

Abstract
Insulin from islet β-cells maintains glucose homeostasis by stimulating peripheral tissues to remove glucose from circulation. Persistent elevation of insulin demand increases β-cell number through self-replication or differentiation (neogenesis) as part of a compensatory response. However, it is not well understood how a persistent increase in insulin demand is detected. We have previously demonstrated that a persistent increase in insulin demand by overnutrition induces compensatory β-cell differentiation in zebrafish. Here, we use a series of pharmacological and genetic analyses to show that prolonged stimulation of existing β-cells is necessary and sufficient for this compensatory response. In the absence of feeding, tonic, but not intermittent, pharmacological activation of β-cell secretion was sufficient to induce β-cell differentiation. Conversely, drugs that block β-cell secretion, including an ATP-sensitive potassium (KATP) channel agonist and an L-type Ca2+ channel blocker, suppressed overnutrition-induced β-cell differentiation. Genetic experiments specifically targeting β-cells confirm existing β-cells as the overnutrition sensor. First, inducible expression of a constitutively active KATP channel in β-cells suppressed the overnutrition effect. Second, inducible expression of a dominant-negative KATP mutant induced β-cell differentiation independent of nutrients. Third, sensitizing β-cell metabolism by transgenic expression of a hyperactive glucokinase potentiated differentiation. Finally, ablation of the existing β-cells abolished the differentiation response. Taken together, these data establish that overnutrition induces β-cell differentiation in larval zebrafish through prolonged activation of β-cells. These findings demonstrate an essential role for existing β-cells in sensing overnutrition and compensating for their own insufficiency by recruiting additional β-cells.
Keywords: diabetes, nutrient sensing, zebrafish
diabetes results from impaired blood glucose regulation due to insufficient insulin secretion. Over the past three decades, the prevalence of diabetes mellitus has more than doubled globally and is predicted to climb in the next two decades (7). The trend is primarily a result of increased type 2 diabetes associated with chronic overnutrition and its resulting obesity. Overnutrition and obesity lead to insulin resistance through multiple mechanisms, resulting in increased insulin demand (48). Normally, the increased insulin demand can be compensated through increasing β-cell number and/or β-cell secretory activity. Because of these compensatory changes, most obese individuals do not develop diabetes; however, defects in compensation likely contribute to diabetes susceptibility and/or progression.
Compensatory increases in β-cell mass in response to increased insulin demand, for example, due to insulin resistance, have been documented in humans and rodents. In humans, evidence from postmortem analyses of β-cell mass has shown a positive correlation between β-cell mass and body mass index in nondiabetic patients. Furthermore, nondiabetic obese individuals have significantly more β-cells than diabetic obese individuals, suggesting a correlation between a failure of compensatory increase in β-cell number and development of diabetes (6, 15, 45). In rodents, both genetic and diet-induced insulin resistance increase β-cell mass (18, 49). β-Cell mass is markedly increased in genetically obese mice (both ob/ob and db/db) and rats (Zucker fa/fa) than control littermates (3, 14, 43). Similarly, mice with genetically induced insulin resistance also have a significant growth in β-cells (11, 41). Together these results suggest that compensatory increases in β-cell mass can be critical to maintain glucose homeostasis.
Compensatory β-cell genesis may arise from self-replication of existing β-cells, differentiation from stem/precursor cells, and/or transdifferentiation from other cell types (5). Although replication of existing β-cells is a major contributor to compensatory expansion, at least in rodents (4, 52), differentiation may also be an important source, especially during overnutrition (5, 22, 43). Where β-cell replication occurs, it is commonly thought that β-cells sense higher concentrations of glucose, insulin, GLP-1, or free fatty acids in the blood and reenter the cell cycle cell autonomously (1, 9). However, noncell autonomous mechanisms are also involved in inducing compensatory β-cell replication. For example, β-cell replication induced by insulin resistance resulting from either high-fat diet, genetic obesity, or liver-specific ablation of insulin receptor involves circulating factors (10, 12) and requires activation of Erk1/2 in liver and an intact vagal nerve (19, 31). Recently, a liver-derived factor, β-trophin, has been identified as a β-cell mitogen induced by insulin resistance (55).
Regardless of the mechanism or mechanisms by which β-cell mass increases, much less is known about how a persistently increased demand for insulin is sensed in the organism to trigger the compensatory β-cell differentiation. This is partly because of the lack of appropriate models to address the molecular mechanisms. Several groups have demonstrated that nutrient infusion in rats induces β-cell differentiation (5, 22, 42, 43, 53), whereas others have demonstrated that β-cell replication occurs in similar experimental paradigms (4, 50). The mixed occurrence of differentiation and replication in the rodent models, together with the low genetic tractability and the inaccessibility of the islet tissue of the rat model, makes it difficult to elucidate the molecular mechanisms of compensatory differentiation. To circumvent some of this experimental difficulty, we have developed a model of compensatory β-cell differentiation in the genetically tractable zebrafish (35). In this model, the number of β-cells increases by 30% from 32 to more than 42 after 8-h culture in nutrient-containing medium by a single mechanism without replication or transdifferentiation. The new β-cells arise from endocrine precursor cells expressing mnx1 or nkx2.2 (35). The genetic and anatomic tractability of the zebrafish should facilitate molecular events underlying compensatory differentiation. This study focuses on determining the cellular and molecular mechanism by which insufficient insulin secretory capacity is sensed. Using a series of pharmacological and genetic analyses, we show that prolonged activation of the existing β-cells is necessary and sufficient for overnutrition-induced differentiation.
MATERIALS AND METHODS
Zebrafish strains and maintenance.
Zebrafish were raised in an Aquatic-Habitats system on a 14:10-h light-dark cycle. Embryos were obtained from natural crossing and raised according to standard methods; animals were staged by hours postfertilization (hpf) and days postfertilization (dpf) (25). Tg(−1.2ins:H2BmCherry) was used to mark β-cells, and β-cells were counted as described (35). All procedures have been approved by the Vanderbilt University Institutional Animal Care and Use Committee.
Establishment and identification of transgenic lines.
