sites, though the Human Methylation27 BeadArray is relatively depleted (275/27 578 CpG loci or 0.99%) of CpG loci within 1 KB of ERα binding sites in T47D cells. Next, we examined the distribution of age-related CpG loci in canonical CpG islands and observed a significant enrichment of CpG island loci among age-related CpGs (P = 8.7E-6, Table 2A). Further, age-related CpG loci were significantly more likely to exhibit increased age-related methylation (P < 0.008), than decreased age-related methylation. Lastly, we tested for enrichment of polycomb group protein target genes (PCGTs) among CpGs with age-related methylation. We observed substantial PCGT methylation enrichment among loci that demonstrated age-related methylation in normal breast tissue (P = 0.028, Table 2B; Table S2).
Table 2. CpGs among the 204 age-related loci in the normal breast are enriched for localization to CpG Islands (A) and polycomb group target genes (PCGT) (B).
| A | |||
|---|---|---|---|
| CpG island | CpG island | Non CpG island | |
| All array CpGs | 20 006 (72%) | 7572 (28%) | |
| Age-related CpGs | 175 (86%) | 29 (14%) | P value = 8.7E-6* |
| *Fisher’s exact test | |||
| B | |||
| Polycomb group target gene (PCGT) | PCGT target | Non PCGT target | |
| All array CpGs | 3610 (13%) | 23 968 (87%) | |
| Age-related CpGs | 38 (19%) | 166 (81%) | P value = 0.028* |
| *Fisher exact test |
Validation of age-related methylation
To confirm observed associations between age and methylation, selected age-related loci were pyrosequenced in an independent set of disease-free breast tissues from the NDRI (n = 20). Two of the seven CpG loci, CSNK1G2 and IGFBP3, reached statistical significance for age-related methylation (P = 0.001, P = 0.002), and age-related methylation was validated for two other loci, RGS17 (P = 0.06) and RBM13 (P = 0.1). Consistent directional age-related methylation was observed for all seven CpG loci in our independent set of normal breast tissues (Table 3; Fig. S1).
Table 3. CpG loci selected for validation of DNA methylation relationship with subject age.
| Discovery | Validation | |||||||
|---|---|---|---|---|---|---|---|---|
| Gene | CpG Island Status | Direction of Association w/ Age | P value GSE32393 | P value GSE31979 | Direction of Association w/ Age NDRI | P value NDRI | Illumina cg ID | Chromosome |
| CSNK1G2 | CpG Island | + | 0.033 | 0.029 | + | 0.001 | cg26776924 | 19 |
| IGFBP3 | CpG Island | + | 0.011 | 0.011 | + | 0.002 | cg22083798 | 7 |
| RGS17 | CpG Island | + | 0.002 | 0.040 | + | 0.06 | cg23651356 | 6 |
| RBM13 | CpG Island | + | 0.009 | 0.025 | + | 0.10 | cg23663332 | 8 |
| MME | Non CpG Island | + | 0.002 | 0.006 | + | 0.18 | cg23273897 | 3 |
| PAX9 | CpG Island | - | 0.047 | 0.035 | - | 0.40 | cg26620157 | 14 |
| PEPP-2 | Non CpG Island | - | 0.028 | 0.017 | - | 0.62 | cg15465321 | X |
We then sought additional validation of selected loci with age-related methylation and accessed DNA methylation data from tumor-adjacent normal breast tissue samples in TCGA (n = 96). Six of the seven CpG loci passed TCGA QA/QC and were available in the TCGA methylation data set. Three of these six CpG loci had significant age-related methylation; CSNK1G2 (P = 0.008), RBM13 (P = 0.045), and MME (P = 0.037). Another CpG locus—associated with RGS17—exhibited age-related methylation (P = 0.07) (Table S3).
Comparing age-related methylation in normal breast with breast tumor methylation
Among CpGs with age-related methylation changes in the normal breast, we next compared methylation at these sites between normal and tumor tissues. Breast tumor methylation data was available from GSE32393 (n = 114) and GSE31979 (n = 103). Logistic regression models for tumor status were fit to arcsine square root transformed β values for each of CpG loci on the arrays separately in the two data sets, adjusting for age. Overall, the two data sets shared 7403 CpG loci that were consistently and significantly (P < 0.05) different between breast tumor and disease-free tissues. As we observed in the age-related loci, there was also a substantial PCGT methylation enrichment among loci that demonstrated significant differences between breast tumor and disease-free tissue (P = 1.4E-28, Table S4; Fig. S2). Among the 204 CpG loci with age-related methylation in normal breast, 24 CpG loci possessed significant differences in DNA methylation between tumor and disease-free breast in a direction consistent with the age-related change (Table S5). Figure 2 shows the comparison of normal breast and breast tumor methylation for the seven CpG loci selected for independent validation and demonstrates hypermethylation in tumors of CpG loci with age-related methylation in normal breast tissue. CpGs associated with IGFBP3 and RBM13 did not exhibit significantly increased methylation in tumors compared with normal breast for both GEO data sets. However, DNA methylation has been shown to be related with molecular subtypes of breast cancer including estrogen receptor (ER) status.21,22 When stratifying tumor methylation by ER status, CpGs associated with IGFBP3 and RBM13 exhibit significantly increased methylation in ER+ breast tumors (P < 0.05), compared with normal breast tissue in both GEO data sets (Fig. 3; Table S6).
