

Abstract
Epithelial cells are key players in the pathobiology of numerous hypoxia-induced lung diseases. The mechanisms mediating such hypoxic responses of epithelial cells are not well characterized. Earlier studies reported that hypoxia stimulates protein kinase C (PKC)δ activation in renal cancer cells and an increase in expression of a heparin-binding growth factor, midkine (MK), in lung alveolar epithelial cells. We reasoned that hypoxia might regulate MK levels via a PKCδ-dependent pathway and hypothesized that PKCδ-driven MK expression is required for hypoxia-induced lung epithelial cell proliferation and differentiation. Replication of human lung epithelial cells (A549) was significantly increased by chronic hypoxia (1% O2) and was dependent on expression of PKCδ. Hypoxia-induced proliferation of epithelial cells was accompanied by translocation of PKCδ from Golgi into the nuclei. Marked attenuation in MK protein levels by rottlerin, a pharmacological antagonist of PKC, and by small interfering RNA-targeting PKCδ, revealed that PKCδ is required for MK expression in both normoxic and hypoxic lung epithelial cells. Sequestering MK secreted into the culture media with a neutralizing antibody reduced hypoxia-induced proliferation demonstrating that an increase in MK release from cells is linked with epithelial cell division under hypoxia. In addition, recombinant MK accelerated transition of hypoxic epithelial cells to cells of mesenchymal phenotype characterized by elongated morphology and increased expression of mesenchymal markers, α-smooth muscle actin, and vimentin. We conclude that PKCδ/MK axis mediates hypoxic proliferation and differentiation of lung epithelial cells. Manipulation of PKCδ and MK activity in epithelial cells might be beneficial for the treatment of hypoxia-mediated lung diseases.
Keywords: hypoxia, PKCδ, midkine, proliferation, differentiation, lung epithelial cells
alveolar hypoxia, a common feature of many respiratory disorders such as pulmonary hypertension and chronic obstructive pulmonary diseases, provokes structural changes in the lung manifested by thickening of the alveolar wall (1, 13, 34). In the early phase of hypoxia, apoptosis of lung epithelial cells has been observed. In contrast, in the later phase of alveolar hypoxia, cell death is no longer dominant and instead epithelial cells undergo reactive hyperplasia, a bellwether for epithelial dysfunction in the lung (34). Therefore, understanding of molecular pathways involved in hypoxia-induced loss of alveolar epithelium integrity is crucial for designing therapies targeting such injurious signaling pathways. Although prolonged exposure to hypoxia stimulates differentiation of lung epithelial cells to cells of mesenchymal phenotype in culture (39), the molecular mechanisms driving chronic hypoxia-stimulated responses in these cells remain unknown.
Based on our earlier studies of vascular cell proliferation (6, 28), one signaling mediator relevant to hypoxia-related proliferative responses is protein kinase C (PKC). Of particular interest is PKCδ, a member of a superfamily of lipid-activated serine/threonine kinases, which plays a critical role in endotoxin-induced lung injury (3). Furthermore, PKCδ controls transcriptional activity of hypoxia-inducible factor (HIF)-1α and expression of vascular endothelial growth factor (VEGF) thereby regulating hypoxia-induced angiogenesis in cancer (18, 25, 36). Even though PKCδ is known as a prosurvival kinase for lung epithelial cells (4), the role of this isozyme in hypoxia-stimulated proliferative responses of lung epithelial cells has not been explored.
Because hypoxia, via HIF-1α activation, also induces midkine (MK), a heparin-binding growth factor, expression in the airway epithelium of the lung (26) and hyperglycemic conditions induce MK expression that promotes accumulation of extracellular matrix through the phosphorylation of PKC in kidney mesangial cells (16), we sought to examine the role of PKC in hypoxic signaling that is relevant to 1) the HIF-1α induction in the lung; 2) PKCδ; and 3) its potential relationship with MK in hypoxic lung epithelial cells. Such interactions are plausible, because in human pulmonary diseases characterized by hypoxia, such as cystic fibrosis, enhanced expression of MK has been reported in the airways (23). Despite the fact that hypoxia is an inducer of MK, how MK affects hypoxia-induced proliferation and differentiation of lung epithelial cells is unknown.
We, therefore, hypothesized that hypoxia through PKCδ-regulated elevation in MK levels will stimulate lung epithelial cell proliferation and differentiation. In the present study, we have demonstrated that human lung epithelial cells, when exposed to chronic hypoxia, have the ability to increase their proliferation in a PKCδ-dependent manner. Most importantly, PKCδ inhibition with small interfering (si)RNA-mediated PKCδ knockdown dramatically reduced MK levels in these cells. The hypoxia-driven increase in MK protein expression and secretion coincided with increased proliferation of lung epithelial cells. In addition to raising proliferation, exogenously added recombinant MK (rMK) promoted differentiation of hypoxic epithelial cells. We conclude that PKCδ is a key regulator of MK expression via which it controls proliferation and differentiation of human lung epithelial cells responding to hypoxia. Our results suggest that attenuation of PKCδ/MK axis should be seriously considered when developing therapeutic strategies for treatment of lung diseases involving hypoxia.
METHODS
Cell culture.
Human lung epithelial cells (A549) were cultured in a humidified atmosphere of 5% CO2-95% air at 37°C in MEM (Invitrogen, Carlsbad, CA) supplemented with 10% FBS, 100 U/ml penicillin, 0.1 mg/ml streptomycin, and 2 mM l-glutamine. Culture medium was changed twice per week. Cells were harvested with trypsin (0.2 g/l) containing EDTA (0.5 g/l) and seeded in culture plates as described below.
Proliferation assay.
A549 cells were seeded at the density of 15 × 103 cells/well in 1.0 ml of 10% FBS/MEM in 24-well plates and allowed to attach overnight before the culture medium was replaced with serum-free medium. After 24 h of starvation in serum-free medium, cells were exposed to either normoxia (21% O2) or hypoxia (1% O2) for 5 days. For hypoxic conditions, cells were placed in sealed humidified gas chambers as described previously (5). The following three methods have been used to examine cell proliferation. First, DNA synthesis was evaluated by the Click-iT EdU (5-ethynyl-2′-deoxyuridine) assay according to manufacturer's protocol (Invitrogen). EdU fluorescence was detected on a SpectraMax Gemini XS microplate spectrofluorometer (Molecular Devices, Sunnyvale, CA) at an excitation of 568 nm and emission of 585 nm. Second, hypoxia-induced proliferation of A549 cells was evaluated by hemocytometrically counting the total number of cells at the end of each experimental period. Third, mitochondrial activity-based CellTiter 96 AQueous One Solution Cell Proliferation Assay (Promega, Madison, WI) was used for the assessment of hypoxia-induced replication responses of A549 cells in the presence or absence of PKCδ-targeted siRNA or MK-neutralizing antibody. For these experiments, cells (2,500/well) were plated in the 96-well plates (Costar, Cambridge, MA) and serum starved as described above. Cells were then either transiently transfected with a control nontargeting siRNA and a siRNA-targeting PKCδ (Dharmacon, Lafayette, CO) or preincubated with a MK-targeting neutralizing antibody and a nonimmunized control IgG (Epitomics, Burlingame, CA).
