

Abstract
Cervical cancer is a major health concern among women in Latin America due to its high incidence and mortality. Therefore, the discovery of molecular markers for cervical cancer screening and triage is imperative. The aim of this study was to use a genome wide DNA methylation approach to identify novel methylation biomarkers in cervical cancer. DNA from normal cervical mucosa and cervical cancer tissue samples from Chile was enriched with Methylated DNA Immunoprecipitation (MeDIP), hybridized to oligonucleotide methylation microarrays and analyzed with a stringent bioinformatics pipeline to identify differentially methylated regions (DMRs) as candidate biomarkers. Quantitative Methylation Specific PCR (qMSP) was used to study promoter methylation of candidate DMRs in clinical samples from two independent cohorts. HPV detection and genotyping were performed by Reverse Line Blot analysis. Bioinformatics analysis revealed GGTLA4, FKBP6, ZNF516, SAP130, and INTS1 to be differentially methylated in cancer and normal tissues in the Discovery cohort. In the Validation cohort FKBP6 promoter methylation had 73% sensitivity and 80% specificity (AUC = 0.80). ZNF516 promoter methylation was the best biomarker, with both sensitivity and specificity of 90% (AUC = 0.92), results subsequently corroborated in a Prevalence cohort. Together, ZNF516 and FKBP6 exhibited a sensitivity of 84% and specificity of 81%, when considering both cohorts. Our genome wide DNA methylation assessment approach (MeDIP-chip) successfully identified novel biomarkers that differentiate between cervical cancer and normal samples, after adjusting for age and HPV status. These biomarkers need to be further explored in case-control and prospective cohorts to validate them as cervical cancer biomarkers.
Keywords: companion diagnostics, biomarkers, cervical cancer, promoter methylation, MeDIP
Introduction
Cervical cancer represents the third most common cancer and the fourth cause of cancer death worldwide among women, despite decreasing incidence and mortality rates in the developed world.1 The implementation of cytology screening using the Papanicolaou (Pap) test for the detection of Low-Grade Squamous Intraepithelial Lesions (LSIL) and High-Grade Squamous Intraepithelial Lesions (HSIL), the precursor lesions of cervical cancer, had an important role in the decrease of cervical cancer incidence seen in developed countries.2 However, in developing nations, lack of infrastructure and resources has been a major obstacle for the effective implementation of routine screening strategies. As a result, around 80% of cervical cancers diagnosed in these countries represent the most common cause of cancer-related deaths among women and the leading cause of death overall.3
Human papillomavirus (HPV), the most common sexually transmitted infection, causes virtually all cases of cervical cancer, with high-risk serotypes HPV 16 and HPV 18 being responsible for 70% of these worldwide.4 The progression from LSIL to HSIL and to invasive carcinoma is usually slow, taking several years, sometimes decades.5 This prolonged course provides an excellent window to implement screening tools, like the Pap smear, to effectively detect, treat, and cure the precursor lesion of cervical carcinoma. Nonetheless, Pap smear is limited by having a low sensitivity (55%) for detection of high-grade cervical lesions and an increased number of false-negative results.3,6 Furthermore, the awareness of the strong correlation between persistent infection with HPV and cervical cancer has prompted the development of HPV-based DNA tests for screening. These tests are more sensitive than Pap smears and appear to be more reproducible from one laboratory to another.7 Co-testing with cytology and HPV at 5-y intervals is now a highly recommended strategy for cervical cancer screening for women aged 30–64 in the US, mainly because HPV-negative/Pap-negative women have very low cervical cancer risk.8-10 Clinical management for HPV-positive/Pap-negative women, however, is not firmly established.11,12 Co-testing with Pap and HPV has higher sensitivity and specificity than by themselves, but cannot predict who will progress to cervical carcinoma.13,14 Furthermore, privacy, cultural, and resources considerations are barriers to the effective implementation of cervical cytology and HPV screening for millions of women worldwide.15 Novel biomarkers with higher sensitivity and specificity to improve detection rates may help reduce the cervical cancer burden in developing countries.
Nowadays, it is well recognized that epigenetic changes play an important role in cancer initiation and progression.16 Epigenetic changes affect gene expression without changing the DNA sequence and they comprise DNA methylation, histone modifications and nucleosome repositioning.17-19 DNA methylation is defined as the addition of a methyl group on a cytosine that precedes a guanosine (known as CpG). These CpGs cluster in regions known as CpG islands, which are usually located in the 5′end of many genes with tumor suppressor function and are commonly unmethylated in normal cells.20 Promoter methylation is a common mechanism leading to gene inactivation,17,21 and has been found to be a potential biomarker for several types of cancer, including cervical cancer.22-25
The aim of this study was to use a global DNA methylation approach for the discovery of novel potential methylation based biomarkers that could differentiate cervical cancer from normal samples. Specifically, we used the Methylated DNA immunoprecipitation (MeDIP) assay, an unbiased and high-throughput method to detect novel differentially methylated regions, to enrich for methylated DNA in seven invasive cervical carcinomas and 12 normal samples (Fig. 1). The differentially methylated candidate genes discovered by this approach, were further validated in two larger independent cohorts of normal, LSIL, HSIL, and cervical cancer samples by quantitative methylation-specific PCR (qMSP). Molecular data was then compared with demographic and clinico-pathological characteristics of the patients. All samples were collected in Chile.
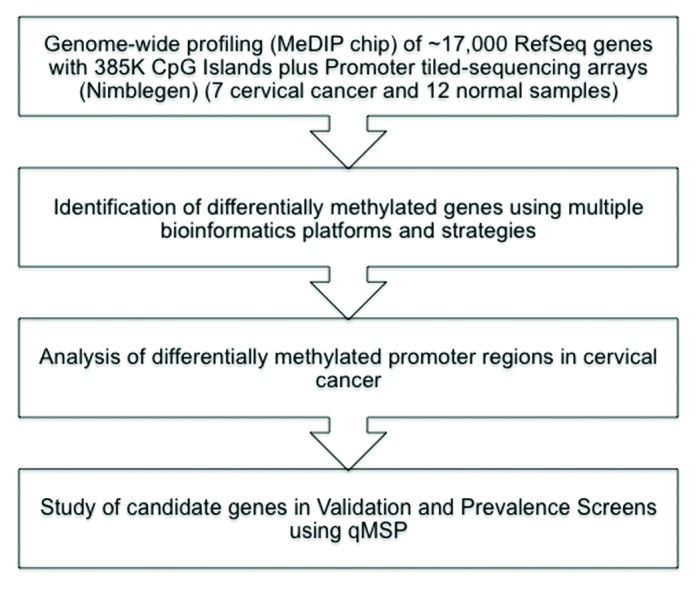
Figure 1. Flowchart of the data analysis and integration tasks performed to identify methylated and downregulated biomarkers in cervical cancer.
