

Abstract
Acute kidney injury (AKI) is a complication of sepsis and leads to a high mortality rate. Human and animal studies suggest that mitochondrial dysfunction plays an important role in sepsis-induced multi-organ failure; however, the specific mitochondrial targets damaged during sepsis remain elusive. We used a clinically relevant cecal ligation and puncture (CLP) murine model of sepsis and assessed renal mitochondrial function using high-resolution respirometry, renal microcirculation using intravital microscopy, and renal function. CLP caused a time-dependent decrease in mitochondrial complex I and II/III respiration and reduced ATP. By 4 h after CLP, activity of manganese superoxide dismutase (MnSOD) was decreased by 50% and inhibition was sustained through 36 h. These events were associated with increased mitochondrial superoxide generation. We then evaluated whether the mitochondria-targeted antioxidant Mito-TEMPO could reverse renal mitochondrial dysfunction and attenuate sepsis-induced AKI. Mito-TEMPO (10 mg/kg) given at 6 h post-CLP decreased mitochondrial superoxide levels, protected complex I and II/III respiration, and restored MnSOD activity by 18 h. Mito-TEMPO also improved renal microcirculation and glomerular filtration rate. Importantly, even delayed therapy with a single dose of Mito-TEMPO significantly increased 96-h survival rate from 40% in untreated septic mice to 80%. Thus, sepsis causes sustained inactivation of three mitochondrial targets that can lead to increased mitochondrial superoxide. Importantly, even delayed therapy with Mito-TEMPO alleviated kidney injury, suggesting that it may be a promising approach to treat septic AKI.
Keywords: sepsis, kidney, mitochondria, oxidative stress, mitochondrial antioxidant
sepsis is characterized by a severe systemic inflammatory response caused by a microbial infection. There are estimates of more than 1,000,000 cases of sepsis in the United States each year (31) leading to over 200,000 deaths and a healthcare cost in the billions of dollars (2, 24). Acute kidney injury (AKI) is a frequent complication of sepsis that dramatically increases mortality (21, 38). Consequently, protecting the kidney could significantly improve survival in septic patients. Currently, there are no specific therapies to treat sepsis-induced AKI and clinicians must rely only on generalized supportive care (27, 44).
Since the kidney is such a highly metabolic organ, it is dependent on stable mitochondrial function to generate the ATP necessary for active tubular transport. The electron transport chain (ETC) located within the mitochondria is composed of five complexes including complex I–IV and ATP synthase. Mitochondria are known to produce small amounts of superoxide radical (O2·−) during the normal respiratory process (52). Normally, superoxide is efficiently scavenged by the major endogenous antioxidant enzyme manganese superoxide dismutase (MnSOD) located in the mitochondrial matrix (16). However, damage to the respiratory chain or MnSOD inactivation can result in superoxide levels that may overwhelm antioxidant defenses.
While there is growing appreciation that mitochondrial damage occurs during sepsis and that the severity of mitochondrial dysfunction correlates with adverse outcomes and increased patient mortality (7, 17, 18), the nature of the mitochondrial defects and their consequences have not been well-characterized, especially in the kidney. There is evidence suggesting that patients who die of severe sepsis have decreased mitochondrial function compared with survivors (7, 10) and in rodent models of sepsis, impaired mitochondrial function in the heart (19, 57), skeletal muscle (8, 42, 59), liver (8), and lung (3) has been reported. However, to our knowledge, only a single report has focused on renal mitochondrial function during sepsis. Tran et al. (50) reported that cytochrome c oxidase activity was decreased at 18 h postsepsis using an immunohistochemistry-based activity measurement method.
In the cecal ligation puncture (CLP) model of severe sepsis using the aged mouse (36), oxidant generation by the renal tubules and renal microvascular failure are early events, which lead to AKI (23, 55, 60). Our recent studies documented increased tubular mitochondrial superoxide generation as early as 4 h post-CLP (55). Oxidative stress is a hallmark of sepsis in humans and suggested to link microvascular failure and parenchymal cell injury (6, 18, 47, 53). It has also been suggested that oxidative stress might play a causal role in mitochondrial dysfunction during sepsis leading to end-organ failure (9, 13).
The goals of our study were to identify specific mitochondrial defects, which could contribute to mitochondrial superoxide generation, and to evaluate the therapeutic potential of the mitochondria-targeted antioxidant Mito-TEMPO to prevent damage using a delayed dosing paradigm. Using high-resolution respirometry in freshly isolated kidney tissue from mice made septic by CLP, we mapped the time-dependent changes in respiratory complex activities as well as MnSOD activity. Functional studies with Mito-TEMPO demonstrated that mitochondrial defects could be targeted pharmacologically to improve renal function and reduce mortality in this model of severe sepsis.
MATERIALS AND METHODS
CLP murine model of sepsis.
CLP was performed in male 40-wk-old C57/BL6 mice (Harlan, Indianapolis, IN), as described previously (54, 60). Following a midline laparotomy under isoflurane anesthesia, the cecum was ligated 1.5 cm from the tip with a 4-0 silk suture and punctured twice with a 21-gauge needle. A 1-mm column of fecal material was expressed from each puncture. The cecum was isolated but neither ligated nor punctured in control sham-operated mice (Sham). All mice received 1 ml of prewarmed saline at the end of surgery and were placed on a heating pad. Mice were given imipenem/cilastatin (14 mg/kg) at 6 h and for studies extending beyond 18 h, animals received additional doses of imipenem/cilastatin (7 mg/kg) at 12-h intervals beginning at 18 h, except during the biotelemetry studies. In those studies, mice received only the single dose at 6 h. All animals were housed and handled in accordance to the National Institutes of Health Guide for the Care and Use of Laboratory Animals with approval by the Institutional Animal Care and Use Committee at the University of Arkansas for Medical Sciences.
Administration of Mito-TEMPO.
Mito-TEMPO (Enzo Lifesciences, Farmingdale, NY) was prepared in normal saline before each experiment and kept in the dark at 4°C. For the dose-response studies, Mito-TEMPO was administered as a single dose of 3, 10, or 30 mg/kg via intraperitoneal injection at the time of CLP. In subsequent experiments, Mito-TEMPO was administered at 6 h post-CLP as indicated in the text.