New transgenic lines were generated using the Tol2 transposon system (51). For constitutive expression of human GCKV91L (23) in β-cells, a transgenic construct consisting of two engineered genes carried by the Tol2 transposon vector was made. Tg(cryaa:tagRFP) marks the lens (referred to as lens red, LR) of transgenic fish while Tg(−1.2ins:GCKV91L) directs β-cell expression of the mutant protein using a 1.2-kb insulin promoter (see Fig. 5A). This vector was designated as Tg(−1.2ins: GCKV91L; LR). For inducible expression of a truncated human BID (tBID) (32), mouse Kir6.2DN-GFP (28), and mouse Kir6.2CA-GFP (27) in β-cells, each transgenic construct consists of three engineered genes: Tg(−1.2ins:rtTA-EcR′) to drive a tertracycline and ecdysone-dependent transcription activator in β-cells (26); either Tg(TRE:tBID), Tg(TRE:Kir6.2DN-GFP), or Tg(TRE:Kir6.2CA-GFP) to express the effector proteins; and Tg(cryaa:tagRFP). For simplicity, rtTA-EcR′ and TRE were collectively designated as TE-ON. Hence, these transgenes were named Tg(−1.2ins:htBidTE-ON; LR), Tg(−1.2ins: Kir6.2DN-GFPTE-ON; LR), and Tg(−1.2ins: Kir6.2CA-GFPTE-ON; LR), respectively. Individual founders with tagRFP expression in the lens at 4 dpf were raised to maturity and outcrossed to the wild-type AB line. F1 progeny were screened for lens tagRFP expression and confirmed by PCR. Initial analyses were performed in at least two independent lines for each transgene, and similar results were obtained. All results reported here were from F1 or F2 fish of these lines.
Fig. 5.

β-Cell expression of a hyperactive GCK potentiates glucose-induced β-cell differentiation. A: schematic representation of the Tg(−1.2ins:GCKV91L; LR) transgene used to express a hyperactive human GCK mutant (GCKV91L) in β-cells. B: RT-PCR analysis of nontransgenic wild-type larvae or Tg(−1.2ins:GCKV91L; LR) transgenic larvae with (RT+) or without (RT−) reverse transcriptase confirmed expression of GCKV91L. C: the number of β-cells was significantly increased in Tg(−1.2ins:GCKV91L; LR);Tg(−1.2ins:H2BmCherry) transgenic larvae incubated in subthreshold 10 mM glucose compared with nontransgenic larvae. All values are means ± SE; n are shown inside of the bars. Groups labeled with different letters are significantly different from each other (P < 0.05).
Feeding and compound treatment.
For glucose feeding, d-glucose (Sigma-Aldrich) was dissolved in Milli-Q water at 200 mmol/l and used at a working concentration of 10 or 20 mmol/l. For egg yolk feeding, chicken eggs were obtained from local grocery stores, and the yolk was separated and diluted to 5% by volume with 0.3× Danieau solution as described (35). All drugs were made in 1,000× stock solution and stored in light-protected Eppendorf tubes at −20°C: compound A (30 mmol/l; EMD Millipore), glibenclamide (20 mmol/l; Sigma-Aldrich), and diazoxide (0.3 mol/l; Sigma-Aldrich) in DMSO and verapamil (10 mmol/l; Enzo) in water.
For induction of transgene expression, larvae were treated with doxycycline hyclate (100 mmol/l in ethanol stored in the dark at −20°C, 2,000×) and tebufenozide (50 mmol/l in DMSO at −20°C, 2,000×; Sigma-Aldrich) for 48 h (from 3 to 5 dpf) before feeding.
RNA extraction and RT-PCR.
Total RNA was extracted from 10 zebrafish embryos using Trizol Reagents (Invitrogen) and digested by the RQ1 RNase-Free DNase (Promega) to remove any genomic DNA contamination. First-strand cDNA was synthesized using Moloney murine leukemia virus reverse transcriptase (Promega) with oligo(dT)16 as first-strand primers following the manufacturer's instructions. PCR primers used were as follows: β-actin, 5′-CTTGCGGTATCCACGAGAC-3′ and GCGCCATACAGAGCAGAA; human glucokinase (hGCK), 5′-GCAGGAGGAGGACCTGAAGAA-3′ and 5′-CCGGGGTTTGCAGAGCTCTC-3′; and mKir6.2, 5′-TGCGTCACAAGCATCCACTCC-3′ and 5′-TGGTGATGCCCGTGGTTTCTA-3′. For β-actin, PCR was under the following conditions: 94°C for 3 min, then 28 cycles of 30 s at 95°C, 30 s at 58°C, 30 s at 72°C, and final extension at 72°C for 5 min. For hGCK and mKir6.2, 35 cycles of PCR with an annealing temperature of 60°C were used.
β-Cell ablation.
Stable F1 Tg(−1.2ins:htBidTE-ON; LR) transgenic fish were crossed to homozygous Tg(−1.2ins:H2BmCherry) transgenic fish. Embryos were sorted based on the red lens fluorescence at 3 dpf and then induced as described above for 48 h, refreshing the media every 24 h. Animals were allowed to recover in drug-free media for 40 h before overnutrition treatment.
The larvae were then fixed in 4% paraformaldehyde and imaged using a Zeiss LSM710 confocal microscope.
Free glucose assay.
Free glucose was determined by a glucose assay kit (BioVision). A pool of 10 larvae was homogenized in 100 μl of sample buffer, cleared by centrifugation, and stored at −80°C. Free glucose in the equivalent of one larva (10 μl of homogenate) was determined according to the manufacturer's instructions. Fluorescence (excitation, 535 nm; emission, 590 nm) was measured using a SpectraMax M5 Microplate Reader (Molecular Devices). At least three pools of each sample were measured.
Immunofluorescence and 5-ethynyl-2-deoxyuridine staining.
The larval zebrafish of Tg(−1.2ins:H2BmCherry) were stained using proliferating cell nuclear antigen (PCNA, 1:2,000; Sigma-Aldrich P8825) using standard techniques. To identify proliferating β-cells, 5 dpf embryos were incubated with 100 μmol/l 5-ethynyl-2-deoxyuridine (EdU) for 24 h labeling. EdU was detected using the Click-iT EdU Alexa Fluor 488 Imaging Kit (C10337; Invitrogen) according to published protocols (35). All images were collected using a Zeiss LSM510 or Zeiss LSM710 (Carl Zeiss).
Statistics.
Data are means ± SE. Data were analyzed by one-way ANOVA followed by Fisher post hoc test or t-test (SPSS, Chicago, IL). Significance was accepted at P < 0.05.
RESULTS
Inhibition of membrane depolarization of nutrient-sensing cells suppresses overnutrition-induced β-cell differentiation.
Hypothalamic neurons and pancreatic β-cells are two major postingestive nutrient sensors. In both cell types, nutrients inhibit the ATP-sensitive potassium (KATP) channels, resulting in membrane depolarization and Ca2+ influx through the voltage-sensitive L-type Ca2+ channel (40). To test whether nutrient inhibition of KATP channel is necessary for the overnutrition-induced β-cell differentiation, we used diazoxide, a KATP channel opener, to inhibit nutrient-induced membrane depolarization. As shown previously (35), sustained exposure of 6-day-old larvae to overnutrition results in a 25–30% increase in β-cell number with chicken egg yolk (Fig. 1, A and C) and an 20–25% increase in β-cell number with 20 mM glucose (Fig. 1, B and D). Diazoxide significantly attenuated the increase in β-cells induced by either egg yolk or glucose treatment (Fig. 1, A and B). Larvae treated with diazoxide had a significant increase of free glucose, indicating insulin secretion was suppressed as expected (Fig. 1E). These data show that the inactivation of KATP channels that occurs during nutrient exposure is necessary for overnutrition-induced β-cell differentiation. Similarly, verapamil, an inhibitor of L-type Ca2+ channels that open after membrane depolarization, also significantly decreased β-cell differentiation induced by either 5% egg yolk or 20 mM glucose (Fig. 1, C and D). As expected, verapamil increased total free glucose levels (Fig. 1F). These data demonstrate that inhibition of KATP channels or activation of L-type Ca2+ channels is necessary for overnutrition-induced β-cell differentiation.
