

Abstract
MicroRNAs (miRNAs) are small, noncoding regulatory RNAs that act as posttranscriptional repressors by binding to the 3′-untranslated region (3′-UTR) of target genes. They require processing by Dicer, an RNase III enzyme, to become mature regulatory RNAs. Previous work from our laboratory revealed critical roles for miRNAs in nephron progenitors at midgestation (Ho J, Pandey P, Schatton T, Sims-Lucas S, Khalid M, Frank MH, Hartwig S, Kreidberg JA. J Am Soc Nephrol 22: 1053–1063, 2011). To interrogate roles for miRNAs in the early metanephric mesenchyme, which gives rise to nephron progenitors as well as the renal stroma during kidney development, we conditionally ablated Dicer function in this lineage. Despite normal ureteric bud outgrowth and condensation of the metanephric mesenchyme to form nephron progenitors, early loss of miRNAs in the metanephric mesenchyme resulted in severe renal dysgenesis. Nephron progenitors are initially correctly specified in the mutant kidneys, with normal expression of several transcription factors known to be critical in progenitors, including Six2, Pax2, Sall1, and Wt1. However, there is premature loss of the nephron progenitor marker Cited1, marked apoptosis, and increased expression of the proapoptotic protein Bim shortly after the initial inductive events in early kidney development. Subsequently, there is a failure in ureteric bud branching and nephron progenitor differentiation. Taken together, our data demonstrate a previously undetermined requirement for miRNAs during early kidney organogenesis and indicate a crucial role for miRNAs in regulating the survival of this lineage.
Keywords: Dicer, microRNA, kidney development
the adult mammalian kidney, the metanephros, is essential not only for its excretory functions but also for its regulatory role in fluid homeostasis and hormone synthesis (reviewed in Ref. 30). In mice, the metanephric kidney begins to develop at embryonic day 10.5 (E10.5) with the outgrowth of the ureteric bud from the Wolffian duct into the metanephric mesenchyme (reviewed in Ref. 24). These two tissues undergo reciprocal inductive events, such that the ureteric bud receives signals from the metanephric mesenchyme to undergo repetitive rounds of branching to give rise to the renal collecting system. In concert, the metanephric mesenchyme establishes two cell fates in response to signals from the ureteric bud: renal stromal cells and a condensed cap of mesenchymal cells, the nephron progenitors, which give rise to the multiple cell types of the nephron. Nephron progenitors express several transcription factors, including sine oculis homeobox homolog 2 (Six2), Sal-like 1 (Drosophila) protein (Sall1), CREB-binding protein/p300-interacting transactivator with Asp/Glu-rich C-terminal domain 1 (Cited1), paired box gene 2 (Pax2), and Wilms tumor-suppressor gene 1 (WT1) (5, 22, 27, 31, 32, 37). These transcription factors are required for the proliferation and survival of nephron progenitors and for subsequent differentiation of the progenitors, beginning with a mesenchymal-to-epithelial transition to form early developing nephrons (the renal vesicle) that express the neural cell adhesion molecule (NCAM) (18). Thus changes in the balance between cell survival and differentiation can result in renal hypoplasia or early renal agenesis in kidney development (21, 39).
To date, much of the emerging information regarding the molecular mechanisms that regulate kidney development has been focused on the function of specific genes. An additional layer of complexity exists, where small noncoding microRNAs (miRNAs) regulate the flow of genetic information via controlling the translation and/or stability of their target mRNAs (reviewed in Ref. 1). Within the developing kidney, >170 miRNA families were recently identified by small RNA deep sequencing (36). The first functional studies for miRNAs in the developing kidney have used a conditional approach to knock down Dicer activity, which is required for the production of mature miRNAs, in specific cell lineages. These studies revealed that miRNAs have crucial roles in nephron progenitors, the ureteric epithelium, podocytes, and juxtaglomerular cells (13–15, 26, 33, 34).
We have previously demonstrated that miRNA loss in Six2-expressing nephron progenitors, and their derivatives, results in premature depletion of this population at midgestation due to increased apoptosis (15). This was associated with increased expression of the proapoptotic protein Bim, a known target of several miRNAs present in the developing kidney (15, 20, 29, 36, 38, 40). Bim is thought to interact with the prosurvival protein Bcl-2 to regulate cell survival, and the gene dosage of Bim appears critical, since loss of a single Bim allele is sufficient to rescue cystic kidneys observed in Bcl2-deficient mice (2). Together, these data suggested a model in which miRNA-mediated regulation of Bim expression modulates nephron progenitor survival. Interestingly, the nephron progenitor population is preserved until midgestation in these embryos, raising the question of whether miRNAs play a role in the early inductive events of kidney organogenesis.
To ask this, we conditionally ablated Dicer in the early metanephric mesenchyme using a Pax3CreTg allele (9, 11, 12). This resulted in severe renal dysgenesis with marked apoptosis and increased expression of Bim in the metanephric mesenchyme by E11.5. Nephron progenitors are initially correctly specified and normal ureteric bud outgrowth occurs; however, subsequent ureteric branching and nephron differentiation do not occur. Taken together, our data suggest that miRNA-mediated regulation of Bim activity and, hence nephron progenitor survival, is essential for normal kidney development.
MATERIALS AND METHODS
Mouse strains.