Results
Patient characteristics
Characteristics of the participants in this study are described in Table S1. The median age of cervical cancer patients was significantly older (51 y) than that of normal patients (41 y), patients with LSIL (40) and patients with HSIL (35) (all P < 0.01). 76% (n = 223) of the patients were of Hispanic descent (non-Mapuche) and 24% (n = 71) were of Mapuche (indigenous inhabitants of south-central Chile) descent. HPV genotyping with PCR and Reverse Line Blot analyses revealed that 80% of the participants (234/294) were HPV positive. As expected, the prevalence of infection from HPV 16 (70%) and HPV 18 (23%) was the highest among cancer patients. In ten of these patients (8%), both HPV 16 and 18 were present. Age, ethnicity and HPV status of the patients selected from MeDIP-chip are listed in Table S2.
Table S3 compares the characteristics of patients selected for the Validation and Prevalence cohorts. There were no demographic differences between the Validation and Prevalence cohorts in the normal samples. Nevertheless, cancer patients in the Prevalence cohort were more from Mapuche (P = 0.02) and indigent (P = 0.02) participants, compared with the Validation cohort.
Genome wide profiles of promoter methylation
Our bioinformatics pipeline identified a total of 444 gene loci significantly methylated, subdivided according to two cutoffs defined for the maximal distance between a methylation peak and the TSS: –1000 to +1000, called the standard cut-off; –500 to +500, called the narrow cut-off. We found 255 unique gene loci that are cancer specific methylated according to the standard cutoff and 189 according to the narrow cutoff. One hundred and sixty two (162) genes were jointly identified by the intersection between both lists. These stringent criteria ensured that all 10 genes selected for biomarker validation had methylated peaks within a CPG island located in the promoter region, 500 base pairs upstream from the TSS in all the hybridized tumor samples and none in the normal samples hybridized to the arrays.
The individual probe methylation values were log-transformed and used to generate a heatmap based on unsupervised hierarchical clustering. The clustering by methylated CpG loci distinguished between normal and cervical cancer samples. A subset of genes showed promoter methylation in cervical cancer tissues compared with normal cervical tissues. Tumor samples also showed evidence of global loss of methylation when compared with normal tissue samples. Gain of promoter methylation and loss of methylation typically in repetitive regions are hallmarks of tumor cells (Fig. 2).26-28 Chromosomal profiles of differentially methylated loci, such as those described in Figure S1A and B, may eventually be used for predictive biomarker discovery and research development.29
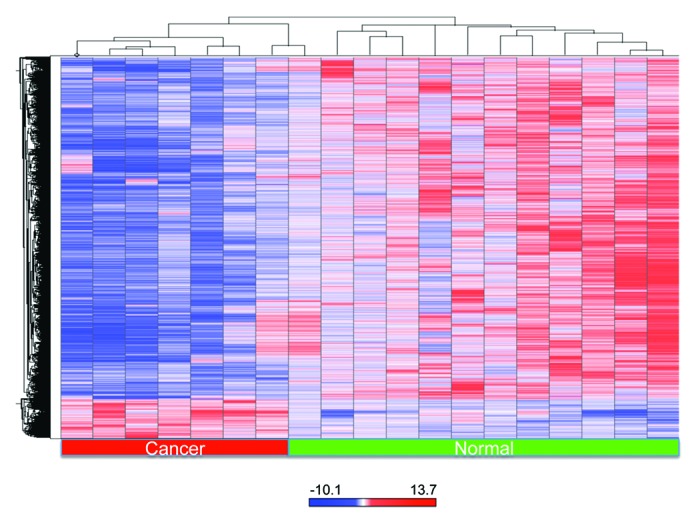
Figure 2. Heatmap of a subset of statistically significant methylated probes with more than 2-fold change differential methylation value when comparing normal to tumor samples (Unsupervised clustering). Because the empirical P values were calculated genome-wide, adjustment for multiple testing was performed. The P values were transformed into q-values, using the Benjamin–Hochberg correction. The probes that were found to have q-values less than 0.05 were deemed to be statistically significant and were included in the final gene list. The red color was selected to represent methylated gene promoters and the blue color to represent unmethylated genes. The red bar at the bottom indicates tumor samples and the green bar indicates normal samples.
Genome-wide evaluation reveals promoter methylation of ZNF516 and FKBP6 as biomarkers in cervical cancer
We identified 2044 differentially methylated probes between tumor and normal samples. More than half of the methylated gene promoters identified by the Nimblegen protocol (60%) were hypermethylated in all cancer samples and not in normal samples. The top five genes in the list that contained CpG islands in their promoter regions were selected for further analysis with MSP and qMSP. These genes were: GGTLA4, FKBP6, ZNF516, SAP130, and INTS1. The promoter region around the TSS site of these five genes (1) and the graphical representations of the bisulfite sequencing results (2) tested in the Discovery cohort are shown in Figure S2A (FKBP6); Figure S2B (GGTLA4); Figure S2C (INTS1); Figure S2D (SAP130); and Figure S2E (ZNF516). The MSP results for the Discovery Samples are shown in Figure S3A. The MSP results for the selected Prevalence and Premalignant samples are shown in Figure S3B.
Promoter methylation analysis of FKBP6, INTS1, ZNF516, SAP130, and GGTLA4 was then quantified by qMSP (Fig. 3A) in the Validation cohort (19 normal and 30 cancer samples). Correlation with clinical diagnosis, Area Under the Curve, methylation cut-off values, sensitivity, specificity, and the percentage of correctly classified patients are shown in Table 1. Promoter methylation of FKBP6, INTS1 and ZNF516, was further evaluated by qMSP (Fig. 3B) in the Prevalence cohort (20 normal samples and 90 cancer samples) using the cutoffs identified in the Validation cohort for each gene promoter.

Figure 3. (A) Scatterplots of qMSP analysis for candidate gene promoters in the Validation cohort (normal n = 19, cancer n = 30). The relative level of methylated DNA for each gene in each sample was determined as a ratio of MSP for the amplified gene to β-actin. Red line denotes cut-off value. (B) Scatterplots of qMSP analysis of FKBP6, INTS1, and ZNF516 in the Prevalence cohort (normal n = 18, cancer n = 90). The relative level of methylated DNA for each gene in each sample was determined as a ratio of MSP for the amplified gene to β-actin. Red line denotes cut-off value.
Table 1. Predictive accuracy of FKBP6, INTS1, ZNF516, SAP130, and GGTLA4 with cervical cancer.