Fig. 1.

Pharmacological suppression of ATP-sensitive potassium (KATP) channels or L-type Ca2+ channels inhibits overnutrition-induced β-cell differentiation. A and B: β-cell number following overnutrition in the presence or absence of the KATP channel activator diazoxide (Diaz, 300 μM). Tg(−1.2ins:H2BmCherry) larvae cultured in 5% egg yolk (Yolk) (A) or 20 mM glucose (Glu) (B) for 8 h at 6 days postfertilization (dpf) induced β-cell differentiation, whereas inclusion of diazoxide in 5% egg yolk (A) or 20 mM glucose (B) suppressed the increase. CTL, control. C and D: β-cell number in 6 dpf Tg(−1.2ins:H2BmCherry) larvae following overnutrition in the presence or absence of the L-type Ca2+ channel blocker verapamil (Vera, 10 μM). Verapamil suppressed β-cell differentiation following 8-h culture in 5% egg yolk (C) or 20 mM glucose (D). E and F: total free glucose levels in 6 dpf Tg(−1.2ins:H2BmCherry) larvae. Diazoxide incubation for 8 h significantly increased free glucose levels, which was exacerbated by 5% egg yolk but not 20 mM glucose (E). Verapamil incubation alone did not increase free glucose levels but significantly increased free glucose levels when coadministered with 5% egg yolk or 20 mM glucose (F). All values are means ± SE; n are shown inside of the bars. Groups labeled with different letters are significantly different from each other (P < 0.05).
Prolonged pharmacological activation of the KATP channel and glucokinase induces β-cell differentiation.
Next, we tested whether inactivation of the KATP channel, as would normally occur in the presence of nutrients, is sufficient to induce β-cell differentiation. When 6-day-old larvae were treated for 8 h with glibenclamide, an inhibitor of KATP channels, β-cell number increased by about 20% even in the absence of nutrients (Fig. 2A). The new β-cells likely arise from differentiation of endocrine precursor cells, similar to those induced by overnutrition (35), since no increase of replicating β-cells was detected by EdU labeling or PCNA staining (Fig. 2C and data not shown). The scarcity of EdU incorporation was not due to technical issues, since it was readily detected in previously described Tg(−1.2ins:CDK4R24C) larvae (33). Because only sustained, not intermittent, feeding results in β-cell differentiation (35), we hypothesized that intermittent glibenclamide treatment should not induce differentiation of new β-cells. Indeed, intermittent exposure to glibenclamide was insufficient to induce β-cell differentiation, demonstrating that prolonged pharmacological inhibition of KATP channel is sufficient to elicit β-cell differentiation (Fig. 2A). These results demonstrate that prolonged KATP channel inhibition is sufficient to drive β-cell differentiation in the absence of nutrients.
Fig. 2.
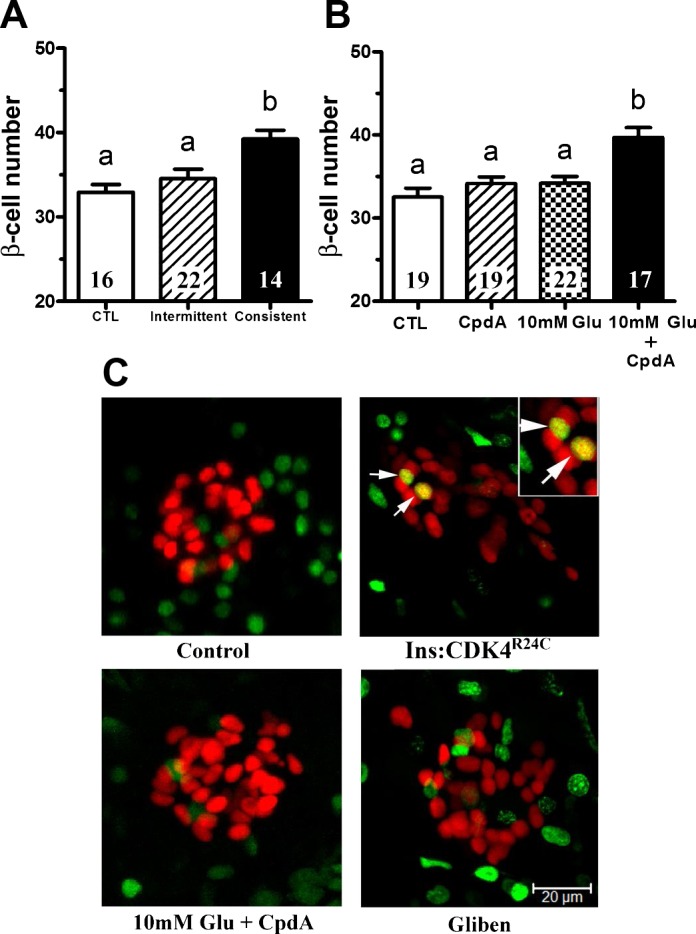
Pharmacological activation of KATP channels and glucokinase (GCK) induces β-cell differentiation. A: β-cell number in 6 dpf Tg(−1.2ins:H2BmCherry) larvae after 8 h of sustained treatment with 20 μM glibenclamide (Consistent), a KATP channel inhibitor, in the absence of overnutrition increased β-cell number significantly, but intermittent glibenclamide treatment (Intermittent) did not. Intermittent treatment was three repetitions of 2 h of glibenclamide incubation followed by 1 h washout. B: β-cell number in 6 dpf Tg(−1.2ins:H2BmCherry) larvae in the presence of the glucokinase activator compound A (CpdA) or in the presence of 10 mM glucose did not significantly change. In contrast, β-cell number increased significantly in the presence of both 10 mM glucose and compound A. C: 5-ethynyl-2-deoxyuridine (EdU) labeling in 6-dpf Tg(−1.2ins:H2BmCherry) larvae untreated or treated with 10 mM glucose plus compound A or glibenclamide (Gliben). EdU (green)-positive β-cells (red) were hardly detected. Dividing β-cells were detected consistently in the positive control Tg(−1.2ins:CDK4R24C; LR);Tg(−1.2ins:H2BmCherry) larvae. The images are confocal projections, and scale bars indicate 20 μm. All values are means ± SE; n are shown inside of the bars. Groups labeled with different letters are significantly different from each other (P < 0.05).