The Pax3Cre transgene (Pax3CreTg) directs Cre-mediated excision in the metanephric mesenchyme of the developing kidney (9, 11, 23) (from Dr. Jonathan Epstein, University of Pennsylvania, Philadelphia, PA). These mice were crossed with a conditionally floxed Dicer allele, which is required for the production of mature miRNAs, to generate Pax3CreTg; Dicerflx/flx embryos (12) (from Dr. Clifford Tabin, Harvard Medical School, Boston, MA). Timed matings were performed, and the day on which plugs were observed was considered E0.5. Embryonic tissue was genotyped by PCR with the following primers as described previously: 1) Pax3CreTg allele, forward primer 5′- AATCTTATGGTCACCTGAGTGTTAAATGTCCAATTTAC-3′ and reverse primer 5′-CATCTTCAGGTTCTGCGGG-3′, yielding a 230-bp band indicating the presence of Cre recombinase; and 2) Dicerflx allele, forward primer 5′-CCTGACAGTGACGGTCCAAAG-3′ and reverse primer 5′-CATGACTCTTCAACTCAAACT-3′, yielding a 350-bp wild-type band and a 400-bp Dicerflx band (11, 12). Cre-negative littermates were used as controls (hereafter, controls refer to Dicerflx/+ or Dicerflx/flx embryos). No difference was observed in the kidneys of Dicerflx/+ or Dicerflx/flx embryos in these experiments. All animals were housed in the vivarium at the Rangos Research Center at the Children's Hospital of Pittsburgh (UPMC, Pittsburgh, PA), and all animal experiments were carried out in accordance with the policies of the Institutional Animal Care and Use Committee at the Children's Hospital of Pittsburgh.
Histopathology and immunohistochemical staining.
For morphological analysis, control and Pax3CreTg; Dicerflx/flx embryos were fixed in 4% paraformaldehyde overnight, embedded in paraffin, sectioned at 7–9 μm, and stained with hematoxylin and eosin at E11.5 and E12.5. For immunohistochemical staining, after deparaffinization, rehydration, and permeabilization in KPBS-BT (phosphate-buffered saline with potassium chloride 200 mg/l, supplemented with 0.25% bovine serum albumin and 0.1% Triton X-100), antigen retrieval was performed using sodium citrate (10 mM, pH 6.0) buffer. Sections were then blocked in 5% bovine serum albumin before incubation overnight with the primary antibody. The next day, sections were washed with KPBS-BT, incubated with secondary antibodies, and washed before visualization with a Leica DM2500 microscope and photographed with a Qimaging QICAM Fast 1394 camera. The primary antibodies used were as follows: rabbit anti-Six2 antibody (1:200, Proteintech, Chicago, IL), rabbit anti-Cited1 antibody (1:80, Thermo Fisher Scientific, Cheshire, UK), rabbit anti-Pax2 antibody (1:200, Invitrogen, Carlsbad, CA), rabbit anti-Sall1 antibody (1:150, Abcam, Cambridge, MA), rabbit anti-Bim antibody (1:70, Cell Signaling Technology, Danvers, MA), mouse anti-neural cell adhesion molecule (NCAM) antibody (1:100, Sigma-Aldrich, St. Louis, MO), monoclonal mouse anti-calbindin-D28k antisera (1:100, Sigma-Aldrich), rabbit anti-Wt1 antibody (1:80, Santa Cruz Biotechnology, Santa Cruz, CA), mouse anti-Bcl-2 (1:100, Santa Cruz Biotechnology), and rabbit anti-phosphorylated histone H3 (PH3) antibody (1:100, Sigma-Aldrich). The secondary antibodies used were donkey anti-rabbit 594 (1:200, Jackson ImmunoResearch Laboratories, West Grove, PA) and donkey anti-mouse 488 (Jackson ImmunoResearch Laboratories).
Apoptosis assays.
Terminal deoxynucleotidyl transferase 2′-deoxyuridine,5′-triphosphate nick-end labeling (TUNEL) assays were performed on control and Pax3CreTg; Dicerflx/flx tissue sections at E11.5 and E12.5 using an ApopTag Plus Fluorescein In Situ Apoptosis Detection kit, per the manufacturer's instructions (EMD Millipore, Billerica, MA).
Quantitative real-time PCR.
Control and Pax3CreTg; Dicerflx/flx embryos were collected at E11.5 from three separate litters. E11.5 kidneys were snap frozen, and the embryos were genotyped as outlined above. RNA was extracted from the kidneys of one mutant and one littermate control per litter using a Qiagen MicroRNeasy kit (Qiagen, Valencia, CA). The RNA was then quantitated using a Nanodrop, and cDNA was synthesized from 100 ng of RNA using an Invitrogen SuperScript First Strand Synthesis kit, as per the manufacturer's directions (Invitrogen, Grand Island, NY). A no reverse-transcriptase reaction was performed as a negative control. Quantitative real-time PCR (qPCR) was then conducted using SsoAdvanced SYBR Green Supermix (Bio-Rad, Hercules, CA) for Bim and Bcl2 (see Table 1 for primers) with a Techne TC-412 thermal cycler (Bibby Scientific US, Burlington, NJ). Each sample was standardized against the housekeeping gene glyceraldehyde-3-phosphate dehydrogenase, and the fold-change between controls and mutants was calculated by comparing the ΔCT in mutants to controls. A two-tailed Student's t-test was used to determine statistical significance.
Table 1.
