

Abstract
Squamous esophageal epithelium adapts to acid reflux-mediated injury by proliferation and differentiation via signal transduction pathways. Induction of the Wnt antagonist Dickkopf-1 (Dkk1) is involved in tissue repair during inflammation and cellular injury. In this study, we aimed to identify the biological role of Dkk1 in human reflux esophagitis with respect to cell growth and regulation of Wnt signaling. Esophageal biopsies from reflux-esophagitis patients (n = 15) and healthy individuals (n = 10) were characterized in terms of Dkk1 expression. The role of Dkk1 in response to acid-mediated epithelial injury was analyzed by cellular assays in vitro utilizing squamous esophageal epithelial cell lines (EPC1-hTERT, EPC2-hTERT, and HEEC). Dkk1 was significantly overexpressed in human reflux-esophagitis tissue compared with healthy esophageal mucosa at transcriptional and translational levels. After acute and chronic acid (pH 4) exposure, esophageal squamous epithelial cell lines expressed and secreted high levels of Dkk1 in response to stress-associated DNA injury. High extracellular levels of human recombinant Dkk1 inhibited epithelial cell growth and induced cellular senescence in vitro, as demonstrated by reduced cell proliferation, G0/G1 cell cycle arrest, elevated senescence-associated β-galactosidase activity, and upregulation of p16. Acid pulsing induced Dkk1-mediated senescence, which was directly linked to the ability of Dkk1 to antagonize the canonical Wnt/β-catenin signaling. In healthy esophageal mucosa, Dkk1 expression was associated with low expression of transcriptionally active β-catenin, while in reflux-esophagitis tissue, Dkk1 overexpression correlated with increased senescence-associated β-galactosidase activity and p16 upregulation. The data indicate that, in human reflux esophagitis, Dkk1 functions as a secreted growth inhibitor by suppressing Wnt/β-catenin signaling and promoting cellular senescence. These findings suggest a significant role for Dkk1 and cellular senescence in esophageal tissue homeostasis during reflux esophagitis.
Keywords: Dickkopf-1, gastroesophageal reflux disease, esophagitis, Wnt
gastroesophageal reflux disease (GERD) is one of the most prevalent gastrointestinal disorders, and its pathogenesis is linked to the mucosal injury caused by prolonged exposure of esophageal squamous epithelium to acidic gastric contents (11). The spectrum of mucosal injury observed in GERD patients is highly variable, ranging from no visible mucosal damage to more severe manifestations, such as erosions, ulcerations, stricture formation, and development of Barrett's esophagus (BE), a metaplastic replacement of the normal squamous epithelium with intestinal-type columnar epithelium, which predisposes to esophageal adenocarcinoma (EAC) (46). The response of the esophageal epithelium to GERD-associated injury relies on the processes of tissue regeneration and adaptation to injury (defense), which are fundamentally orchestrated by proliferation, apoptosis, differentiation, and migration (38). These functions, although preprogrammed in the genome, are continuously under the influence of several signal transduction pathways (22), which, in the case of GERD, remain largely undefined.
Canonical Wnt/β-catenin signaling is among the signaling pathways that promote cell proliferation and differentiation during embryonic development and tissue homeostasis (32). In adult self-renewing tissues, such as intestinal epithelium, Wnt signaling persists as a key regulator of tissue repair, while its mutational deregulation is responsible for malignant transformation (17). Wnt signaling is known to participate in esophageal development during foregut morphogenesis (21), while components of the pathway have been found to be differentially expressed within the esophageal mucosa (3). However, the role of Wnt/β-catenin signaling in normal adult esophagus, especially with respect to the pathogenesis of GERD, is yet to be defined.
Dickkopf-1 (Dkk1) is a gene that encodes a secreted glycoprotein that regulates cell fate and growth during embryogenesis, tissue homeostasis, and cancer (35). The function of Dkk1 has been tightly associated with its ability to specifically antagonize canonical Wnt/β-catenin signaling by binding to the extracellular domain of the Wnt coreceptor LDL receptor-related protein 6 (LRP-6), thus preventing the Wnt-induced stabilization of β-catenin (33). Induction of Dkk1 has been strongly associated with tissue inflammation, cellular stress, and cellular injury (26, 44, 49). Its activation has been reported as a cellular response to proapoptotic stimuli induced by different types of genotoxic stress (44, 17, 52). At the transcriptional level, Dkk1 is a direct target of the β-catenin/T-cell factor (TCF) complex, suggesting a negative-feedback loop in Wnt signaling (36). Our previous report demonstrated increased Dkk1 gene expression in the noninflamed esophageal squamous mucosa of GERD patients compared with healthy individuals and BE patients (2). This finding may suggest a significant role for Dkk1-mediated inhibition of Wnt/β-catenin signaling in the pathogenesis of GERD. Yet the ability of Dkk1 to block Wnt signals and its biological impact on esophageal epithelial cell biology have not been examined.
Responses of tissues, such as esophageal epithelium, that continually encounter stress and damage from exogenous and endogenous sources range from complete recovery to self-destruction (apoptosis). Cellular senescence, a state of permanent cell cycle arrest, has been increasingly recognized as an additional cellular response to various stressors, such as oxidative stress, DNA damage, and oncogene activation (5). The senescent cell is arrested and incapable of responding to mitogens, although it is viable and metabolically active. The senescent state is characterized by large flat cell morphology and expression of senescence-associated β-galactosidase (SA-β-Gal) activity and cyclin-dependent kinase inhibitor p16INK4a (p16) (16). Although increasing evidence places p16/retinoblastoma protein (pRb) and p53/21 pathways within the heart of the senescence program as its downstream effectors (5), the upstream signaling pathways are not fully identified. Emerging evidence supports a direct interaction between senescence and Wnt/β-catenin signaling (1). Repression of Wnt signaling has been shown to trigger early onset of the senescence program, while its activation delays oncogene-induced senescence (12, 53). Whether acid reflux induces cellular senescence in esophageal epithelium has not been addressed.
The present study was undertaken to investigate the biological role of Dkk1 in human esophageal epithelium with respect to acid-mediated epithelial injury. We hypothesized that Dkk1 overexpression is implicated in the regeneration of reflux-damaged esophageal squamous epithelium by suppression of Wnt/β-catenin signaling and induction of epithelial cell growth arrest. By analyzing human esophageal specimens and squamous esophageal cell lines in vitro, we present evidence suggesting that acid-mediated Dkk1 overexpression in reflux esophagitis suppresses Wnt/β-catenin signaling and promotes epithelial cell growth arrest via cellular senescence.
MATERIALS AND METHODS
Human esophageal specimens.
Human esophageal biopsy specimens were obtained from patients ≥18 yr of age undergoing endoscopy for active reflux esophagitis (n = 15) and healthy individuals undergoing endoscopy for nonesophageal indications (n = 10). The diagnosis of reflux esophagitis was set macroscopically (erosions) and pathologically. Two biopsies were obtained from each side (distal and proximal) of the esophagus in each patient. The distal biopsies were obtained from the site of inflamed mucosa in the distal esophagus in esophagitis patients and 1 cm above the Z line in healthy controls. Proximal biopsies were taken 5 cm below the upper esophageal sphincter in both groups (Fig. 1A). Patients with eosinophilic esophagitis or previous history of Barrett's metaplasia and associated dysplasia or EAC were excluded. Studies were approved by the Human Research Review Committee of the Medical College of Wisconsin, and study participants gave written informed consent prior to their studies. Patients' data are presented in Table 1.
Fig. 1.

Dkk1 expression in healthy esophageal mucosa and reflux esophagitis tissue. A: topography of biopsy acquisition. UES and LES, upper and lower esophageal sphincter. B: scatter graph of quantitative RT-PCR data for Dickkopf-1 (Dkk1) mRNA expression in distal and proximal esophageal mucosa of healthy individuals (n = 10) and esophagitis patients (n = 15). Data were normalized to β-actin expression. Top: Dkk1 mRNA expression in proximal biopsies of the 2 groups is not different. Middle: Dkk1 mRNA expression is greater in biopsies from esophagitis patients than healthy controls. *P ≤ 0.05 (by Mann-Whitney test). Horizontal lines indicate means. Bottom: fold change in expression of Dkk1 between proximal and distal mucosa. Biopsies from esophagitis patients show greater differential expression between proximal and distal mucosa than biopsies from healthy controls. *P ≤ 0.05 (by Mann-Whitney test). C: Western blot analysis of Dkk1 protein expression in human esophageal biopsies. Top: representative blots. Bottom: Dkk1 protein expression relative to β-actin expression in biopsies from healthy controls (n = 3) and esophagitis patients (n = 8). Values are means ± SE. *P ≤ 0.05 (by paired t-test); ns, not significant. D: immunohistochemistry for Dkk1 in esophageal biopsies. Sections were stained with hematoxylin-eosin (H&E) for histological confirmation of esophagitis. Images represent results from 4 experiments. E: ELISA of Dkk1 protein secretion in organ culture medium from distal mucosa relative to paired proximal mucosa in healthy controls (n = 3) and esophagitis patients (n = 3). Values are means ± SE. *P ≤ 0.05 (by paired t-test).