Glucokinase (GCK) controls the glucose sensitivity of a subset of cells, including β-cells and hepatocytes as well as neurons and glia of the hypothalamus (21). Compound A (CpdA) is an allosteric activator of GCK that increases the sensitivity of GCK-expressing cells to circulating glucose (13, 37). We have previously shown that culturing larvae in 20 mM glucose elicits a compensatory β-cell increase but that 10 mM glucose is insufficient to induce this response (35). We reasoned that, if GCK functioned as a component of the nutrient sensor, then, in the absence of glucose, CpdA should have no effect, whereas, in the sustained presence of subthreshold 10 mM glucose, CpdA should induce β-cell differentiation. As expected, in the presence of either CpdA alone or 10 mM glucose alone, no change in β-cell number was observed in 6-day-old larvae (Fig. 2B). However, in the presence of both CpdA and 10 mM glucose, β-cell number increased significantly (Fig. 2, B and C). As well, the new β-cells are likely from differentiation since no increase of EdU incorporation was detected in β-cells during the 8-h period (Fig. 2C). The data suggest that glucose metabolism in GCK-expressing cells is necessary to initiate β-cell differentiation.
Genetic suppression of KATP channel in β-cells leads to supernumerary β-cells.
Results from the pharmacological analyses suggest prolonged membrane depolarization of certain nutrient-sensing cells is necessary and sufficient to induce β-cell differentiation. We hypothesized that β-cells might be a critical component of the sensor for overntrition. To determine whether β-cells act as a component of overnutrition sensor, we genetically manipulated KATP channels in the β-cells. The transgenic line Tg(−1.2ins: Kir6.2DN-GFPTE-ON; LR) allows inducible expression of a dominant-negative form of Kir6.2, Kir6.2(AAA)-GFP (28), in β-cells (Fig. 3A). Expression of Kir6.2DN-GFP should maintain the channel in the closed state, depolarizing the cells independent of intracellular ATP production (28). Transgenic and control larvae were exposed to doxycycline and tebufenozide for 48 h from 3 to 5 dpf to induce Kir6.2DN-GFP. Transgene expression was confirmed by RT-PCR analysis at 6 dpf (Fig. 3B). Expression of Kir6.2DN is expected to result in constitutive secretion of insulin, resulting in lowered baseline glucose levels (27). Indeed, the transgene was shown to be functional by the decrease in free glucose levels measured in larvae at 6 dpf (Fig. 3C). There were significantly more β-cells in unfed Kir6.2DN-GFP-expressing larvae than sibling control larvae at 6 dpf (Fig. 3D). The additional β-cells were not derived from β-cell replication, since no increase of EdU-labeled β-cells was detected, similar to those induced by overnutrition (Fig. 3E). Interestingly, overnutrition did not further increase the number of β-cells in Tg(−1.2ins: Kir6.2DN-GFPTE-ON; LR) larvae (Fig. 3D), either because the pool of available undifferentiated cells is fully induced or because the KATP channels in transgenic β-cells are no longer ATP-sensitive. The results demonstrate that genetically induced constitutive depolarization of β-cells is sufficient to induce differentiation.
Fig. 3.

β-Cell expression of Kir6.2DN induces β-cell differentiation. A: schematic representation of the Tg(−1.2ins:Kir6.2DN-GFPTE-ON; LR) transgene used to express a dominant-negative murine Kir6.2, a KATP channel subunit, in β-cells under the regulation of the dually inducible tetracycline and ecdysone-dependent transcription factor (TE-ON) system. Transgenic fish were marked with an α-crystalline-driven tagRFP. B: RT-PCR of nontransgenic larvae and Tg(−1.2ins:Kir6.2DN-GFPTE-ON; LR) larvae either without or with doxycycline (Dox) and tebufenozide (Tbf) induction. Expression of Kir6.2DN was only observed after induction. C: total free glucose level of 6 dpf Tg(−1.2ins:H2BmCherry) (Kir6.2DN−) and Tg(−1.2ins:Kir6.2DN-GFPTE-ON; LR); Tg(−1.2ins:H2BmCherry) (Kir6.2DN+) larvae treated with or without doxycycline and tebufenozide. Significantly decreased free glucose levels were observed in Dox + Tbf-induced Tg(−1.2ins:Kir6.2DN-GFPTE-ON; LR); Tg(−1.2ins:H2BmCherry) larvae. D: β-cell number in 6 dpf Tg(−1.2ins:Kir6.2DN-GFPTE-ON; LR);Tg(−1.2ins:H2BmCherry) larvae increased significantly in animals induced to express Kir6.2DN in β-cells in the absence of feeding; however, β-cell number in these induced double-transgenic larvae was not significantly different from sibling controls following overnutrition either using 20 mM glucose or 5% egg yolk. All values are means ± SE; n are shown inside of the bars. Groups labeled with different letters are significantly different from each other (P < 0.05). E: transgenic larvae were incubated and labeled with EdU-containing medium for 24 h starting at 5 dpf. EdU-positive cells are revealed with green fluorescence, and β-cells are marked by red fluorescence by nuclear mCherry. Double-transgenic Tg(−1.2ins: Kir6.2DN-GFPTE-ON; LR); Tg(−1.2ins:H2BmCherry) larvae (b) and double-transgenic Tg(−1.2ins: GCKV91L; LR);Tg(−1.2ins:H2BmCherry) (c) larvae at 6 dpf showed little evidence of overlapping colabeling. The images are confocal projections, and scale bars indicate 20 μm.
Genetic activation of KATP channel in β-cells suppresses overnutrition-induced β-cell differentiation.
To determine whether depolarization of β-cells is necessary for overnutrition-induced β-cell differentiation, we generated a transgenic zebrafish Tg(−1.2ins:Kir6.2CA-GFPTE-ON; LR) that inducibly and β-cell specifically expresses Kir6.2ΔN2-30/K185Q-GFP, a constitutively active form of Kir6.2 (Kir6.2CA-GFP) (27) (Fig. 4A). Kir6.2CA-GFP locks KATP in the open state, preventing β-cell depolarization. Transgenic expression of Kir6.2CA in β-cells in mice results in profound neonatal diabetes, since the β-cell can no longer respond to nutritional cues by secreting insulin (27). After 48 h of induction starting at 3 dpf, Tg(−1.2ins:Kir6.2CA-GFPTE-ON; LR) larvae showed robust Kir6.2CA expression as measured by RT-PCR at 6 dpf (Fig. 4B) and had significantly increased free glucose at 6 dpf (Fig. 4C). When cultured for an additional 8 h with or without 5% egg yolk at 6 dpf, induction of Kir6.2CA-GFP expression significantly reduced compensatory β-cell differentiation (Fig. 4D). Similarly, induction of Kir6.2CA-GFP expression significantly reduced compensatory β-cell differentiation induced by 20 mM glucose (Fig. 4D). Expression of Kir6.2CA-GFP had no effect on basal β-cell number compared with sibling controls (Fig. 4D). These data indicate that depolarization of existing β-cells resulting from ATP-dependent inhibition of KATP channels is necessary for overnutrition-induced β-cell differentiation.