Primers for the genes that were assessed by real-time PCR
| Gene Name | Accession No. | Primers (5′-3′) | Fragment Size, bp |
|---|---|---|---|
| GAPDH | NM_008084 | F-AACCTGCCAAGTATGATGA | 119 |
| R-GGAGTTGCTGTTGAAGTC | |||
| Bcl-2 | NM_177410 | F-GAGCAAGAATCCAGCATT | 180 |
| R-TTGTTAGTCCACCAGAGG | |||
| Bim | NM_207680 | F-GTAGTGGCTGGTTCTCTT | 247 |
| R-CATCTGCTGCTAATACTTCTC |
F, forward primer; R, reverse primer.
qPCR for miRNA expression was performed using total RNA extracted from control and Pax3CreTg; Dicerflx/flx E11.5 kidneys from three separate litters using a Direct-zol RNA miniprep kit (Zymo Research, Irvine, CA). Taqman miRNA assays (Life Technologies, Grand Island, NY) were conducted to determine relative expression levels of mmu-miR-17-5p (miRBase ID mmu-miR-17-5p) and mmu-miR-10a (miRBase ID mmu-miR-10a), per the manufacturer's instructions. Expression levels were normalized to that of the endogenous control, snoRNA-234, using the cycle threshold value (Ct). The two-tailed Student's t-test was used to determine statistical significance.
Locked nucleic acid in situ hybridization.
Locked nucleic acid (LNA) in situ hybridization was conducted on 10-μm cryosections of E11.5 embryos using an LNA probe (Exiqon) complementary to the miR-17-5p and miR-10a mature miRNA sequence, as previously described (15). The LNA probe was digoxigenin (DIG) labeled using an oligonucleotide tailing kit (Roche, Indianapolis, IN). In brief, sections were dried at room temperature for 1 h and fixed in 4% PFA for 10 min. After three 3-min washes in diethylpyrocarbonate (DEPC)-treated PBS, sections were allowed to permeabilize in 15 μg/ml proteinase K for 2 min, washed three times in DEPC-PBS, fixed again in 4% PFA for 5 min, washed three times for 3 min, and acetylated for 10 min in a glass histology jar (200 ml H2O, 2.66 ml triethanolamine, 0.35 ml 37% HCl, and 0.75 ml acetic anhydride added dropwise). Slides were subsequently washed again in PBS for 3 × 5 min and prehybridized at 42°C for 2 h in hybridization buffer (50% formamide, 5× SSC, 1% SDS, 50 μg/ml yeast tRNA, and 50 μg/ml heparin). Hybridization was subsequently carried out at 40°C overnight with 0.5 μM probe concentration. Slides were washed twice for 30 min in 2× SSC at 40°C. Slides were then placed in 1% Roche blocking reagent, 1% heat-inactivated sheep serum (HISS) in NTT (0.15 M sodium chloride, 0.1 M Tris, pH 7.5, 0.1% Tween) for 1 h for blocking. This was followed by 1:1,000 anti-DIG antibody in 1% Roche blocking reagent, 1% HISS in NTT overnight at 4°C. After three 30-min washes with NTT, three 5-min washes were performed with staining buffer (0.1 M Tris, pH 9.5, 0.05 M MgCl2, 0.1 M NaCl, and 0.2% Tween 20), and the color reaction was developed with BM Purple (Roche) over 3 days.
RESULTS
Removal of Dicer activity in early metanephric mesenchyme results in severe renal dysgenesis.
To demonstrate a requirement for miRNAs in the early metanephric mesenchyme, we conditionally ablated Dicer in the metanephric mesenchyme using a floxed Dicer allele (12) and a Pax3CreTg allele (9, 11, 23). Mice from the Pax3CreTg and Dicerflx/flx transgenic mouse lines have no reported renal anomalies and are viable and fertile with no reported or observed anomalies in our laboratory (9, 11, 12, 23). Excision of the floxed Dicer allele by the Pax3CreTg allele would be expected to result in loss of miRNAs in the early metanephric mesenchyme of the developing kidney. To verify this, LNA in situ hybridization was performed on E11.5 kidneys from control and mutant (Pax3CreTg; Dicerflx/flx) embryos (Figure 1, A–D). In control E11.5 kidneys, both mmu-miR-10a and mmu-miR-17-5p are expressed in the ureteric bud and condensing metanephric mesenchyme (Fig. 1, A and C). As expected, there is a loss of mmu-miR-10a and mmu-miR-17-5p expression in the metanephric mesenchyme, but not the ureteric bud in Pax3CreTg; Dicerflx/flx embryos (Fig. 1, B and D). In keeping with this finding, qPCR for mmu-miR-10a and mmu-miR-17-5p demonstrated a significant decrease in expression for both miRNAs in mutant kidneys (P < 0.05) (Fig. 1E).
Fig. 1.

Cre-mediated excision of the conditionally floxed Dicer allele via the Pax3CreTg allele results in loss of microRNA (miRNA) expression in the metanephric mesenchyme at embryonic day 11.5 (E11.5). A–D: locked nucleic acid (LNA) in situ hybridization demonstrates loss of mmu-miR-10a and mmu-miR-17-5p expression in the metanephric mesenchyme of Pax3CreTg, Dicerflx/flx kidneys (B and D) compared with controls (A and C). Black dashed lines, metanephric mesenchyme; yellow dashed lines, ureteric bud. The magnification is ×20. E: quantitative real-time PCR performed on total RNA isolated from E11.5 kidneys confirms decreased expression of mmu-miR-10a and mmu-miR-17-5p (*P < 0.05, paired t-test). Relative expression of mmu-miR-10a and mmu-miR-17-5p denotes the fold-change in ΔCT in mutants normalized to controls. Bars represent SE.