Table 1.
Demographic, endoscopic, and histological findings in esophagitis patients and healthy controls
| Subject/Patient No. | Age, yr | Sex | BMI | Indication for Upper GI Endoscopy | Endoscopy Report | Pathology Report |
|---|---|---|---|---|---|---|
| Healthy subjects | ||||||
| 1 | 61 | M | 27 | Abdominal pain | Normal-looking squamous mucosa | No abnormalities |
| 2 | 46 | F | 21 | Weight loss, diarrhea | Normal-looking squamous mucosa | No abnormalities |
| 3 | 23 | M | 26 | Abdominal pain | Normal-looking squamous mucosa | No abnormalities |
| 4 | 37 | M | 32 | Vitamin B deficiency | Normal-looking squamous mucosa | No abnormalities |
| 5 | 60 | F | 47 | Abdominal pain | Normal-looking squamous mucosa | No abnormalities |
| 6 | 24 | F | 22 | Abdominal pain | Normal-looking squamous mucosa | No abnormalities |
| 7 | 25 | M | 22 | Crohn's disease | Normal-looking squamous mucosa | No abnormalities |
| 8 | 46 | F | 21 | Family history of esophageal cancer | Normal-looking squamous mucosa | No abnormalities |
| 9 | 39 | F | 25 | Severe iron deficiency anemia | Normal-looking squamous mucosa | No abnormalities |
| 10 | 54 | M | 26 | Not reported | Normal-looking squamous mucosa | No abnormalities |
| 41 | 5/5 | 27 | ||||
| Esophagitis patients | ||||||
| 1 | 60 | F | 29 | History of GERD | Irregular Z line | Mild esophagitis |
| 2 | 35 | F | 25 | History of GERD with supraesophageal symptoms | Few whitish plaques in distal esophagus | Stratified squamous esophageal mucosa with mild spongiosis and focal chronic inflammation |
| 3 | 45 | M | 30 | Odynophagia, history of GERD | Irregular Z line with short tongue of reddish colored mucosa | Mild esophagitis |
| 4 | 58 | M | 35 | History of GERD, hiatal hernia | Slightly irregular Z line, without obvious erosions | Mild esophagitis |
| 5 | 30 | M | 29 | History of GERD/heartburn | Los Angeles classification grade B erosive esophagitis | Mild Esophagitis |
| 6 | 43 | F | 28 | History of GERD | Normal-looking squamous mucosa | Stratified squamous mucosa with slight basal cell hyperplasia and slight increase in chronic inflammatory cells |
| 7 | 77 | M | 19 | History of GERD/odynophagia | Long 3-cm complex distal stricture | Ulceration and inflamed granulation |
| 8 | 84 | F | 26 | History of GERD/heartburn | Los Angeles classification grade C erosive esophagitis | Ulcerative esophagitis |
| 9 | 42 | M | 25 | History of GERD, hiatal hernia | Irregular Z line | Mild esophagitis |
| 10 | 63 | M | 33 | Screening | Irregular Z line | Mild esophagitis |
| 11 | 55 | F | 27 | Abdominal pain/recurrent dysphagia | Corrugated esophagus | Mild esophagitis |
| 12 | 45 | M | 28 | Dysphagia, history of erosive esophagitis | Los Angeles classification grade B erosive esophagitis | Mild esophagitis |
| 13 | 74 | M | 27 | History of GERD | Reddish distal mucosa | Mild esophagitis |
| 14 | 77 | F | 27 | History of GERD | Ring-type stenosis above the Z line with friable mucosa | Mild esophagitis |
| 15 | 77 | F | 30 | History of GERD | Distal esophageal stricture | Mild esophagitis |
| 58 | 8/7 | 28 |
GERD, gastroesophageal reflux disease; GI, gastrointestinal.
Cell culture.
Human squamous esophageal telomerase-immortalized cells, EPC1-hTERT (EPC1) and EPC2-hTERT (EPC2) with intact p53 and p16 genes (18), were the generous gifts of Dr. Hiroshi Nakagawa (Gastroenterology Division, University of Pennsylvania, Philadelphia, PA). Cells were grown in monolayers according to a standard protocol (http://www.med.upenn.edu/molecular/documents/EPCcellprotocol032008.pdf). Primary human esophageal epithelial cells (HEEC) were obtained from ScienCell Research Laboratories (Carlsbad, CA) and grown according to the manufacturer's protocol.
Reagents and antibodies.
Recombinant human Dkk1 (rhDkk1, 100 or 500 ng/ml), recombinant human Wnt3a (rhWnt3a, 200 ng/ml), and neutralizing anti-Dkk1 monoclonal antibody (anti-Dkk1 Ab, 10 μg/ml) were obtained from R & D Systems (Minneapolis, MN). Human IgG (10 μg/ml; Innovative Research, Novi, MI) served as proper control for treatment with anti-Dkk1 Ab. At 10 μg/ml, anti-Dkk1 Ab will block ≥90% of Dkk1 binding to the LRP-6 receptor (R & D Systems). Antibodies were used against Dkk1 (Abcam, Cambridge, MA), active β-catenin (ABC; Millipore, Billerica, MA), Ki-67 (Millipore), p16 (BD Pharmigen, San Jose, CA), p53 (Santa Cruz Biotechnology, Santa Cruz, CA), p21 (Santa Cruz Biotechnology), β-catenin (Cell Signaling Technology, Danvers, MA), β-actin (Sigma, St. Louis, MO), and horseradish peroxidase-conjugated secondary antibodies (Santa Cruz Biotechnology). Unless otherwise indicated, all other chemicals were purchased from Sigma-Aldrich.
Acid pulsing.
Cell monolayers at 60–70% confluence were exposed to acidified full growth medium (brought to pH 4.0 using HCl), either acutely for 5 and 30 min as single exposures or chronically for 1 min, three times per day with 3-h intervals between acid pulses. Between acid exposures, acidified medium was removed, and cells were washed twice with PBS and reincubated in their respective neutral (pH 7.4) growth medium, alone or with the indicated supplementation, for the remainder of the experiment. The 60–70% confluence was preferred to avoid overconfluent cell monolayers in the control/untreated cells due to the prolonged period of acid pulsing. The pH level and durations of acid exposure were chosen to simulate typical episodes of gastroesophageal reflux in GERD patients.
Cellular assays.
Proliferation was assessed by [3H]thymidine uptake, as described elsewhere (39), or by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay, as suggested by the manufacturer (Roche Diagnostics, Indianapolis, IN). Cells were seeded into 96-well plates (2 × 104/well) and, on the following day (day 0), treated with rhDkk1 or anti-Dkk1 Ab at the indicated concentrations.
Apoptosis was determined by annexin V-FITC/propidium iodide (PI) according to the manufacturer's protocol (BD Biosciences, San Jose, CA).
Cell cycle analysis.
The fraction of cells in the G1/G0, S, G2/M, and sub-G1 phases after nuclear staining with PI was used to quantify cell cycle changes. Briefly, cells were equally seeded in 12-well plates (1 × 105/well) and incubated with rhDkk1 (500 ng/ml) for 48 h. Stained cells were measured by flow cytometric analysis [fluorescein-activated cell sorting (FACS)] by a FACSCalibur (BD Biosciences). Appropriate windows were selected to differentiate between cells with sub-G1 DNA content (hypodiploid) and cells in different phases of cell cycle distribution. Histograms were produced using CellQuest software.
SA-β-Gal activity was assessed using a cellular senescence assay kit (Chemicon/Millipore, Billerica, MA), as described previously (10). In addition to the samples described above, SA-β-Gal activity was also examined in tissues obtained from esophagitis patients, BE patients, and healthy controls (n = 3 in each group). Briefly, the biopsies were incubated immediately after acquisition in freshly prepared β-Gal staining solution for 24 h at 37°C. The samples were then embedded in optimal cutting temperature compound (OCT, Sakura Finetek, Torrance, CA) and prepared for cryostat sections. The sections were photographed and finally stained with hematoxylin-eosin.
RNA isolation and real-time quantitative PCR.
RNA was isolated from biopsies and cell cultures using Arcturus PicoPure RNA (Life Technologies, Grand Island, NY) and the RNeasy kit (Qiagen, Valencia, CA), respectively. cDNA was synthesized from 1 μg of total RNA using the iScript cDNA synthesis kit according to the manufacturer's protocol (Bio-Rad, Hercules, CA). Real-time PCR was performed with SsoFast EvaGreen Supermix (Bio-Rad), with 250 nM primer and 1 μl of cDNA per 20-μl reaction. Normalized gene expression was analyzed with iQ5 software (Bio-Rad). The primers are listed in Table 2.
Table 2.