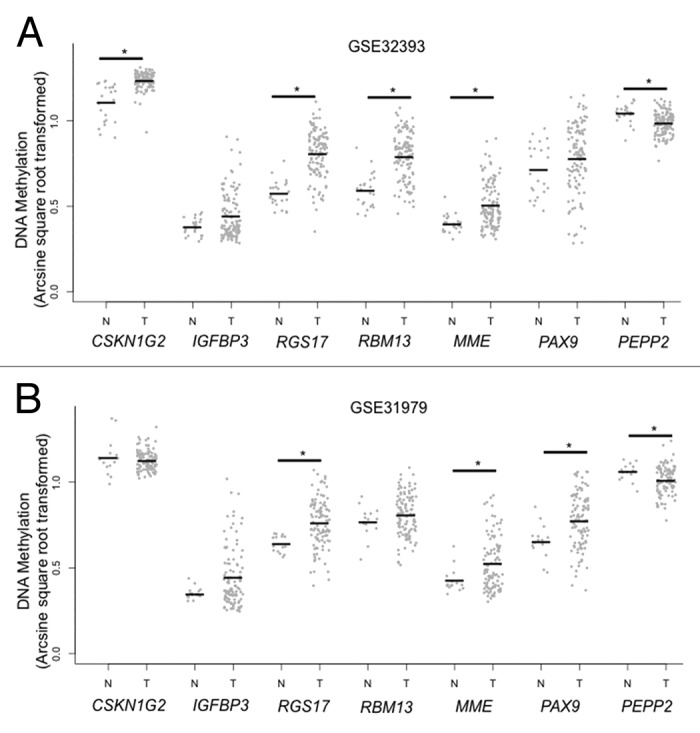
Figure 2. Age-related CpG loci in disease-free vs. tumor tissue for CpG loci selected for validation. Comparison of normal breast (N) with breast tumor (T) DNA methylation among CpG loci selected for validation. Frozen breast cancer tissues that were excised from patients prior to treatment and confirmed to have more than 50% epithelial cells from GSE32393 (n = 114) (A) and GSE31979 (n = 103) (B) were tested for a difference against normal tissues using logistic regression analyses for tumor status and arcsine square root transformed β-values adjusting for age (*P < 0.05).
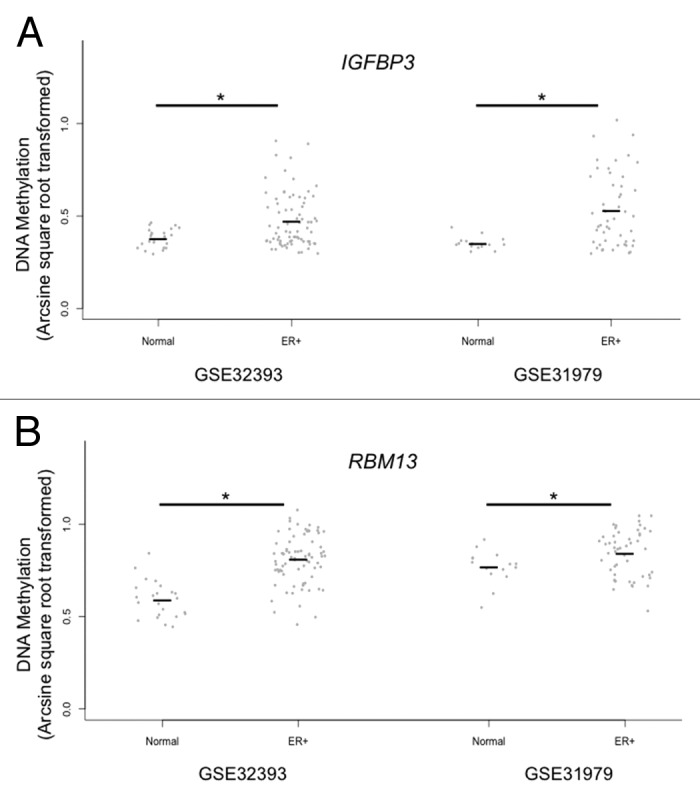
Figure 3. Tumor subtype specific age-related CpG loci in disease-free vs. ER positive tumor tissue. Comparison of estrogen receptor positive breast tumor DNA methylation to normal breast tissues at the IGFBP3 (A) and RBM13 (B) associated CpG loci in GSE32393 and GSE31979. Logistic regression analyses for tumor status, using only estrogen receptor (ER) positive tumors (GSE32393: ER+ = 76 tumors, GSE31979: ER+ = 50 tumors), were tested for a difference against normal tissues modeling arcsine square root transformed β-values (*P < 0.05).