Histological analysis of the Pax3CreTg; Dicerflx/flx embryos demonstrates that kidney development begins normally with the outgrowth of the ureteric bud from the Wolffian duct and the presence of the metanephric mesenchyme around the ureteric bud at E11.5 (Fig. 2, A–D). However, mutants have poorly condensed metanephric mesenchyme, with pyknotic-appearing cells that are most prominent at the periphery compared with controls (= Dicerflx/+ or Dicerflx/flx) (Fig. 2D). By E12.5, the mutant metanephros is markedly abnormal, with failure of the ureteric bud to branch and increasing numbers of pyknotic metanephric mesenchymal cells relative to controls (Fig. 2, E–H). The mutant metanephros undergoes essentially complete regression by E14.5, with no identifiable renal structures (Fig. 2I). In general, the failure of the metanephric mesenchyme to appropriately condense and grow could be due to several possible mechanisms: 1) impaired ability to be normally specified into nephron progenitors; 2) decreased proliferation of the metanephric mesenchyme (and thus an inability to self-renew during kidney development); and 3) decreased survival (increased apoptosis) of the metanephric mesenchyme. These possibilities are addressed in the following experiments.
Fig. 2.

Loss of mature miRNAs in the early metanephric mesenchyme results in decreased mesenchymal condensation and an arrest in ureteric branching. A–D: at E11.5, the metanephric mesenchyme does not normally condense around the ureteric bud in Pax3CreTg, Dicerflx/flx kidneys (B and D) compared with control kidneys (A and C). E–H: at E12.5, the ureteric bud fails to undergo branching in Pax3CreTg, Dicerflx/flx kidneys (F and H) compared with control kidneys (E and G). I: at E14.5, there are no kidney elements present in the mutants compared with controls. Arrowheads, Wolffian duct; black dashed lines, metanephric mesenchyme; yellow dashed lines, ureteric bud; K, kidney; B, bladder. The magnification is ×10 (A, B, E, and F) or ×20 (C, D, G, and H).
Normal initial specification of nephron progenitors occurs in Pax3CreTg; Dicerflx/flx kidneys.
To determine whether the ureteric bud and metanephric mesenchyme are normally specified in Pax3CreTg; Dicerflx/flx metanephroi, immunofluorescence was performed for several markers of these cell lineages at E11.5 (Fig. 3). As in controls, the mutant metanephric mesenchyme expresses several transcription factors found in nephron progenitors, including Six2, Sall1, Wt1, and Pax2, as well as NCAM (Fig. 3, A and B, E–L) (18). However, unlike controls, there is an almost complete abolishment of Cited1 in the mutant metanephric mesenchyme (Fig. 3, M and N). In contrast, markers found in the ureteric bud, including Cited1, Pax2, and calbindin, are normally expressed in mutants at E11.5 (Fig. 3, K–P). These data are consistent with the normal initial specification of nephron progenitors in the absence of miRNAs in the early metanephric mesenchyme, although the progenitors themselves may not be completely functionally normal (given the decrease in Cited1 expression).
Fig. 3.
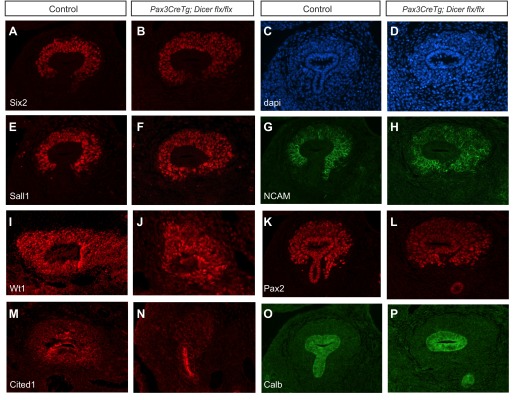
Immunofluorescent staining of E11.5 kidneys demonstrates initial specification of nephron progenitors and normal ureteric bud markers in mutant kidneys. (A, B, E–L) Expression of the nephron progenitor markers Six2, Sall1, NCAM, Wt1, and Pax2 is preserved in Pax3CreTg, Dicerflx/flx kidneys. C and D: labeling of nuclei with DAPI confirms the histological presence of nephron progenitors and the ureteric bud. M and N: Cited1 expression is decreased in Pax3CreTg, Dicerflx/flx nephron progenitors compared with controls. O and P: calbindin staining is normal in ureteric buds of Pax3CreTg, Dicerflx/flx kidneys vs. controls. Pax2 and Cited1 staining also mark the ureteric buds and is unchanged between controls and mutants. The magnification is ×20.
At E12.5, there is an ongoing decrease in Cited1 expression in Pax3CreTg; Dicerflx/flx kidneys (Fig. 4, M and N). In contrast, there is still persistent expression of many nephron progenitor markers, including Six2, Sall1, NCAM, WT1, and Pax2; however, there are far fewer nephron progenitors present in Pax3CreTg; Dicerflx/flx kidneys than in littermate controls (Fig. 4, A and B, E–N). In addition, there is no evidence of early developing nephrons in E12.5 Pax3CreTg; Dicerflx/flx kidneys, with no NCAM-positive renal vesicles compared with controls (Fig. 4, G and H). Finally, Pax2 and calbindin staining reveals a failure of ureteric bud branching in E12.5 mutant kidneys compared with controls (Fig. 4, K and L, O and P). Together, these results suggest that there is an ongoing loss of nephron progenitors, a failure of progenitors to differentiate normally, and a lack of ureteric bud branching, resulting in a lack of nephron formation in Pax3CreTg; Dicerflx/flx kidneys by E12.5.
Fig. 4.