High-resolution respirometry.
Mitochondrial respiratory complex activity was measured by the high-resolution respirometry (HRR) technique using Oxygraph-2K (Oroboros Instruments) as we recently described (39). Briefly, a representative section of kidney (5–8 mg wt) containing both cortex and medulla was freshly isolated from Sham or CLP mice at 6, 18, or 36 h. The renal section was minced and permeabilized using 100 μg/ml of saponin by gentle shaking at 4°C. Complex activities were measured using with the substrate-inhibitor-titration protocol (SIT) (39). Complex I substrates, malate (2 mM) and glutamate (10 mM), were added to initiate the respiration followed by addition of 2.5 mM ADP to achieve maximum active respiration. The stimulated complex I respiration was then completely inhibited by addition of 0.2 mM rotenone. Complex II/III respiration was then stimulated by addition of 10 mM succinate (complex II substrate) followed by addition of 10 μM antimycin A (complex III respiration inhibitor). Lastly, complex IV respiration was monitored by addition of 1 mM N,N,N9,N9-tetramethyl-p-phenylene-diamine (substrate for complex IV) made in 0.8 M ascorbate (pH 6.0) and respiration of complex IV was inhibited by using 800 mM sodium azide. Data analysis was performed following normalization to weight of the permeabilized kidney tissue using DATLAB 4.2 software (Oroboros).
ATP assay.
An ATP-luciferase-based bioluminescence assay kit (Sigma, St. Louis, MO) was used to measure ATP levels in the Sham and septic kidney tissue lysates. Luminescence was measured using a TD 20/20 luminometer (Turner Designs, Sunnyvale, CA).
MnSOD activity and protein expression analyses.
Enzymatic activity of MnSOD was determined in kidney tissue lysates by the cytochrome c reduction method in the presence of 1 mM KCN to inhibit Cu, Zn SOD activity, as previously described (33). MnSOD protein expression was analyzed by Western blot using a polyclonal anti-MnSOD antibody (1:1,000) and GADPH was detected using a polyclonal anti-GAPDH (1:1,000) served as a loading control, as described previously (41). Anti-MnSOD and anti-GAPDH antibodies were purchased from EMD Millipore (Billerica, MA).
Measurement of systemic mean arterial pressure and heart rate.
Biotelemetry was used to measure mean arterial pressure (MAP) and heart rate (HR) in conscious mice as described previously (22). Under isoflurane anesthesia, telemetry transmitters (Data Sciences International, St. Paul, MN) were implanted into the carotid artery of mice. After a week of recovery, mice underwent Sham or the CLP surgery and received antibiotic and fluid resuscitation at 6 h post-CLP. MAP and HR were then recorded every 5 min through 24 h.
Intravital video microscopy to measure renal capillary perfusion and mitochondrial superoxide generation.
Renal capillary perfusion and mitochondrial superoxide generation were analyzed using intravital video microscopy (IVVM) as previously described (55). At 10 min before IVVM and while under isoflurane anesthesia, mice received an injection containing FITC-dextran (500,000 Da, 2 μmol/kg; Sigma) and MitoSox (1.67 mg/kg; Life Technologies, Grand Island, NY) via the penile vein. The left kidney of the mouse was then exposed through a flank incision and positioned on a glass stage above an Axiovert 200 inverted fluorescence microscope equipped with an Axiocam HSm digitizing camera (Carl Zeis AG, Jena, Germany). FITC-dextran was used to visualize capillary flow. For each mouse, five 10-s videos were captured at ∼30 frames/s from five randomly selected fields of view. In brief, ∼150 randomly selected vessels per kidney were classified into three categories of blood perfusion: “continuous flow,” in which red blood cell movement in the vessel was not interrupted during the video; “intermittent flow,” in which red blood cell movement stopped or reversed at any time during the video; and “no flow,” in which no red blood cell movement was detected. Data were expressed as the percentage of vessels in each of the three categories. To quantitate mitochondrial superoxide levels, images of MitoSOX fluorescence were captured from the five randomly selected fields of view (×200 magnification) used to determine perfusion. ImageJ software (National Institutes of Health, Bethesda, MD) was used to quantify fluorescence intensity of each image after subtracting background fluorescence intensity. Data were expressed as arbitrary units per square micrometer (55).
Measurement of glomerular filtration rate.
Glomerular filtration rate (GFR) was measured using FITC-inulin clearance as previously described (22). A 5% FTIC-inulin solution at a dose of 3.74 μl/g was administered via the penile vein. Blood (25 μl) was collected at specific time intervals and FITC-inulin was measured via fluorometry using a standard curve. A two-phase decay nonlinear regression analysis was used to calculate inulin clearance. GFR was calculated using a two-phase nonlinear regression analysis and normalizing the values to the combined weight of both kidneys.
Nitrotyrosine staining.
Nitrotyrosine immunohistochemical analysis was performed as previously described (40). Briefly, following antigen retrieval (citrate buffer; 20 min) and protein blocking, the kidney sections were incubated with anti-nitrotyrosine antibody (Millipore; 1:6,000) and incubated overnight at 4°C. The specificity of nitrotyrosine antibody binding in the renal tissue was confirmed by blocking the antibody with 3-nitrotyrosine (10 mM; data not shown). Immunoreactivity was detected by Dako Envision+ System-HRP (Dako, Carpinteria, CA). Semiquantitative evaluation of nitrotyrosine staining was performed based on the intensity in 10–12 high-power fields (×400) from the cortex and medulla using the following scores: 0 - null/negative; 1 - weak staining; 2 - mild staining; 3 - high staining; 4 - very high staining.
Measurement of blood urea nitrogen.
Blood urea nitrogen (BUN) was measured using the Quantichrom urea assay kit (BioAssay Systems, Hayward, CA). Data were expressed as serum BUN concentration in milligrams per deciliter.
Survival study.
Core body temperature was used as an indicator of pending death (56). It was measured every 6 h using a rectal probe. Mice were considered nonsurvivors if they died or had to be euthanized when two consecutive readings of core body temperature were <28.0°C.
Statistical analysis.