| Gene | Spearman correlation coefficient | P value | AUC | Methylationcut-off | Sensitivity | Specificity | Correctly classified |
|---|---|---|---|---|---|---|---|
| Discovery cohort (n = 49) |
|||||||
| FKBP6 | 0.506 | <0.001 | 0.800 | 59.58 | 73% | 79% | 76% |
| INTS1 | 0.255 | 0.077 | 0.651 | 61.34 | 50% | 74% | 59% |
| ZNF516 | 0.752 | <0.001 | 0.946 | 198.68 | 90% | 95% | 92% |
| SAP130 | –0.552 | <0.001 | 0.289 | 6.94 | 0% | 84% | 33% |
| GGTLA4 | –0.059 | 0.686 | 0.465 | 90.78 | 47% | 47% | 47% |
| Prevalence cohort (n = 108) | |||||||
| FKBP6 | 0.361 | <0.001 | 0.768 | 59.58 | 58% | 83% | 73% |
| INTS1 | 0.220 | 0.035 | 0.664 | 61.34 | 41% | 76% | 48% |
| ZNF516 | 0.418 | <0.001 | 0.828 | 198.68 | 60% | 100% | 66% |
Using the most optimal cut-off as determined by the Area Under the Curve (AUC) of the Receiver Operator Characteristics (ROC) curve,30 FKBP6 methylation (cut-off 59.58) had a sensitivity of 73%, a specificity of 80% and an AUC of 0.80 in the Validation cohort (Fig. S4A); INTS1 (cut-off 61.34) had a sensitivity of 50%, specificity of 70% and an AUC of 0.63 (Fig. S4B); ZNF516 methylation (cut-off 198.68) was found to be the most accurate for cancer detection with sensitivity of 90%, specificity of 90% and an AUC of 0.92 in the Validation cohort (Fig. S4C). ROC analysis results similar to the Validation cohort were noted in the Prevalence cohort, with a sensitivity and specificity of 60% and 80% for FKBP6, 43% and 72% for INTS1, and 61% and 90% for ZNF516 respectively, indicating that ZNF516 methylation has the best predictive value. ZNF516 had an AUC of 0.76, FKBP6 0.74 and INTS1 0.64 (Fig. S4D–F). Together, ZNF516 and FKBP6 achieved sensitivity of 84% and specificity of 81%, considering both cohorts (Fig. S4G).
FKBP6, INTS1, and ZNF516 were further evaluated by qMSP in 137 premalignant lesions tissue samples (LSIL = 53, HSIL = 84) and compared with the normal (n = 37) and cervical samples (n = 120), combining the Validation and Prevalence cohorts. Figure S5 shows scatter plots for all normal, LSIL, HSIL, and cancer samples. Table S3 compares median methylation values for the three genes in normal, premalignant and cervical cancer lesions. FKBP6 median methylation levels for normal samples (median 32.69) were significantly lower (all P < 0.01) than those for LSIL (median 121.50), HSIL (median 79.65), and cancer (median 74.54). Interestingly, LSIL median methylation values were significantly higher than HSIL (P < 0.01) and cancer (P < 0.01) values. HSIL and cancer median methylation values were not significantly different. INTS1 methylation levels were similar in cancer (median 55.01) and LSIL (median 55.78, P = 0.84) and HSIL (median 48.07, P = 0.18) tissues but significantly higher compared with the normal cervical samples (median 40.35, P < 0.01). ZNF516 methylation levels in normal samples (median 84.94) were significantly lower than LSIL (median 235.94), HSIL (median 136.42), and cancer (median 273.75) (all P < 0.01). Interestingly, as in INTS1 and FKBP6, ZNF516 median methylation in LSIL was higher than in HSIL (P < 0.01). Absolute numbers of patients with absence or presence of methylation according to the cut-offs defined in the Validation cohort are shown in Table S4.
Promoter methylation is associated with HPV status, age and ethnicity
Logistic regression analysis of various clinical characteristics in all 37 normal and 120 cancer samples revealed that methylation of FKBP6 was related to the presence of HPV infection (OR = 4.51, 95% C.I. = 2.04–9.97, P < 0.001) (Table 2). ZNF516 methylation was associated with older age (OR = 1.02, 95% C.I. = 1.00–1.05, P = 0.03) and HPV infection (OR = 11.84, 95% C.I. = 4.59–30.57, P < 0.001). A borderline significant association was found between methylation of ZNF516 and ethnicity: promoter methylation was less frequently observed in Mapuche than in non-Mapuche participants (OR = 0.50, 95% C.I. = 0.25–1.01, P = 0.05).
Table 2. Logistic regression results showing the association of promoter methylation with HPV status and socio-demographic variables.
| FKBP6 methylation present | OR | (95% C.I.) | P value |
|---|---|---|---|
| Age (continuous) | 1.02 | (1.00–1.04) | 0.09 |
| Ethnicity (Mapuche) | 0.66 | (0.32–1.36) | 0.26 |
| Socio-economic status (non-indigent) | 0.72 | (0.37–1.38) | 0.32 |
| HPV infection (present) | 4.51 | (2.04–9.97) | < 0.01 |
| INTS1 methylation present | |||
| Age (continuous) | 1.00 | (0.98–1.03) | 0.78 |
| Ethnicity (Mapuche) | 0.78 | (0.37–1.64) | 0.51 |
| Socio-economic status (non-indigent) | 0.94 | (0.48–1.86) | 0.87 |
| HPV infection (present) | 1.82 | (0.84–3.98) | 0.13 |
| ZNF516 methylation present | |||
| Age (continuous) | 1.02 | (1.00–1.05) | 0.03 |
| Ethnicity (Mapuche) | 0.50 | (0.25–1.01) | 0.05 |
| Socio-economic status (non-indigent) | 1.48 | (0.78–2.78) | 0.23 |
| HPV infection (present) | 11.84 | (4.59–30.57) | <0.01 |
OR, Odds Ratio; 95% C.I., 95% Confidence Interval.
We subsequently examined whether promoter methylation of FKBP6, ZNF516, and INTS1 could discriminate between HPV positive and HPV negative, normal and cancer samples. During bivariate analysis we found a significant association between the clinical diagnosis of cancer and both age (OR = 1.05, 95% C.I. = 1.02–1.08, P < 0.01), and presence of HPV infection (OR = 139.78, 95% C.I. = 35.81–545.66, P < 0.01). Therefore, we then fitted independent unadjusted and adjusted logistic regression models to evaluate the association between clinical diagnosis of cancer and promoter methylation of FKBP6, INTS1 and ZNF516 to assess the potential confounding effect of age and HPV status. This analysis revealed that methylation of FKBP6 (OR = 7.15, 95% C.I. = 1.45–35.34, P = 0.01) and ZNF516 (OR = 26.72, 95% C.I. = 2.61–273.05, P < 0.01) were associated with cervical cancer diagnosis, independently of age and HPV infection (Table 3).
Table 3. Logistic regression results showing the association of promoter methylation with tumor status.
| Cervical cancer present (unadjusted) | OR | (95% C.I.) | P value |
|---|---|---|---|
| FKBP6 methylated | 7.11 | (2.86–17.65) | <0.01 |
| INTS1 methylated | 2.34 | (1.00–5.46) | 0.05 |
| ZNF516 methylated | 71.79 | (9.48–543.54) | <0.01 |
| Cervical cancer present (adjusted for age and HPV status) | |||
| FKBP6 methylated | 7.15 | (1.45–35.34) | 0.02 |
| INTS1 methylated | 3.56 | (0.75–16.87) | 0.11 |
| ZNF516 methylated | 26.72 | (2.61–273.05) | <0.01 |
OR, Odds Ratio; 95% C.I., 95% Confidence Interval.