To evaluate the effects of MK-neutralizing antibody on proliferation, normoxic growth of A549 cells was continued for 72 h after a single treatment with either MK-targeting neutralizing antibody or control rabbit IgG at the concentrations of 0.01, 0.1, and 1.0 μg/ml. Because a single treatment with the antibodies was insufficient to produce an antiproliferative effect in the hypoxic A549 cells, to achieve such effect, hypoxic cultures were treated with anti-MK antibody or control IgG (both at 1.0 μg/ml) every 24 h for 4 days.
To examine whether MK can stimulate proliferation, serum-starved epithelial cells were treated with rMK (R&D Systems, Minneapolis, MN) for 24 h. Proliferation was evaluated by incubating cells with a CellTiter 96 AQueous One Solution Reagent (1–2 h) and spectrophotometrically measuring absorbance at 490 nm. Each experiment was repeated independently three times.
siRNA transfection.
Transfection of A549 cells with siRNA duplexes targeting PKCδ (5′-AAGATCATCGGCAGATGCACT-3′) and nontargeting negative control siRNA (both at the concentration of 25 nM) was carried out for 6 h using TransIT-TKO transfection reagent (Mirus Bio, Madison, WI) according to the manufacturer's recommendations. Transfected cells were allowed to recover for additional 48 h before being exposed to either normoxia or hypoxia for another 48 h. At the end of the experimental period, cells were processed for either proliferation assay or Western immunoblot analyses for detection of PKCδ and MK.
Immunofluorescent staining.
A549 cells were prepared for immunofluorescent staining according to the previously described method (29). Briefly, quiescent cells grown in eight-well chamber-slides were exposed to hypoxia (1% O2) for different lengths of time. At the end of the exposure, cells were fixed with cold 4% paraformaldehyde and then sequentially incubated with anti-PKCδ antibody (BD Biosciences, San Jose, CA), biotin-conjugated anti-rabbit IgG (Dako, Carpinteria, CA), and streptavidin-conjugated Alexa 488 (Invitrogen Life Technologies, Grand Island, NY). To detect PKCδ localization within specific subcellular organelles, Golgi and early endosomes were labeled with green fluorescent protein (GFP)-tagged probes, the CellLight Golgi-GFP BacMam2.0 and CellLight Early Endosomes-GFP BacMam2.0, respectively, according to the manufacturer's instructions (Invitrogen). Labeled cells were exposed to either normoxia or hypoxia for different lengths of time, fixed with 4% paraformaldehyde, and processed for double immunofluorescent staining for PKCδ and the GFP label indicating either Golgi or early endosomes. The images were captured with ×63 oil immersion objective using Zeiss 710 laser scanning confocal microscope equipped with an AxioCam HR camera (Zeiss) and ZEN 2009 software.
To characterize the mesenchymal phenotype of the cells, fixed normoxic and hypoxic cells were incubated with either anti-α-smooth muscle actin (α-SMA) or anti-vimentin antibodies and then with Alexa 594-conjugated anti-mouse IgG. Nuclei were stained with Hoechst 33342, and Prolong Gold Anti-fade reagent (Invitrogen) was placed on slides before coverslipping. Phase contrast and fluorescence images were captured with ×40 objective using Leica DMI 3000B microscope (Leica, Bannockburn, IL) and SPOT CCD camera (SPOT Diagnostics, Sterling Heights, MI).
MK expression analysis by Western immunoblot.
To determine whether PKCδ plays an important role in MK expression, rottlerin, a PKC antagonist (Calbiochem, La Jolla, CA) was used. Cells were preincubated with rottlerin (3 μM) for 1 h and then exposed to either normoxia or hypoxia for 48 h. Equal protein amounts from whole cell lysates were separated by SDS-PAGE electrophoresis, transferred onto PVDF membranes (Amersham Pharmacia Biotechnologies, Piscataway, NJ), and incubated with rabbit polyclonal anti-MK antibody (PeproTech, Rocky Hill, NJ).
PKCδ-targeting and control siRNAs were also used to examine the role of this PKC isozyme in MK expression in A549 cells. Transfected cells were exposed to either normoxia or hypoxia for 48 h and harvested in lysis buffer, and the lysates were subjected to Western immunoblot analyses for the evaluation of PKCδ, MK, and β-actin levels. Bands were visualized with LumiPhos reagents (Pierce, Southfield, MI), digitized, and quantified according to the previously described method (6, 28).
MK ELISA.
Epithelial cells were plated in 10% FBS/MEM and then incubated in serum-free medium according to the above-mentioned protocol. After 24 h in serum-free medium, cultures were supplemented with 40 μg/ml heparin and exposed to either normoxia or hypoxia for different lengths of time. Conditioned media (CM) were collected from normoxic and hypoxic cells for the analysis of secreted MK levels. The MK concentration in the CM was determined using an ELISA kit according to manufacturer's protocol (PeproTech) and quantified against a standard curve established using recombinant human MK (32 to 2,000 pg/ml). All the experiments were repeated three times independently.
RT-PCR.
RNA was extracted from normoxia- and hypoxia-exposed A549 cells with TRIzol reagent (Invitrogen, Frederick, MD) and purified using RNeasy Mini Kit (Qiagen, Valencia, CA), and RT-PCR was performed in an iCycler thermal cycler (Bio-Rad Laboratories, Hercules, CA) with Taq DNA polymerase (Invitrogen). The PCR products were resolved by electrophoresis on ethidium bromide-stained agarose gels and visualized by ultraviolet illumination. All the experiments were repeated at least three times. Primers were as follows: MK forward, 5′-ATGCAGCACCGAGGCTTCCT-3′, and reverse, 5′-ATCCAGGCTTGGCGTCTAGT-3′; and β-actin (loading control) forward, 5′-AACTGGGACGACATGGAGAA-3′, and reverse, 5′-GGTAGTCAGTCAGGTCCC-3′.
Data analysis.
The data were analyzed by one-way ANOVA followed by the Student-Newman-Keuls post hoc test for multiple comparisons. Data are reported as means ± SE and considered significantly different if P ≤ 0.05.
RESULTS
Hypoxia stimulates proliferation of human lung epithelial cells.
Knowing that in vivo acute hypoxia induces apoptosis in lung epithelial cells, whereas chronic hypoxia leads to increased proliferation of these cells (34), we examined whether prolonged hypoxia stimulates human lung epithelial cell replication. We modeled chronic hypoxia by exposing A549 cells to 1% O2 in serum-free medium for 5 days and assessed cell proliferation by two independent techniques. First, proliferation was determined by EdU incorporation (Fig. 1A). EdU, which can be detected spectrofluorometrically, is a nucleoside analog to thymidine that is incorporated into DNA during DNA synthesis. In A549 cells, hypoxia induced a steady increase in DNA synthesis resulting in cumulative 88-fold upregulation (relative to time 0) in EdU incorporation after 5 days of hypoxia exposure (Fig. 1A). Although DNA synthesis in normoxic cells was increased until day 3, the rate of replication was significantly lower compared with hypoxic cells and by day 5, in dramatic contrast to hypoxic cells, normoxic DNA synthesis sharply declined (Fig. 1A). These data unequivocally demonstrate that prolonged hypoxia induces a lasting increase in DNA synthesis in human lung epithelial cells.
Fig. 1.