Sequence of primers used for RT-PCR analysis
| Gene Name | Gene ID No. | Sequence | |
|---|---|---|---|
| β-Actin | ACTB | NM_001101.3 | Forward: 5′-CAC TCT TCC AGC CTT CCT TC-3′ |
| Reverse: 5′-GGT GTA ACG CAA CTA AGT CAT AG-3′ | |||
| Cyclin D1 | CCND1 | NM_053056.2 | Forward: 5′-CAT CTA CAC CGA CAA CTC CAT C-3′ |
| Reverse: 5′-TCT GGC ATT TTG GAG AGG AGG-3′ | |||
| β-Catenin | CTNNB1 | NM_001098209.1 | Forward: 5′-GTT CAG TTG CTT GTT CGT GC-3′ |
| Reverse: 5′-GTT GTG AAC ATC CCG AGC TAG-3′ | |||
| Axin2 | AXIN2 | NM_004655.3 | Forward: 5′-CCA CTG GCC GAT TCT TCC TT-3′ |
| Reverse: 5′-TAC CGG AGG ATG CTG AAG GC-3′ | |||
| Dickkopf-1 | DKK1 | NM_012242.2 | Forward: 5′-AGC GTT GTT ACT GTG GAG AAG-3′ |
| Reverse: 5′-GTG TGA AGC CTA GAA GAA TTA CTG-3′ | |||
| p21 | CDKN1A | NM_000389 | Forward: 5′-GGA AGA CCA TGT GGA CCT GT-3′ |
| Reverse: 5′-GGC GTT TGG AGT GGT AGA AA-3′ | |||
| Ki-67 | MKI67 | NM_001145966 | Forward: 5′-ATT ATC GTT CCT TCA GGT ATG-3′ |
| Reverse: 5′-TCA TCA GGG TCA GAA GAG AA-3′ | |||
| p16 | CDKN2A, p16Ink4A | NM_000077 | Forward: 5′-CAA CGC ACC GAA TAG TTA CG-3′ |
| Reverse: 5′-CAG CTC CTC AGC CAG GTC-3′ |
Western blot analysis.
Total cell extracts were prepared and subjected to Western blotting, as previously described (40). Cytoplasmic and nuclear fractions were prepared using the NE-PER kit according to the manufacturer's protocol (Pierce/Thermo Scientific, Rockford, IL).
Organ culture and ELISA.
Mucosal biopsies of equal size were cultured in 0.5 ml of RPMI medium for 24 h, as previously described (40). Dkk1 secretion was assessed in tissue and cell culture media by ELISA (R & D Systems) according to the manufacturer's protocol.
Immunohistochemistry.
Mucosal esophageal biopsy specimens were embedded in paraffin blocks and cut into 5-μm sections. The sections were routinely stained with hematoxylin-eosin for histological diagnosis, and additional sequential sections were subjected to immunohistochemistry, as described previously (19).
Immunofluorescence staining.
Antibodies against Ki-67 (Millipore), Dkk1 (Abcam), and active β-catenin (Millipore), as well as Alexa Fluor 488 and 594 as secondary antibodies (Invitrogen, Carlsbad, CA), were used. 4′,6-Diamidino-2-phenylindole staining was performed to ensure nuclear localization. Coverslips were mounted on Superfrost slides (Thermo Fisher Scientific, Lafayette, CO) with ProLong antifade mounting medium (Invitrogen) and visualized using a fluorescence microscope (model BX-40, Olympus) and a digital camera (model DFC 300FX, Leica). Labeling index for Ki-67 was determined by counting ≥500 cells in ≥5 different random fields.
Luciferase reporter assay.
Cells at 60–70% confluence were transfected with DNA plasmids of β-catenin-lymphoid enhancer factor (LEF)/TCF-sensitive (TOP-flash) or β-catenin-LEF/TCF-insensitive (FOP-flash) reporter vectors (Addgene, Cambridge, MA) using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. TOP-flash or FOP-flash (1.5 μg) was added to each well of a 24-well plate. phRL-TK plasmid (Promega, Madison, WI) was cotransfected as control for transfection efficiency. Experimentation followed at 48 h posttransfection as indicated. Reporter assay was performed using a dual luciferase reporter system (Promega). Luciferase activity was measured using a luminometer (GLOMAX 20/20, Promega). Values for each reporter were normalized to phRL-TK values. TOP-flash, but not FOP-flash, is responsive to coactivation of TCF/LEF by β-catenin.
Dkk1 gene silencing.
EPC2 cells were transfected with predesigned short interfering RNA (siRNA) targeting human Dkk1 [ON-TARGETplus SMARTpool, human DKK1 (22943), Dharmacon, Lafayette, CO] and a control negative siRNA targeting a sequence not sharing homology with the human genome (ON-TARGETplus nontargeting siRNA, Dharmacon) using DharmaFECT transfection reagent according to the manufacturer's protocol (Dharmacon). The ratio of siRNA to DharmaFECT reagent was 25 nM siRNA to 1.5 μl of transfection reagent.
Statistical analysis.
IBM SPSS Statistics 19 software was used for statistical analysis. All experiments were performed in triplicate, and values are means ± SE. Gene expressions were compared between esophagitis patients and healthy controls using the Mann-Whitney test. Gene or protein expressions between proximal and distal esophagus within the same patient's group were compared using paired t-test. Gene expressions were correlated using Pearson's correlation test. Significant differences in cellular assays were evaluated by Student's t-test. P < 0.05 was considered statistically significant.
RESULTS
Dkk1 is overexpressed in human reflux esophagitis.
We previously demonstrated higher levels of Dkk1 mRNA expression in distal normal-looking squamous esophageal mucosa of GERD patients than BE patients and healthy individuals (2). In this study, we expanded our investigation to the lesion of reflux esophagitis (erosion) and quantitatively compared Dkk1 mRNA levels between distal and proximal esophageal mucosa in reflux esophagitis patients and healthy controls. Real-time PCR demonstrated significantly higher Dkk1 gene expression in the distal inflamed mucosa of esophagitis patients than healthy controls [1.93 ± 0.97 vs. 0.55 ± 0.15 (SE), P ≤ 0.05 by Mann-Whitney test; Fig. 1B]. Dkk1 mRNA expression was not different in the proximal mucosa of the two groups. The fold change in Dkk1 expression between distal and proximal esophagus was significantly greater in reflux esophagitis patients than healthy individuals (4.84 ± 0.97 vs. 1.18 ± 0.82, P ≤ 0.05 by Mann-Whitney test).
Dkk1 overexpression in esophagitis tissue was confirmed at the protein level by Western blot analysis and immunohistochemistry. In healthy individuals, there was no difference in Dkk1 protein expression between distal and paired proximal mucosa. In esophagitis patients, Dkk1 protein expression was markedly higher in the esophagitis lesion (distal) than the paired proximal mucosa (P ≤ 0.05 by paired t-test; Fig. 1C). Immunohistochemical analysis of Dkk1 expression in healthy esophageal epithelium was weak and mostly defined at the basal mucosal layer, while a strong and diffuse pattern throughout the inflamed mucosa was observed in esophagitis patients (Fig. 1D). Given that Dkk1 is a secreted glycoprotein, using ELISA, we quantified Dkk1 protein secretion in organ culture media. Dkk1 secretion was markedly elevated in the esophagitis lesions compared with the proximal mucosa (P ≤ 0.05 by paired t-test; Fig. 1E).
Taken together, these findings demonstrate increased Dkk1 expression and secretion in esophageal squamous mucosa from reflux-esophagitis patients.
Acidic pH induces Dkk1 overexpression in squamous esophageal epithelial cell lines in vitro.
Next, we determined whether acid, as a major cellular stressor of gastric refluxate, can induce transcription of Dkk1 by stressing EPC1 and EPC2 squamous esophageal cell lines with acute or chronic acid exposure. Dkk1 protein expression and secretion were confirmed in both cell lines at rest by Western blotting and ELISA, respectively (Fig. 2, A and B). After acute acid (pH 4.0) exposure for 5 and 30 min, Dkk1 mRNA transcription was increased in both cell lines, as determined by real-time PCR (Fig. 2C). Similarly, chronic acid pulsing for up to 9 days (1 min of exposure, 3 times per day) upregulated Dkk1 gene expression in both cell lines (Fig. 2D). In agreement with the acid-induced Dkk1 transcriptional activation, Dkk1 protein levels were markedly elevated in growth medium and cytosol of the acid-stimulated cells compared with unstimulated cells, as determined by ELISA and Western blotting (Fig. 2, E and F), respectively. Cell death induced by pH 4.0 following short-term pulsing for 1 min was ≤5%, as tested by Trypan blue exclusion, and monolayers remained intact, as examined using an inverted microscope (data not shown). These data suggest that acidic pH induces upregulation of Dkk1 in esophageal epithelial cells in vitro.
Fig. 2.

Effect of acidic pH on Dkk1 expression in esophageal cell lines. A: Western blot analysis of baseline Dkk1 protein expression in whole cell lysates of EPC1 and EPC2 cells. β-Actin served as a loading control. B: ELISA of Dkk1 protein secretion in equally seeded EPC1 and EPC2 cells at rest after 24 h of incubation. Values are means ± SE of 3 independent experiments. C and D: quantitative RT-PCR analysis of Dkk1 mRNA expression in EPC1 and EPC2 cells following acute acid exposure (C) and chronic acid pulsing (D). Total RNA was collected 3 h after acid exposure. E: ELISA of Dkk1 protein secretion following chronic acid pulsing. Data represent relative mean Dkk1 secretion. *P ≤ 0.05, **P ≤ 0.01 vs. no acid on the respective day. F: Western blot analysis of Dkk1 protein expression in whole lysates of EPC1 cells following chronic acid pulsing.