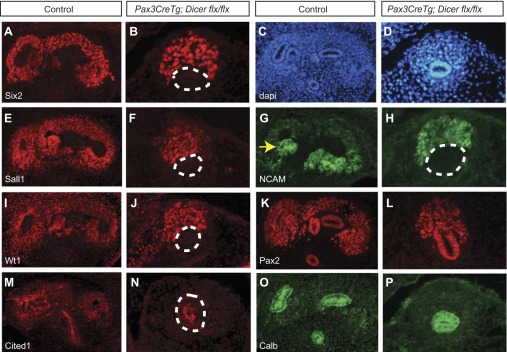
Immunofluorescent staining of E12.5 kidneys shows a failure in ureteric branching and fewer nephron progenitors in Pax3CreTg, Dicerflx/flx kidneys. A, B, E–N: although Six2, Sall1, NCAM, Wt1, Pax2, and Cited1 continues to be present in Pax3CreTg, Dicerflx/flx nephron progenitors, there are far fewer positive cells than in controls (A, E, G, I, K, M, O). C and D: labeling of nuclei with DAPI confirms the histological presence of nephron progenitors and the ureteric bud. O and P: calbindin staining reveals unbranched ureteric buds in Pax3CreTg, Dicerflx/flx kidneys compared with branches seen in controls. Yellow arrow, renal vesicle; white dashed lines, ureteric bud. The magnification is ×20.
Dicer ablation in the metanephric mesenchyme results in increased apoptosis and Bim expression.
Given that mutant metanephric mesenchymal cells become largely pyknotic, TUNEL staining was performed to determine whether this indicated excessive apoptosis. TUNEL staining reveals markedly elevated apoptosis in the E11.5 and E12.5 Pax3CreTg; Dicerflx/flx metanephric mesenchyme compared with low levels in controls (Fig. 5, A–H). In contrast, staining for the mitotic marker PH3 demonstrates no gross differences in proliferation between control and mutant kidneys at these time points (data not shown).
Fig. 5.
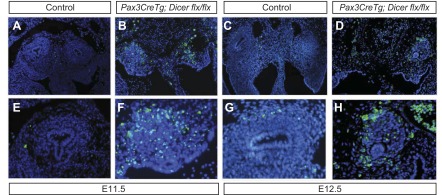
Increased apoptosis occurs in E11.5 and E12.5 Pax3CreTg, Dicerflx/flx metanephric mesenchyme. A, B, E, and F: terminal deoxynucleotidyl transferase 2′-deoxyuridine,5′-triphosphate nick-end labeling (TUNEL) staining of E11.5 kidneys demonstrates increased apoptosis in Pax3CreTg, Dicerflx/flx metanephric mesenchyme vs. controls. C, D, G, and H: TUNEL staining of E12.5 kidneys reveals continued excessive apoptosis in mutant kidneys compared with controls. The magnification is ×10 (A–D) and ×20 (E–H).
Since there is increased apoptosis in Pax3CreTg; Dicerflx/flx metanephric mesenchyme, immunofluorescence was performed for the proapoptotic protein Bim and its interacting prosurvival protein Bcl2. There is a marked increase in Bim expression in Pax3CreTg; Dicerflx/flx kidneys at both E11.5 and E12.5 compared with controls by immunofluorescence (Fig. 6, E–H). Furthermore, real-time PCR confirms that Bim transcripts are significantly elevated compared with controls (2.67-fold higher, P < 0.05) at E11.5 (Fig. 6I). In contrast, immunostaining and real-time PCR for Bcl2 is unchanged in mutants vs. controls (Fig. 6, A–D). Thus the loss of miRNAs in the early metanephric mesenchyme leads to excessive apoptosis that is associated with increased Bim expression.
Fig. 6.
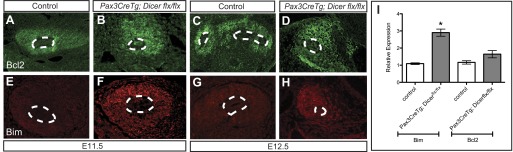
Immunofluorescent staining demonstrates increased Bim and unchanged Bcl2 expression in the metanephric mesenchyme of Pax3CreTg, Dicerflx/flx E11.5 and E12.5 kidneys. A–D: Bcl2 staining in control (A and C) and Pax3CreTg, Dicerflx/flx kidneys (B and D) is unchanged at E11.5 and E12.5. E–H: Bim staining in Pax3CreTg, Dicerflx/flx kidneys (F and H) is increased at E11.5 and E12.5 compared with controls (E and G). The magnification is ×20. I: quantitative real-time PCR performed on total RNA isolated from E11.5 kidneys confirms increased expression of Bim (*P < 0.05, paired t-test) and no significant difference in Bcl2 expression (P > 0.05, paired t-test). The relative expression of Bim and Bcl2 denotes the fold-change in ΔCT in mutants normalized to controls. Bars represent SE.
DISCUSSION
Our previous work demonstrated that loss of miRNAs in nephron progenitors resulted in premature depletion of progenitors due to increased apoptosis (15). Interestingly, the nephron progenitor population remained preserved until midgestation, raising the question of whether miRNAs play a role in early renal development. In this study, we demonstrate that conditional ablation of Dicer in the early metanephric mesenchyme with a Pax3cre transgenic line results in severe renal dysgenesis, despite normal ureteric bud outgrowth and initial specification of the metanephric mesenchyme to form nephron progenitors. Several transcription factors known to be critical in nephron progenitors are expressed normally, including Six2, Pax2, Sall1, and Wt1. However, there is premature loss of the transcription factor Cited1, marked apoptosis, and increased expression of the proapoptotic protein Bim shortly after the early inductive events in early kidney development. Subsequently, there is a failure in ureteric bud branching and nephron progenitor differentiation. Taken together, our data demonstrate a previously undetermined requirement for miRNAs in the early metanephric mesenchyme and indicate a crucial role for miRNAs in regulating the survival of this lineage.