Hypoxia stimulates proliferation of A549 cells. A: DNA synthesis was evaluated in A549 cells by the Click-iT EdU Assay on days 0, 1, 3, and 5 of exposure to normoxia or hypoxia (1% O2). Fresh 5-ethynyl-2′-deoxyuridine (EdU; 10 μm) was added to the culture medium 24 h before each time point/EdU fluorescence measurement. EdU incorporation values are expressed as means ± SE from 3 independent experiments with 8 wells per condition. *P < 0.001, compared with day 0; **P < 0.001, compared with day 3 normoxia and day 1 hypoxia; #P < 0.001, compared with day 5 normoxia and day 3 hypoxia. B: cells exposed to either normoxia or hypoxia (1% O2) for up to 5 days, were trypsinized, and enumerated hemocytometrically on days 0, 1, 3, and 5 of exposure. *P < 0.05, compared with say 0; **P < 0.05, compared with day 3 normoxia; #P < 0.05, compared with day 5 normoxia.
The second method by which hypoxia-induced proliferation of lung epithelial cells was demonstrated involved hemocytometric cell counts. Hypoxic cells divided at a steady rate as evidenced by continuous increase in cell numbers reaching a twofold increase in cell count after 5 days of exposure (Fig. 1B). Under normoxic conditions, cell division was halted by day 3 of normoxic exposure and from that point cell counts declined further so that at the end of 5 days, the reduction in normoxic cell numbers paralleled the reduction in normoxic DNA synthesis (Fig. 1, A and B). Collectively, these data demonstrate that prolonged hypoxia is a major driver of proliferation of human lung epithelial cells.
PKCδ mediates hypoxia-induced proliferation of A549 cells.
Since PKCδ isozyme responds to nonhypoxic stimuli by modifying growth and survival responses of epithelial cells (4, 8, 9), we began by examining the potential role of PKCδ in driving hypoxia-induced lung epithelial cell proliferation. To that end, we first examined the effects of inhibitors of PKC, GF109203X and rottlerin, on A549 cell proliferation. The inhibitory effects of rottlerin (3 μM) on serum-stimulated growth of epithelial cells were significantly greater than the effects of GF109203X (3 μM; Fig. 2A). In addition, both inhibitors at a higher concentration (10 μM) further reduced the growth rate of A549 cells (Fig. 2A). From these antagonistic properties of GF109203X and rottlerin on proliferation, we conclude that under hypoxic conditions PKC controls lung epithelial cell replication.
Fig. 2.

Hypoxia-induced proliferation of A549 cells is dependent on PKCδ isozyme. A: maximal inhibition of serum-stimulated growth of A549 cells was achieved in the presence of rottlerin. Cultures received a single treatment with GF109203X or rottlerin. Cells were grown in serum-containing medium for 5 days, trypsinized, and enumerated hemocytometrically. Data are means ± SE from 3 independent experiments. *P < 0.001, compared with control; **P < 0.001, compared with control and GF109203X (3 μM); ***P < 0.001, compared with control and GF109203X (10 μM). B: PKCδ-targeting small interfering (si)RNA selectively reduced PKCδ protein levels in A549 cells as detected by Western immunoblot. Cells were transiently transfected with either PKCδ-targeting siRNA or nontargeting siRNA using TransIT TKO. Cell lysates were collected 48 h posttransfection and immunoblotted for PKCδ and α-tubulin. C: effects of PKCδ siRNA on hypoxia-induced proliferation of A549 cells. Transfected A549 cells were stimulated with either normoxia or hypoxia for 48 h before a mitochondrial activity-based proliferation assay was performed using CellTiter 96 AQueous One Solution Reagent. Values are means ± SE of 3 independent experiments. *P < 0.001, compared with normoxia; **P < 0.001, compared with hypoxia control, hypoxic TKO, and hypoxic nontargeting siRNA.
To evaluate the role of PKCδ in hypoxia-stimulated proliferative responses of epithelial cells we employed a genetic approach using PKCδ-specific siRNA. As evaluated by Western immunoblot analysis, PKCδ protein levels were reduced only in cells transfected with PKCδ-targeting siRNA, whereas transfection reagent (TKO) and nontargeting siRNA failed to affect PKCδ levels confirming the selectivity and efficiency of PKCδ siRNA against its target (Fig. 2B). In the presence of PKCδ-targeting siRNA, hypoxia-induced upregulation of lung epithelial cell proliferation was selectively attenuated to a degree similar to that observed under normoxia (Fig. 2C). Consistent with the specificity of the PKCδ-targeting siRNA, hypoxic epithelial cell proliferation was unaffected by either TKO or nontargeting siRNA (Fig. 2C). In addition, and in further support for the selectivity of the PKCδ siRNA with respect to hypoxic proliferation, none of the treatments (TKO, nontargeting siRNA, or PKCδ-targeting siRNA) have had any effect on normoxic proliferation (Fig. 2C). Taken together, these data demonstrate that hypoxia-induced proliferation of human lung epithelial cells is dependent on expression of the PKCδ isozyme.
PKCδ localizes in the Golgi of human lung epithelial cells.
Since translocation of PKC isozymes between subcellular compartments is a hallmark of enzyme activation (32), we evaluated distribution of PKCδ to specific organelles and asked whether hypoxia induces translocation of this PKC isozyme in A549 cells. Under normoxic conditions, immunofluorescent detection demonstrated that PKCδ was concentrated outside the nuclear membrane and restricted to a discrete locus along the long axis of the nucleus (Fig. 3A, red fluorescence). One hour of exposure to hypoxia was sufficient to increase the intensity of PKCδ fluorescent signal relative to signal intensity in cells grown under normoxic conditions, i.e., before initiation of hypoxic exposure (Fig. 3A). After 3 h, hypoxia-exposed cells demonstrated a prominent localization of PKCδ to distinct punctuate perinuclear structures as well as an increase in PKCδ signal within nuclei themselves (Fig. 3A). At 24 h of hypoxia exposure, PKCδ localization pattern revealed continued presence of PKCδ signal in the nucleus, which is also characteristic of cells at 3 h of hypoxia (Fig. 3A). These data suggest that 3-h hypoxic exposure is sufficient for formation of punctuate PKCδ localization pattern within a selective perinuclear region as well as its translocation into the nuclei of A549 cells.
Fig. 3.

In A549 cells, Golgi-localized PKCδ translocates to nuclei with hypoxia. A: cells transfected with CellLight Golgi-GFP BacMam 2.0 were exposed to either normoxia or hypoxia for 0, 1, 3, and 24 h and then immunodetected for PKCδ (red) and green fluorescent protein (GFP; green); nuclei are blue. Representative images show PKCδ/Golgi colocalization. B: A549 cells were transfected with CellLight Early Endosomes-GFP BacMam2.0 to label early endosomes and processed for double immunofluorescent staining for PKCδ (red) and for early endosomes-GFP (green). Scale bars = 10 μm.
To indentify which specific organelle in the perinuclear region accumulates PKCδ, A549 cells exposed to hypoxia for 30 min were evaluated for the localization patterns of organelle-specific markers by immunofluorescent staining to visually ascertain potential similarities to the pattern of PKCδ isozyme expression (data not shown). This procedure of staining pattern comparison indicated that PKCδ is not localized to either mitochondria, or endosomes, or endoplasmic reticulum, or centromere suggesting that PKCδ in lung epithelial cells must localize to yet another subcellular compartment.