Acid-induced DNA damage underlies Dkk1 upregulation.
Upregulation of Dkk1 has been associated with activation of DNA damage-signaling pathways in cells undergoing different types of stress (44). Therefore, we examined whether acid-induced DNA damage of esophageal epithelial cell lines underlies Dkk1 activation in esophagitis. Histone H2AX phosphorylation (γH2AX) has been found to be a sensitive marker for genotoxically mediated DNA double-strand breaks (43). As shown in EPC2 cells by immunofluorescence staining, chronic acid pulsing resulted in increased expression of γH2AX in a time-dependent manner, indicating DNA damage (Fig. 3A). To test whether EPC2 cells respond to DNA damage by upregulating Dkk1, we stressed EPC2 cells with UV-B radiation or camptothecin, both of which are well established inducers of cellular DNA damage (41). As shown in Fig. 3B, both stressors induced a high (40-fold) elevation of Dkk1 gene transcription. These observations suggest that Dkk1 overexpression is the cellular response of esophageal epithelial cells to DNA damage triggered by acidic injury.
Fig. 3.

Acid-induced DNA damage underlies Dkk1 overexpression. A: phosphorylated histone H2AX (γH2AX) nuclear foci formation in EPC2 cells following chronic acid pulsing. Note increase in γH2AX-containing nuclear foci in acid-treated, but not untreated control, cells. 4′6-Diamidino-2-phenylindole (DAPI) staining demonstrates total number of cell nuclei in the same field. Cells treated with camptothecin served as positive control. Images represent results from 3 experiments. B: quantitative RT-PCR analysis of Dkk1 mRNA expression in EPC2 cells 24 h after treatment with camptothecin (1 μM) or exposure to UV-B radiation (100 J/m2). Values are means ± SE of 3 experiments. **P ≤ 0.001 vs. (untreated) control.
Dkk1 inhibits esophageal epithelial cell proliferation.
To define how Dkk1 overexpression affects esophageal epithelial cell growth, rhDkk1 protein was utilized to test the effect of Dkk1 gain-of-function on cell proliferation. Proliferation rates of both esophageal epithelial cell lines, assessed by [3H]thymidine uptake, were significantly decreased following rhDkk1 (500 ng/ml) administration for 48 h (Fig. 4A). As determined by MTT assay, the absorbance values of rhDkk1-treated cells were markedly decreased, in a dose-dependent manner, compared with untreated cells (Fig. 4B), confirming Dkk1-mediated cell growth inhibition. The antiproliferative effect of Dkk1 was further verified by assessing expression of the Ki-67 proliferative marker. EPC2 cells treated with rhDkk1 demonstrated a significant decrease in the Ki-67 labeling index compared with untreated cells (Fig. 4C). Furthermore, when endogenous Dkk1 was blocked by anti-Dkk1 blocking Ab, cell proliferation was elevated after 48 and 96 h compared with control cells and human IgG-treated cells (Fig. 4D). These data demonstrate that Dkk1 is a negative regulator of esophageal epithelial cell growth.
Fig. 4.

Dkk1 inhibits esophageal epithelial cell proliferation. A: [3H]thymidine uptake assay in EPC1 and EPC2 cells following treatment with recombinant human Dkk1 (rhDkk1). Cells were pulsed for 24 h with 1 μCi/ml [3H]thymidine. Values are means ± SE of 3 independent experiments. *P ≤ 0.05 vs. untreated. B: 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay following treatment with 100 and 500 ng/ml rhDkk1 in EPC1 and EPC2 cells. Values are means ± SE of 3 biological replicates performed in sextuplicate and presented as percentage of untreated controls on day 0. *P ≤ 0.05, untreated vs. 100 ng/ml rhDkk1; #P ≤ 0.05, untreated vs. 500 ng/ml rhDkk1. C: immunofluorescence staining of Ki-67 in EPC2 cells following treatment with 500 ng/ml rhDkk1 for 48 h. LI, labeling index. Values are means ± SE of 3 independent experiments. *P ≤ 0.05. D: MTT assay following treatment with anti-Dkk1 Ab or human IgG in EPC2 cells. Values are means ± SE of 3 experiments performed in triplicate and presented as percentage of untreated controls on day 0. *P ≤ 0.05 vs. untreated and human IgG.
Dkk1 induces cell cycle arrest, but not apoptosis, in esophageal epithelial cells in vitro.
Dkk1 overexpression has been associated with activation of the apoptotic program in various tumor cell models (44, 51). Thus we first examined whether Dkk1's negative effect on esophageal epithelial cell growth is the result of apoptosis activation. Annexin V-FITC apoptosis assay revealed that rhDkk1 (500 ng/ml) application for 48 h did not induce early cell apoptosis in EPC2 cells (1.68%) compared with control cells (1.85%). In contrast, application of 1 mM camptothecin (positive control) markedly induced cell apoptosis (8.82%; Fig. 5A). Similar results were also generated in EPC1 cells and verified by terminal deoxynucleotidyl transferase dUTP-mediated nick end-labeling apoptosis assay (data not shown). We also confirmed the absence of Dkk1-mediated apoptosis in a primary esophageal epithelial cell line (HEEC; Fig. 5B) to exclude the possibility that the inability of rhDkk1 to induce apoptosis in EPC2 and EPC1 cell lines is due to telomerase protection against cell apoptosis (15).
Fig. 5.

Dkk1 induces cell cycle arrest, but not apoptosis. A and B: annexin V-FITC apoptosis assay for detection of early apoptotic EPC2 cells and human esophageal epithelial cells (HEEC) following treatment with rhDkk1. Camptothecin served as positive control. C and D: representative graphs and quantitative analysis of flow cytometric cell cycle assessment in EPC1 and EPC2 cells following treatment with rhDkk1. Values are means ± SE of ≥3 independent experiments performed in triplicate. *P ≤ 0.05, G0/G1 phase in untreated vs. rhDkk1.
The inhibition of cell proliferation without induction of apoptosis raised the following question: Does Dkk1 promote cell cycle arrest? Using flow cytometry, we analyzed the cell cycle by measuring DNA content in cells treated with rhDkk1 (500 ng/ml) and in control cells (Fig. 5C). The quantitative data show that rhDkk1 significantly increased the fraction of cells in the G0/G1 phase in both cell lines (Fig. 5D). In parallel, rhDkk1 decreased the fraction of cells in the S and G2/M phases, verifying its antiproliferative effect. These data demonstrate that Dkk1 inhibits growth by delaying the cell cycle in the G0/G1 phase and not via apoptosis.
Dkk1 is a mediator of esophageal epithelial cellular senescence in vitro.
Next, to address whether Dkk1 promotes irreversible growth arrest via cellular senescence, we performed SA-β-Gal assay. As shown in Fig. 6A, administration of rhDkk1 (500 ng/ml) for 48 h induced a senescent phenotype in EPC2 cells. Dkk1-mediated cellular senescence was further verified by concurrent examination of senescence-associated markers. Expression of p16 mRNA, but not p21 or cyclin D1, was increased following treatment with rhDkk1, while Ki-67 gene expression was markedly suppressed (Fig. 6B). Treatment of EPC2 cells with rhDkk1 resulted in dephosphorylation of retinoblastoma protein (Rb) and upregulation of p16 protein expression, while expression of p53 and its transcriptional target p21 remained unchanged (Fig. 6C). This suggests that Dkk1 promotes senescence by activating the p16/Rb pathway, and not the p53/p21 pathway. Similar results were obtained in EPC1 cells (data not shown). Taken together, these findings suggest that Dkk1 promotes growth arrest in esophageal epithelial cells via premature senescence.
Fig. 6.

Dkk1 promotes cellular senescence. A: percentage of senescence-associated β-galactosidase (SA-β-Gal)-positive EPC2 cells before (untreated) and after treatment with rhDkk1 (left). Values are means ± SE of ≥500 cells of ≥3 independent biological replicates. *P ≤ 0.05. Right: SA-β-Gal staining in EPC2 cells before and after treatment with rhDkk1. Arrowheads, SA-β-Gal-positive cells. B: quantitative RT-PCR analysis of p16, p21, Ki-67, and cyclin D1 mRNA levels in EPC2 cells treated with rhDkk1. Data were normalized to β-actin expression. Values are means ± SE of 3 independent experiments. *P ≤ 0.05 vs. untreated. C: Western blot analysis of phosphorylated (Ser780) retinoblastoma (Rb), p53, p21, and p16 protein expression in untreated and rhDkk1-treated EPC2 cells. β-Actin served as loading control. Blots represent results from 3 experiments.