Discussion
Epigenetic control of gene expression (and gene expression potential) is essential for cell specificity and function. Deregulation of epigenetic control mechanisms, such as DNA methylation have been observed in both age and tissue-dependent manners, and associated with disease states such as cancer.7,23 Evidence suggests that DNA methylation alterations are involved with aging-related processes and contribute to an increase in age-related disease susceptibility. Thus, characterizing age-dependent DNA methylation changes that occur in non-pathologic breast tissue and comparing them with alterations present in tumors may expose novel links between the aging process and breast carcinogenesis. We identified CpG loci whose methylation varies with age in disease-free breast tissues. Among CpG sites with age-related methylation, we observed enrichment for CpG islands and polycomb group gene targets (PCGTs). Importantly, CpG loci with age-related methylation increases in normal breast tissue were hypermethylated in breast tumors, suggesting that age-related methylation may contribute to increased risk of cellular transformation.
It is now recognized that factors such as exposure to carcinogens, inflammation, and diet are related with altered DNA methylation.24-26 However, the mechanisms contributing to DNA methylation alterations are unclear. Changes to DNA methylation patterns credited to aging may reflect the total of an accumulation of exposures, stochastic events, reduced methyltransferase enzyme fidelity, and truly age-specific alterations. DNA methylation patterns of disease-free breast tissue would thus be expected to vary among individuals, especially with a wide range of subject ages. The identification of age-related CpG loci and mechanisms that direct the specific patterning of age-related DNA methylation in disease-free breast tissue is useful for understanding changes that result from aging, but that have not yet resulted in a pathologic phenotype. To expose age-related alterations, we first characterized variations in CpG methylation that are consistently associated with aging through analysis of two separate genome-wide data sets. Our data suggests that there may be common genomic regions that are particularly susceptible to changes in DNA methylation over time in disease-free breast tissue and in cancer. Alternatively, our results could indicate that certain regions and genes may be enriched for age-related methylation due to cellular selection that preserves particular patterns of stochastically acquired methylation alterations.
Enrichment for age-related DNA methylation in CpG islands and PCGTs in breast tissues is consistent with prior investigations in other tissue types.7,12,16,17 In the independent disease-free breast population we observed that both our statistically validated CpG loci, IGFBP3 and CSNK1G2, are located within CpG islands and near to transcriptional start sites (CSNK1G2 = 53 bp and IGFBP3 = 530 bp to the TSS), in addition, IGFBP3 is also a PCGT. It remains unclear whether there are additional regulatory features of the IGFBP3 and CSNK1G2 loci that may be responsible for the robust relationship between methylation and age across populations and future investigations are needed to more precisely identify the genomic regions that may be preferentially deregulated with age. Methylation of PCGTs with age and in cancers has been observed across other tissue types. Given that PCGTs are epigenetically regulated during differentiation and are deregulated in cancer, enrichment of PCGTs among our age-related loci suggests that aging may contribute to cancer risk through methylation-induced dysregulation of cell fate transitions, allowing increased cellular plasticity, adaptability, and survival potential.