Data presented as means ± SE were analyzed using Prism 6.0 (GraphPad Software, San Diego, CA). Data with three or more groups were analyzed using a one-way ANOVA followed by the Newman-Keuls or Dunnett's post hoc tests. Survival cures were analyzed using a Mantel-Cox log rank test. P values <0.05 were considered statistically significant.
RESULTS
Sepsis caused a progressive decline in both renal mitochondrial complex respiration and ATP levels.
The mouse model of sepsis used in this study mimics a number of the pathogenic features of human sepsis including the use of fluids and antibiotics to resemble the current standard of care used for septic patients (27, 46). These older mice rapidly develop hypotension, decreased renal blood flow, decreased GFR, and decreased peritubular microcirculation, hypoxia, and oxidative stress (36, 55, 60, 64). To assess the development of mitochondrial dysfunction, mitochondrial complex respiration was measured using HRR in freshly isolated kidney tissue (Fig. 1A). The earliest defect detected was in complex II/III respiration, which was significantly decreased as early as 6 h after sepsis compared with the Sham group and continued to decline significantly through 36 h. Complex I respiration was significantly decreased at 18 h and declined even further at 36 h post-CLP. In contrast, complex IV respiration was unaffected by CLP at any time point studied. Since the major function of the ETC is to generate ATP, defects in mitochondrial respiration can result in decreased ATP synthesis. At 18 h post-CLP, kidney levels of ATP were unaffected. However, at 36 h post-CLP, the time when both complex I and complex II/III activities were at their lowest, ATP levels were significantly decreased to near 50% of the levels in the sham group (Fig. 1B).
Fig. 1.

Sepsis caused a decline in renal mitochondrial complex respiration and ATP production. A: high-resolution respirometry (HRR) was used to assess the status of mitochondrial complexes [I, II/III, and IV of the electron transport chain (ETC)] of fresh renal biopsies harvested 6–36 h post-cecal ligation and puncture (CLP). Values are oxygen flux expressed as a percentage of sham levels (means ± SE, n = 4–8 mice/group). B: ATP levels in the kidney at 18 and 36 h are presented as percentage of sham levels (means ± SE, n = 7–8 mice/group). *P < 0.05 compared with Sham and #P < 0.05 compared with CLP 18 h.
Sepsis caused renal MnSOD inactivation and increased mitochondrial superoxide generation.
Inhibition of complex I and III is known to generate increased superoxide and downstream oxidants (25, 26, 37). Therefore, mitochondrial superoxide was measured using MitoSOX Red and the IVVM technique (55). As shown in Fig. 2A, superoxide levels were significantly increased at both 6 and 18 h post-CLP. Normally, mitochondrial superoxide is scavenged by mitochondrial MnSOD (34), a homotetrameric protein (96 kDa) located in the mitochondrial matrix, which scavenges the superoxide radical (5, 43, 58). MnSOD activity was significantly reduced by ∼50% relative to the activity in the Sham group at 4 h post-CLP (Fig. 2B) and remained decreased through 36 h. Since decreased MnSOD activity could be due to reduced expression, MnSOD protein was measured using Western blot analysis and did not change following CLP (Fig. 2C).
Fig. 2.

Sepsis increased renal mitochondrial superoxide levels and reduced manganese superoxide dismutase (MnSOD) activity. A: renal mitochondrial superoxide levels were measured by MitoSox Red fluorescence using the intravital video microscopy (IVVM) technique (see materials and methods). Values are presented means ± SE, n = 5–9 mice/group. B: renal MnSOD activity was measured at 4, 6, and 18 h post-CLP. Values are expressed as percentage of sham activity (means ± SE, n = 4–8 mice/group). C: representative Western blot showing no change in the expression of MnSOD protein during sepsis. GAPDH was used as a loading control. Image is representative of 3 separate experiments. D: quantification band intensity from C by densitometry. *P < 0.05 compared with Sham.
Dose-response effects of Mito-TEMPO.
Given the evidence for mitochondrial oxidative stress shown in Fig. 2, we examined whether the mitochondria-targeted antioxidant Mito-TEMPO could ameliorate mitochondrial dysfunction during sepsis. We showed previously that tubular superoxide generation is associated with renal microcirculatory failure (23, 55). Initial experiments were carried out to determine the minimally effective dose of Mito-TEMPO needed to reduce superoxide generation during sepsis. Animals were treated with Mito-TEMPO at doses of 3, 10, or 30 mg/kg ip at the time of CLP. Cortical tubular MitoSOX fluorescence (measure of superoxide) and peritubular capillary perfusion were measured using IVVM 6 h later. The 6-h time point was selected since our prior studies showed significant superoxide generation as well as impaired capillary perfusion at this time point (55). Only the 10-mg/kg dose of Mito-TEMPO reduced mitochondrial superoxide generation compared with the CLP group (Fig. 3A). Also, only the 10-mg/kg dose of Mito-TEMPO preserved peritubular capillary perfusion (Fig. 3B) supporting a link between tubular mitochondrial superoxide generation and microcirculatory failure. The lack of protection observed with the higher (30 mg/kg) dose of Mito-TEMPO may be related to accumulation of this cationic agent within the mitochondria, which could depolarize mitochondria. Based on these dose-finding results, Mito-TEMPO was used at 10 mg/kg ip for all subsequent experiments.
Fig. 3.

Dose-response effects of Mito-TEMPO (MT). Renal capillary perfusion (A) and MitoSox Red fluorescence (superoxide; B) were measured by IVVM at 6 h post-CLP. Three doses of Mito-TEMPO were assessed when administered (ip) at the time of CLP (0 h). The 10-mg/kg dose prevented the increase in superoxide generation and prevented the decline in peritubular capillary perfusion. Values are expressed as means ± SE, n = 4–6 mice/group. *P < 0.05 compared with Sham.
Delayed dosing with Mito-TEMPO protected renal mitochondrial complex respiration and MnSOD activity.