Dkk1 mediates acid-induced senescence in esophageal epithelial cell lines in vitro.
Whether the upregulation of Dkk1 mediates acid-induced senescence was tested by SA-β-Gal staining. Chronic acid pulsing for 5 days markedly increased the senescent phenotype in EPC2 cells (Fig. 7, A and B), while use of anti-Dkk1 Ab to block Dkk1 between the acid pulses diminished the acid-induced senescence, indicating that Dkk1 is a secreted mediator of cellular senescence in acid-damaged esophageal epithelial cells (Fig. 7, A and B). This notion was further supported by a direct correspondence of the mRNA levels of p16 senescent marker and the SA-β-Gal staining findings following chronic acid pulsing without or with anti-Dkk1 Ab (Fig. 7C).
Fig. 7.

Dkk1 mediates acid-induced senescence in esophageal epithelial cells. A: quantitative analysis of SA-β-Gal staining following chronic acid pulsing for 5 days in EPC2 cells (left). Between acid pulses, cells recovered in normal growth medium or in medium containing anti-Dkk1 Ab or human IgG. Values are means ± SE of 3 independent experiments. *P ≤ 0.05, **P ≤ 0.01. Right: SA-β-Gal staining in EPC2 cells before and after treatment with rhDkk1. Arrowheads, SA-β-Gal-positive cells. B and C: SA-β-Gal staining and quantitative RT-PCR analysis of p16 mRNA levels in EPC2 cells subjected to acid pulsing as described in A. D: MTT assay following acid pulsing in EPC2 cells. No acid, cells not exposed to acid (control); acid, cells subjected to acid pulsing and allowed to recover in normal growth medium between acid pulses and during postacid period; acid + anti-Dkk1 Ab, cells subjected to acid pulsing and allowed to recover in growth medium containing anti-Dkk1 Ab between acid pulses and during postacid period; acid + human IgG, cells exposed to acid pulsing and allowed to recover in growth medium containing human IgG between acid pulses and during the postacid period. MTT assay was performed 0, 24, 48 and 72 h postacid. For 0 h, MTT reagent was added 1 h after seeding the cells. Values are means ± SD of 3 independent experiments performed in sextuplicate. *P ≤ 0.05. E: MTT assay of EPC2 cells following acid pulsing in the absence of Dkk1 blocking. Anti-Dkk1 Ab was added only in the postacid period.
In agreement with increased senescent phenotype, chronic acid pulsing suppressed cell proliferation of EPC2 cells, as assessed by MTT assay at 24, 48, and 72 h after 5 days of chronic acid pulsing (Fig. 7D). However, the rate of cell proliferation was rescued compared with control cells (untreated or treated with human IgG) when cells were incubated during and after the acid pulsing with anti-Dkk1 Ab, underscoring the significant impact of Dkk1 on the acid-induced growth inhibition (Fig. 7D). Acid-suppressed proliferation was not rescued when anti-Dkk1 Ab was added after, and not during, the acid pulsing, suggesting that the acid-induced elevation of Dkk1 levels caused a growth arrest during the acid pulses that cannot be reversed by blocking Dkk1 only in the postacid period (Fig. 7E). In summary, these results highlight the finding that Dkk1 is a mediator of cellular senescence in acid-damaged esophageal epithelial cells in vitro.
Dkk1 regulates the levels of transcriptionally active β-catenin.
Suppression of Wnt/β-catenin signaling is an early-activated trigger of the senescence program (53). Thus we addressed whether Dkk1, as a specific Wnt inhibitor, promotes senescence by suppressing Wnt/β-catenin signaling.
We used a specific antibody against ABC to verify the ability of Dkk1 to suppress β-catenin transcriptional activity. ABC, dephosphorylated β-catenin specifically at Ser37/Thr41, is not susceptible to ubiquitination and degradation, and its cytoplasmic/nuclear amounts are considered to be highly transcriptionally active (7, 47). Incubation of EPC2 cells with rhDkk1 reduced intracellular levels of ABC protein (Fig. 8A). On the other hand, siRNA-mediated Dkk1 knockdown increased the baseline levels of intracellular ABC in EPC2 cells (Fig. 8B), signifying that Dkk1 suppresses ABC protein levels and, subsequently, β-catenin transcriptional activity. Dkk1-mediated suppression of Wnt/β-catenin signaling was further supported when Dkk1 protein secretion that was efficiently suppressed by gene silencing (Fig. 8C) increased the mRNA levels of AXIN2, a sensitive Wnt target gene (23) (Fig. 8D). With respect to senescent markers, Dkk1 gene silencing increased Ki-67 mRNA levels, while it suppressed p16 gene expression twofold (Fig. 8D). Negative regulation of Dkk1 over ABC was confirmed in vivo by costaining of human esophageal specimens for Dkk1 and ABC protein expression (Fig. 8E). As shown by fluorescence microscopy, ABC expression was markedly lower in the basal than the superficial mucosal layer, whereas Dkk1 demonstrated a reverse pattern of expression.
Fig. 8.

Dkk1 downregulates transcriptional active β-catenin. A: Western blot analysis of active β-catenin (ABC dephosphorylated at Ser37/Thr41) and total β-catenin expression in whole cell lysates of EPC2 cells following treatment with rhDkk1. B: Western blot analysis of ABC protein following efficient Dkk1 short interfering RNA (siRNA)-mediated knockdown. β-Actin served as loading control. Blots represent results from 3 experiments. *P ≤ 0.05. C: ELISA of Dkk1 secretion in culture medium following transfection with Dkk1 siRNA. Values are means ± SE of 3 independent biological replicates. *P ≤ 0.05 vs. control siRNA. D: quantitative RT-PCR analysis of mRNA levels for Dkk1, Axin2, Ki-67, and p16 genes in EPC2 cells following Dkk1 siRNA-mediated gene silencing. Gene expression was normalized to β-actin expression. Values (means ± SE of 3 biological replicates) are relative to mock control. *P ≤ 0.05, #P ≤ 0.01. &Dkk1 knockdown ≥80%. E: immunofluorescence microscopy for colocalization of Dkk1 and ABC protein in esophageal mucosa. Arrowheads, basal layer of esophageal epithelium. Images represent results from ≥3 different tissues.
Taken together, these findings confirm that Dkk1 is a negative regulator of β-catenin transcriptional activity in esophageal epithelium.
Wnt-mediated β-catenin signaling activation attenuates acid-induced senescence in esophageal epithelial cells in vitro.
Next, to determine whether Wnt/β-catenin activation can attenuate acid-induced senescence in reverse, we used rhWnt3a (200 ng/ml) to stimulate canonical Wnt/β-catenin signaling in EPC2 cells undergoing chronic acid pulsing for 5 days. As shown in Fig. 9A, fewer cells stained positive for SA-β-Gal in the presence than absence of rhWnt3a between acid pulses, implying that Wnt-mediated β-catenin signaling activation suppresses acid-induced senescence.
Fig. 9.

Wnt/β-catenin signaling activation counteracts acid-induced senescence and is inhibited by Dkk1. A: quantitative analysis of SA-β-Gal staining following chronic acid pulsing in the presence of recombinant human Wnt3a (rhWnt3a) in EPC2 cells. Values are means ± SE of 3 independent experiments. *P ≤ 0.05. B: quantitative RT-PCR analysis of AXIN2 gene expression (a) and TOP/FOP-flash reporter assay (b) in EPC2 cells following 24 h of stimulation with rhDkk1, rhWnt3a, anti-Dkk1 Ab, and human IgG. TOP/FOP-flash activity was normalized to Renilla activity. AXIN2 gene expression was normalized to β-actin expression. TOP/FOP-flash activity values are relative to unstimulated cells of 3 independent experiments. *P ≤ 0.05.
Given the known antagonism between Dkk1 and Wnt agonists in suppressing and activating β-catenin signaling, respectively, we investigated whether Dkk1 is able to counteract Wnt-mediated β-catenin signaling. We assessed AXIN2 mRNA levels and β-catenin transcriptional activity (TOP-flash) in EPC2 cells treated with rhWnt3a (200 ng/ml) and rhDkk1 (500 ng/ml), alone or in combination (Fig. 9B). TOP-flash activity did not differ from FOP-flash activity in control EPC2 cells, indicating the absence of appreciable TCF/β-catenin-mediated transcriptional activity in resting conditions. rhDkk1 alone did not alter AXIN2 mRNA levels or TOP-flash activity, whereas rhWnt3a increased both, suggesting Wnt3a-mediated β-catenin signaling activation. Interestingly, when cells were incubated with both recombinant proteins, rhDkk1 abolished the effect of rhWnt3a on activating TOP-flash activity and AXIN2 transcription, verifying the Dkk1 antagonism over Wnt agonists. Coincubation with rhWnt3a and anti-Dkk1 Ab resulted in enhanced induction of AXIN2 gene transcription and TOP-flash activity, suggesting that even the endogenous amounts of Dkk1 protein impair Wnt-mediated β-catenin signaling. The latter is also supported by significantly higher (up to 6-fold) AXIN2 gene transcription in Dkk1 siRNA knockdown cells stimulated with rhWnt3a compared with nontarged siRNA control cells stimulated with rhWnt3A (data not shown). Collectively, these results imply that Dkk1 overexpression promotes cellular senescence by antagonizing canonical Wnts.