Discussion
In the present study, we used a comprehensive genome wide DNA methylation profiling approach to identify differentially methylated regions in cervical cancer compared with normal cervical tissue. After a detailed bioinformatics analysis, we selected five genes for validation in independent cohorts using qMSP. ZNF516 and FKBP6 demonstrated higher methylation frequencies and levels in cancer when compared with normal tissue. Promoter methylation of ZNF516 showed sensitivity of 90% and specificity of 95% in the Validation cohort and 60% and 100% in the prevalence cohort. FKBP6 methylation presented a sensitivity of 73% and specificity of 79% in the Validation cohort and 41% and 79% in the Prevalence cohort. Considering all the samples, ZNF516 and FKBP6 as a panel achieved a higher predictive power. When either of these genes was methylated, sensitivity was 84% and specificity 81%. We also found a significant association between promoter methylation and positive HPV status. Importantly, however, methylation of either of these genes was associated with cancer, independently of HPV status, which renders them as possible additional markers along with Pap and HPV co-testing.
Promoter methylation of these two genes was also analyzed in LSIL and HSIL lesions. FKBP6 presented similar levels of methylation in high-grade lesions and cancer, but intriguingly low-grade lesions showed higher levels of methylation than both, and this pattern was similar to the one found for ZNF516. Promoter methylation of both genes could perhaps be a driver or a passenger mark of the inflammatory process associated to cervical oncogenesis and progression, given the fact that a fraction of HSIL progress to cancer and some regress.31 Trimble et al.32 showed that around 30% of HSIL regress spontaneously, those infected with HPV16 being less likely to regress. Meanwhile, the majority of LSIL ultimately regresses. That is why these lesions are generally not treated and a follow-up PAP test is performed. However, there are no specific markers of progression from LSIL to HSIL or from HSIL to carcinoma.
The fact that promoter methylation of ZNF516 and FKBP6 distinguishes malignant lesions from normal cervical tissue and is also present in LSIL and HSIL lesions, enhances their potential use as biomarkers of progression to cervical cancer. They can possibly be used in combination with other molecular markers, such as HPV methylation33,34 in HPV positive patients. Therefore, HPV positive patients with LSIL and HSIL could be followed longitudinally, to evaluate whether promoter methylation of ZNF516 and FKBP6 together with other potential biomarkers of progression, are associated with progression of the lesion to carcinoma. However, our results should be further validated in case-control and prospective studies that focus on premalignant lesions, to assess and evaluate in which context they could be complementary to HPV testing, before we can establish which lesions from the ones harboring these epigenetic alterations are most likely to progress to a malignant state.
The approach we followed in this project was divided in two phases: (1) an unbiased genome-wide evaluation of the methylome for candidate gene selection in a Discovery cohort and; (2) confirmation of candidate genes by MSP and qMSP in Prevalence and Validation cohorts. Candidate genes identified with this approach may serve as potential biomarkers for positive identification and progression monitoring of premalignant cervical cancer lesions, either independently or in combination with HPV testing and cytological examination.35,36
This approach is appropriate in this era of genomic high-throughput analyses, because validation techniques in patient samples are needed to consolidate genome-wide findings. Using a similar approach, Lendvai et al.23 showed evidence that COL25A1 and KATNAL2 promoter methylation could distinguish between low-grade and high-grade lesions. Methylation was present in high-grade lesions and cancer but not in low-grade lesions and normal samples, demonstrating their potential for early diagnosis and possible role in cancer progression. When compared with our results, our differential methylation study identified regions that could significantly distinguish between premalignant and malignant lesions vs. normal tissue, but not between low- and high-grade premalignant lesions. In another study conducted by Huang et al.,37 a MeDIP-array was used to profile the methylation status of genes in cervical cancer tissues and normal cervical scrapings. ZNF582 was found to be hypermethylated in HSIL lesions and cervical cancer samples, after validation in an independent cohort. More recently Farkas et al. identified 24 potential biomarkers for cervical cancer using the 450K Illumina Infinium Human Methylation BeadChip assay. Fifteen of the 24 candidate biomarkers have not yet been correlated with any cancer type, while eight of them have been identified in tumor sites, other than cervical.38 These studies have demonstrated that global DNA methylation assays, like MeDIP-arrays, are powerful tools for the genome-wide study of methylated genes in cervical cancer. The fact that we did not find any correlation between the candidate genes reported by these studies and the ones we are reporting may be due to differences in sample source, sample collection, microarray kinetics, bioinformatics pipelines, genetic and/or cultural factors, all of which can affect the end results of these complex experiments.
In our study, the normal, premalignant and 20% of the tumor samples were obtained by cytobrush, which provides a heterogeneous population of cells, inevitably leading to misrepresentation of cell types and therefore of methylation status. This can possibly explain the reason we didn’t observe significant differences in methylation between low and high-grade premalignant lesions using qMSP.
FKBP6 (FK-506-binding protein 6) is a member of the immunophilins FKBP family located on chromosome 7q11.23 and is expressed in various tissues with the highest expression levels observed in testis.39 Mutations in FKBP6 have been associated to male infertility both in mice and humans.40FKBP6 is deleted in Williams-Beuren syndrome, a developmental disorder.39 To our knowledge, this is the first study reporting an association of FKBP6 with cancer, and specifically with cervical cancer. Nevertheless, further genetic and epigenetic studies are needed to decipher its role in carcinogenesis.
ZNF516 (Zinc finger protein 516) is located on chromosome 18q23. Burrell et al.41 identified three cancer suppressor genes on 18q (being ZNF516 one of them), and also demonstrated that gene silencing of these genes led to DNA replication stress, structural chromosome abnormalities, and chromosome disaggregation, all characteristics of chromosomal instability. Our data on ZNF516 being hypermethylated in cervical cancer when compared with normal cervical mucosa reinforces its potential as a tumor suppressor gene in cervical cancer.
In Latin America, cervical cancer has high incidence and mortality rates, in part related to the aggressive nature of the disease and the late diagnosis at presentation that is frequently seen, making it one of the most important women health issues. The findings reported in this manuscript could potentially have clinical implications for the studied population from Chile, which has one of the highest cervical cancer mortality rates in the world, and the highest mortality-to-incidence ratios for cervical cancer in Latin America.42 An assay that quantifies promoter methylation of ZNF516 and FKBP6, validated as a cervical cancer screening biomarker in larger cohorts with well-known follow up, demographic and clinicopathological data, could potentially reduce the mortality-to-incidence ratio for this neoplasm in Chile and other countries worldwide.
The utility of additional biomarkers, as for example the promoter methylation of ZNF516 and FKBP6, in conjunction with joint Pap smear and HPV-DNA testing warrants further investigation in women of different ethnic and geographic backgrounds.22,43 Comprehensive prospective population-based studies using standardized methylation assays are needed before these promoter methylated sequences can be translated into useful cervical cancer biomarkers that could eventually be adopted by the clinical community.22
Methods
Clinical samples
Tissue samples were collected from 2004 to 2008; at the high-risk cervical cancer clinic of Doctor Hernán Henríquez Aravena (HHA) tertiary care regional hospital, in Temuco, Chile. The diagnosis was confirmed by histological examination (biopsy) performed by a team of three pathologists from HHA. A random set of pathology slides from the study samples was sent for diagnostic confirmatory review to a pathologist at Johns Hopkins School of Medicine. The Institutional Review Boards of the HHA and the Johns Hopkins School of Medicine approved the protocol for this study. Normal, LSIL and HSIL samples used in this study were collected by cytobrush. Tumor samples were mostly obtained from formalin-fixed paraffin-embedded (FFPE) blocks, with the exception of a fraction that were collected by cytobrush during surgery.