To evaluate Mito-TEMPO using a more clinically relevant dosing paradigm, animals were treated with Mito-TEMPO (10 mg/kg) at 6 h post-CLP, a time when MnSOD activity is inhibited (Fig. 2B), mitochondrial superoxide is at the highest, peritubular capillary perfusion is the lowest, and GFR is reduced by 60% (23, 55). As shown in Fig. 4A, delayed dosing with Mito-TEMPO prevented the inhibition of complex I respiration at 18 h and significantly improved complex I respiration at 36 h post-CLP. Mito-TEMPO also improved complex II/III respiration at 18 h but not at 36 h. Although CLP did not affect complex IV respiration, Mito-TEMPO transiently increased complex IV respiration in CLP mice at 18 h. Mito-TEMPO treatment had no effect on any of the respiratory complex activities in Sham mice (data not shown). Despite some improvements in complex I respiration, delayed dosing with Mito-TEMPO did not increase ATP levels (Fig. 4B). Importantly, delayed dosing with Mito-TEMPO actually reversed the inactivation of MnSOD (Fig. 4C).
Fig. 4.

Delayed treatment with Mito-TEMPO blunted sepsis-induced decreases in renal mitochondrial complex respiration and restored MnSOD activity. Mito-TEMPO was administered (10 mg/kg ip) at 6 h post-CLP. At 18 and 36 h post-CLP, the activities of mitochondrial complex I, II/III, and IV (A), ATP levels in the kidney (B), and MnSOD activity (C) were measured. Values are expressed as percentage of sham levels (means ± SE, n = 4–8 mice/group). Values for the Sham and CLP groups are the same as presented in Figs. 1 and 2. *P < 0.05 compared with Sham and #P < 0.05 compared with CLP 18 h.
Delayed dosing with Mito-TEMPO restored renal capillary perfusion and improved GFR.
At 6 h (Fig. 3A) and 18 h (Fig. 5A) post-CLP renal peritubular capillary perfusion is significantly reduced. Mito-TEMPO (10 mg/kg) given at 6 h post-CLP restored the renal microcirculation by 18 h. CLP also causes a dramatic fall in GFR (23, 55). Delayed treatment with Mito-TEMPO significantly improved GFR (Fig. 5B). Since our CLP model is one of severe sepsis associated with hypotension (23, 55), agents that increase MAP could improve the renal microcirculation and GRF without acting directly on the kidney. To help address this, the effects of Mito-TEMPO on MAP were evaluated using biotelemetry. Mito-TEMPO (10 mg/kg) produced no effects on systemic MAP (Fig. 5C).
Fig. 5.

Effects of delayed treatment with Mito-TEMPO on renal capillary perfusion, glomerular filtration rate (GFR), and mean arterial pressure (MAP). Mito-TEMPO was administered (10 mg/kg ip) at 6 h post-CLP. A: Mito-TEMPO completely restored renal capillary perfusion. Values are expressed as means ± SE, n = 4–5 mice/group. B: Mito-TEMPO significantly improved GFR as measured by inulin clearance (see materials and methods). Values are expressed as means ± SE, n = 4 mice/group. C: Mito-TEMPO did not alter MAP. Values are expressed as means ± SE, n = 5–8 mice/group. *P < 0.05 compared with Sham and #P < 0.05 compared with CLP 18 h.
Mito-TEMPO reduced renal oxidative stress.
Superoxide and nitric oxide react to form the RNS peroxynitrite, which increases in renal tubules rapidly following induction of sepsis (23, 55, 60–62). Since Mito-TEMPO reduced mitochondrial superoxide generation, nitrotyrosine was measured using immunohistochemistry as an indicator of oxidative stress (Fig. 6, A and B). Relatively low levels of nitrotyrosine staining were observed in the kidneys of these aged mice. At 18 h post-CLP, the levels of nitrotyrosine staining were significantly higher in both the renal cortex and medulla at 18 h post-CLP compared with the basal levels seen in the sham-treated group. Mito-TEMPO significantly decreased the levels of nitrotyrosine in the CLP mice back to levels observed in the Sham group. Therefore, Mito-TEMPO significantly decreases the level of oxidative stress in the kidney during sepsis even when administered at a delayed time of 6 h post-CLP.
Fig. 6.

Delayed treatment with Mito-TEMPO decreased sepsis-induced oxidative stress in the kidney. A: oxidative stress was assessed by immunohistochemistry for protein-nitrotyrosine adducts (brown staining). Representative micrographs (×400 original magnification) showing cortex, medulla, and papillary regions are shown for the Sham, CLP at 18 h, and CLP + Mito-TEMPO (MT) at 18 h groups treated. Mito-TEMPO (10 mg/kg ip) was administered at 6 h post-CLP. B: semiquantitative evaluation of nitrotyrosine staining was performed as described in materials and methods. Values are as means ± SE, n = 5 mice/group. *P < 0.05 compared with Sham.
Delayed dosing with Mito-TEMPO improved core body temperature, serum BUN, and survival rate of septic mice.
CLP caused a significant decrease in core body temperature and increase in serum BUN (Fig. 7, A and B). Delayed therapy with Mito-TEMPO (10 mg/kg) significantly improved core body temperature and reduced serum BUN. Importantly, delayed therapy with Mito-TEMPO (10 mg/kg) significantly improved survival rate from 40% in septic control mice to ∼83% at 96 h post-CLP (Fig. 7).
Fig. 7.
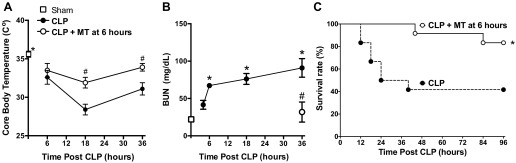
Effects of delayed treatment with Mito-TEMPO on body temperature, serum blood urea nitrogen (BUN), and survival. Mito-TEMPO was administered (10 mg/kg ip) at 6 h post-CLP. Mito-TEMPO improved core body temperature (A) and serum BUN levels (B). Values are expressed as means ± SE, n = 12–18 mice/group in A and n = 3–5 mice/group in B. *P < 0.05 compared with Sham and #P < 0.05 compared with CLP. C: Mito-TEMPO increased survival of septic mice, n = 12 mice/group. *P < 0.05 compared with CLP; Mantel-Cox log-rank test, n = 12 mice/group.