In pursuit of such a PKCδ-harboring compartment in A549 cells, we attempted to elucidate the subcellular localization of PKCδ within an additional organelle known for its proximity to the nuclear membrane, the Golgi apparatus. Cells virally transduced with either CellLight Golgi-GFP that labels Golgi or with CellLight Early Endosomes-GFP that labels early endosomes, a negative control for PKCδ localization, were exposed to hypoxia and evaluated for the colocalization with PKCδ. Examination of subcellular compartments across a full cell depth (Z-stack analysis) from confocal microscopy images depicting immunofluorescent staining for PKCδ (red) and Golgi (green) revealed that PKCδ colocalizes with Golgi (yellow) under both normoxia and hypoxia (1 and 3 h; Fig. 3A). In contrast, PKCδ did not colocalize with early endosomes as evidenced by the lack of overlap between PKCδ staining (red) and early endosome staining (green; Fig. 3B). These images demonstrate that under normoxia PKCδ resides in the Golgi and upon detection of the hypoxic stimulus it reorganizes within Golgi and translocates into nuclei representing a signature of hypoxia-induced activation of the isozyme.
PKCδ regulates MK expression in A549 cells.
Regulation of proliferation-stimulating cytokines, such as VEGF (25, 36) and interleukin-6 (IL-6), by PKCδ (30) together with the isozyme localization in the Golgi and its translocation into nuclei of A549 cells described above suggests that PKCδ might regulate proliferation of epithelial cells by controlling growth factor expression. We initiated these studies by examining whether PKCδ will affect expression of MK, a known hypoxia-regulated mitogenic growth factor in the lung (23, 26). We began by treating A549 cells with rottlerin, a potential PKCδ-targeting antagonist. In both normoxic and hypoxic cells, rottlerin treatment markedly reduced intracellular MK protein expression, a finding consistent with the role of PKCδ as a regulator of MK levels in epithelial cells stimulated by hypoxia as well as its role in the maintenance of MK levels under normoxic conditions (Fig. 4, A and B).
Fig. 4.

Midkine (MK) expression is regulated by PKCδ. A: treatment with rottlerin results in reduction of MK protein levels in lung epithelial cells. Rottlerin (3 μM) and DMSO, the vehicle, were added to the cells, preincubated at 37°C for 1 h and then exposed to either normoxia (Nor) or hypoxia (Hyp; 1% O2) for 48 h. At the end of the exposure, cells were lysed with lysis buffer and the lysates were used for the determination of MK levels. B: quantification of MK levels from 3 different immunoblots (e.g., Fig. 4A) in control- and rottlerin-treated cells. Values are means ± SE from 3 independent experiments. *P < 0.05, compared with normoxia/DMSO; **P < 0.05, compared with hypoxia/DMSO. C: MK protein levels in A549 cells are attenuated in the presence of PKCδ-targeting siRNA. TransIT-TKO reagent was used to transfect lung epithelial cells with either nontargeting negative control or PKCδ-targeting siRNA. Transfected cells were exposed to either normoxia or hypoxia (1% O2) for 48 h before Western immunoblot analyses for detection of PKCδ, MK, and β-actin. D: Quantification of MK protein levels from 3 different immunoblots (e.g., Fig. 4C) in the presence of PKCδ-targeting siRNA. Values are means ± SE of 3 independent experiments; shown are representative immunoblots. *P < 0.05, compared with control siRNA under normoxia; **P < 0.05, compared with control siRNA under hypoxia.
Since the specificity of rottlerin as an antagonist of PKCδ has come under scrutiny (31), we employed PKCδ-targeting siRNA to directly attenuate PKCδ expression and thereby inhibit its actions. PKCδ-specific siRNA selectively reduced PKCδ protein levels in both normoxic and hypoxic epithelial cells whereas neither control siRNA nor transfection reagent alone affected PKCδ expression (Fig. 4C). In both normoxic and hypoxic cells, intracellular MK levels were significantly reduced upon attenuation of PKCδ expression with PKCδ-targeting siRNA (Fig. 4, C and D). These findings establish a link between PKCδ expression/activation and MK expression, an interaction that is necessary for the maintenance of intracellular MK levels in lung epithelial cells.
Secreted MK stimulates A549 cell proliferation.
Because MK is a hypoxia-inducible mitogenic factor in the lung (23, 26), an increase in MK protein levels was expected, but none was observed when intracellular MK levels were quantified by Western immunoblot analysis in A549 cells exposed to hypoxia (Fig. 4, A and B). We hypothesized that the absence of an increase in MK protein levels in whole cell lysates of hypoxic A549 cells is consistent with hypoxia stimulating MK secretion into culture medium in an effort to maintain a steady-state levels of intracellular MK. To explore this possibility, we opted to first examine whether hypoxia regulates transcription of MK and observed that mRNA encoding for MK was significantly increased in hypoxic epithelial cells compared with normoxic cells (Fig. 5, A and B).
Fig. 5.

Secreted MK stimulates epithelial cell proliferation. A: MK mRNA levels are higher in hypoxia-exposed cells compared with normoxic cells. Serum-deprived cells were exposed to either normoxia or hypoxia (1% O2) for 72 h before RT-PCR for MK and β-actin. B: quantification of MK mRNA levels. Values are means ± SE of 3 independent experiments. *P < 0.05, compared with normoxia. C: MK secretion into conditioned media (CM) is significantly upregulated by hypoxia-exposed A549 cells. CM collected from normoxic and hypoxic cells were quantified for MK protein levels by ELISA. The quantity of MK in the CM was evaluated with respect to a standard curve using recombinant human MK (32–2,000 pg/ml). Values are means ± SE of 3 independent experiments. *P < 0.05, compared with days 1 and 3 normoxia MK; **P < 0.05, compared with day 5 normoxia and day 3 hypoxia. D: normoxic proliferation of A549 cells is significantly reduced in the presence of MK neutralizing antibody. Serum-deprived cells were treated with nonimmunized control IgG and MK neutralizing antibody (both at the concentrations of 0.01, 0.1, and 1 μg/ml) and cultured for 72 h. Cell proliferation was evaluated by a CellTiter 96 AQueous One Solution Reagent. *P < 0.05, compared with control IgG (1 μg/ml). E: in hypoxic cultures, daily addition of MK neutralizing antibody attenuates replication of A549 cells. MK neutralizing antibody and nonimmunized IgG were added to serum-deprived cells and cultures were exposed to hypoxia (1% O2) for 96 h. *P < 0.05, compared with control IgG. F: recombinant MK (rMK) stimulates proliferation of A549 cells. Serum-deprived cells were treated with rMK (100 ng/ml) for 24 h and cell proliferation was evaluated. *P < 0.05, compared with control. Data are from 3 independent experiments.