Association of Dkk1 overexpression with cellular senescence in esophagitis tissue.
To investigate how the above-described findings apply in human reflux esophagitis in vivo, we analyzed p16 mRNA expression by real-time PCR in the same esophageal biopsies of esophagitis patients in which we analyzed Dkk1 mRNA expression. As shown in Fig. 10A, gene expression of p16 gene was higher in the distal inflamed mucosa than the corresponding proximal healthy mucosa in esophagitis patients (P ≤ 0.05 by paired t-test). Remarkably, p16 mRNA expression in inflamed mucosa with esophagitis correlated positively with the respective Dkk1 mRNA expression (r = 0.958, P ≤ 0.0001 by Pearson's test; Fig. 10B). Immunohistochemical analysis revealed colocalization of the two proteins in the mucosal compartments of esophagitis tissue, while Western blot analysis verified the positive correlation of p16 and Dkk1 protein expression in human reflux esophagitis (Fig. 10C). Moreover, SA-β-Gal assay in representative esophageal biopsies from esophagitis patients and healthy controls demonstrated positive SA-β-Gal activity in inflamed mucosa, in contrast to healthy mucosa. SA-β-Gal staining in Barrett's intestinal metaplasia was used as positive control (13). These results confirm the association of acid-induced Dkk1 overexpression with the process of cellular senescence in human reflux esophagitis in vivo.
Fig. 10.

Dkk1 overexpression associates with cellular senescence in vivo. A: scatter graph of quantitative RT-PCR data for p16 mRNA expression in distal and proximal esophageal mucosa of esophagitis patients (n = 15). Results were normalized to β-actin expression (P ≤ 0.05 by paired t-test). B: correlation of Dkk1 mRNA expression with p16 mRNA expression in distal esophageal mucosa of esophagitis patients. (r = 0.958, P ≤ 0.00001 by Pearson's correlation test). C and D: immunohistochemistry for colocalization and Western blot analysis for coexpression of Dkk1 and p16 protein expression in esophageal biopsies of 3 esophagitis patients (1, 2, and 3). E: SA-β-Gal staining in healthy human esophageal mucosa and reflux esophagitis. Arrows, β-Gal-positive cells on the basal layer of esophageal mucosa with esophagitis. Barrett's metaplasia serves as control for positive SA-β-Gal activity. Images represent results from ≥3 different patients in each group.
DISCUSSION
In this study, we have shown that Dkk1, the specific Wnt antagonist, is induced by acidic pH and functions as a secreted mediator of epithelial cellular senescence in human reflux esophagitis. In an enhancement of previous findings of high Dkk1 gene expression in the normal-looking squamous mucosa of esophagitis patients (2), Dkk1 was found to be upregulated in the inflamed squamous mucosa of esophagitis patients at the transcriptional and translational levels. In vitro, esophageal squamous epithelial cell lines were shown to express and secrete high levels of Dkk1 in response to acid-induced DNA injury. Utilizing recombinant Dkk1 protein, we have shown that high extracellular levels of Dkk1 inhibit epithelial cell growth in vitro by inducing cell cycle arrest via cellular senescence. Furthermore, acidic pH induced cellular senescence, which was directly linked to the ability of Dkk1 to antagonize the canonical Wnt/β-catenin signaling. Finally, an association between Dkk1 overexpression and induction of cellular senescence was verified in human esophagitis tissue. Taken together, these findings reveal a novel mechanism of esophageal epithelial response to acidic injury that involves upregulation of Dkk1 with subsequent modulation of Wnt/β-catenin signaling and induction of cellular senescence.
Induction of Dkk1 has been linked to cellular stress, causing DNA damage, and to activation of proinflammatory mediators. Characteristically, brain tumor cells respond to DNA damage by elevating Dkk1 expression (44), while activation of IFN-γ and TNF-α signaling pathways has been reported upstream of Dkk1 overexpression in colitis and rheumatoid arthritis (26, 49). In reflux esophagitis, the cellular injury is triggered by intraluminal stressors, such as acid and bile, and results in DNA damage (14) and activation of multifaceted inflammatory mechanisms (42). In this study, we emphasized the effect of acid, based on the high Dkk1 levels reported in the normal-looking mucosa of esophagitis patients, away from the margins of the reflux-induced lesions/erosions, suggesting that Dkk1 is more likely affected by intraluminal stressors than by inflammatory mediators (2). Indeed, acid-mediated cellular injury induced Dkk1 overexpression in vitro. This effect was directly linked to acid-mediated DNA damage (14), just as the esophageal epithelial cells respond to alternative inducers of DNA injury such as camptothecin by upregulating Dkk1. In favor of this observation, Dkk1 transcriptional activation has been associated with the p53 tumor suppressor, the activation of which has been reported upon cellular stress accompanied by DNA damage (30, 50, 52). Nevertheless, a chemokine-mediated regulation of Dkk1 transcription, following acid injury, cannot be ruled out at this point and is under investigation by our group.
With respect to the impact of Dkk1 overexpression on epithelial functions, we demonstrated that high levels of Dkk1 promote growth arrest via cellular senescence in vitro. We subsequently showed that Dkk1 is a secreted mediator of an actual acid-induced cellular senescence in the setting of acidic cellular injury. These findings are in agreement with published reports supporting an antiproliferative profile for Dkk1 in response to proapoptotic stimuli (35), as well as a senescence-inducing activity for Wnt antagonists in primary fibroblasts (12). Of interest, ectopic Dkk1 expression has been shown to induce severe intestinal mucosal injury in mice via inhibition of intestinal epithelial cell proliferation (29), indicating that the high Dkk1 levels in esophagitis tissue might correlate with the magnitude of mucosal injury. Such a correlation in this study was, however, not possible, because of the limited number of patients and the lack of all grades of esophagitis.
At the molecular level, we demonstrated that the Dkk1-mediated antagonism of canonical Wnt signaling underlies the induction of senescence in acid-damaged esophageal epithelial cells. Acid-induced senescence was markedly alleviated when, between acid exposures, Wnt signaling was enhanced directly by Wnt stimulation or indirectly by blocking Dkk1's ability to antagonize Wnt. The excessive amounts of exogenous Wnt3a very likely overwhelmed the acid-induced increases in amounts of Dkk1. In reverse, excessive presence of Dkk1 attenuated the Wnt3a-mediated β-catenin transcriptional activation, verifying the ability of Dkk1 to dampen canonical Wnt signaling. In support of the latter, the Dkk1 expression levels correlated negatively with the intracellular amounts of transcriptionally active β-catenin in vitro and in vivo. However, the actual level of Wnt/β-catenin signaling activation in esophageal epithelial cells has not been identified. As shown by luciferase assay in vitro, esophageal cells at rest do not demonstrate an appreciable level of β-catenin signaling. Interestingly, the depletion of Dkk1 increased β-catenin signaling, implying that the inactivated status of Wnt signaling in resting conditions may rely on the desensitization to Wnt signals by the baseline secretion of endogenous Dkk1. Given that Dkk1 is a main downstream target gene of β-catenin, functioning in a negative-feedback loop (36), the following question arises: Is the Dkk1 upregulation during esophagitis aiming to counteract an actual Wnt/β-catenin signaling activation? This notion may justify the highly proliferative epithelial response addressed often in reflux esophagitis, despite the Dkk1 overexpression; however, this idea remains speculative and merits further study.
Our in vitro findings further support a link between the Dkk1-mediated Wnt antagonism upstream and the Rb/p16 signaling activation downstream in esophageal epithelial cells undergoing cellular senescence in response to acidic injury. Interaction between the Rb pathway and Wnt/β-catenin signaling has been reported during the initiation of senescence (12). Characteristically, repression of Wnt signaling via β-catenin RNA interference (RNAi) knockdown induced senescence characterized by Rb dephosphorylation, while simultaneous Rb knockdown abolished β-catenin RNAi-induced senescence (12). Curiously, senescence due to DNA damage would be expected to be more dependent on p53 activation (8). However, p16 also provides a distinct barrier to prevent growth of cells with severely damaged DNA (48). Interestingly, Dkk1 expression correlated directly with p16 expression and SA-β-Gal activity also in vivo, underscoring a significant role for cellular senescence in the pathogenesis of human reflux esophagitis.