The plasticity of the epigenome, in the absence of disease state, is highlighted by the observation that DNA methylation patterns can be altered in people without cancer as a consequence of aging.7,27 These methylation changes with age, that have yet to result in disease, may further perpetuate the aging process in the breast by disrupting the activity of critical proteins. For instance, the deregulation of the IGF-1 axis has been implicated in the aging process and the IGFBP3 protein, whose gene is subjected to an age-related increase in methylation in the disease-free breast, is known to regulate IGF activity.28 Several studies have also demonstrated that circulating levels of IGFBP3 decrease with age in both male and female populations.28-30 Methylation of IGFBP3 with aging may, in part, be responsible for reduced levels of the IGFBP3 protein in older individuals. As a result, this age-related methylation alteration might have a significant contribution to the aging process and longevity through disruption of the IGF-1 axis. Accordingly, in the examination of non-pathologic tissues there are likely to be DNA methylation changes that occur as a function of age that may or may not increase risk of carcinogenesis. In contrast to bystander events, candidate gene studies have exposed disease-relevant methylation variations for women with higher breast cancer risk. Disease-free women with higher risk of breast cancer (based on the Gail model), had higher levels of methylation in normal breast tissue at selected tumor suppressor genes compared with disease-free women with low risk.15 In our study, to discern whether regions of the epigenome with age-related DNA methylation in normal breast are relevant to disease etiology, we compared DNA methylation at these regions between normal breast and breast tumors. Indeed, our results demonstrate that CpG loci with age-related methylation alterations in disease-free breast are hypermethylated in breast tumors, suggesting that age-related DNA methylation alterations may precede carcinogenesis or indicate an increased susceptibility to cellular transformation. In disease-free breast tissues one would not expect genomic instability or extensive aberrant gene expression. Consistent with our results, the age-related methylation changes we observed in disease-free breast tissues were relatively moderate and likely reflect an increase in a population of cells in normal tissue that have become poised for neoplastic transformation. We postulate that disruption in the regulation of disease-relevant genes could represent common epigenetic changes in a subpopulation of normal cells that contribute to increase the risk of breast carcinogenesis with aging. In fact, among the 24 CpG loci that possessed significant differences in DNA methylation between tumor and disease-free breast in a direction consistent with the age-related change there were several genes whose deregulation have previously been shown to play crucial roles in carcinogenesis. For example, hypermethylation has been shown in the promoter region of the putative tumor-suppressors ADAMTS18, EDNRB, PLA2R1 in several human tumors;31-33 copy number loss of MME and KCNK12 genes are frequent events in sporadic breast cancer;34,35 and PEPP2, which experienced a loss of methylation with increasing age and further loss in tumors, is a purported cancer-promoting gene.36 Additionally, we observed that there is specificity for some DNA methylation alterations among ER positive tumors compared with disease-free breast tissue. For instance, insulin-like growth factor binding protein 3 (IGFBP3) encodes a protein that binds insulin-like growth factor-I to block its antiapoptotic actions, and the aberrant methylation of this putative tumor suppressor gene has been observed in ER positive breast cancer.37,38 It is therefore reasonable to suggest that age-related shifts in methylation may not only confer greater cancer risk, but some shifts may confer a stronger predisposition to a particular tumor subtype depending upon tissue context and patterning of age-related alterations.
The precise mechanisms underlying age-related alterations to the breast epigenome remain incompletely clear and most investigations to probe the contribution of DNA methylation alterations in breast biology have predominantly focused on tumors. There is a resulting paucity of genome-wide methylation data that has been published on non-diseased breast tissue. Hence, one potential limitation in this analysis is the low statistical power due to small sample sizes. In an attempt to mitigate this limitation, we analyzed two separate data sets in our discovery step and validated selected loci with consistent results in an independent set of normal breast tissues and using TCGA tumor-adjacent normal breast tissues. Together, these data indicate that the CpG sites we observed to exhibit age-related methylation have consistent methylation changes across several populations. Another consideration when studying breast tissue is modest age-related increases in adipose tissue,39 as cell-type-specific DNA methylation could influence results. However, we observed further increases in methylation among breast tumors compared with loci with age-related methylation disease-free breast tissue, suggesting that the observed age-related methylation in normal breast is not adipocyte-specific. Finally, although age may reflect an accumulation of exposures, in this study we were not able to examine the relation of methylation with other known breast cancer risk factors or exposures. Future population-based studies are needed to investigate the relation of DNA methylation with breast cancer risk factors such as reproductive history, body mass index, and family history of disease in normal breast tissue and their relation to methylation alterations in breast tumors.
We have provided evidence for variation in disease-free breast methylation that is related to aging. We extended our investigation of these methylation alterations to breast tumor tissues, and observed common biology of epigenetic alterations that exist between cancer and aging. Cancer and aging are tightly interconnected processes, and our approach identified methylation alterations present in the normal aging breast methylome that may increase disease risk in cancer-free individuals. Age-related methylation changes in disease-free breast that also are present in tumors offer additional insights into the relation among epigenetic alterations, aging, and breast carcinogenesis.
Materials and Methods
Tissues and study subjects
We accessed two genome-wide DNA methylation data sets from the Gene Expression Omnibus that included normal breast tissues (GEO, http://www.ncbi.nlm.nih.gov/geo/, Accession numbers GSE32393 and GSE31979), consisting of 23 (GSE32393) and 15 (GSE31979) reduction mammoplasty specimens from disease-free women. For independent validation, fresh-frozen disease-free breast tissue was obtained from 20 subjects sourced from the National Disease Research Interchange (NDRI). For additional in silico validation, level 3 DNA methylation data for 96 tumor-adjacent normal breast samples were downloaded from TCGA repository (http://cancergenome.nih.gov).40 GSE32393 and GSE31979 also contain breast tumor methylation data, for 114 and 103 tumors respectively, these data were used to compare tumor and normal breast DNA methylation.