Fig. 4.

β-Cell expression of Kir6.2CA inhibits β-cell differentiation. A: schematic representation of Tg(−1.2ins:Kir6.2CA-GFPTE-ON; LR) used to inducibly express a murine Kir6.2CA in β-cells. B: RT-PCR of nontransgenic larvae and Tg(−1.2ins:Kir6.2CA(TE-ON); LR) larvae either without or with doxycycline and tebufenozide induction. Expression of Kir6.2CA was only observed after induction. C: total free glucose level of 6 dpf Tg(−1.2ins:H2BmCherry) (Kir6.2CA−) and Tg(−1.2ins:Kir6.2CA-GFPTE-ON; LR); Tg(−1.2ins:H2BmCherry) (Kir6.2CA+) larvae treated with or without doxycycline and tebufenozide. Dox + Tbf-induced Tg(−1.2ins:Kir6.2CA-GFPTE-ON; LR); Tg(−1.2ins:H2BmCherry) larvae had significantly increased free glucose levels. D: β-cell number of 6 dpf Tg(−1.2ins:Kir6.2CA-GFPTE-ON; LR); Tg(−1.2ins:H2BmCherry) larvae was not significantly changed by induction of Kir6.2CA expression in unfed larvae. In contrast, the increase in β-cell number was significantly smaller following overnutrition with either 5% egg yolk or 20 mM glucose. All values are means ± SE; n are shown inside of the bars. Groups labeled with different letters are significantly different from each other (P < 0.05).
Targeted expression of hyperactive GCK enhances glucose-induced β-cell differentiation.
To further verify that prolonged activation of β-cells is necessary and sufficient to induce β-cell differentiation, we expressed a human GCK mutant (GCKV91L) that has a >8.5-fold higher affinity for glucose than wild-type GCK (23) in β-cells (Fig. 5, A and B). The glucose affinity of GCK in β-cells determines the threshold of glucose-triggered KATP depolarization. At 6 dpf, unfed Tg(−1.2ins:GCKV91L; LR) larvae had an unchanged β-cell number compared with nontransgenic larvae (Fig. 5C). However, when cultured in subthreshold doses of 10 mM glucose, GCKV91L-expressing animals had significantly more β-cells than sibling controls (Fig. 5C). Again, no increase of EdU-positive β-cells was detected, consistent with these cells arising from differentiation and not β-cell replication (Fig. 3E). These results are similar to what we observed using CpdA to activate GCK (Fig. 2B). Taken together, these data demonstrate that persistent activation of glucose metabolism in β-cells drives β-cell differentiation.
Overnutrition-induced β-cell differentiation requires β-cells.
To confirm the hypothesis that β-cells are required for sensing insufficient insulin secretory capacity, we generated a transgenic line that can be used to specifically ablate β-cells. In this line, Tg(−1.2ins:htBidTE-ON; LR), the proapoptotic protein tBID (32), is expressed under the control of the tetracycline- and ecdysone-inducible system (26) (Fig. 6A). Expression of tBID was induced for 48 h starting at 72 hpf and followed by 40 h of washout. The β-cell number was reduced to 7.4 following this treatment (Fig. 6, B and D), similar to the 7.6 β-cells in MTZ-treated Tg(ins:CFP-NTR)s892 larvae after a 48-h recovery (2). Most of the remaining β-cells have lower levels of mCherry signal (Fig. 6B), suggesting that they are likely immature, either newly regernerated as in the in MTZ-treated Tg(ins:CFP-NTR)s892 larvae (2) or had low insulin promoter activity that allowed them to survive the ablation. The larvae with β-cell ablation had increased free glucose compared with controls, even 48 h after recovery, further suggesting insufficient β-cell function (Fig. 6C). To determine whether overnutrition-induced β-cell differentiation was impaired in the β-cell-ablated larvae, ablated and control larvae were treated with 5% egg yolk for 8 h after the 40 h of recovery. At the end of the experiment (7 dpf), the control animals had an increased β-cell count by ∼20–30% (Fig. 6D), consistent with our previous findings (35); however, the number of β-cells did not increase in ablated animals (Fig. 6D). It is unlikely that overnutrition-induced new β-cells were killed since the tBID inducers doxycycline and tebufenozide had been washed out for 40 h before overnutrition treatment. Whereas a complete ablation of the β-cells has not proven possible in our hands or others (2), these data demonstrate that reducing β-cell number by ∼75% prevents an overnutrition-induced increase in β-cell differentiation. These results demonstrate that more than eight mature, functional β-cells are necessary for compensatory β-cell differentiation upon overnutrition.
Fig. 6.

Overnutrition-induced β-cell differentiation requires existing β-cells. A: schematic representation of the transgene Tg(−1.2ins:htBidTE-ON; LR) used to inducibly ablate insulin-expressing cells in the presence of doxycycline and tebufenozide. In the transgene, the proapoptotic gene truncated BID (tBID) is under the control of a TRE-based promoter that is activated by a TE-ON driven by the zebrafish insulin promoter. An α-crystalline-driven tagRFP was used to mark transgene carriers. B: Tg(−1.2ins:H2BmCherry) (Control) or double transgenic Tg(−1.2ins:htBidTE-ON; LR); Tg(−1.2ins:H2BmCherry) (Ablation) larvae were incubated for 48 h with doxycycline and tebufenozide or drug-free medium and allowed to recover for 40 h in drug-free medium. Islets were then imaged with equivalent imaging parameters for β-cells. Note the mCherry+ acellular debris (arrows) that remained 48 h after drug washout. The images are confocal projections, and scale bars indicate 10 μm. C: total free glucose level of 6 dpf Tg(−1.2ins:H2BmCherry) (tBID−) and Tg(−1.2ins:htBidTE-ON; LR); Tg(−1.2ins:H2BmCherry) (tBID+) larvae treated with or without doxycycline and tebufenozide. Significantly increased free glucose levels were observed in Dox + Tbf-induced Tg(−1.2ins:htBidTE-ON; LR); Tg(−1.2ins:H2BmCherry) larvae. D: Dox- and Tbf-treated 6 dpf Tg(−1.2ins:H2BmCherry) (−) and double-transgenic Tg(−1.2ins:htBidTE-ON; LR); Tg(−1.2ins:H2BmCherry) (+), and control double-transgenic Tg(−1.2ins:htBidTE-ON; LR); Tg(−1.2ins:H2BmCherry) (+) larvae were cultured in 5% egg yolk or nutrient-free medium for 8 h. Overnutrition did not significantly affect β-cell number following β-cell ablation in unfed and yolk-fed double-transgenic larvae, but control animals responded normally. All values are means ± SE; n are shown inside of the bars. Groups labeled with different letters are significantly different from each other (P < 0.05).