DISCUSSION
The objectives of this study were to determine the effects sepsis has on renal mitochondrial function and then to evaluate the mitochondria as a potential therapeutic target. The key findings were that sepsis leads to a decrease in renal complex I and II/III respiration, MnSOD activity, and ATP levels. This was associated with increased mitochondrial superoxide levels, impaired renal microcirculation, and impaired renal function. Strikingly, a single dose of the mitochondria-targeted antioxidant Mito-TEMPO protected improved renal mitochondrial function, renal microcirculation, GFR, and increased survival of septic mice even with delayed administration.
Mitochondria generate ATP through sequential electron transfer through the ETC (complexes I–IV). The status of renal mitochondrial ETC function has not been well-studied during sepsis. In this study, we employed the SIT protocol using the HRR to measure the activities of complexes I, II/III, and IV as described in materials and methods. HRR is considered superior to other methods, which require isolation of mitochondria from injured tissues and can yield inconsistent populations of mitochondria, while HRR uses fresh tissue. Others have showed impaired mitochondrial respiration during sepsis in organs including the heart, liver, and skeletal muscle tissues (8, 19, 42, 57, 59). Tran et al. (50) showed a decline in renal complex IV activity using snap-frozen tissue and an immunohistochemistry method. We observed no defect in complex IV, perhaps due to the different methods employed, as we used freshly isolated renal biopsies and HRR. Our time course studies revealed a very early decline in complex II/III activity followed by a decline in complex I activity. Interestingly, the lack of effect on complex IV at any time point studied suggests unique susceptibilities of the individual respiratory complexes. Moreover, the time course studies suggest there is a critical threshold of inhibition of renal mitochondrial respiration before ATP levels are reduced.
To our knowledge, this is the first report to show inactivation of renal MnSOD during sepsis. Moreover, the data suggest inactivation is through a posttranslational modification of MnSOD. One such modification could be tyrosine nitration, which our laboratory has shown leads to MnSOD inactivation (29, 30). However, we were not able to show tyrosine nitration of MnSOD during sepsis (data not shown). In more recent studies, we showed that a thiol modulator, S-nitrosoglutathione, leads to MnSOD inactivation in proximal tubule cells (41). Thus, it is possible that a similar mechanism of thiol modification may be responsible for sepsis-mediated MnSOD inactivation and studies are underway to specifically address the mechanistic basis of MnSOD inactivation in the kidney during sepsis.
In contrast to our current findings showing no change in MnSOD protein expression during CLP, two studies focusing on the brain (12) and liver (4) reported increased MnSOD protein. Numerous studies employing different models of sepsis (including CLP, lipopolysaccharide, Escherichia coli, and S. aureus) have reported induction of MnSOD mRNA in the liver (1, 11, 14, 20, 28, 48, 49), but we are unaware of any studies showing MnSOD alterations in the kidney after sepsis. Consistent with our findings, a study by Yang et al. (63) found that MnSOD protein levels remain unchanged in the kidney after LPS treatment. Additional studies are needed to establish whether renal MnSOD is regulated differently than it is in other organs or whether the differences noted are model-specific. Nonetheless, this is the first report showing that MnSOD activity is reduced in the kidney during sepsis.
We established previously that sepsis causes a significant increase in mitochondrial superoxide and oxidative stress in the kidney (23, 55, 60). The current findings suggest a possible mechanism for superoxide generation and its relationship to renal microvascular failure and the development of AKI. Previous studies by our laboratory did show that the generalized antioxidants resveratrol (23) and MnTmPyP (55) could protect the kidney in this model. However, antioxidant therapies have not been highly successful in critically ill patients (35, 45) perhaps because general antioxidants are not specifically targeted to the mitochondria (17). To help explore this, we evaluated Mito-TEMPO using a delayed dosing paradigm to more closely mimic the clinical setting wherein therapy in the septic patient usually begins only after the onset of symptoms (46). The precise mechanism of the antioxidant activity of Mito-TEMPO remains unclear but is likely related to mitochondria-targeted superoxide scavenging (15). Mito-TEMPO is a nitroxide linked to the positively charged triphenyl phosphonium cation, which targets Mito-TEMPO specifically into the mitochondria (51). In an LPS-induced sepsis model, Mito-TEMPO was shown to protect against mitochondrial dysfunction and oxidative stress in the liver; however, this study delivered Mito-TEMPO 30 min before LPS treatment (11). Using a more clinically relevant delayed dosing paradigm, we found that delayed therapy with Mito-TEMPO not only partially restored complex II/III activity but prevented the inhibition of complex I activity at 18 h. The reason for the significant increase in complex IV activity at 18 h post-CLP with Mito-TEMPO treatment remains unknown, but may be related to an attempt to repair mitochondria via mitochondrial biogenesis. At 36 h post-CLP, Mito-TEMPO significantly improved complex I respiration, but not complex II/II respiration, which may help explain why Mito-TEMPO did not improve renal ATP levels at 36 h post-CLP. The biological half-life of Mito-TEMPO is unknown so it is possible that the single dose used in these studies was unable afford protection through 36 h because of its elimination. Additional studies are needed to address this possibility.
Interestingly, delayed therapy with Mito-TEMPO completely reversed MnSOD inactivation and significantly reduced renal oxidative stress. We do not know why Mito-TEMPO afforded complete protection against MnSOD inactivation, while only partially restored complex I activity, but this could be related to its site of action. MnSOD resides in the mitochondrial matrix, while the complexes are embedded in mitochondrial membranes. Thus, it is possible that a more lipid-soluble mitochondrial antioxidant may provide more protection against complex inactivation. Alternatively, the mechanism of inactivation may be different.
Regardless of the exact mechanism(s) of action, Mito-TEMPO produced profound protective effects on the renal microcirculation and renal function. Mito-TEMPO protected the renal microcirculation and, with delayed therapy, even reversed renal microcirculatory failure without affecting systemic blood pressure. In this model, GFR falls more than 50% by 6 h (55) so it is remarkable that delayed treatment with Mito-TEMPO also significantly improved GFR. These findings support the growing realization of the importance of the peritubular microenvironment to overall renal function (32). Given the complex and rapid development of sepsis-induced AKI in this model (55), it is not surprising that delayed therapy with Mito-TEMPO did not completely restore GFR. Targeting renal vascular resistance (22) or oxidative stress (23, 55) with delayed therapy also only partially restored GFR. These findings support the view that combination therapy will likely be the most efficacious in the septic patient.