To determine the methylation status of promoter regions across the genome, 12 normal and 7 cervical cancer tissue samples, all 19 collected by cytobrush to diminish tissue collection-bias, were enriched for methylated DNA with MeDIP and hybridized to oligonucleotide tiled-sequencing arrays (385K CpG Islands plus Promoter arrays, Nimblegen). After detailed bioinformatics analysis, a list of genes was generated and MSP (including an initial bisulfite sequencing step) was used to evaluate their methylation status in the same samples hybridized to the array. We used qMSP to examine in Validation and Prevalence cohorts the promoter methylation of candidate genes discovered by MeDIP-chip. The Validation and Prevalence cohorts were created by randomly choosing DNA samples isolated from 37 normal and 120 cancer patients. The 37 normal samples were collected by cytobrush and the 120 tumor samples were obtained from FFPE blocks. The Validation cohort consisted of 19 normal and 30 cancer patients. The remaining 18 normal and 90 cancer samples formed the Prevalence cohort. As a final experiment we compared the qMSP results obtained in these two cohorts (Validation and Prevalence) with cervical brush biopsies from 53 LSIL and 84 HSIL patients to evaluate the usefulness of these markers as progression markers in cervical cancer.
HPV genotyping
HPV detection and genotyping were performed as previously described.44 Reverse Line Blot42 analysis was performed using 38 modified oligoprobes for the analysis. A panel of 36 HPV viral types was used as positive control. HPV 16, 18, 31, and 33 were commercial plasmid clones (ATCC) and the remaining HPV types were provided by Dr Peter Snijders (VU University Medical Center, Amsterdam, The Netherlands). Negative controls consisted of commercial genomic DNA (Promega) and non-template controls consisted of molecular grade water.
DNA extraction
DNA was extracted from tissue samples obtained from cytobrush and FFPE blocks. Samples were digested with 1% SDS and 20 μg/mL proteinase K (Sigma) at 48° C for 48 h, followed by phenol/chloroform extraction and ethanol precipitation (standard methods).
MeDIP Discovery workflow
Design, implementation, and validation of the MeDIP-chip experiment workflow were performed at Johns Hopkins University. DNA samples were sent to Johns Hopkins School of Medicine for MeDIP enrichment and then shipped to Iceland for sample labeling, array hybridization, and methylation array scanning in Nimblegen’s laboratories.
Methylated DNA enrichment and array hybridization
DNA from normal cervical mucosa (n = 12) and cervical cancer tissue (n = 7) samples obtained by cytobrush, were enriched with MeDIP, labeled and hybridized to the 385K CpG Islands plus Promoter oligonucleotide tiling arrays (Nimblegen). The single array design covers all 28 ,226 UCSC Genome Browser-annotated CpG islands and the promoter regions for all RefSeq genes. The promoter region covered is one kilobase (–800 to +200 relative to the transcription start sites).
The MagMeDIP kit (Diagenode) was used to enrich DNA with methylated cytosines according to the manufacturer’s protocol. Genomic DNA (500 ng) was sheared using a water bath sonicator (Bioruptor UCD-200, Diagenode) at “LOW” power setting in the following cycles: (alternating 5 min sonication and 2 min on ice) for a total sonication time of 15 min. Sonicated DNA was then analyzed on a 1.5% agarose gel to ensure that sonicated fragments had an optimal size of 200–1000 bp. Sonicated DNA was denatured for 10 min at 95 °C and immunoprecipitated with monoclonal antibody against 5-methylcytidine. The immunoprecipitated methylated DNA (IP) and the input genomic DNA was amplified and purified with the GenomePlex Complete Whole Genome Amplification (WGA) Kit (Sigma-Aldrich) and the QIAquick PCR Purification Kit (Qiagen). IP DNA (2 μg) was labeled with Cy5 fluorophore and the input genomic DNA was labeled with Cy3 fluorophore. Labeled DNAs were combined and hybridized to the 385K Human CpG Island-Plus-Promoter Array (Nimblegen).
Differential methylation bioinformatics
The standard Nimblegen algorithms were used to compute the normalized data and identify peaks of enrichment, coinciding with methylated regions. The methylation peak scores for each probe in the methylation arrays were calculated and ranked using the ACME algorithm as implemented by default.45 Non-unique probes were not filtered from the analysis. In the Peak Identification workflow step, NimbleGen uses a permutation-based algorithm to find statistically significant peaks likely to be representative of methylation events. This analysis estimates the false discovery rate (FDR) for each peak. A cutoff for significance of 90% was utilized. This value is the percentage of a hypothetical maximum log2 ratio (mean + 6 standard deviation). The percent step interval for decreasing cutoff values was set to 1 and the number of cutoff steps was set to 76. The sliding window was set to 500 base pairs. The minimum number of probes required to call a peak when not all probes in the window were above cutoff was set to 4. The minimum number of probes required to call a peak when all probes in the window were above cutoff was set to 2. The data was permuted within each chromosome. The FDR was calculated across all data, optimizing for strong peaks of varied widths, stratified on number of probes greater than cutoff in peak.
Next, the data were transformed into a more usable format, i.e., the peaks near known transcription start sites (TSSs) were identified, according to two different cut-offs for the maximal distance between a peak and a TSS: –1000 to +1000, called the standard cut-off; –500 to +500, called the narrow cut-off.
In a first pass analysis at the probe-set level, the cancer specific methylated gene promoters were identified as those genes that had a methylated probe-set in at least one of the primary cancer samples and in none of the normal samples. To maximize the amount of informative loci, this condition was set at a slightly more stringent level: the cancer specific methylated gene promoters were identified as those genes that had a methylated probe-set in 20% or more of the cancer cases. Practically, this is equivalent to at least two samples with methylated probe-sets for a particular gene, out of a total of seven tumor samples. A third, more stringent inclusion criterion was implemented to identify cancer specific methylated gene promoters: genes needed to have methylated probe-sets in 100% of cancer and in none of the normal tissues to satisfy this criterion. This was the criterion we used to select differentially methylated candidates.
Methylated genes in this project had methylated probe-sets in 100% of cancer and in none of the normal tissues. We then excluded the probes, within the candidate gene probe-sets, that mapped to chromosomal regions outside of an 800 base pairs window upstream from the transcription start site (TSS), a region that lies within the standard cutoff. Finally, we used the methylation peak scores to rank the methylated probes. The genes with the top ten scoring probes were selected for validation with qMSP. All bioinformatics analyses were performed using R version 2.11.1.
Hierarchical clustering analysis and heatmap creation
The log2 ratio value of all probes on the Nimblegen arrays was used to generate a heatmap based on unsupervised hierarchical clustering with Spotfire DecisionSite (Somerville, MA). An ANOVA was performed between Normal and Tumor samples on mean subtracted log2 ratio values for each probe. False Discovery Rate was calculated using the Benjamini-Hochberg correction on probes with a p-value less than 0.05. This clustering was based on the unweighted average method using correlation as the similarity measure and ordering by average values (q < 0.001). The red color was selected to represent methylated gene promoters and the blue color to represent unmethylated gene promoters.