DNA isolation, bisulfite modification, PCR, and pyrosequencing analysis
DNA was extracted from fresh-frozen NDRI subject breast tissue using the DNeasy Blood and Tissue Kit according to the manufacturer’s protocol (Qiagen, 69504) and quantified via a Nanodrop spectrophotometer (Thermo Scientific, ND-2000). Genomic DNA was bisulfite modified using the EZ DNA Methylation Kit (Zymo Research, D5002) in adherence with the manufacturer’s protocol. CpG targets for pyrosequencing analysis were selected from CpG loci rank-ordered by magnitude of association with age in both GEO data sets. First, the 204 age-related loci were sorted by regression coefficient value, and CpGs whose range of methylation among samples exceeded Δβ > 0.1 were considered for validation. The top ten CpG loci with increased age-related methylation and the top five CpG loci with decreased age-related methylation that met these criteria were selected for pyrosequencing assay design using the PyroMark Assay Design software 2.0.1.15 (Qiagen). Pyrosequencing assays whose design scores were >70 (five with age-related increases, and two with age-related decreases) were pursued for independent validation. PCR primer sequences and sequencing primer sequences are listed in Table S7. Methylation data from bisulfite pyrosequencing of selected CpGs in the NDRI samples were processed using the PyroMark CpG software (Qiagen) under the default analysis parameters and the data was exported for additional analyses in the R software environment.
Statistical analysis
All methylation data were analyzed using the R statistical package, version 2.15.2 (www.r-project.org). To identify age-related CpG loci linear regression models were applied array-wide in locus-by-locus analyses separately in the two GEO data sets by modeling arcsine square root transformed methylation as the response for variance stabilization and normality considerations41 and subject age as the independent variable. An association was considered significant where P < 0.05. To reduce type I error in these smaller subject populations, the intersection of CpG loci between the two data sets that were directionally consistent and significantly associated with age were used in subsequent analyses. Linear regression models were also used for the validation analyses of the select CpG loci in the NDRI population, modeling percent methylation from pyrosequencing as the response and NDRI subject age as the independent variable. For in silico validation of select CpG loci, level 3 normalized Illumina Infinium Human Methylation450 BeadChip data from TCGA was used modeling arcsine square root transformed methylation as the response variable and subject age as the independent variable. The Database for Annotation, Visualization, and Integrated Discovery was used for an analysis of molecular pathways.42,43 In the DAVID analysis, the set of genes represented on the Illumina HumanMethylation27 array was used as the referent set, and the set of genes associated with age-related CpG loci was the gene set tested. Enrichment analyses of sequence and genomic features among age-related CpG loci were performed using two-tailed Fisher’s exact tests. CpG Island status was taken from the Illumina HumanMethylation27 annotation file and the polycomb group (PcG) target status for a given CpG loci was based on whether the gene associated with that CpG was described as a PcG target in previously published works.44-47 Estrogen Receptor α transcription factor binding sites were identified from ERα ChIP-Seq experiments in T47D cells and presence of a CpG within 1 KB of these regions was queried.48 To compare breast tumor and normal breast DNA methylation, tumor methylation data from the same GEO data sets was modeled using logistic regression where tumor status was the response, fit to arcsine square root transformed β-values and adjusting for subject age. A stratified analysis of estrogen receptor positive tumors vs. normal breast methylation also was performed. Associations where P < 0.05 were considered to be statistically significant.
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Financial Disclosures
This work was supported by NIH grants P20GM104416 (BCC), R25CA134286 (DCK), and American Cancer Society Research Grant IRG-82-003-27 (CC).