The preserved expression of Pax2, Wt1, Sall1, and Six2 supports the notion that most of the earliest inductive events in mammalian kidney development occur normally in Pax3CreTg; Dicerflx/flx mutant kidneys. Pax2 is required for the normal specification of the intermediate mesoderm, from which the metanephric mesenchyme is derived, and Pax2 mutant mice lack kidneys, ureters, and genital tracts (37). Wt1 is expressed broadly in the early metanephric mesenchyme and functions to promote the survival of the metanephric mesenchyme (22). Unlike in the Pax3CreTg; Dicerflx/flx mutants, the global loss of Wt1 results in failure of ureteric bud outgrowth, earlier apoptosis in the metanephric mesenchyme, and renal agenesis (22). In contrast, Sall1 is required for complete ureteric bud invasion and metanephric mesenchyme induction, and loss of Sall1 results in a phenotype reminiscent of that seen with the Pax3CreTg; Dicerflx/flx mutants (27). Thus the loss of miRNAs in the early mesenchyme does not impair the initial specification of the metanephric mesenchyme or induction of ureteric bud outgrowth from the Wolffian duct.
The transcription factors Six2 and Cited1 mark a population of nephron-committed, self-renewing, multipotent population of progenitors derived from the metanephric mesenchyme during kidney development (4, 19). While the loss of Cited1 results in no overt renal defects, the removal of Six2 activity results in premature differentiation of nephron progenitors and early cessation of nephrogenesis due to a depletion of nephron progenitors (5, 32). Cited1 has previously been reported to be expressed at low levels in the early metanephric mesenchyme and ureteric bud stalk at E10.5 and subsequently becomes more robustly expressed in nephron progenitors at E12.5 (5). Recent evidence suggests that Cited1 marks a subpopulation of more “undifferentiated” Six2-positive cells, excluding mesenchymal Six2-positive cells that have already been induced to undergo a mesenchymal-to-epithelial transition (25). Furthermore, other studies support the concept of a heterogeneous nephron progenitor population, with subpopulations of cells that are self-renewing and others that are committed to differentiation into nephrons (7, 17). Cited1 is thought to function via repression of Wnt/β-catenin signaling and activation of Smad4-dependent transcription in nephron progenitors (28). Taken together, the decreased Cited1 expression and lack of developing nephrons in Pax3CreTg; Dicerflx/flx mutant kidneys may therefore reflect an early defect in nephron progenitors, subsequent to specification and before differentiation into renal vesicles.
The marked increase in apoptosis observed in the metanephric mesenchyme of Pax3CreTg; Dicerflx/flx mutant kidneys is in accord with our prior observation of increased apoptosis in nephron progenitors that lack miRNAs in Six2-TGCTg; Dicerflx/flx kidneys (15). Unlike the severe renal dysgenesis phenotype in Pax3CreTg; Dicerflx/flx kidneys that results in renal agenesis, the nephron progenitor population is preserved until midgestation, and hypodysplastic kidneys form, in Six2-TGCTg; Dicerflx/flx animals. Given that both the Six2-TGC and Pax3CreTg alleles have been reported to direct Cre excision as early as E11.5 in the developing kidney, the earlier and more severe phenotype in Pax3CreTg; Dicerflx/flx embryos likely reflects miRNA loss from both renal stroma and nephron progenitors, as driven by the Pax3CreTg transgene (in contrast to the Six2-TGC allele, which drives expression in the nephron progenitors) (9, 11, 19, 23). Thus the current data support an earlier requirement for miRNAs in the metanephric mesenchyme (which subsequently gives rise to both the renal stroma and nephron progenitors) than previously described.
In broad terms, the balance between proapoptotic and prosurvival factors determines whether a cell undergoes apoptosis or survives. Bim is a member of the Bcl-2 family of proapoptotic proteins and is thought to release the apoptotic effector proteins Bax or Bak from their interaction with prosurvival proteins such as Bcl-2 (6, 8). While Bim null mice have been described to have structural kidney defects, loss of a single Bim allele in Bcl-2 null mice is sufficient to rescue the renal cystic phenotype, suggesting that the gene dosage of Bim is critical (2, 3, 35). Bim is a known miRNA target of several miRNAs expressed in the developing kidney, including the miR-17∼92 and miR-24 (15, 20, 29, 36, 38, 40). The loss of mmu-miR-17–5p expression in the metanephric mesenchyme of Pax3CreTg; Dicerflx/flx embryos would be expected to result in increased Bim expression. In addition, Bim is also predicted to be a target of mmu-miR-10a via TargetScan (10). Thus the elevated Bim expression observed in the metanephric mesenchyme of Pax3CreTg; Dicerflx/flx mutant kidneys is consistent with the concept that miRNAs regulate the survival of the metanephric mesenchyme by repressing Bim activity.
In summary, we demonstrate a previously undetermined requirement for miRNAs during early kidney organogenesis. Dicer ablation in the early metanephric mesenchyme results in severe renal dysgenesis despite normal initial specification of nephron progenitors and ureteric bud outgrowth. The renal dysgenesis is due to marked apoptosis that is associated with increased expression of Bim, shortly after the initial inductive events in metanephric kidney development. Taken together, our data suggest a model where miRNAs modulate the balance between survival and apoptosis in the metanephric mesenchyme by targeting the proapoptotic protein Bim.