Validation with quantitative Methylation Specific PCR (qMSP)
qMSP was used to validate the candidate genes identified with the MeDIP-chip Validation workflow on a separate cohort of tissue samples from normal and cervical cancer patients. Bisulfite converted DNA was used as template for fluorescence-based real-time PCR, as previously described.46 Briefly, bisulfite sequencing was used to verify that we can amplify the promoter region of interest and that it contains CpG islands whose methylation can be quantified with qMSP. Subsequently, MSP primers and qMSP probes were designed and optimized as follows.
The Nimblegen probe sequences for the 10 genes selected for this study were used to identify the chromosomal regions selected to design bisulfite sequencing and MSP primers. Bisulfite sequencing (BS) was performed to determine the methylation status of the normal and tumor tissues prior to MSP, on the same samples hybridized to Nimblegen arrays. Bisulfite-treated DNA was amplified using BS primer sets for a 5′ region within 800 bp of the TSS that included at least part of a CpG Island. The primer sequences did not contain CpG dinucleotides in order to obtain unbiased sequencing PCR products. Each amplified DNA sample was sequenced using nested, forward, or reverse primers.
After verifying with bisulfite sequencing that we had located a suitable area in the promoter region for qMSP validation, MSP primers and qMSP probes were designed to specifically amplify this region in the candidate gene promoters. Primers and probes were tested on positive (in vitro methylated bisulfite converted DNA) and negative controls (genomic unmethylated bisulfite converted DNA) to ensure amplification of the desired product and non-amplification of unmethylated DNA, respectively. Primer and probe sequences are provided in Table S6.
Fluorogenic PCR reactions were performed in duplicates in a reaction volume of 20 μL that contained 3 μL of bisulfite-modified DNA; 600 nM concentrations of forward and reverse primers; 200 nM probe; 0.6 U of platinum Taq polymerase (Invitrogen, Frederick, MD); 200 μM concentrations each of dATP, dCTP, dGTP and dTTP; and 6.7 mM MgCl2. Amplifications were performed using the reaction profile: 95 °C for 3 min, followed by 50 cycles at 95 °C for 15 s and 60 °C for 1 min in a 7900HT sequence detector (Applied Biosystems, Beverly, MA) and were analyzed by a sequence detector system (SDS 2.4; Applied Biosystems).
Each plate included patient DNA samples, positive controls (100% Methylated Bisulfite converted DNA, ZymoResearch) and multiple water blanks as non-template controls. Serial dilutions (90–0.0009 ng) of this DNA were used to construct a standard curve for each plate. The relative level of methylated DNA for each gene in each sample was determined as a ratio of the amplified gene quantity to the quantity of β-actin multiplied by 100.
Statistical analysis
All analyses were performed using Stata 11 and SPSS statistics version 19. The age differences in the Validation and Prevalence cohorts were compared using the Mann-Whitney U test; differences between socio-economic status, ethnicity and HPV status were analyzed using the chi-square test or the Fisher’s exact test. The samples were categorized as unmethylated or methylated based on detection of methylation above a threshold set for each gene. Thresholds were determined by ROC curves. To determine predictive accuracy of the methylated genes Spearman Correlation Coefficients, scatter plots, specificity, sensitivity, and Area Under the Curve47 were used. The Mann-Whitney U test was used to compare methylation levels of different groups. Finally, logistic regression analysis was used to determine the relation between methylation and clinical characteristics. Presence of methylation was used as dependent factor and the various clinical factors were used as independent factors. The association between methylation and clinical diagnosis was also assessed by logistic regression, where clinical diagnosis was used as a response variable, and methylation as a predictive variable. To adjust for age and HPV status, multivariate logistic regression analysis was performed, with clinical diagnosis as dependent and methylation, age, and HPV status as independent factors. Results with a P value < 0.05 were considered statistically significant.
Supplementary Material
Disclosure of Potential Conflicts of Interest
Sidransky D owns MDxHealth (formerly Oncomethylome Sciences, SA) stock, which is subject to certain restrictions under University policy. Sidransky D is a paid consultant to MDxHealth, and is a paid member of the company's Scientific Advisory Board. The Johns Hopkins University in accordance with its conflict of interest policies is managing the terms of this arrangement.
Grant Support
This research was supported in part by National Cancer Institute grants K01-CA164092 and U01-CA84986; and CONICYT, Proyecto de Atracción de Capital Humano -Modalidad de Estadía Corta de la Comisión Nacional de Ciencia y Tecnología, Chile (Guerrero-Preston R.); CORFO, Centro de Excelencia en Estudios Genéticos e Inmunológicos (CEGIN) 09CN14–5960, Scientific and Technological Bioresource Nucleus (BIOREN); Brebi P is recipient of grants from the FONDECYT Proyecto Postdoctorado 3120141. Noordhuis MG is recipient of grants from the Dutch Cancer Society and Jo Kolk studiefonds. Ili C is recipient of grants from FONDECYT Proyecto Postdoctorado 3130630. Soudry E is recipient of a fellowship grant from the American Physicians Fellowship for Medicine in Israel.
Acknowledgments
This research used a web database application provided by Research Information Technology Systems (https://www.rits.onc.jhmi.edu/).48 The funding agencies had no role in the design of the study, data collection or analysis, the interpretation of the results, the preparation of the manuscript, or the decision to submit the manuscript for publication. The authors wish to thank Connover Talbot, from Johns Hopkins Medical Institutions Deep Sequencing and Microarray Core (http://www.microarray.jhmi.edu/member.cgi), for his assistance in microarray and pathways data visualization, and integration.
Glossary
Abbreviations:
- Pap
Papanicolaou
- CIN
cervical intraepithelial neoplasia
- LSIL
low-grade squamous intraepithelial lesion
- HSIL
high-grade squamous intraepithelial lesion
- LGL
low-grade lesion
- HGL
high-grade lesion
- HPV
human papilloma virus
- MeDIP
methylated DNA immunoprecipitation
- HHHA
Doctor Hernán Henríquez Aravena
- MSP
methylation specific PCR
- qMSP
quantitative methylation specific PCR
- RT-PCR
real-time reverse transcriptase-PCR
- IP
immunoprecipitated methylated DNA
- TSSs
transcription start sites
- BS
bisulfite sequencing analysis
- IPA
Ingenuity Pathway Analysis
- Mapuche
native Chilean people
- AUC
area under the curve
- OR
odds ratio
- 95%C.I.