Most importantly, a single delayed dose of Mito-TEMPO improved morbidity and significantly increased the survival septic mice from 40 to 83% through 96 h post-CLP. Together, these data show that Mito-TEMPO offers overall systemic protection during the course of this severe sepsis model and supports the notion that mitochondrial dysfunction is a generalized consequence of sepsis (7, 17, 18).
In conclusion, this study has identified three mitochondrial targets (complex I and II/III, and MnSOD) that are damaged during sepsis, and we showed that Mito-TEMPO was able to attenuate sepsis-induced mitochondrial dysfunction and oxidative stress helping to improve renal function and survival. These data show that mitochondrial dysfunction, a previously unrecognized mediator of septic AKI, can be targeted using a mitochondrial antioxidant to attenuate septic AKI, even with delayed treatment.
GRANTS
This work was supported by National Institutes of Health (NIH) Grants R01DK075991 and RO1GM106419 and N. K. Patil was supported by a predoctoral fellowship from the American Heart Association (12PRE12040174). Additional support was provided by the UAMS Translational Research Institute supported by NIH Grant UL1TR000039.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: N.K.P., L.A.M.-C., and P.R.M. conception and design of research; N.K.P. and N.P. performed experiments; N.K.P., N.P., L.A.M.-C., and P.R.M. analyzed data; N.K.P., L.A.M.-C., and P.R.M. interpreted results of experiments; N.K.P., N.P., and P.R.M. prepared figures; N.K.P., L.A.M.-C., and P.R.M. drafted manuscript; N.K.P., L.A.M.-C., and P.R.M. edited and revised manuscript; L.A.M.-C. and P.R.M. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank the University of Arkansas for Medical Sciences (UAMS) Experimental Pathology Core and the Biotelemetry and Ultrasound Core for their services. A portion of these studies was presented in abstract form during the American Society of Nephrology Kidney Week 2013 Annual Meeting.
REFERENCES
- 1.Alexander HR, Sheppard BC, Jensen JC, Langstein HN, Buresh CM, Venzon D, Walker EC, Fraker DL, Stovroff MC, Norton JA. Treatment with recombinant human tumor necrosis factor-alpha protects rats against the lethality, hypotension, and hypothermia of gram-negative sepsis. J Clin Invest 88: 34–39, 1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med 29: 1303–1310, 2001 [DOI] [PubMed] [Google Scholar]
- 3.Aslami H, Pulskens WP, Kuipers MT, Bos AP, van Kuilenburg AB, Wanders RJ, Roelofsen J, Roelofs JJ, Kerindongo RP, Beurskens CJ, Schultz MJ, Kulik W, Weber NC, Juffermans NP. Hydrogen sulfide donor NaHS reduces organ injury in a rat model of pneumococcal pneumosepsis, associated with improved bio-energetic status. PLos One 8: e63497, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bartz RR, Suliman HB, Fu P, Welty-Wolf K, Carraway MS, MacGarvey NC, Withers CM, Sweeney TE, Piantadosi CA. Staphylococcus aureus sepsis and mitochondrial accrual of the 8-oxoguanine DNA glycosylase DNA repair enzyme in mice. Am J Respir Crit Care Med 183: 226–233, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Borgstahl GE, Parge HE, Hickey MJ, Beyer WF, Jr, Hallewell RA, Tainer JA. The structure of human mitochondrial manganese superoxide dismutase reveals a novel tetrameric interface of two 4-helix bundles. Cell 71: 107–118, 1992 [DOI] [PubMed] [Google Scholar]
- 6.Boueiz A, Hassoun PM. Regulation of endothelial barrier function by reactive oxygen and nitrogen species. Microvasc Res 77: 26–34, 2009 [DOI] [PubMed] [Google Scholar]
- 7.Brealey D, Brand M, Hargreaves I, Heales S, Land J, Smolenski R, Davies NA, Cooper CE, Singer M. Association between mitochondrial dysfunction and severity and outcome of septic shock. Lancet 360: 219–223, 2002 [DOI] [PubMed] [Google Scholar]
- 8.Brealey D, Karyampudi S, Jacques TS, Novelli M, Stidwill R, Taylor V, Smolenski RT, Singer M. Mitochondrial dysfunction in a long-term rodent model of sepsis and organ failure. Am J Physiol Regul Integr Comp Physiol 286: R491–R497, 2004 [DOI] [PubMed] [Google Scholar]
- 9.Brealey D, Singer M. Mitochondrial dysfunction in sepsis. Curr Infect Dis Rep 5: 365–371, 2003 [DOI] [PubMed] [Google Scholar]
- 10.Carre JE, Orban JC, Re L, Felsmann K, Iffert W, Bauer M, Suliman HB, Piantadosi CA, Mayhew TM, Breen P, Stotz M, Singer M. Survival in critical illness is associated with early activation of mitochondrial biogenesis. Am J Respir Crit Care Med 182: 745–751, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Choumar A, Tarhuni A, Letteron P, Reyl-Desmars F, Dauhoo N, Damasse J, Vadrot N, Nahon P, Moreau R, Pessayre D, Mansouri A. Lipopolysaccharide-induced mitochondrial DNA depletion. Antioxid Redox Signal 15: 2837–2854, 2011 [DOI] [PubMed] [Google Scholar]
- 12.Correa F, Ljunggren E, Patil J, Wang X, Hagberg H, Mallard C, Sandberg M. Time-dependent effects of systemic lipopolysaccharide injection on regulators of antioxidant defence Nrf2 and PGC-1alpha in the neonatal rat brain. Neuroimmunomodulation 20: 185–193, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Crouser ED. Mitochondrial dysfunction in septic shock and multiple organ dysfunction syndrome. Mitochondrion 4: 729–741, 2004 [DOI] [PubMed] [Google Scholar]
- 14.De Leo ME, Landriscina M, Palazzotti B, Borrello S, Galeotti T. Iron modulation of LPS-induced manganese superoxide dismutase gene expression in rat tissues. FEBS Lett 403: 131–135, 1997 [DOI] [PubMed] [Google Scholar]