DISCUSSION
Whereas it is well established that mammals have the ability to change β-cell number and synthetic capacity in response to changes in insulin demand, the sensing mechanisms of the mismatch between demand and capacity that drive these changes remain incompletely understood. Defects in the ability to increase β-cell number to compensate for increased demand for insulin may account for the susceptibility of type 2 diabetes. We have found that, similar to compensatory differentiation of β-cells in rodents under conditions of nutrient infusion, overnutrition induces compensatory β-cell differentiation in zebrafish larvae (35). In zebrafish larvae, this compensation is rapid, occurring in <24 h; easily produced by soaking the fish in nutrients; and robust, facilitating mechanistic analysis. We show here that the compensatory response requires prolonged activation of existing β-cells, suggesting that β-cells are the sensor of overnutrition and function non-cell autonomously to promote differentiation of endocrine precursor cells.
Our conclusion that overnutrition-induced β-cell differentiation requires glucosensing cells is based on several lines of evidence. First, suppression of β-cell depolarization by activating KATP either pharmacologically or genetically in the β-cells inhibits the induction (Figs. 1 and 4). Second, activation of β-cells by inhibiting KATP either pharmacologically or genetically in the β-cell is sufficient for induction of β-cell genesis in the absence of nutrients (Figs. 2 and 3). Third, modulation of β-cell metabolism by manipulation of GCK activity or β-cell exocytosis by manipulation of L-type Ca2+ channel activity also lead to similar results (Figs. 1, 2, and 5). Fourth, intermittent inhibition of KATP, as for meal-style feeding, fails to elicit the induction, demonstrating the importance of prolonged activation of the sensor (Fig. 2A). Last, after genetic ablation of β-cells, there is a failure to activate overnutrition-induced compensatory β-cell genesis (Fig. 6).
These results strongly implicate the β-cells as the primary sensor mediating compensatory β-cell genesis and that the molecular components of this sensing apparatus are shared with the glucose-stimulated insulin secretion system. Other glucosensing cells are known to modulate β-cell number and express these same molecular components. For instance, hypothalamic neurons and glia express these components and are implicated in β-cell mass regulation (8, 30, 34). Our study, however, demonstrates that the β-cells emit a sufficient signal for induction of the compensatory β-cell genesis, since sustained depolarization of the β-cells through the Kir6.2DN was sufficient for the induction. Likewise, β-cell-specific expression of a hypersensitive GCK that triggers membrane depolarization at lower glucose threshold confers the compensatory response at a suboptimal level of glucose. Furthermore, β-cell ablation and β-cell-specific expression of the Kir6.2CA inhibit overnutrition-driven β-cell genesis. Although other sensors may be involved, these data suggest that β-cell is the primary sensor in our assay.
The identity of the neogenic signals released by β-cells after prolonged activation is of great interest for future research. Although insulin may be an integral part of the neogenic signal, the signal likely consists of other factors. This is because that pulsatile activation of β-cell secretion by either intermittent feeding (35) or intermittent inhibition of KATP (this study) fails to induce β-cell differentiation and differentiation occurs only when β-cell activation is sustained. One possible explanation is that only persistently high insulin levels drive β-cell differentiation. A precedent for differential effects of transient and prolonged insulin exposure comes from insulin's known mitogenic effects (20). Prolonged insulin exposure initiates signal transduction events that are distinct from transient incubation in cultured hepatocytes (29). However, the time scale in our in vivo experiments (2 vs. 8 h) is very different from that of the in vitro system (30 vs. 120 min), making direct comparisons difficult. Alternatively, extended secretion may cause β-cells to release other factors to induce β-cell differentiation. In addition to insulin, β-cells secrete many other molecules that are capable of inducing differentiation, such as biogenic monoamines (54), γ-aminobutyric acid (46), and fibroblast growth factors (16). Recently, it has been reported that inhibition of 5-hydroxytryptamine synthesis or action abolishes β-cell genesis during pregnancy in mice (24). Other neurotransmitters may also influence differentiation (38, 56). The prolonged activity of the β-cells may be needed to accumulate enough of the minor secretory products or to trigger the secretion of the neogenic signal. Finally, it is possible that it is the cellular stress caused by prolonged β-cell synthesis and secretion of insulin that drives the generation of the neogenic signal.
It is interesting to speculate how this model system may correlate with human disease and mammalian physiology. There are several clear differences in our system compared with studies of β-cell mass changes in the adult mouse and rat. In addition to the potential difference in β-cell physiology between fish and mammals, our system uses an immature, but not embryonic stage, the larval stage zebrafish as the model organism. At this stage the animal is dependent for all its nutrient and metabolic needs on its ability to identify and consume food. It therefore likely possesses more mature physiology and metabolism than neonatal mammals. Consistent with this, its β-cell number is relatively stable in normal rearing conditions (17, 35). The large increase only occurs after continuous culture in nutrient solutions (35) or prolonged stimulation of β-cells (this study), consistent with a compensatory response. Perhaps more important than the developmental stage of the animal is the unusual replication independent nature of the β-cell genesis observed in our system. The advantage of this system, over and above the advantage of a large and robust compensatory response that occurs within 8 h, lies in the reduction of the complexity of the experimental system. This reductionist model allowed unambiguous demonstration of the non-cell autonomous nature of the response as well as a functional dissection of the underlying cellular and molecular processes. These results provide models that must then be tested in the more complicated and generally used systems. Because zebrafish also generate β-cells through replication at later stages (36, 39), it will be of great interest to explore these systems to determine if the sensor system identified here functions universally in β-cell genesis or whether this is model specific.
The relevance to β-cell physiology in adult mammals notwithstanding, it is worth noting that the β-cell secretion pathway has been shown to play a critical role in β-cell replication in adult mice. For example, acute administration (24 h or less) of a GCK activator, a KATP inhibitor, or an L-type channel blocker promotes β-cell replication (44, 47). Although it is proposed that the hyperactivated β-cells replicate cells autonomously (9), existing data cannot rule out a paracrine mechanism. By manipulating membrane depolarization at a stage when β-cells are refractory to replication (33, 39), we revealed a cell nonautonomous role of hyperactivated β-cells in β-cell genesis. It is tantalizing to speculate that the same factor(s) may be secreted from hyperactivated β-cells in adult mammals and contribute to the ensuing β-cell replication.