95% confidence interval
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/epigenetics/article/27120
Footnotes
Previously published online: www.landesbioscience.com/journals/epigenetics/article/27120
References
- 1.Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127:2893–917. doi: 10.1002/ijc.25516. [DOI] [PubMed] [Google Scholar]
- 2.Arbyn M, Raifu AO, Weiderpass E, Bray F, Anttila A. Trends of cervical cancer mortality in the member states of the European Union. Eur J Cancer. 2009;45:2640–8. doi: 10.1016/j.ejca.2009.07.018. [DOI] [PubMed] [Google Scholar]
- 3.Lowy D. Human papillomavirus, cervical cancer prevention, and more. Vaccine. 2008;26(Suppl 10):iii–iv. doi: 10.1016/j.vaccine.2008.06.032. [DOI] [PubMed] [Google Scholar]
- 4.Schiffman M, Castle PE, Jeronimo J, Rodriguez AC, Wacholder S. Human papillomavirus and cervical cancer. Lancet. 2007;370:890–907. doi: 10.1016/S0140-6736(07)61416-0. [DOI] [PubMed] [Google Scholar]
- 5.Holowaty P, Miller AB, Rohan T, To T. Natural history of dysplasia of the uterine cervix. J Natl Cancer Inst. 1999;91:252–8. doi: 10.1093/jnci/91.3.252. [DOI] [PubMed] [Google Scholar]
- 6.Arbyn M, Bergeron C, Klinkhamer P, Martin-Hirsch P, Siebers AG, Bulten J. Liquid compared with conventional cervical cytology: a systematic review and meta-analysis. Obstet Gynecol. 2008;111:167–77. doi: 10.1097/01.AOG.0000296488.85807.b3. [DOI] [PubMed] [Google Scholar]
- 7.Boone JD, Erickson BK, Huh WK. New insights into cervical cancer screening. J Gynecol Oncol. 2012;23:282–7. doi: 10.3802/jgo.2012.23.4.282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Woodman CB, Collins SI, Young LS. The natural history of cervical HPV infection: unresolved issues. Nat Rev Cancer. 2007;7:11–22. doi: 10.1038/nrc2050. [DOI] [PubMed] [Google Scholar]
- 9.Dillner J, Rebolj M, Birembaut P, Petry KU, Szarewski A, Munk C, de Sanjose S, Naucler P, Lloveras B, Kjaer S, et al. Joint European Cohort Study Long term predictive values of cytology and human papillomavirus testing in cervical cancer screening: joint European cohort study. BMJ. 2008;337:a1754. doi: 10.1136/bmj.a1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Saslow D, Solomon D, Lawson HW, Killackey M, Kulasingam SL, Cain J, Garcia FA, Moriarty AT, Waxman AG, Wilbur DC, et al. ACS-ASCCP-ASCP Cervical Cancer Guideline Committee American Cancer Society, American Society for Colposcopy and Cervical Pathology, and American Society for Clinical Pathology screening guidelines for the prevention and early detection of cervical cancer. CA Cancer J Clin. 2012;62:147–72. doi: 10.3322/caac.21139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Katki HA, Schiffman M, Castle PE, Fetterman B, Poitras NE, Lorey T, Cheung LC, Raine-Bennett T, Gage JC, Kinney WK. Five-year risks of CIN 3+ and cervical cancer among women who test Pap-negative but are HPV-positive. J Low Genit Tract Dis. 2013;17(Suppl 1):S56–63. doi: 10.1097/LGT.0b013e318285437b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Massad LS, Einstein MH, Huh WK, Katki HA, Kinney WK, Schiffman M, Solomon D, Wentzensen N, Lawson HW, 2012 ASCCP Consensus Guidelines Conference 2012 updated consensus guidelines for the management of abnormal cervical cancer screening tests and cancer precursors. Obstet Gynecol. 2013;121:829–46. doi: 10.1097/AOG.0b013e3182883a34. [DOI] [PubMed] [Google Scholar]
- 13.Katki HA, Schiffman M, Castle PE, Fetterman B, Poitras NE, Lorey T, Cheung LC, Raine-Bennett T, Gage JC, Kinney WK. Five-year risk of recurrence after treatment of CIN 2, CIN 3, or AIS: performance of HPV and Pap cotesting in posttreatment management. J Low Genit Tract Dis. 2013;17(Suppl 1):S78–84. doi: 10.1097/LGT.0b013e31828543c5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schiffman M, Wentzensen N. Human papillomavirus infection and the multistage carcinogenesis of cervical cancer. Cancer Epidemiol Biomarkers Prev. 2013;22:553–60. doi: 10.1158/1055-9965.EPI-12-1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Flores YN, Bishai DM, Lorincz A, Shah KV, Lazcano-Ponce E, Hernández M, Granados-García V, Pérez R, Salmerón J. HPV testing for cervical cancer screening appears more cost-effective than Papanicolau cytology in Mexico. Cancer Causes Control. 2011;22:261–72. doi: 10.1007/s10552-010-9694-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wolffe AP, Matzke MA. Epigenetics: regulation through repression. Science. 1999;286:481–6. doi: 10.1126/science.286.5439.481. [DOI] [PubMed] [Google Scholar]
- 17.Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002;3:415–28. doi: 10.1038/nrg816. [DOI] [PubMed] [Google Scholar]
- 18.Brait M, Sidransky D. Cancer epigenetics: above and beyond. Toxicol Mech Methods. 2011;21:275–88. doi: 10.3109/15376516.2011.562671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maldonado L, Hoque MO. Epigenomics and ovarian carcinoma. Biomark Med. 2010;4:543–70. doi: 10.2217/bmm.10.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Esteller M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat Rev Genet. 2007;8:286–98. doi: 10.1038/nrg2005. [DOI] [PubMed] [Google Scholar]
- 21.Herman JG, Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med. 2003;349:2042–54. doi: 10.1056/NEJMra023075. [DOI] [PubMed] [Google Scholar]
- 22.Lai HC, Lin YW, Huang TH, Yan P, Huang RL, Wang HC, Liu J, Chan MW, Chu TY, Sun CA, et al. Identification of novel DNA methylation markers in cervical cancer. Int J Cancer. 2008;123:161–7. doi: 10.1002/ijc.23519. [DOI] [PubMed] [Google Scholar]
- 23.Lendvai Á, Johannes F, Grimm C, Eijsink JJ, Wardenaar R, Volders HH, Klip HG, Hollema H, Jansen RC, Schuuring E, et al. Genome-wide methylation profiling identifies hypermethylated biomarkers in high-grade cervical intraepithelial neoplasia. Epigenetics. 2012;7:1268–78. doi: 10.4161/epi.22301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shivapurkar N, Sherman ME, Stastny V, Echebiri C, Rader JS, Nayar R, Bonfiglio TA, Gazdar AF, Wang SS. Evaluation of candidate methylation markers to detect cervical neoplasia. Gynecol Oncol. 2007;107:549–53. doi: 10.1016/j.ygyno.2007.08.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang N, Eijsink JJ, Lendvai A, Volders HH, Klip H, Buikema HJ, van Hemel BM, Schuuring E, van der Zee AG, Wisman GB. Methylation markers for CCNA1 and C13ORF18 are strongly associated with high-grade cervical intraepithelial neoplasia and cervical cancer in cervical scrapings. Cancer Epidemiol Biomarkers Prev. 2009;18:3000–7. doi: 10.1158/1055-9965.EPI-09-0405. [DOI] [PubMed] [Google Scholar]