- 15.Dikalova AE, Bikineyeva AT, Budzyn K, Nazarewicz RR, McCann L, Lewis W, Harrison DG, Dikalov SI. Therapeutic targeting of mitochondrial superoxide in hypertension. Circ Res 107: 106–116, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fridovich I. Superoxide radical and superoxide dismutases. Annu Rev Biochem 64: 97–112, 1995 [DOI] [PubMed] [Google Scholar]
- 17.Galley HF. Bench-to-bedside review: targeting antioxidants to mitochondria in sepsis. Crit Care 14: 230, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Galley HF. Oxidative stress and mitochondrial dysfunction in sepsis. Br J Anaesth 107: 57–64, 2011 [DOI] [PubMed] [Google Scholar]
- 19.Gellerich FN, Trumbeckaite S, Hertel K, Zierz S, Muller-Werdan U, Werdan K, Redl H, Schlag G. Impaired energy metabolism in hearts of septic baboons: diminished activities of Complex I and Complex II of the mitochondrial respiratory chain. Shock 11: 336–341, 1999 [PubMed] [Google Scholar]
- 20.Haden DW, Suliman HB, Carraway MS, Welty-Wolf KE, Ali AS, Shitara H, Yonekawa H, Piantadosi CA. Mitochondrial biogenesis restores oxidative metabolism during Staphylococcus aureus sepsis. Am J Respir Crit Care Med 176: 768–777, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heemskerk S, Masereeuw R, Russel FG, Pickkers P. Selective iNOS inhibition for the treatment of sepsis-induced acute kidney injury. Nat Rev Nephrol 5: 629–640, 2009 [DOI] [PubMed] [Google Scholar]
- 22.Holthoff JH, Wang Z, Patil NK, Gokden N, Mayeux PR. Rolipram improves renal perfusion and function during sepsis in the mouse. J Pharmacol Exp Ther 347: 357–364, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Holthoff JH, Wang Z, Seely KA, Gokden N, Mayeux PR. Resveratrol improves renal microcirculation, protects the tubular epithelium, and prolongs survival in a mouse model of sepsis-induced acute kidney injury. Kidney Int 81: 370–378, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis. N Engl J Med 348: 138–150, 2003 [DOI] [PubMed] [Google Scholar]
- 25.Ksenzenko M, Konstantinov AA, Khomutov GB, Tikhonov AN, Ruuge EK. Effect of electron transfer inhibitors on superoxide generation in the cytochrome bc1 site of the mitochondrial respiratory chain. FEBS Lett 155: 19–24, 1983 [DOI] [PubMed] [Google Scholar]
- 26.Kussmaul L, Hirst J. The mechanism of superoxide production by NADH:ubiquinone oxidoreductase (complex I) from bovine heart mitochondria. Proc Natl Acad Sci USA 103: 7607–7612, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Levy MM, Dellinger RP, Townsend SR, Linde-Zwirble WT, Marshall JC, Bion J, Schorr C, Artigas A, Ramsay G, Beale R, Parker MM, Gerlach H, Reinhart K, Silva E, Harvey M, Regan S, Angus DC. The Surviving Sepsis Campaign: results of an international guideline-based performance improvement program targeting severe sepsis. Crit Care Med 38: 367–374, 2010 [DOI] [PubMed] [Google Scholar]
- 28.Macgarvey NC, Suliman HB, Bartz RR, Fu P, Withers CM, Welty-Wolf KE, Piantadosi CA. Activation of mitochondrial biogenesis by heme oxygenase-1-mediated NF-E2-related factor-2 induction rescues mice from lethal Staphylococcus aureus sepsis. Am J Respir Crit Care Med 185: 851–861, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.MacMillan-Crow LA, Crow JP, Kerby JK, Beckman JS. Nitration and inactivation of manganese superoxide dismutase in chronic rejection of human renal allografts. Proc Natl Acad Sci USA 93: 11853–11858, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.MacMillan-Crow LA, Crow JP, Thompson JA. Peroxynitrite-mediated inactivation of manganese superoxide dismutase involves nitration and oxidation of critical tyrosine residues. Biochemistry 37: 1613–1622, 1998 [DOI] [PubMed] [Google Scholar]
- 31.Martin GS. Sepsis, severe sepsis and septic shock: changes in incidence, pathogens and outcomes. Expert Rev Mol Med 10: 701–706, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mayeux PR, Macmillan-Crow LA. Pharmacological targets in the renal peritubular microenvironment: implications for therapy for sepsis-induced acute kidney injury. Pharmacol Ther 134: 139–155, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McCord JM, Fridovich I. Superoxide dismutase: an enzymic function for erythrocuprein (hemocuprein). J Biol Chem 244: 6049–6055, 1969 [PubMed] [Google Scholar]
- 34.McCord JM, Fridovich I. The utility of superoxide dismutase in studying free radical reactions. I. Radicals generated by the interaction of sulfite, dimethyl sulfoxide, and oxygen. J Biol Chem 244: 6056–6063, 1969 [PubMed] [Google Scholar]
- 35.Mishra V. Oxidative stress and role of antioxidant supplementation in critical illness. Clin Lab 53: 199–209, 2007 [PubMed] [Google Scholar]
- 36.Miyaji T, Hu X, Yuen PS, Muramatsu Y, Iyer S, Hewitt SM, Star RA. Ethyl pyruvate decreases sepsis-induced acute renal failure and multiple organ damage in aged mice. Kidney Int 64: 1620–1631, 2003 [DOI] [PubMed] [Google Scholar]
- 37.Muller FL, Liu Y, Van Remmen H. Complex III releases superoxide to both sides of the inner mitochondrial membrane. J Biol Chem 279: 49064–49073, 2004 [DOI] [PubMed] [Google Scholar]