GRANTS
This work was supported by the Vanderbilt Diabetes Research and Training Centers and National Institutes of Health (NIH) Grant DK-088686 (W. Chen) and by the American Diabetes Association Grant 1-13-BS-027 (W. Chen). We used the core(s) of the Vanderbilt Diabetes Research and Training Center funded by Grant DK-02593 from NIH, and confocal imaging was performed in the VUMC Cell Imaging Shared Resource (supported by NIH Grants CA-68485, DK-20593, DK-58404, HD-15052, DK-59637, and EY-08126).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: M.L. and W.C. conception and design of research; M.L., L.A.M., and W.C. performed experiments; M.L., L.A.M., P.P.-M., and W.C. analyzed data; M.L., L.A.M., and W.C. interpreted results of experiments; M.L. and L.A.M. prepared figures; M.L. drafted manuscript; M.L., L.A.M., P.P.-M., and W.C. edited and revised manuscript; M.L., L.A.M., P.P.-M., and W.C. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Colin G. Nichols (Washington University) for providing the Kir6.2DN-GFP and Kir6.2CA-GFP constructs.
REFERENCES
- 1.Ahren B, Pacini G. Islet adaptation to insulin resistance: mechanisms and implications for intervention. Diabetes Obes Metab 7: 2–8, 2005 [DOI] [PubMed] [Google Scholar]
- 2.Andersson O, Adams BA, Yoo D, Ellis GC, Gut P, Anderson RM, German MS, Stainier DY. Adenosine signaling promotes regeneration of pancreatic beta cells in vivo. Cell Metab 15: 885–894, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bock T, Pakkenberg B, Buschard K. Increased islet volume but unchanged islet number in ob/ob mice. Diabetes 52: 1716–1722, 2003 [DOI] [PubMed] [Google Scholar]
- 4.Bonner-Weir S, Deery D, Leahy JL, Weir GC. Compensatory growth of pancreatic beta-cells in adult rats after short-term glucose infusion. Diabetes 38: 49–53, 1989 [DOI] [PubMed] [Google Scholar]
- 5.Bonner-Weir S, Li WC, Ouziel-Yahalom L, Guo L, Weir GC, Sharma A. Beta-cell growth and regeneration: replication is only part of the story. Diabetes 59: 2340–2348, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 52: 102–110, 2003 [DOI] [PubMed] [Google Scholar]
- 7.Chen L, Magliano DJ, Zimmet PZ. The worldwide epidemiology of type 2 diabetes mellitus–present and future perspectives. Nat Rev Endocrinol 8: 228–236, 2012 [DOI] [PubMed] [Google Scholar]
- 8.Choudhury AI, Heffron H, Smith MA, Al-Qassab H, Xu AW, Selman C, Simmgen M, Clements M, Claret M, Maccoll G, Bedford DC, Hisadome K, Diakonov I, Moosajee V, Bell JD, Speakman JR, Batterham RL, Barsh GS, Ashford ML, Withers DJ. The role of insulin receptor substrate 2 in hypothalamic and beta cell function. J Clin Invest 115: 940–950, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dadon D, Tornovsky-Babaey S, Furth-Lavi J, Ben-Zvi D, Ziv O, Schyr-Ben-Haroush R, Stolovich-Rain M, Hija A, Porat S, Granot Z, Weinberg-Corem N, Dor Y, Glaser B. Glucose metabolism: key endogenous regulator of beta-cell replication and survival Diabetes Obes Metab 14, Suppl 3: 101–108, 2012 [DOI] [PubMed] [Google Scholar]
- 10.El Ouaamari A, Kawamori D, Dirice E, Liew CW, Shadrach JL, Hu J, Katsuta H, Hollister-Lock J, Qian WJ, Wagers AJ, Kulkarni RN. Liver-derived systemic factors drive beta cell hyperplasia in insulin-resistant states. Cell Rep 3: 401–410, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fernandez AM, Kim JK, Yakar S, Dupont J, Hernandez-Sanchez C, Castle AL, Filmore J, Shulman GI, Le Roith D. Functional inactivation of the IGF-I and insulin receptors in skeletal muscle causes type 2 diabetes. Genes Dev 15: 1926–1934, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Flier SN, Kulkarni RN, Kahn CR. Evidence for a circulating islet cell growth factor in insulin-resistant states. Proc Natl Acad Sci USA 98: 7475–7480, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Futamura M, Hosaka H, Kadotani A, Shimazaki H, Sasaki K, Ohyama S, Nishimura T, Eiki J, Nagata Y. An allosteric activator of glucokinase impairs the interaction of glucokinase and glucokinase regulatory protein and regulates glucose metabolism. J Biol Chem 281: 37668–37674, 2006 [DOI] [PubMed] [Google Scholar]
- 14.Gapp DA, Leiter EH, Coleman DL, Schwizer RW. Temporal changes in pancreatic islet composition in C57BL/6J-db/db (diabetes) mice. Diabetologia 25: 439–443, 1983 [DOI] [PubMed] [Google Scholar]
- 15.Hanley SC, Austin E, Assouline-Thomas B, Kapeluto J, Blaichman J, Moosavi M, Petropavlovskaia M, Rosenberg L. β-Cell mass dynamics and islet cell plasticity in human type 2 diabetes. Endocrinology 151: 1462–1472, 2010 [DOI] [PubMed] [Google Scholar]
- 16.Hart AW, Baeza N, Apelqvist A, Edlund H. Attenuation of FGF signalling in mouse beta-cells leads to diabetes. Nature 408: 864–868, 2000 [DOI] [PubMed] [Google Scholar]
- 17.Hesselson D, Anderson RM, Beinat M, Stainier DY. Distinct populations of quiescent and proliferative pancreatic beta-cells identified by HOTcre mediated labeling. Proc Natl Acad Sci USA 106: 14896–14901, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hull RL, Kodama K, Utzschneider KM, Carr DB, Prigeon RL, Kahn SE. Dietary-fat-induced obesity in mice results in beta cell hyperplasia but not increased insulin release: evidence for specificity of impaired beta cell adaptation. Diabetologia 48: 1350–1358, 2005 [DOI] [PubMed] [Google Scholar]
- 19.Imai J, Katagiri H, Yamada T, Ishigaki Y, Suzuki T, Kudo H, Uno K, Hasegawa Y, Gao J, Kaneko K, Ishihara H, Niijima A, Nakazato M, Asano T, Minokoshi Y, Oka Y. Regulation of pancreatic beta cell mass by neuronal signals from the liver. Science 322: 1250–1254, 2008 [DOI] [PubMed] [Google Scholar]
- 20.Ish-Shalom D, Christoffersen CT, Vorwerk P, Sacerdoti-Sierra N, Shymko RM, Naor D, De Meyts P. Mitogenic properties of insulin and insulin analogues mediated by the insulin receptor. Diabetologia 40, Suppl 2: S25–S31, 1997 [DOI] [PubMed] [Google Scholar]