- 26.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 27.Ehrlich M. DNA hypomethylation in cancer cells. Epigenomics. 2009;1:239–59. doi: 10.2217/epi.09.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Feinberg AP, Ohlsson R, Henikoff S. The epigenetic progenitor origin of human cancer. Nat Rev Genet. 2006;7:21–33. doi: 10.1038/nrg1748. [DOI] [PubMed] [Google Scholar]
- 29.Walter K, Holcomb T, Januario T, Du P, Evangelista M, Kartha N, Iniguez L, Soriano R, Huw L, Stern H, et al. DNA methylation profiling defines clinically relevant biological subsets of non-small cell lung cancer. Clin Cancer Res. 2012;18:2360–73. doi: 10.1158/1078-0432.CCR-11-2635-T. [DOI] [PubMed] [Google Scholar]
- 30.Sun XW, Kuhn L, Ellerbrock TV, Chiasson MA, Bush TJ, Wright TC., Jr. Human papillomavirus infection in women infected with the human immunodeficiency virus. N Engl J Med. 1997;337:1343–9. doi: 10.1056/NEJM199711063371903. [DOI] [PubMed] [Google Scholar]
- 31.Melnikow J, Nuovo J, Willan AR, Chan BK, Howell LP. Natural history of cervical squamous intraepithelial lesions: a meta-analysis. Obstet Gynecol. 1998;92:727–35. doi: 10.1016/S0029-7844(98)00245-2. [DOI] [PubMed] [Google Scholar]
- 32.Trimble CL, Piantadosi S, Gravitt P, Ronnett B, Pizer E, Elko A, Wilgus B, Yutzy W, Daniel R, Shah K, et al. Spontaneous regression of high-grade cervical dysplasia: effects of human papillomavirus type and HLA phenotype. Clin Cancer Res. 2005;11:4717–23. doi: 10.1158/1078-0432.CCR-04-2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oka N, Kajita M, Nishimura R, Ohbayashi C, Sudo T. L1 gene methylation in high-risk human papillomaviruses for the prognosis of cervical intraepithelial neoplasia. Int J Gynecol Cancer. 2013;23:235–43. doi: 10.1097/IGC.0b013e31827da1f6. [DOI] [PubMed] [Google Scholar]
- 34.Wentzensen N, Sun C, Ghosh A, Kinney W, Mirabello L, Wacholder S, Shaber R, LaMere B, Clarke M, Lorincz AT, et al. Methylation of HPV18, HPV31, and HPV45 genomes and cervical intraepithelial neoplasia grade 3. J Natl Cancer Inst. 2012;104:1738–49. doi: 10.1093/jnci/djs425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vucic EA, Wilson IM, Campbell JM, Lam WL. Methylation analysis by DNA immunoprecipitation (MeDIP) Methods Mol Biol. 2009;556:141–53. doi: 10.1007/978-1-60327-192-9_10. [DOI] [PubMed] [Google Scholar]
- 36.Apostolidou S, Hadwin R, Burnell M, Jones A, Baff D, Pyndiah N, Mould T, Jacobs IJ, Beddows S, Kocjan G, et al. DNA methylation analysis in liquid-based cytology for cervical cancer screening. Int J Cancer. 2009;125:2995–3002. doi: 10.1002/ijc.24745. [DOI] [PubMed] [Google Scholar]
- 37.Huang RL, Chang CC, Su PH, Chen YC, Liao YP, Wang HC, Yo YT, Chao TK, Huang HC, Lin CY, et al. Methylomic analysis identifies frequent DNA methylation of zinc finger protein 582 (ZNF582) in cervical neoplasms. PLoS One. 2012;7:e41060. doi: 10.1371/journal.pone.0041060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Farkas SA, Milutin-Gašperov N, Grce M, Nilsson TK. Genome-wide DNA methylation assay reveals novel candidate biomarker genes in cervical cancer. Epigenetics. 2013;8:8. doi: 10.4161/epi.26346. [DOI] [PubMed] [Google Scholar]
- 39.Meng X, Lu X, Morris CA, Keating MT. A novel human gene FKBP6 is deleted in Williams syndrome. Genomics. 1998;52:130–7. doi: 10.1006/geno.1998.5412. [DOI] [PubMed] [Google Scholar]
- 40.Zhang W, Zhang S, Xiao C, Yang Y, Zhoucun A. Mutation screening of the FKBP6 gene and its association study with spermatogenic impairment in idiopathic infertile men. Reproduction. 2007;133:511–6. doi: 10.1530/REP-06-0125. [DOI] [PubMed] [Google Scholar]
- 41.Burrell RA, McClelland SE, Endesfelder D, Groth P, Weller MC, Shaikh N, Domingo E, Kanu N, Dewhurst SM, Gronroos E, et al. Replication stress links structural and numerical cancer chromosomal instability. Nature. 2013;494:492–6. doi: 10.1038/nature11935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Goss PE, Lee BL, Badovinac-Crnjevic T, Strasser-Weippl K, Chavarri-Guerra Y, St Louis J, Villarreal-Garza C, Unger-Saldaña K, Ferreyra M, Debiasi M, et al. Planning cancer control in Latin America and the Caribbean. Lancet Oncol. 2013;14:391–436. doi: 10.1016/S1470-2045(13)70048-2. [DOI] [PubMed] [Google Scholar]
- 43.Lie AK, Kristensen G. Human papillomavirus E6/E7 mRNA testing as a predictive marker for cervical carcinoma. Expert Rev Mol Diagn. 2008;8:405–15. doi: 10.1586/14737159.8.4.405. [DOI] [PubMed] [Google Scholar]
- 44.van den Brule AJ, Pol R, Fransen-Daalmeijer N, Schouls LM, Meijer CJ, Snijders PJ. GP5+/6+ PCR followed by reverse line blot analysis enables rapid and high-throughput identification of human papillomavirus genotypes. J Clin Microbiol. 2002;40:779–87. doi: 10.1128/JCM.40.3.779-787.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Scacheri PC, Crawford GE, Davis S. Statistics for ChIP-chip and DNase hypersensitivity experiments on NimbleGen arrays. Methods Enzymol. 2006;411:270–82. doi: 10.1016/S0076-6879(06)11014-9. [DOI] [PubMed] [Google Scholar]
- 46.Hoque MO, Kim MS, Ostrow KL, Liu J, Wisman GB, Park HL, Poeta ML, Jeronimo C, Henrique R, Lendvai A, et al. Genome-wide promoter analysis uncovers portions of the cancer methylome. Cancer Res. 2008;68:2661–70. doi: 10.1158/0008-5472.CAN-07-5913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Naucler P, Ryd W, Törnberg S, Strand A, Wadell G, Elfgren K, Rådberg T, Strander B, Johansson B, Forslund O, et al. Human papillomavirus and Papanicolaou tests to screen for cervical cancer. N Engl J Med. 2007;357:1589–97. doi: 10.1056/NEJMoa073204. [DOI] [PubMed] [Google Scholar]
- 48.Astor BC, Matsushita K, Gansevoort RT, van der Velde M, Woodward M, Levey AS, Jong PE, Coresh J, Astor BC, Matsushita K, et al. Chronic Kidney Disease Prognosis Consortium Lower estimated glomerular filtration rate and higher albuminuria are associated with mortality and end-stage renal disease. A collaborative meta-analysis of kidney disease population cohorts. Kidney Int. 2011;79:1331–40. doi: 10.1038/ki.2010.550. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.