GRANTS
This work was supported by Pennsylvania Department of Health Grant CON001250. S. Sims-Lucas is supported by National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) Grant K01 DK096996, and J. A. Kreidberg is supported by NIDDK Grant R01 DK087794. J. Ho's laboratory is supported in part by a Norman S. Coplon Extramural Grant from Satellite Healthcare (for work unrelated to this current manuscript). J. Ho is also supported by NIDDK Grant R00 DK087922.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: J.Y.C., S.S.-L., J.A.K., and J.H. provided conception and design of research; J.Y.C., S.S.-L., D.S.B., A.J.B., and J.H. performed experiments; J.Y.C., S.S.-L., D.S.B., A.J.B., and J.H. analyzed data; J.Y.C., S.S.-L., D.S.B., A.J.B., J.A.K., and J.H. interpreted results of experiments; J.Y.C. and J.H. prepared figures; J.Y.C. and J.H. drafted manuscript; J.Y.C., S.S.-L., D.S.B., J.A.K., and J.H. edited and revised manuscript; J.Y.C., S.S.-L., D.S.B., A.J.B., J.A.K., and J.H. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Carlton Bates, Kenneth Walker, and Valeria DiGiovanni for thoughtful discussion and review of the manuscript.
REFERENCES
- 1.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell 136: 215–233, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bouillet P, Cory S, Zhang LC, Strasser A, Adams JM. Degenerative disorders caused by Bcl-2 deficiency prevented by loss of its BH3-only antagonist Bim. Dev Cell 1: 645–653, 2001 [DOI] [PubMed] [Google Scholar]
- 3.Bouillet P, Metcalf D, Huang DC, Tarlinton DM, Kay TW, Kontgen F, Adams JM, Strasser A. Proapoptotic Bcl-2 relative Bim required for certain apoptotic responses, leukocyte homeostasis, and to preclude autoimmunity. Science 286: 1735–1738, 1999 [DOI] [PubMed] [Google Scholar]
- 4.Boyle S, Misfeldt A, Chandler KJ, Deal KK, Southard-Smith EM, Mortlock DP, Baldwin HS, de Caestecker M. Fate mapping using Cited1-CreERT2 mice demonstrates that the cap mesenchyme contains self-renewing progenitor cells and gives rise exclusively to nephronic epithelia. Dev Biol 313: 234–245, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boyle S, Shioda T, Perantoni AO, de Caestecker M. Cited1 and Cited2 are differentially expressed in the developing kidney but are not required for nephrogenesis. Dev Dyn 236: 2321–2330, 2007 [DOI] [PubMed] [Google Scholar]
- 6.Danial NN. BCL-2 family proteins: critical checkpoints of apoptotic cell death. Clin Cancer Res 13: 7254–7263, 2007 [DOI] [PubMed] [Google Scholar]
- 7.Das A, Tanigawa S, Karner CM, Xin M, Lum L, Chen C, Olson EN, Perantoni AO, Carroll TJ. Stromal-epithelial crosstalk regulates kidney progenitor cell differentiation. Nat Cell Biol 15: 1035–1044, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ewings KE, Wiggins CM, Cook SJ. Bim and the pro-survival Bcl-2 proteins: opposites attract, ERK repels. Cell Cycle 6: 2236–2240, 2007 [DOI] [PubMed] [Google Scholar]
- 9.Grieshammer U, Cebrian C, Ilagan R, Meyers E, Herzlinger D, Martin GR. FGF8 is required for cell survival at distinct stages of nephrogenesis and for regulation of gene expression in nascent nephrons. Development 132: 3847–3857, 2005 [DOI] [PubMed] [Google Scholar]
- 10.Grimson A, Farh KK, Johnston WK, Garrett-Engele P, Lim LP, Bartel DP. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol Cell 27: 91–105, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hains D, Sims-Lucas S, Kish K, Saha M, McHugh K, Bates CM. Role of fibroblast growth factor receptor 2 in kidney mesenchyme. Pediatr Res 64: 592–598, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harfe BD, McManus MT, Mansfield JH, Hornstein E, Tabin CJ. The RNaseIII enzyme Dicer is required for morphogenesis but not patterning of the vertebrate limb. Proc Natl Acad Sci USA 102: 10898–10903, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harvey SJ, Jarad G, Cunningham J, Goldberg S, Schermer B, Harfe BD, McManus MT, Benzing T, Miner JH. Podocyte-specific deletion of dicer alters cytoskeletal dynamics and causes glomerular disease. J Am Soc Nephrol 19: 2150–2158, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ho J, Ng KH, Rosen S, Dostal A, Gregory RI, Kreidberg JA. Podocyte-specific loss of functional microRNAs leads to rapid glomerular and tubular injury. J Am Soc Nephrol 19: 2069–2075, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ho J, Pandey P, Schatton T, Sims-Lucas S, Khalid M, Frank MH, Hartwig S, Kreidberg JA. The pro-apoptotic protein Bim is a microRNA target in kidney progenitors. J Am Soc Nephrol 22: 1053–1063, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Karner CM, Das A, Ma Z, Self M, Chen C, Lum L, Oliver G, Carroll TJ. Canonical Wnt9b signaling balances progenitor cell expansion and differentiation during kidney development. Development 138: 1247–1257, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Klein G, Langegger M, Goridis C, Ekblom P. Neural cell adhesion molecules during embryonic induction and development of the kidney. Development 102: 749–761, 1988 [DOI] [PubMed] [Google Scholar]