To substantiate our prediction that transcriptional upregulation of MK mRNA by hypoxia is associated with secretion of MK protein from A549 cells, we quantified MK secreted from cells by measuring MK in CM by ELISA (Fig. 5C). CM from A549 cells exposed to 3 days of hypoxia contained significantly greater amounts of MK protein (1.2-fold increase) than were found in the CM from normoxic cells (Fig. 5C). After 5 days of hypoxia, secreted MK levels in the CM were increased further (2.0-fold increase) compared with MK levels in normoxic CM (Fig. 5C). CM studies revealed that hypoxia-induced increases in MK mRNA expression that parallel elevation in secreted MK are consistent with the idea that hypoxia balances the production of MK with its secretion in a way that assures constancy of intracellular MK protein levels under hypoxia (Fig. 4). Therefore, our data support the idea that hypoxia is a strong inducer of de novo MK expression and is associated with MK secretion from lung epithelial cells.
To examine the functional role of secreted MK in proliferation, a monoclonal anti-MK antibody was used to neutralize secreted MK in the CM of cultured A549 cells. To examine the effects of MK neutralization in control normoxic cells, the neutralizing antibody or a nonimmunized control rabbit IgG was added to the normoxic cultures at the following concentrations: 0.01, 0.1, and 1.0 μg/ml. Marked reduction in normoxic replication was observed after 3 days in culture supplemented with 1 μg/ml of MK neutralizing antibody, whereas control rabbit IgG did not affect proliferation of the cells (Fig. 5D). In contrast, to achieve blockade of hypoxia-stimulated proliferation with MK-neutralizing antibody, the antibody (1 μg/ml) was added daily for 4 days (Fig. 5E). Analogous daily treatment with control IgG (1 μg/ml) had no effect on proliferation of hypoxic A549 cells (Fig. 5E).
rMK was then used to confirm that MK is a mitogen for A549 cells. The replication rate was greater in rMK-treated cells compared with that of the control cells (Fig. 5F). Taken together, these data strongly suggest that hypoxia induces MK expression and secretion and that secreted MK is a growth-promoting factor for human lung epithelial cells.
rMK accelerates morphological changes of hypoxic epithelial cells to mesenchymal-like cells.
To determine whether MK promotes epithelial mesenchymal transition (EMT) under hypoxic conditions, we added exogenous rMK in the presence or absence of hypoxia and monitored cell morphology. As shown in Fig. 6, exposure to hypoxia or treatment with rMK altered epithelial cell morphology from round, characteristic of epithelial cells to elongated mesenchymal-like cells indicating that hypoxia and rMK, when applied singly, initiate EMT in A549 cells. However, rMK in combination with hypoxia resulted in appearance of most strikingly elongated A549 cells, a finding consistent with an additive effect of these two EMT inducers in our tissue culture model of EMT, a process that in vivo essentially involves a combination of multiple stimuli including hypoxia and MK. Since transforming growth factor-β1 (TGF-β1) is a well-known mediator of EMT, we included TGF-β1 as a positive control in our experiments (Fig. 6). Morphological differentiation patterns of epithelial cells induced by TGF-β1 were highly similar to morphological changes induced by a combination of hypoxia and rMK treatment. These data illustrate that MK accelerates differentiation of hypoxic epithelial cells to cells exhibiting mesenchymal phenotype.
Fig. 6.

rMK accelerates phenotypic transition of hypoxic epithelial cells to cells with mesenchymal morphology. A549 cells were plated and serum-starved according to the above-mentioned method. rMK (100 ng/ml) was added to cells and then cultures were exposed to either normoxia or hypoxia (1% O2) for up to 72 h. Transforming growth factor-β1 (TGF-β1; 2 ng/ml) was used as a positive control for the epithelial mesenchymal transition process. Representative bright-field photomicrographs from 3 independent experiments are presented. Scale bar = 10 μm.
rMK stimulates α-SMA and vimentin expression in hypoxic epithelial cells.
Since the evidence of morphological changes alone is inadequate to support a definite identification of differentiation, in addition to the morphological studies, we evaluated A549 cultures for the presence of two mesenchymal markers, α-SMA and vimentin. First, we determined the protein levels of α-SMA, a well-known indicator of mesenchymal phenotype. Immunofluorescent staining revealed that α-SMA expression is maximally upregulated in the presence of both hypoxia and rMK (Fig. 7A). Significant upregulation of α-SMA in epithelial cells by a combination of hypoxia and rMK was confirmed by Western immunoblot analysis (Fig. 7, B and C). Collectively, these data demonstrate that MK, in the presence of hypoxia, accelerates human lung epithelial cell differentiation to cells of mesenchymal phenotype characterized by increased α-SMA protein levels.
Fig. 7.
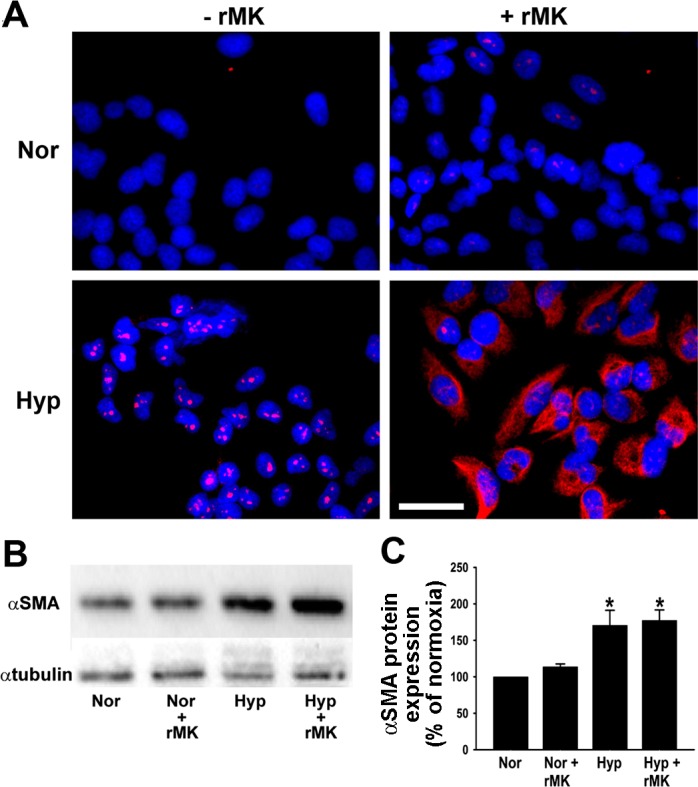
Expression of α-smooth muscle actin (α-SMA), a mesenchymal cell marker, is increased in hypoxic A549 cells by rMK. A: serum-starved A549 cells were treated with rMK (100 ng/ml) and exposed to either normoxia or hypoxia for 72 h. At the end of the exposure, α-SMA (red) was immunodetected. Nuclei are blue. Representative micrographs from 1 of the 3 independent experiments are shown. Scale bar = 10 μm. B: increase in α-SMA protein levels as observed on a representative Western immunoblot. Serum-starved epithelial cells were treated with rMK (100 ng/ml) and then exposed to either normoxia or hypoxia for 72 h before Western immunoblotting for detection of α-SMA and α-tubulin. C: quantification of α-SMA levels from 3 different immunoblots (e.g., Fig. 7B) in response to rMK treatment of normoxic and hypoxic cells. *P < 0.01, compared with Nor and Nor + rMK results.