- 38.Murugan R, Kellum JA. Acute kidney injury: what's the prognosis? Nat Rev Nephrol 7: 209–217, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Parajuli N, Campbell LH, Marine A, Brockbank KG, Macmillan-Crow LA. MitoQ blunts mitochondrial and renal damage during cold preservation of porcine kidneys. PLos One 7: e48590, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Parajuli N, Marine A, Simmons S, Saba H, Mitchell T, Shimizu T, Shirasawa T, Macmillan-Crow LA. Generation and characterization of a novel kidney-specific manganese superoxide dismutase knockout mouse. Free Radic Biol Med 51: 406–416, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Patil NK, Saba H, Macmillan-Crow LA. Effect of S-nitrosoglutathione on renal mitochondrial function: a new mechanism for reversible regulation of manganese superoxide dismutase activity? Free Radic Biol Med 56: 54–63, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Peruchi BB, Petronilho F, Rojas HA, Constantino L, Mina F, Vuolo F, Cardoso MR, Goncalves CL, Rezin GT, Streck EL, Dal-Pizzol F. Skeletal muscle electron transport chain dysfunction after sepsis in rats. J Surg Res 167: e333–e338, 2011 [DOI] [PubMed] [Google Scholar]
- 43.Ravindranath SD, Fridovich I. Isolation and characterization of a manganese-containing superoxide dismutase from yeast. J Biol Chem 250: 6107–6112, 1975 [PubMed] [Google Scholar]
- 44.Ricci Z, Polito A, Ronco C. The implications and management of septic acute kidney injury. Nat Rev Nephrol 7: 218–225, 2011 [DOI] [PubMed] [Google Scholar]
- 45.Rinaldi S, Landucci F, De Gaudio AR. Antioxidant therapy in critically septic patients. Curr Drug Targets 10: 872–880, 2009 [DOI] [PubMed] [Google Scholar]
- 46.Russell JA. Management of sepsis. N Engl J Med 355: 1699–1713, 2006 [DOI] [PubMed] [Google Scholar]
- 47.Spanos A, Jhanji S, Vivian-Smith A, Harris T, Pearse RM. Early microvascular changes in sepsis and severe sepsis. Shock 33: 387–391, 2010 [DOI] [PubMed] [Google Scholar]
- 48.Suliman HB, Welty-Wolf KE, Carraway MS, Schwartz DA, Hollingsworth JW, Piantadosi CA. Toll-like receptor 4 mediates mitochondrial DNA damage and biogenic responses after heat-inactivated E. coli. FASEB J 19: 1531–1533, 2005 [DOI] [PubMed] [Google Scholar]
- 49.Sweeney TE, Suliman HB, Hollingsworth JW, Piantadosi CA. Differential regulation of the PGC family of genes in a mouse model of Staphylococcus aureus sepsis. PLos One 5: e11606, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tran M, Tam D, Bardia A, Bhasin M, Rowe GC, Kher A, Zsengeller ZK, Akhavan-Sharif MR, Khankin EV, Saintgeniez M, David S, Burstein D, Karumanchi SA, Stillman IE, Arany Z, Parikh SM. PGC-1alpha promotes recovery after acute kidney injury during systemic inflammation in mice. J Clin Invest 121: 4003–4014, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Trnka J, Blaikie FH, Smith RA, Murphy MP. A mitochondria-targeted nitroxide is reduced to its hydroxylamine by ubiquinol in mitochondria. Free Radic Biol Med 44: 1406–1419, 2008 [DOI] [PubMed] [Google Scholar]
- 52.Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol 552: 335–344, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tyml K. Critical role for oxidative stress, platelets, and coagulation in capillary blood flow impairment in sepsis. Microcirculation 18: 152–162, 2011 [DOI] [PubMed] [Google Scholar]
- 54.Wang Z, Herzog C, Kaushal GP, Gokden N, Mayeux PR. Actinonin, a meprin A inhibitor, protects the renal microcirculation during sepsis. Shock 35: 141–147, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang Z, Holthoff JH, Seely KA, Pathak E, Spencer HJ, Gokden N, Mayeux PR. Development of oxidative stress in the peritubular capillary microenvironment mediates sepsis-induced renal microcirculatory failure and acute kidney injury. Am J Pathol 180: 505–516, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Warn PA, Brampton MW, Sharp A, Morrissey G, Steel N, Denning DW, Priest T. Infrared body temperature measurement of mice as an early predictor of death in experimental fungal infections. Lab Anim 37: 126–131, 2003 [DOI] [PubMed] [Google Scholar]
- 57.Watts BA, 3rd, George T, Sherwood ER, Good DW. A two-hit mechanism for sepsis-induced impairment of renal tubule function. Am J Physiol Renal Physiol 304: F863–F874, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Weisiger RA, Fridovich I. Mitochondrial superoxide simutase. Site of synthesis and intramitochondrial localization. J Biol Chem 248: 4793–4796, 1973 [PubMed] [Google Scholar]
- 59.Welty-Wolf KE, Simonson SG, Huang YC, Fracica PJ, Patterson JW, Piantadosi CA. Ultrastructural changes in skeletal muscle mitochondria in gram-negative sepsis. Shock 5: 378–384, 1996 [DOI] [PubMed] [Google Scholar]
- 60.Wu L, Gokden N, Mayeux PR. Evidence for the role of reactive nitrogen species in polymicrobial sepsis-induced renal peritubular capillary dysfunction and tubular injury. J Am Soc Nephrol 18: 1807–1815, 2007 [DOI] [PubMed] [Google Scholar]
- 61.Wu L, Mayeux PR. Effects of the inducible nitric oxide synthase inhibitor l-N6-(1-iminoethyl)-lysine on microcirculation and reactive nitrogen species generation in the kidney following lipopolysaccharide administration in mice. J Pharmacol Exp Ther 320: 1061–1067, 2007 [DOI] [PubMed] [Google Scholar]
- 62.Wu L, Tiwari MM, Messer KJ, Holthoff JH, Gokden N, Brock RW, Mayeux PR. Peritubular capillary dysfunction and renal tubular epithelial cell stress following lipopolysaccharide administration in mice. Am J Physiol Renal Physiol 292: F261–F268, 2007 [DOI] [PubMed] [Google Scholar]
- 63.Yang CC, Ma MC, Chien CT, Wu MS, Sun WK, Chen CF. Hypoxic preconditioning attenuates lipopolysaccharide-induced oxidative stress in rat kidneys. J Physiol 582: 407–419, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yasuda H, Yuen PS, Hu X, Zhou H, Star RA. Simvastatin improves sepsis-induced mortality and acute kidney injury via renal vascular effects. Kidney Int 69: 1535–1542, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]