Supplemental Materials
Supplemental materials may be found here: http://www.landesbioscience.com/journals/epigenetics/article/27015/
Footnotes
Previously published online: www.landesbioscience.com/journals/epigenetics/article/27015
References
- 1.Benz CC, Yau C. Ageing, oxidative stress and cancer: paradigms in parallax. Nat Rev Cancer. 2008;8:875–9. doi: 10.1038/nrc2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Edwards BK, Howe HL, Ries LA, Thun MJ, Rosenberg HM, Yancik R, Wingo PA, Jemal A, Feigal EG. Annual report to the nation on the status of cancer, 1973-1999, featuring implications of age and aging on U.S. cancer burden. Cancer. 2002;94:2766–92. doi: 10.1002/cncr.10593. [DOI] [PubMed] [Google Scholar]
- 3.Tlsty TD, Crawford YG, Holst CR, Fordyce CA, Zhang J, McDermott K, Kozakiewicz K, Gauthier ML. Genetic and epigenetic changes in mammary epithelial cells may mimic early events in carcinogenesis. J Mammary Gland Biol Neoplasia. 2004;9:263–74. doi: 10.1023/B:JOMG.0000048773.95897.5f. [DOI] [PubMed] [Google Scholar]
- 4.Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet. 2003;33(Suppl):245–54. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- 5.Widschwendter M, Jones PA. DNA methylation and breast carcinogenesis. Oncogene. 2002;21:5462–82. doi: 10.1038/sj.onc.1205606. [DOI] [PubMed] [Google Scholar]
- 6.Issa JP, Ottaviano YL, Celano P, Hamilton SR, Davidson NE, Baylin SB. Methylation of the oestrogen receptor CpG island links ageing and neoplasia in human colon. Nat Genet. 1994;7:536–40. doi: 10.1038/ng0894-536. [DOI] [PubMed] [Google Scholar]
- 7.Christensen BC, Houseman EA, Marsit CJ, Zheng S, Wrensch MR, Wiemels JL, Nelson HH, Karagas MR, Padbury JF, Bueno R, et al. Aging and environmental exposures alter tissue-specific DNA methylation dependent upon CpG island context. PLoS Genet. 2009;5:e1000602. doi: 10.1371/journal.pgen.1000602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liang P, Song F, Ghosh S, Morien E, Qin M, Mahmood S, Fujiwara K, Igarashi J, Nagase H, Held WA. Genome-wide survey reveals dynamic widespread tissue-specific changes in DNA methylation during development. BMC Genomics. 2011;12:231. doi: 10.1186/1471-2164-12-231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, Ballestar ML, Heine-Suñer D, Cigudosa JC, Urioste M, Benitez J, et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci U S A. 2005;102:10604–9. doi: 10.1073/pnas.0500398102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kwabi-Addo B, Chung WB, Shen L, Ittmann M, Wheeler T, Jelinek J, Issa JPJ. Age-related DNA methylation changes in normal human prostate tissues. Clin Cancer Res. 2007;13:3796–802. doi: 10.1158/1078-0432.CCR-07-0085. [DOI] [PubMed] [Google Scholar]
- 11.Rakyan VK, Down TA, Maslau S, Andrew T, Yang TP, Beyan H, Whittaker P, McCann OT, Finer S, Valdes AM, et al. Human aging-associated DNA hypermethylation occurs preferentially at bivalent chromatin domains. Genome Res. 2010;20:434–9. doi: 10.1101/gr.103101.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Teschendorff AE, Menon U, Gentry-Maharaj A, Ramus SJ, Weisenberger DJ, Shen H, Campan M, Noushmehr H, Bell CG, Maxwell AP, et al. Age-dependent DNA methylation of genes that are suppressed in stem cells is a hallmark of cancer. Genome Res. 2010;20:440–6. doi: 10.1101/gr.103606.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Euhus DM, Bu D, Milchgrub S, Xie XJ, Bian A, Leitch AM, Lewis CM. DNA methylation in benign breast epithelium in relation to age and breast cancer risk. Cancer Epidemiol Biomarkers Prev. 2008;17:1051–9. doi: 10.1158/1055-9965.EPI-07-2582. [DOI] [PubMed] [Google Scholar]
- 14.Fackler MJ, Rivers A, Teo WW, Mangat A, Taylor E, Zhang Z, Goodman S, Argani P, Nayar R, Susnik B, et al. Hypermethylated genes as biomarkers of cancer in women with pathologic nipple discharge. Clin Cancer Res. 2009;15:3802–11. doi: 10.1158/1078-0432.CCR-08-1981. [DOI] [PubMed] [Google Scholar]
- 15.Lewis CM, Cler LR, Bu DW, Zöchbauer-Müller S, Milchgrub S, Naftalis EZ, Leitch AM, Minna JD, Euhus DM. Promoter hypermethylation in benign breast epithelium in relation to predicted breast cancer risk. Clin Cancer Res. 2005;11:166–72. [PubMed] [Google Scholar]