Since upregulation of vimentin, an intermediate filament protein, is considered to be a prerequisite for the induction of EMT (27), in addition to α-SMA, we evaluated the effect of hypoxia and rMK on the expression of this second mesenchymal marker. Hypoxia induced an increase in vimentin expression in A549 cells as demonstrated by immunofluorescent and immunoblotting techniques (Fig. 8, A and B). The highest levels of vimentin expression, however, were achieved by a combination of hypoxia and rMK (Fig. 8, B and C). Together these data suggest that, in lung epithelial cells, MK cooperates with hypoxia toward the most effective acceleration of the EMT.
Fig. 8.
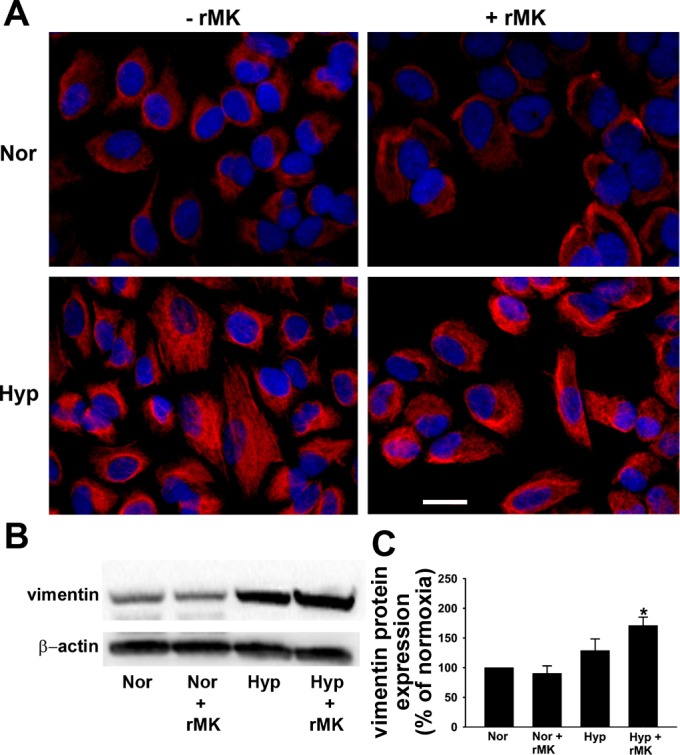
rMK induces vimentin expression in hypoxic A549 cells. A: immunofluorescent staining for vimentin (red). A549 cells were grown with or without rMK and exposed to either normoxia or hypoxia for 72 h. Representative photomicrographs from 3 independent experiments are shown. Scale bar = 10 μm. B: Western immunoblot showing vimentin expression. Serum-starved A549 cells were exposed to either normoxia or hypoxia in the absence or presence of rMK (100 ng/ml). After 72 h of exposure, Western immunoblots for detection of vimentin and β-actin were performed. A representative immunoblot from 3 independent experiments is shown. C: quantification of vimentin levels from 3 Western immunoblots (e.g., Fig. 8B) in normoxic and hypoxic cells in the presence or absence of rMK. *P < 0.01, compared with Nor and Nor + rMK.
DISCUSSION
We report that prolonged hypoxia stimulates proliferation of human lung epithelial cells and that such hypoxic proliferative responses are mediated by a PKCδ isozyme and are associated with translocation of PKCδ from Golgi into nuclei. In addition, we describe here that PKCδ regulates MK protein levels in human lung epithelial cells as the blockade of the isozyme by various approaches results in marked reduction in MK expression. Most importantly, hypoxia-induced upregulation of MK expression and secretion increases proliferation and differentiation of hypoxic epithelial cells. We conclude that the PKCδ/MK axis is a key regulator of epithelial cell phenotype in conditions involving hypoxia.
The responses of lung epithelial cells to hypoxia are dependent on the severity and duration of the hypoxic exposure (1, 13). Here, we report enhanced proliferation of human lung epithelial cells in response to prolonged hypoxia (1% O2 for 5 days). In contrast, primary rat alveolar epithelial type II cells respond to subacute hypoxia (0.5% O2 for 2 days) with enhanced apoptosis and cell cycle arrest (17). At the first glance, differences between the two studies appear to be related to examination of a human cell line (our study) vs. primary epithelial cells (rodent study) and slight differences between oxygen concentrations tested. However, a more attractive explanation for such differences in epithelial cell responses in the two studies involves a potential for an initial apoptotic response of epithelial cells experiencing acute hypoxia that with time creates a trophic microenvironment engendering conditions that favor long-term cell division as seen in prolonged hypoxia. One study in rat examining the effects of early (3 days) vs. late phase (10 or 30 days) of hypoxia on the epithelium of the lung, reporting that late phase of hypoxia stimulates proliferation in lung epithelial cells (34), provides direct support for our findings of hypoxia-induced proliferation of human lung epithelial cells.
While PKCδ participates in many cellular functions including proliferation, differentiation, apoptosis, and angiogenesis (2, 20), the functional roles of this isozyme must be viewed in the context of pairing specific stimuli with specific cell types. Although PKCδ is activated by hypoxia and functions as a key regulator of hypoxia-induced angiogenesis (18, 25, 36), its role in the proliferative responses of hypoxic epithelial cells remained unexplored until our current report. We identified PKCδ to be a key regulator of hypoxic proliferation of human lung epithelial cells using selective RNAi-mediated knockdown of PKCδ that blocked hypoxia-induced proliferation. Other models of epithelial cell injury, such as asbestos toxicity, confirm involvement of PKC in lung injury as the toxin induces PKCδ-dependent proliferation of lung epithelial cells (19). Therefore, our current studies provide evidence in support of the idea that PKCδ is a pro-proliferative kinase in the setting of hypoxic lung epithelium.
Subcellular localization of PKCδ appears to be another determinant of the balance between its pro- and antiapoptotic functions and in the PKCδ-mediated activation of downstream signaling pathways (7, 14). In our study, under normoxic conditions, PKCδ was present in the Golgi and the hypoxia-induced translocation of PKCδ into nucleus might provide clues as to its potential role in transcriptional events during proliferative responses of lung epithelial cells. PKCδ-mediated phosphorylation of a transcription factor, HIF-1α, provides an example of PKCδ regulation of transcription factors mediating responses to hypoxia (18). Localization of PKCδ to perinuclear structures positive for a Golgi marker in adult rat ventricular myocytes (12) is consistent with our finding of PKCδ localization in the Golgi of human epithelial cells. Such pattern of PKCδ distribution in the Golgi of myocytes and epithelial cells relates not only to hypoxic stimuli as we demonstrate, but, in myocytes, it also correlates with inotropic responses (12). Taken together, subcellular PKCδ distribution studies suggest that translocation of PKCδ between Golgi and nuclei engender adaptive cell responses to hypoxia.
Because MK is a hypoxia-inducible growth factor, the transcription of which is regulated by HIF-1α (26), and because PKCδ is an activator of HIF-1α (18), we examined a possible link between PKCδ and MK. Our studies demonstrate that hypoxic activation of PKCδ is involved in the regulation of MK levels. A link between MK expression and PKC has also been made by Kosugi et al. (15), who reported that cultured MK-deficient murine kidney mesangial cells, compared with cells from the wild-type mice, exhibit reduced PKC phosphorylation following a glucose challenge. Although these studies in mice correlate MK expression with PKC phosphorylation, they do not identify any specific PKC isozyme as a controller of MK protein levels. We filled this knowledge gap by demonstrating that, in human lung epithelial cells, the reduction in MK levels occurs upon blocking PKCδ signaling with siRNA, the first study to report a role for a selective PKC isozyme in regulation of MK levels in relation to hypoxia.