- 19.Kobayashi A, Valerius MT, Mugford JW, Carroll TJ, Self M, Oliver G, McMahon AP. Six2 defines and regulates a multipotent self-renewing nephron progenitor population throughout mammalian kidney development. Cell Stem Cell 3: 169–181, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koralov SB, Muljo SA, Galler GR, Krek A, Chakraborty T, Kanellopoulou C, Jensen K, Cobb BS, Merkenschlager M, Rajewsky N, Rajewsky K. Dicer ablation affects antibody diversity and cell survival in the B lymphocyte lineage. Cell 132: 860–874, 2008 [DOI] [PubMed] [Google Scholar]
- 21.Koseki C, Herzlinger D, al-Awqati Q. Apoptosis in metanephric development. J Cell Biol 119: 1327–1333, 1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kreidberg JA, Sariola H, Loring JM, Maeda M, Pelletier J, Housman D, Jaenisch R. WT-1 is required for early kidney development. Cell 74: 679–691, 1993 [DOI] [PubMed] [Google Scholar]
- 23.Li J, Chen F, Epstein JA. Neural crest expression of Cre recombinase directed by the proximal Pax3 promoter in transgenic mice. Genesis 26: 162–164, 2000 [DOI] [PubMed] [Google Scholar]
- 24.Little MH, McMahon AP. Mammalian kidney development: principles, progress, and projections. Cold Spring Harb Perspect Biol 4: a008300, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mugford JW, Yu J, Kobayashi A, McMahon AP. High-resolution gene expression analysis of the developing mouse kidney defines novel cellular compartments within the nephron progenitor population. Dev Biol 333: 312–323, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nagalakshmi VK, Ren Q, Pugh MM, Valerius MT, McMahon AP, Yu J. Dicer regulates the development of nephrogenic and ureteric compartments in the mammalian kidney. Kidney Int 79: 317–330, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nishinakamura R, Matsumoto Y, Nakao K, Nakamura K, Sato A, Copeland NG, Gilbert DJ, Jenkins NA, Scully S, Lacey DL, Katsuki M, Asashima M, Yokota T. Murine homolog of SALL1 is essential for ureteric bud invasion in kidney development. Development 128: 3105–3115, 2001 [DOI] [PubMed] [Google Scholar]
- 28.Plisov S, Tsang M, Shi G, Boyle S, Yoshino K, Dunwoodie SL, Dawid IB, Shioda T, Perantoni AO, de Caestecker MP. Cited1 is a bifunctional transcriptional cofactor that regulates early nephronic patterning. J Am Soc Nephrol 16: 1632–1644, 2005 [DOI] [PubMed] [Google Scholar]
- 29.Qian L, Van Laake LW, Huang Y, Liu S, Wendland MF, Srivastava D. miR-24 inhibits apoptosis and represses Bim in mouse cardiomyocytes. J Exp Med 208: 549–560, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Quigley R. Developmental changes in renal function. Curr Opin Pediatr 24: 184–190, 2012 [DOI] [PubMed] [Google Scholar]
- 31.Rothenpieler UW, Dressler GR. Pax-2 is required for mesenchyme-to-epithelium conversion during kidney development. Development 119: 711–720, 1993 [DOI] [PubMed] [Google Scholar]
- 32.Self M, Lagutin OV, Bowling B, Hendrix J, Cai Y, Dressler GR, Oliver G. Six2 is required for suppression of nephrogenesis and progenitor renewal in the developing kidney. EMBO J 25: 5214–5228, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sequeira-Lopez ML, Weatherford ET, Borges GR, Monteagudo MC, Pentz ES, Harfe BD, Carretero O, Sigmund CD, Gomez RA. The microRNA-processing enzyme dicer maintains juxtaglomerular cells. J Am Soc Nephrol 21: 460–467, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shi S, Yu L, Chiu C, Sun Y, Chen J, Khitrov G, Merkenschlager M, Holzman LB, Zhang W, Mundel P, Bottinger EP. Podocyte-selective deletion of dicer induces proteinuria and glomerulosclerosis. J Am Soc Nephrol 19: 2159–2169, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sorenson CM, Rogers SA, Korsmeyer SJ, Hammerman MR. Fulminant metanephric apoptosis and abnormal kidney development in bcl-2-deficient mice. Am J Physiol Renal Fluid Electrolyte Physiol 268: F73–F81, 1995 [DOI] [PubMed] [Google Scholar]
- 36.Thiagarajan RD, Cloonan N, Gardiner BB, Mercer TR, Kolle G, Nourbakhsh E, Wani S, Tang D, Krishnan K, Georgas KM, Rumballe BA, Chiu HS, Steen JA, Mattick JS, Little MH, Grimmond SM. Refining transcriptional programs in kidney development by integration of deep RNA-sequencing and array-based spatial profiling. BMC Genomics 12: 441, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Torres M, Gomez-Pardo E, Dressler GR, Gruss P. Pax-2 controls multiple steps of urogenital development. Development 121: 4057–4065, 1995 [DOI] [PubMed] [Google Scholar]
- 38.Ventura A, Young AG, Winslow MM, Lintault L, Meissner A, Erkeland SJ, Newman J, Bronson RT, Crowley D, Stone JR, Jaenisch R, Sharp PA, Jacks T. Targeted deletion reveals essential and overlapping functions of the miR-17 through 92 family of miRNA clusters. Cell 132: 875–886, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Woolf AS, Welham SJ. Cell turnover in normal and abnormal kidney development. Nephrol Dial Transplant 17, Suppl 9: 2–4, 2002 [DOI] [PubMed] [Google Scholar]
- 40.Xiao C, Srinivasan L, Calado DP, Patterson HC, Zhang B, Wang J, Henderson JM, Kutok JL, Rajewsky K. Lymphoproliferative disease and autoimmunity in mice with increased miR-17–92 expression in lymphocytes. Nat Immunol 9: 405–414, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]