- 16.Maegawa S, Hinkal G, Kim HS, Shen L, Zhang L, Zhang J, Zhang N, Liang S, Donehower LA, Issa JP. Widespread and tissue specific age-related DNA methylation changes in mice. Genome Res. 2010;20:332–40. doi: 10.1101/gr.096826.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ohm JE, McGarvey KM, Yu X, Cheng LZ, Schuebel KE, Cope L, Mohammad HP, Chen W, Daniel VC, Yu W, et al. A stem cell-like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing. Nat Genet. 2007;39:237–42. doi: 10.1038/ng1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Widschwendter M, Fiegl H, Egle D, Mueller-Holzner E, Spizzo G, Marth C, Weisenberger DJ, Campan M, Young J, Jacobs I, et al. Epigenetic stem cell signature in cancer. Nat Genet. 2007;39:157–8. doi: 10.1038/ng1941. [DOI] [PubMed] [Google Scholar]
- 19.Zhuang J, Jones A, Lee SH, Ng E, Fiegl H, Zikan M, Cibula D, Sargent A, Salvesen HB, Jacobs IJ, et al. The dynamics and prognostic potential of DNA methylation changes at stem cell gene loci in women’s cancer. PLoS Genet. 2012;8:e1002517. doi: 10.1371/journal.pgen.1002517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carroll JS, Meyer CA, Song J, Li W, Geistlinger TR, Eeckhoute J, Brodsky AS, Keeton EK, Fertuck KC, Hall GF, et al. Genome-wide analysis of estrogen receptor binding sites. Nat Genet. 2006;38:1289–97. doi: 10.1038/ng1901. [DOI] [PubMed] [Google Scholar]
- 21.Fang F, Turcan S, Rimner A, Kaufman A, Giri D, Morris LG, Shen R, Seshan V, Mo Q, Heguy A, et al. Breast cancer methylomes establish an epigenomic foundation for metastasis. Sci Transl Med. 2011;3:75ra25. doi: 10.1126/scitranslmed.3001875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Holm K, Hegardt C, Staaf J, Vallon-Christersson J, Jönsson G, Olsson H, Borg A, Ringnér M. Molecular subtypes of breast cancer are associated with characteristic DNA methylation patterns. Breast Cancer Res. 2010;12:R36. doi: 10.1186/bcr2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baylin SB, Jones PA. A decade of exploring the cancer epigenome - biological and translational implications. Nat Rev Cancer. 2011;11:726–34. doi: 10.1038/nrc3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Christensen BC, Kelsey KT, Zheng S, Houseman EA, Marsit CJ, Wrensch MR, Wiemels JL, Nelson HH, Karagas MR, Kushi LH, et al. Breast cancer DNA methylation profiles are associated with tumor size and alcohol and folate intake. PLoS Genet. 2010;6:e1001043. doi: 10.1371/journal.pgen.1001043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Issa JP. Age-related epigenetic changes and the immune system. Clin Immunol. 2003;109:103–8. doi: 10.1016/S1521-6616(03)00203-1. [DOI] [PubMed] [Google Scholar]
- 26.Schernhammer ES, Giovannucci E, Kawasaki T, Rosner B, Fuchs CS, Ogino S. Dietary folate, alcohol and B vitamins in relation to LINE-1 hypomethylation in colon cancer. Gut. 2010;59:794–9. doi: 10.1136/gut.2009.183707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hannum G, Guinney J, Zhao L, Zhang L, Hughes G, Sadda S, Klotzle B, Bibikova M, Fan JB, Gao Y, et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol Cell. 2013;49:359–67. doi: 10.1016/j.molcel.2012.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Junnila RK, List EO, Berryman DE, Murrey JW, Kopchick JJ. The GH/IGF-1 axis in ageing and longevity. Nat Rev Endocrinol. 2013;9:366–76. doi: 10.1038/nrendo.2013.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Benbassat CA, Maki KC, Unterman TG. Circulating levels of insulin-like growth factor (IGF) binding protein-1 and -3 in aging men: relationships to insulin, glucose, IGF, and dehydroepiandrosterone sulfate levels and anthropometric measures. J Clin Endocrinol Metab. 1997;82:1484–91. doi: 10.1210/jc.82.5.1484. [DOI] [PubMed] [Google Scholar]
- 30.Waters DL, Yau CL, Montoya GD, Baumgartner RN. Serum Sex Hormones, IGF-1, and IGFBP3 Exert a Sexually Dimorphic Effect on Lean Body Mass in Aging. J Gerontol A Biol Sci Med Sci. 2003;58:648–52. doi: 10.1093/gerona/58.7.M648. [DOI] [PubMed] [Google Scholar]
- 31.Li Z, Zhang W, Shao Y, Zhang C, Wu Q, Yang H, Wan X, Zhang J, Guan M, Wan J, et al. High-resolution melting analysis of ADAMTS18 methylation levels in gastric, colorectal and pancreatic cancers. Med Oncol. 2010;27:998–1004. doi: 10.1007/s12032-009-9323-8. [DOI] [PubMed] [Google Scholar]