MK is constitutively expressed at low levels in adult tissues, but MK levels rise in pathological conditions, such as carcinogenesis (22, 24, 35), inflammation (11, 21), and diabetic nephropathy (15). MK responses are also relevant to lung diseases as MK expression is increased in epithelium of the large and small airways and in alveoli and cells of submucosa of cystic fibrosis lung tissue (23). These responses of the epithelial cells are likely related to tissue hypoxia, a low oxygen condition that coincides with an increase in MK protein levels in cystic fibrosis (23). Such increases in MK seen in cystic fibrosis patients demonstrate the importance of hypoxia/MK interactions in human pulmonary diseases and underscore the relevance of our observations that hypoxia upregulates expression and secretion of MK and accelerates differentiation of human lung epithelial cells toward mesenchymal phenotype. The responsiveness of MK to hypoxia might represent a universal response among various cell types as hypoxia-induced MK expression and release have been also reported in human umbilical vein endothelial cells (37). MK production in the respiratory epithelium is also regulated by hypoxia and serves as a paracrine signal that selectively enhances muscularization of small pulmonary arteries (26). Therefore, studies in endothelial and smooth muscle cells provide further support to our idea that hypoxia is an important inducer of MK expression and secretion in the lung.
In addition to effects on cell proliferation, hypoxia is known to be a modifier of cell phenotype by affecting differentiation signaling pathways (33). Lung epithelial cells are intrinsically plastic and capable of responding to environmental signals by changing their size, shape, and state of differentiation. Although hypoxia induces EMT in alveolar epithelial cells (39), the role of MK in these hypoxic responses was previously unknown. We now demonstrate that hypoxia initiates an EMT characterized by cellular changes involving a shift from cells with epithelial morphology to elongated mesenchymal-like cells that is accompanied by increased levels of mesenchymal markers, α-SMA and vimentin. This EMT process is accelerated by exogenously added rMK. One study in lung adenocarcinoma cells reporting that MK accelerates estradiol-induced EMT (38) provides additional credence to our finding demonstrating the role of MK in promoting hypoxia-induced EMT in lung epithelial cells.
The vital role of MK in tissue remodeling is further underscored by studies in MK-deficient mice that have defects in neointima formation, in which restoration of MK function was achieved by administration of exogenous rMK resulting in resumption of neointima formation (10). Taken together, our observations of the role of PKCδ and MK in generating a hypoxic phenotype in human lung epithelial cells raise the possibility that PKCδ/MK axis plays a critical role in the structural remodeling of lung tissues in hypoxia-mediated lung diseases. We conclude that the PKCδ/MK axis should be considered as a novel target when developing therapeutic interventions for lung diseases involving hypoxia.
GRANTS
This work was supported by National Institutes of Health Grants HL-64917 (to M. Das) and AG-012411 and AA-016654 (to W. M. Zawada).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: H.Z., M.O., E.P., and M.D. performed experiments; H.Z., M.O., E.P., and M.D. analyzed data; H.Z., M.O., E.P., W.M.Z., and M.D. interpreted results of experiments; H.Z., M.O., E.P., W.M.Z., and M.D. prepared figures; H.Z., M.O., E.P., W.M.Z., and M.D. approved final version of manuscript; M.O., W.M.Z., and M.D. conception and design of research; M.O., W.M.Z., and M.D. drafted manuscript; W.M.Z. and M.D. edited and revised manuscript.
ACKNOWLEDGMENTS
We thank Dr. Jonathan Dranoff (University of Arkansas for Medical Sciences) for comments on the manuscript and Rupa Idate, Gavin B. Lawlis, and Andy M. Poczobutt for technical assistance. A549 human lung epithelial cells were provided by Dr. Samir E. Witta (University of Colorado Denver).
REFERENCES
- 1.Adamson IY, Young L, Bowden DH. Relationship of alveolar epithelial injury and repair to the induction of pulmonary fibrosis. Am J Pathol 130: 377–383, 1988 [PMC free article] [PubMed] [Google Scholar]
- 2.Basu A, Pal D. Two faces of protein kinase Cδ: the contrasting roles of PKCδ in cell survival and cell death. Scientific World Journal 10: 2272–2284, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chichger H, Grinnell KL, Casserly B, Chung CS, Braza J, Lomas-Neira J, Ayala A, Rounds S, Klinger JR, Harrington EO. Genetic disruption of protein kinase Cδ reduces endotoxin-induced lung injury. Am J Physiol Lung Cell Mol Physiol 303: L880–L888, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Clark AS, West KA, Blumberg PM, Dennis PA. Altered protein kinase C (PKC) isoforms in non-small cell lung cancer cells: PKCδ promotes cellular survival and chemotherapeutic resistance. Cancer Res 63: 780–786, 2003 [PubMed] [Google Scholar]
- 5.Das M, Bouchey DM, Moore M, Hopkins D, Nemenoff R, Stenmark KR. Hypoxia-induced proliferative response of pulmonary artery adventitial fibroblasts is dependent on G-protein mediated activation of MAP kinases. J Biol Chem 276: 15631–15640, 2001 [DOI] [PubMed] [Google Scholar]
- 6.Das M, Burns N, Wilson SJ, Zawada WM, Stenmark KR. Hypoxia exposure induces the emergence of fibroblasts lacking replication repressor signals of PKCzeta in the pulmonary artery adventitia. Cardiovasc Res 78: 440–448, 2008 [DOI] [PubMed] [Google Scholar]
- 7.Gomel R, Xiang C, Finniss S, Lee HK, Lu W, Okhrimenko H, Brodie C. The localization of protein kinase Cdelta in different subcellular sites affects its proapoptotic and antiapoptotic functions and the activation of distinct downstream signaling pathways. Mol Cancer Res 5: 627–639, 2007 [DOI] [PubMed] [Google Scholar]
- 8.Griner EM, Kazanietz MG. Protein kinase C and other diacylglycerol effectors in cancer. Nat Rev Cancer 7: 281–294, 2007 [DOI] [PubMed] [Google Scholar]
- 9.Grossoni VC, Falbo KB, Kazanittz MG, de Kier Joffe ED, Urtreger AJ. Protein kinase C delta enhances proliferation and survival of murine mammary cells. Mol Carcinog 46: 381–390, 2007 [DOI] [PubMed] [Google Scholar]
- 10.Horiba M, Kadomatsu K, Nakamura E, Muramatsu H, Ikematsu S, Sakuma S, Hayashi K, Yuzawa Y, Matsuo S, Kuzuya M, Kaname T, Hirai M, Saito H, Muramatsu T. Neointima formation in a restenosis model is suppressed in midkine-deficient mice. J Clin Invest 105: 489–495, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kadomatsu K. The midkine family in cancer, inflammation and neural development. Nagoya J Med Sci 67: 71–82, 2005 [PubMed] [Google Scholar]