Little is known about the physiological relevance of senescence in vivo, especially during tissue repair. Nevertheless, emerging evidence supports the notion that senescence is an important mechanism for preservation of tissue homeostasis in the presence of detrimental stimuli by preventing accumulation of damage and functional decline (16). In response to injury, cells will undergo senescence, or they will self-destruct (apoptosis), if the damage cannot be easily repaired. In GERD, squamous epithelial cells are continuously injured by the gastric refluxate, which induces oxidative stress and DNA damage (14, 24). As the GERD injury persists, more surface epithelial cells are damaged, and the injury progresses to the deeper layers of the epithelium. In response, an increased cell replication of basal mucosal layers ensues to replace the reflux-damaged epithelial cells, with different growth factors, mitogens, and cytokines/chemokines identified as possible stimulators (45). As a result of this highly proliferative response, the basal layer and papillae of esophageal mucosa appear hyperplastic in severe reflux esophagitis (20). However, with respect to the continuous mucosal injury and the subsequent proliferative response, it becomes apparent that cellular mechanisms that are able to counteract cell proliferation and secure DNA repair are essential for proper esophageal regeneration and protection against genotoxic events (25). Therefore, we propose that Dkk1-mediated cellular senescence may be among those mechanisms that aim to counteract hyperproliferation in reflux esophagitis in an effort to facilitate regeneration and preserve homeostasis.
The significance of our current findings is associated with a deeper understanding of the molecular pathways underlying the epithelial homeostasis in the setting of reflux esophagitis. Modulation of Wnt/β-catenin signaling and subsequent induction of cellular senescence by Dkk1 as shown here consist of novel mechanisms of esophageal epithelial response to GERD injury. The direct link between Dkk1 expression and acidic mucosal injury underscores the notion that Dkk1 may highlight a novel marker that may be able to facilitate better evaluation of antiacid therapies, prediction of mucosal injuries along with patient stratification for follow-up purposes, and development of novel therapies. With respect to the tumorigenic nature of aberrant Wnt-signaling activation (37) and the senescence-associated tumor suppression, Dkk1 may also play a pivotal role in esophageal carcinogenesis. Although high levels of Dkk1 expression have been reported in EAC (9) and aberrant activation of Wnt/β-catenin signaling has been found to promote esophageal intestinal metaplasia (27) and neoplastic progression toward EAC (6, 4, 34), the exact role of Dkk1 during esophageal carcinogenesis remains unknown and requires further study.
An acknowledged limitation of this study is that our conclusions about the Dkk1 antagonism over Wnt signals are based on findings generated mainly in vitro. However, those conclusions do not contradict already reported findings arising from in vivo models of intestinal epithelial injury and inflammation (26), providing a level of confidence in human reflux esophagitis. Moreover, the utilization of recombinant Wnt3a in our experiments raises questions about the actual source of Wnts in the esophageal microenvironment. Positive Wnt3a expression has been reported in human esophageal mucosa (3), indicating that it functions in an autocrine manner, while mesenchymal tissue stem cells, located in the submucosal layer of the gastrointestinal tract, are expected also to secrete Wnts (31). However, how Wnts really participate in esophageal biology remains largely unknown.
In conclusion, we have described a mechanism of esophageal epithelial cellular response to acid-mediated injury in which Dkk1 plays a central role. In response to acid-mediated epithelial injury, Dkk1 is induced and functions as a secreted mediator of cellular senescence via suppression of Wnt/β-catenin signaling (Fig. 11). The high levels of Dkk1 may be crucial in counteracting increased proliferative responses addressed also in esophagitis and, in this way, facilitate a proper regeneration of the damaged esophageal epithelium. In all, our current findings support a homeostatic role for Dkk1 in esophageal mucosa and highlight the need for further investigation of the role of Wnt/β-catenin signaling, as well as the mechanism of senescence, in esophageal mucosal biology and pathobiology.
Fig. 11.
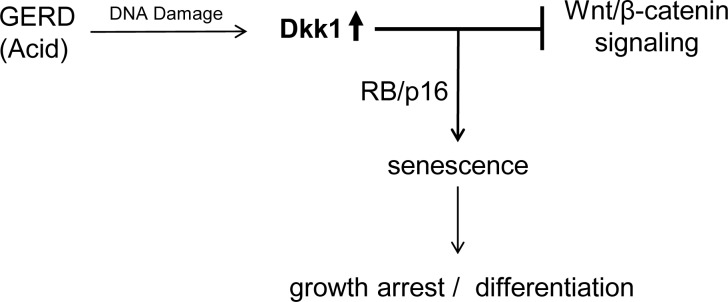
Schematic illustration of the proposed working model demonstrating induction of Dkk1 and its effects on promoting senescence and modulating Wnt/β-catenin signaling in human reflux esophagitis. GERD, gastroesophageal reflux disease.
GRANTS
This study was supported in part by Laura Gralton on behalf of the Daniel and Laura Gruber Charitable Lead Trust #3 and National Center for Advancing Translational Sciences Grant 8UL1 TR-000055.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
O.L., P.R., and R.S. are responsible for conception and design of the research; O.L., L.N., R.M., N.J., J.S., A.C.M., and N.V. performed the experiments; O.L., L.N., J.S., and N.V. analyzed the data; O.L., P.R., and R.S. interpreted the results of the experiments; O.L., P.R., and A.C.M. prepared the figures; O.L. drafted the manuscript; O.L., P.R., A.C.M., and R.S. edited and revised the manuscript; O.L., P.R., L.N., R.M., N.J., A.C.M., N.V., and R.S. approved the final version of the manuscript.
ACKNOWLEDGMENTS
The authors thank Tracy Kaczanowski and Megan DeMara for significant help and support.
REFERENCES
- 1.Adams PD, Enders GH. Wnt-signaling and senescence: a tug of war in early neoplasia? Cancer Biol Ther 7: 1706–1711, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ali I, Rafiee P, Hogan WJ, Jacob HJ, Komorowski RA, Haasler GB, Shaker R. Dickkopf homologs in squamous mucosa of esophagitis patients are overexpressed compared with Barrett's patients and healthy controls. Am J Gastroenterol 101: 1437–1448, 2006 [DOI] [PubMed] [Google Scholar]
- 3.Ali I, Rafiee P, Zheng Y, Johnson C, Banerjee B, Haasler G, Jacob H, Shaker R. Intramucosal distribution of WNT signaling components in human esophagus. J Clin Gastroenterol 43: 327–337, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bian YS, Osterheld MC, Bosman FT, Fontolliet C, Benhattar J. Nuclear accumulation of β-catenin is a common and early event during neoplastic progression of Barrett esophagus. Am J Clin Pathol 114: 583–590, 2000 [DOI] [PubMed] [Google Scholar]
- 5.Campisi J, d'Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol 8: 729–740, 2007 [DOI] [PubMed] [Google Scholar]
- 6.Clevers H. Wnt/β-catenin signaling in development and disease. Cell 127: 469–480, 2006 [DOI] [PubMed] [Google Scholar]
- 7.Cong F, Schweizer L, Chamorro M, Varmus H. Requirement for a nuclear function of β-catenin in Wnt signaling. Mol Cell Biol 23: 8462–8470, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.d'Adda di Fagagna F, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, Von Zglinicki T, Saretzki G, Carter NP, Jackson SP. A DNA damage checkpoint response in telomere-initiated senescence. Nature 426: 194–198, 2003 [DOI] [PubMed] [Google Scholar]
- 9.Darlavoix T, Seelentag W, Yan P, Bachmann A, Bosman FT. Altered expression of CD44 and DKK1 in the progression of Barrett's esophagus to esophageal adenocarcinoma. Virchows Arch 454: 629–637, 2009 [DOI] [PubMed] [Google Scholar]
- 10.Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA 92: 9363–9367, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dodds WJ, Dent J, Hogan WJ, Helm JF, Hauser R, Patel GK, Egide MS. Mechanisms of gastroesophageal reflux in patients with reflux esophagitis. N Engl J Med 307: 1547–1552, 1982 [DOI] [PubMed] [Google Scholar]
- 12.Elzi DJ, Song M, Hakala K, Weintraub ST, Shiio Y. Wnt antagonist SFRP1 functions as a secreted mediator of senescence. Mol Cell Biol 32: 4388–4399, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Going JJ, Stuart RC, Downie M, Fletcher-Monaghan AJ, Keith WN. “Senescence-associated” β-galactosidase activity in the upper gastrointestinal tract. J Pathol 196: 394–400, 2002 [DOI] [PubMed] [Google Scholar]