- 32.Menschikowski M, Platzbecker U, Hagelgans A, Vogel M, Thiede C, Schönefeldt C, Lehnert R, Eisenhofer G, Siegert G. Aberrant methylation of the M-type phospholipase A(2) receptor gene in leukemic cells. BMC Cancer. 2012;12:576. doi: 10.1186/1471-2407-12-576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roh JL, Wang XV, Manola J, Sidransky D, Forastiere AA, Koch WM. Clinical correlates of promoter hypermethylation of four target genes in head and neck cancer: a cooperative group correlative study. Clin Cancer Res. 2013;19:2528–40. doi: 10.1158/1078-0432.CCR-12-3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boberg DR, Batistela MS, Pecharki M, Ribeiro EM, Cavalli IJ, Lima RS, Urban CA, Furtado-Alle L, Souza RL. Copy number variation in ACHE/EPHB4 (7q22) and in BCHE/MME (3q26) genes in sporadic breast cancer. Chem Biol Interact. 2013;203:344–7. doi: 10.1016/j.cbi.2012.09.020. [DOI] [PubMed] [Google Scholar]
- 35.Riener MO, Nikolopoulos E, Herr A, Wild PJ, Hausmann M, Wiech T, Orlowska-Volk M, Lassmann S, Walch A, Werner M. Microarray comparative genomic hybridization analysis of tubular breast carcinoma shows recurrent loss of the CDH13 locus on 16q. Hum Pathol. 2008;39:1621–9. doi: 10.1016/j.humpath.2008.02.021. [DOI] [PubMed] [Google Scholar]
- 36.Shibata-Minoshima F, Oki T, Doki N, Nakahara F, Kageyama SI, Kitaura J, Fukuoka J, Kitamura T. Identification of RHOXF2 (PEPP2) as a cancer-promoting gene by expression cloning. Int J Oncol. 2012;40:93–8. doi: 10.3892/ijo.2011.1173. [DOI] [PubMed] [Google Scholar]
- 37.Zeng L, Jarrett C, Brown K, Gillespie KM, Holly JM, Perks CM. Insulin-like growth factor binding protein-3 (IGFBP-3) plays a role in the anti-tumorigenic effects of 5-Aza-2′-deoxycytidine (AZA) in breast cancer cells. Exp Cell Res. 2013;319:2282–95. doi: 10.1016/j.yexcr.2013.06.011. [DOI] [PubMed] [Google Scholar]
- 38.Tomii K, Tsukuda K, Toyooka S, Dote H, Hanafusa T, Asano H, Naitou M, Doihara H, Kisimoto T, Katayama H, et al. Aberrant promoter methylation of insulin-like growth factor binding protein-3 gene in human cancers. Int J Cancer. 2007;120:566–73. doi: 10.1002/ijc.22341. [DOI] [PubMed] [Google Scholar]
- 39.Brooksby B, Pogue BW, Jiang S, Dehghani H, Srinivasan S, Kogel C, Tosteson TD, Weaver J, Poplack SP, Paulsen KD. Imaging breast adipose and fibroglandular tissue molecular signatures by using hybrid MRI-guided near-infrared spectral tomography. Proc Natl Acad Sci U S A. 2006;103:8828–33. doi: 10.1073/pnas.0509636103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cancer Genome Atlas Network Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rocke DM. On the Beta-Transformation Family. Technometrics. 1993;35:72–81. doi: 10.1080/00401706.1993.10484995. [DOI] [Google Scholar]
- 42.Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 43.Huang W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bracken AP, Dietrich N, Pasini D, Hansen KH, Helin K. Genome-wide mapping of Polycomb target genes unravels their roles in cell fate transitions. Genes Dev. 2006;20:1123–36. doi: 10.1101/gad.381706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee TI, Jenner RG, Boyer LA, Guenther MG, Levine SS, Kumar RM, Chevalier B, Johnstone SE, Cole MF, Isono K, et al. Control of developmental regulators by Polycomb in human embryonic stem cells. Cell. 2006;125:301–13. doi: 10.1016/j.cell.2006.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schlesinger Y, Straussman R, Keshet I, Farkash S, Hecht M, Zimmerman J, Eden E, Yakhini Z, Ben-Shushan E, Reubinoff BE, et al. Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat Genet. 2007;39:232–6. doi: 10.1038/ng1950. [DOI] [PubMed] [Google Scholar]
- 47.Squazzo SL, O’Geen H, Komashko VM, Krig SR, Jin VX, Jang SW, Margueron R, Reinberg D, Green R, Farnham PJ. Suz12 binds to silenced regions of the genome in a cell-type-specific manner. Genome Res. 2006;16:890–900. doi: 10.1101/gr.5306606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gerstein MB, Kundaje A, Hariharan M, Landt SG, Yan KK, Cheng C, Mu XJ, Khurana E, Rozowsky J, Alexander R, et al. Architecture of the human regulatory network derived from ENCODE data. Nature. 2012;489:91–100. doi: 10.1038/nature11245. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.