- 12.Kang M, Walker JW. Protein kinase Cδ and ε mediate positive inotrophy in adult ventricular myocytes. J Mol Cell Cardiol 38: 753–764, 2005 [DOI] [PubMed] [Google Scholar]
- 13.Kasper M, Haroske G. Alterations in the alveolar epithelium after injury leading to pulmonary fibrosis. Histol Histopathol 11: 463–483, 1996 [PubMed] [Google Scholar]
- 14.Kikkawa U, Matsuzaki H, Yamamoto T. Protein kinase C (delta): activation mechanisms and functions. J Biochem (Tokyo) 132: 831–839, 2002 [DOI] [PubMed] [Google Scholar]
- 15.Kosugi T, Yuzawa Y, Sato W, Kawai H, Matsuo S, Takei Y, Muramatsu T, Kadomatsu K. Growth factor midkine is involved in the pathogenesis of diabetic nephropathy. Am J Pathol 168: 9–19, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kosugi T, Sato W. Midkine and the kidney: health and diseases. Nephrol Dial Transplant 27: 16–21, 2012 [DOI] [PubMed] [Google Scholar]
- 17.Krick S, Eul BG, Hanze J, Saval R, Grimminger F, Seeger W. Role of hypoxia-inducible factor-1α in hypoxia-induced apoptosis of primary alveolar epithelial type II cells. Am J Respir Cell Mol Biol 32: 395–403, 2005 [DOI] [PubMed] [Google Scholar]
- 18.Lee JW, Park JA, Kim SH, Seo JH, Lim KJ, Jeong JW, Jeong CH, Chun KH, Lee SK, Kwon YG, Kim KW. Protein Kinase Cδ regulates the stability of hypoxia-inducible factor-1α under hypoxia. Cancer Sci 98: 1476–1481, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lounsbury KM, Stern M, Taatjes D, Jaken S, Mossman BT. Increased localization and substrate activation of protein kinase Cδ in lung epithelial cells following exposure to asbestos. Am J Pathol 160: 1991–2000, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mellor H, Parker PJ. The extended protein kinase C superfamily. Biochem J 332: 281–292, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Muramatsu T. Midkine and pleiotrophin: two related proteins involved in development, survival, inflammation and tumorigenesis. J Biochem 132: 359–371, 2002 [DOI] [PubMed] [Google Scholar]
- 22.Nakagawara A, Milbrandt J, Muramatsu T, Deuel TF, Zhao H, Cnaan A, Brodeur GM. Differential expression of pleiotrophin and midkine in advanced neuroblastomas. Cancer Res 55: 1792–1797, 1995 [PubMed] [Google Scholar]
- 23.Nordin SL, Jovic S, Kurut A, Andersson C, Gela A, Bjartell A, Morgelin M, Olin AI, Lund M, Egesten A. High expression of midkine in the airways of patients with cystic fibrosis. Am J Respir Cell Mol Biol 49: 935–942, 2013 [DOI] [PubMed] [Google Scholar]
- 24.O'Brien T, Cranston D, Fuggle S, Bicknell R, Harris AL. The angiogenic factor midkine is expressed in bladder cancer, and overexpression correlates with a poor outcome in patients with invasive cancers. Cancer Res 56: 2515–2518, 1996 [PubMed] [Google Scholar]
- 25.Pal S, Claffey KP, Dvorak HF, Mukhopadhyay D. The von Hippel-Lindau gene product inhibits vascular permeability factor/vascular endothelial growth factor expression in renal cell carcinoma by blocking protein kinase C pathway. J Biol Chem 272: 27509–27512, 1997 [DOI] [PubMed] [Google Scholar]
- 26.Reynolds PR, Mucenski ML, Le Cras TD, Nichols WC, Whitsett JA. Midkine is regulated by hypoxia and causes pulmonary vascular remodeling. J Biol Chem 279: 37124–37132, 2004 [DOI] [PubMed] [Google Scholar]
- 27.Satelli A, Li S. Vimentin in cancer and its potential as a molecular target for cancer therapy. Cell Mol Life Sci 68: 3033–3046, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Short MD, Fox SM, Lam CF, Stenmark KR, Das M. PKCζ attenuates hypoxia-induced proliferation of fibroblasts by regulating MAP kinase phosphatase-1 expression. Mol Biol Cell 17: 1995–2008, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Short M, Nemenoff RA, Zawada WM, Stenmark KR, Das M. Hypoxia induces differentiation of pulmonary artery adventitial fibroblasts into myofibroblasts. Am J Physiol Cell Physiol 286: C416–C425, 2004 [DOI] [PubMed] [Google Scholar]
- 30.Shukla A, Lounsbury KM, Barrett TF, Gell J, Rincon M, Butnor KJ, Taatjes DJ, Davis GS, Vacek P, Nakayama KI, Nakayama K, Steele C, Mossman Regulation of interleukin-6 secretion by the two-pore-domain potassium channel Trek-1 in alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol 304: L276–L286, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Soltoff SP. Rottlerin: an inappropriate and ineffective inhibitor of PKCδ. Trends Pharmacol Sci 28: 453–458, 2007 [DOI] [PubMed] [Google Scholar]
- 32.Steinberg SF. Distinctive activation mechanisms and functions for protein kinase Cδ. Biochem J 383: 449–59, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stenmark KR, Davie NJ, Reeves JT, Frid MG. Hypoxia, leukocytes, and the pulmonary circulation. J Appl Physiol 98: 715–721, 2005 [DOI] [PubMed] [Google Scholar]
- 34.Sulkowska M. Morphological studies of the lungs in chronic hypobaric hypoxia. Pol J Pathol 48: 225–234, 1997 [PubMed] [Google Scholar]
- 35.Tsutsui J, Kadomatsu K, Matsubara S, Nakagawara A, Hamanoue M, Takao S, Shimazu H, Ohi Y, Muramatsu T. A new family of heparin-binding growth/differentiation factors: increased midkine expression in Wilms' tumor and other human carcinomas. Cancer Res 53: 1281–1285, 1993 [PubMed] [Google Scholar]
- 36.Young TA, Wang H, Munk S, Hammoudi DS, Young DS, Mandelcorn MS, Whiteside CI. Vascular endothelial growth factor expression and secretion by retinal pigment epithelial cells in high glucose and hypoxia is protein kinase C-dependent. Exp Eye Res 80: 651–662, 2005 [DOI] [PubMed] [Google Scholar]
- 37.Weckbach LT, Groesser L, Borgolte J, Pagel JI, Pogoda F, Schymeinsky J, Muller-Hocker J, Shakibaei M, Muramatsu T, Deindl E, Walzog B. Midkine acts as proangiogenic cytokine in hypoxia-induced angiogenesis. Am J Physiol Heart Circ Physiol 303: H429–H438, 2012 [DOI] [PubMed] [Google Scholar]
- 38.Zhao G, Nie Y, Lv M, He L, Wang T, Hou Y. ERβ-mediated estradiol enhances epithelial mesenchymal transition of lung adenocarcinoma through increasing transcription of midkine. Mol Endocrinol 26: 1304–1315, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhou G, Dada LA, Wu M, Kelly A, Trejo H, Zhou Q, Varga J, Sznajder JI. Hypoxia-induced alveolar epithelial-mesenchymal transition requires mitochondrial ROS and hypoxia-inducible factor 1. Am J Physiol Lung Cell Mol Physiol 297: L1120–L1130, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]