- 14.Goldman A, Shahidullah M, Goldman D, Khailova L, Watts G, Delamere N, Dvorak K. A novel mechanism of acid and bile acid-induced DNA damage involving Na+/H+ exchanger: implication for Barrett's oesophagus. Gut 59: 1606–1616, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gorbunova V, Seluanov A, Pereira-Smith OM. Expression of human telomerase (hTERT) does not prevent stress-induced senescence in normal human fibroblasts but protects the cells from stress-induced apoptosis and necrosis. J Biol Chem 277: 38540–38549, 2002 [DOI] [PubMed] [Google Scholar]
- 16.Gorgoulis VG, Halazonetis TD. Oncogene-induced senescence: the bright and dark side of the response. Curr Opin Cell Biol 22: 816–827, 2010 [DOI] [PubMed] [Google Scholar]
- 17.Gregorieff A, Clevers H. Wnt signaling in the intestinal epithelium: from endoderm to cancer. Genes Dev 19: 877–890, 2005 [DOI] [PubMed] [Google Scholar]
- 18.Harada H, Nakagawa H, Oyama K, Takaoka M, Andl CD, Jacobmeier B, von Werder A, Enders GH, Opitz OG, Rustgi AK. Telomerase induces immortalization of human esophageal keratinocytes without p16INK4a inactivation. Mol Cancer Res 1: 729–738, 2003 [PubMed] [Google Scholar]
- 19.Heidemann J, Maaser C, Lügering A, Spahn TW, Zimmer KP, Herbst H, Rafiee P, Domschke W, Krieglstein CF, Binion DG, Kucharzik TF. Expression of vascular cell adhesion molecule-1 (CD 106) in normal and neoplastic human esophageal squamous epithelium. Int J Oncol 28: 777–785, 2006 [PubMed] [Google Scholar]
- 20.Ismail-Beigi F, Horton PF, Pope CE., 2nd Histological consequences of gastroesophageal reflux in man. Gastroenterology 58: 163–174, 1970 [PubMed] [Google Scholar]
- 21.Jacobs IJ, Ku WY, Que J. Genetic and cellular mechanisms regulating anterior foregut and esophageal development. Dev Biol 369: 54–64, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jankowski JA, Wright NA. Epithelial stem cells in gastrointestinal morphogenesis, adaptation and carcinogenesis. Semin Cell Biol 3: 445–456, 1992 [DOI] [PubMed] [Google Scholar]
- 23.Jho EH, Zhang T, Domon C, Joo CK, Freund JN, Costantini F. Wnt/β-catenin/Tcf signaling induces the transcription of Axin2, a negative regulator of the signaling pathway. Mol Cell Biol 22: 1172–1183, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jürgens S, Meyer F, Spechler SJ, Souza R. The role of bile acids in the neoplastic progression of Barrett's esophagus—a short representative overview. Z Gastroenterol 50: 1028–1034, 2012 [DOI] [PubMed] [Google Scholar]
- 25.Katada N, Hinder RA, Smyrk TC, Hiki Y, Kakita A. Duodenoesophageal reflux induces apoptosis in rat esophageal epithelium. Dig Dis Sci 44: 301–310, 1999 [DOI] [PubMed] [Google Scholar]
- 26.Koch S, Nava P, Addis C, Kim W, Denning TL, Li L, Parkos CA, Nusrat A. The Wnt antagonist Dkk1 regulates intestinal epithelial homeostasis and wound repair. Gastroenterology 141: 259–268, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kong J, Crissey MA, Stairs DB, Sepulveda AR, Lynch JP. Cox2 and β-catenin/T-cell factor signaling intestinalize human esophageal keratinocytes when cultured under organotypic conditions. Neoplasia 13: 792–805, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kong J, Nakagawa H, Isariyawongse BK, Funakoshi S, Silberg DG, Rustgi AK, Lynch JP. Induction of intestinalization in human esophageal keratinocytes is a multistep process. Carcinogenesis 30: 122–130, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kuhnert F, Davis CR, Wang HT, Chu P, Lee M, Yuan J, Nusse R, Kuo CJ. Essential requirement for Wnt signaling in proliferation of adult small intestine and colon revealed by adenoviral expression of Dickkopf-1. Proc Natl Acad Sci USA 101: 266–271, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Latonen L, Laiho M. Cellular UV damage responses—functions of tumor suppressor p53. Biochim Biophys Acta 1755: 71–89, 2005 [DOI] [PubMed] [Google Scholar]
- 31.Ling L, Nurcombe V, Cool SM. Wnt signaling controls the fate of mesenchymal stem cells. Gene 433: 1–7, 2009 [DOI] [PubMed] [Google Scholar]
- 32.Logan CY, Nusse R. The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol 20: 781–810, 2004 [DOI] [PubMed] [Google Scholar]
- 33.Mao B, Wu W, Davidson G, Marhold J, Li M, Mechler BM, Delius H, Hoppe D, Stannek P, Walter C, Glinka A, Niehrs C. Kremen proteins are Dickkopf receptors that regulate Wnt/β-catenin signalling. Nature 417: 664–667, 2002 [DOI] [PubMed] [Google Scholar]
- 34.Moyes LH, McEwan H, Radulescu S, Pawlikowski J, Lamm CG, Nixon C, Sansom OJ, Going JJ, Fullarton GM, Adams PD. Activation of Wnt signalling promotes development of dysplasia in Barrett's oesophagus. J Pathol 228: 99–112, 2012 [DOI] [PubMed] [Google Scholar]
- 35.Niehrs C. Function and biological roles of the Dickkopf family of Wnt modulators. Oncogene 25: 7469–7481, 2006 [DOI] [PubMed] [Google Scholar]
- 36.Niida A, Hiroko T, Kasai M, Furukawa Y, Nakamura Y, Suzuki Y, Sugano S, Akiyama T. DKK1, a negative regulator of Wnt signaling, is a target of the β-catenin/TCF pathway. Oncogene 23: 8520–8526, 2004 [DOI] [PubMed] [Google Scholar]
- 37.Nusse R. Wnt signaling in disease and in development. Cell Res 15: 28–32, 2005 [DOI] [PubMed] [Google Scholar]
- 38.Orlando RC. Why is the high grade inhibition of gastric acid secretion afforded by proton pump inhibitors often required for healing of reflux esophagitis? An epithelial perspective. Am J Gastroenterol 91: 1692–1696, 1996 [PubMed] [Google Scholar]
- 39.Otterson MF, Nie L, Schmidt JL, Link BJ, Jovanovic N, Lyros O, Rafiee P. EUK-207 protects human intestinal microvascular endothelial cells (HIMEC) against irradiation-induced apoptosis through the Bcl2 pathway. Life Sci 91: 771–782, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rafiee P, Nelson VM, Manley S, Wellner M, Floer M, Binion DG, Shaker R. Effect of curcumin on acidic pH-induced expression of IL-6 and IL-8 in human esophageal epithelial cells (HET-1A): role of PKC, MAPKs, and NF-κB. Am J Physiol Gastrointest Liver Physiol 296: G388–G398, 2009 [DOI] [PubMed] [Google Scholar]
- 41.Rastogi RP, Richa Kumar A, Tyagi MB, Sinha RP. Molecular mechanisms of ultraviolet radiation-induced DNA damage and repair. J Nucleic Acids 2010: 592980, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rieder F, Biancani P, Harnett K, Yerian L, Falk GW. Inflammatory mediators in gastroesophageal reflux disease: impact on esophageal motility, fibrosis, and carcinogenesis. Am J Physiol Gastrointest Liver Physiol 298: G571–G581, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem 273: 5858–5868, 1998 [DOI] [PubMed] [Google Scholar]
- 44.Shou J, Ali-Osman F, Multani AS, Pathak S, Fedi P, Srivenugopal KS. Human Dkk-1, a gene encoding a Wnt antagonist, responds to DNA damage and its overexpression sensitizes brain tumor cells to apoptosis following alkylation damage of DNA. Oncogene 21: 878–889, 2002 [DOI] [PubMed] [Google Scholar]
- 45.Souza RF. The role of acid and bile reflux in oesophagitis and Barrett's metaplasia. Biochem Soc Trans 38: 348–352, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Spechler SJ, Goyal RK. The columnar-lined esophagus, intestinal metaplasia, and Norman Barrett. Gastroenterology 110: 614–621, 1996 [DOI] [PubMed] [Google Scholar]
- 47.Staal FJ, Noort MV, Strous GJ, Clevers HC. Wnt signals are transmitted through N-terminally dephosphorylated β-catenin. EMBO Rep 3: 63–68, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stein GH, Drullinger LF, Soulard A, Dulić V. Differential roles for cyclin-dependent kinase inhibitors p21 and p16 in the mechanisms of senescence and differentiation in human fibroblasts. Mol Cell Biol 19: 2109–2117, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Walsh NC, Crotti TN, Goldring SR, Gravallese EM. Rheumatic diseases: the effects of inflammation on bone. Immunol Rev 208: 228–251, 2005 [DOI] [PubMed] [Google Scholar]
- 50.Wang J, Shou J, Chen X. Dickkopf-1, an inhibitor of the Wnt signaling pathway, is induced by p53. Oncogene 19: 1843–1848, 2000 [DOI] [PubMed] [Google Scholar]
- 51.Weng LH, Wang CJ, Ko JY, Sun YC, Su YS, Wang FS. Inflammation induction of Dickkopf-1 mediates chondrocyte apoptosis in osteoarthritic joint. Osteoarthritis Cartilage 17: 933–943, 2009 [DOI] [PubMed] [Google Scholar]
- 52.Yang G, Zhang G, Pittelkow MR, Ramoni M, Tsao H. Expression profiling of UVB response in melanocytes identifies a set of p53-target genes. J Invest Dermatol 126: 2490–2506, 2006 [DOI] [PubMed] [Google Scholar]
- 53.Ye X, Zerlanko B, Kennedy A, Banumathy G, Zhang R, Adams PD. Downregulation of Wnt signaling is a trigger for formation of facultative heterochromatin and onset of cell senescence in primary human cells. Mol Cell 27: 183–196, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]