- 21.Iynedjian PB. Molecular physiology of mammalian glucokinase. Cell Mol Life Sci 66: 27–42, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jetton TL, Everill B, Lausier J, Roskens V, Habibovic A, LaRock K, Gokin A, Peshavaria M, Leahy JL. Enhanced β-cell mass without increased proliferation following chronic mild glucose infusion. Am J Physiol Endocrinol Metab 294: E679–E687, 2008 [DOI] [PubMed] [Google Scholar]
- 23.Kassem S, Bhandari S, Rodriguez-Bada P, Motaghedi R, Heyman M, Garcia-Gimeno MA, Cobo-Vuilleumier N, Sanz P, Maclaren NK, Rahier J, Glaser B, Cuesta-Munoz AL. Large islets, beta-cell proliferation, and a glucokinase mutation. N Engl J Med 362: 1348–1350, 2010 [DOI] [PubMed] [Google Scholar]
- 24.Kim H, Toyofuku Y, Lynn FC, Chak E, Uchida T, Mizukami H, Fujitani Y, Kawamori R, Miyatsuka T, Kosaka Y, Yang K, Honig G, van der Hart M, Kishimoto N, Wang J, Yagihashi S, Tecott LH, Watada H, German MS. Serotonin regulates pancreatic beta cell mass during pregnancy. Nat Med 16: 804–808, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kimmel CB, Ballard WW, Kimmel SR, Ullmann B, Schilling TF. Stages of embryonic development of the zebrafish. Dev Dyn 203: 253–310, 1995 [DOI] [PubMed] [Google Scholar]
- 26.Knopf F, Schnabel K, Haase C, Pfeifer K, Anastassiadis K, Weidinger G. Dually inducible TetON systems for tissue-specific conditional gene expression in zebrafish. Proc Natl Acad Sci USA 107: 19933–19938, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koster JC, Marshall BA, Ensor N, Corbett JA, Nichols CG. Targeted overactivity of beta cell K(ATP) channels induces profound neonatal diabetes. Cell 100: 645–654, 2000 [DOI] [PubMed] [Google Scholar]
- 28.Koster JC, Remedi MS, Flagg TP, Johnson JD, Markova KP, Marshall BA, Nichols CG. Hyperinsulinism induced by targeted suppression of beta cell KATP channels. Proc Natl Acad Sci USA 99: 16992–16997, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kubota H, Noguchi R, Toyoshima Y, Ozaki Y, Uda S, Watanabe K, Ogawa W, Kuroda S. Temporal coding of insulin action through multiplexing of the AKT pathway. Mol Cell 46: 820–832, 2012 [DOI] [PubMed] [Google Scholar]
- 30.Kubota N, Terauchi Y, Tobe K, Yano W, Suzuki R, Ueki K, Takamoto I, Satoh H, Maki T, Kubota T, Moroi M, Okada-Iwabu M, Ezaki O, Nagai R, Ueta Y, Kadowaki T, Noda T. Insulin receptor substrate 2 plays a crucial role in beta cells and the hypothalamus. J Clin Invest 114: 917–927, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lausier J, Diaz WC, Roskens V, LaRock K, Herzer K, Fong CG, Latour MG, Peshavaria M, Jetton TL. Vagal control of pancreatic ss-cell proliferation. Am J Physiol Endocrinol Metab 299: E786–E793, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 94: 491–501, 1998 [DOI] [PubMed] [Google Scholar]
- 33.Li M, Maddison LA, Crees Z, Chen W. Targeted overexpression of CKI-insensitive cyclin-dependent kinase 4 increases functional beta-cell number through enhanced self-replication in zebrafish. Zebrafish 10: 170–176, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lin X, Taguchi A, Park S, Kushner JA, Li F, Li Y, White MF. Dysregulation of insulin receptor substrate 2 in beta cells and brain causes obesity and diabetes. J Clin Invest 114: 908–916, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maddison LA, Chen W. Nutrient excess stimulates beta-cell neogenesis in zebrafish. Diabetes 61: 2517–2524, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moss JB, Koustubhan P, Greenman M, Parsons MJ, Walter I, Moss LG. Regeneration of the pancreas in adult zebrafish. Diabetes 58: 1844–1851, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nakamura A, Terauchi Y, Ohyama S, Kubota J, Shimazaki H, Nambu T, Takamoto I, Kubota N, Eiki J, Yoshioka N, Kadowaki T, Koike T. Impact of small-molecule glucokinase activator on glucose metabolism and beta-cell mass. Endocrinology 150: 1147–1154, 2009 [DOI] [PubMed] [Google Scholar]
- 38.Nedergaard J, Herron D, Jacobsson A, Rehnmark S, Cannon B. Norepinephrine as a morphogen?: its unique interaction with brown adipose tissue. Int J Dev Biol 39: 827–837, 1995 [PubMed] [Google Scholar]
- 39.Ninov N, Hesselson D, Gut P, Zhou A, Fidelin K, Stainier DY. Metabolic regulation of cellular plasticity in the pancreas. Curr Biol 23: 1242–1250, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nolan CJ, Prentki M. The islet beta-cell: fuel responsive and vulnerable. Trends Endocrinol Metab 19: 285–291, 2008 [DOI] [PubMed] [Google Scholar]
- 41.Okada T, Liew CW, Hu J, Hinault C, Michael MD, Krtzfeldt J, Yin C, Holzenberger M, Stoffel M, Kulkarni RN. Insulin receptors in beta-cells are critical for islet compensatory growth response to insulin resistance. Proc Natl Acad Sci USA 104: 8977–8982, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Paris M, Bernard-Kargar C, Berthault MF, Bouwens L, Ktorza A. Specific and combined effects of insulin and glucose on functional pancreatic beta-cell mass in vivo in adult rats. Endocrinology 144: 2717–2727, 2003 [DOI] [PubMed] [Google Scholar]
- 43.Pick A, Clark J, Kubstrup C, Levisetti M, Pugh W, Bonner-Weir S, Polonsky KS. Role of apoptosis in failure of beta-cell mass compensation for insulin resistance and beta-cell defects in the male Zucker diabetic fatty rat. Diabetes 47: 358–364, 1998 [DOI] [PubMed] [Google Scholar]
- 44.Porat S, Weinberg-Corem N, Tornovsky-Babaey S, Schyr-Ben-Haroush R, Hija A, Stolovich-Rain M, Dadon D, Granot Z, Ben-Hur V, White P, Girard CA, Karni R, Kaestner KH, Ashcroft FM, Magnuson MA, Saada A, Grimsby J, Glaser B, Dor Y. Control of pancreatic beta cell regeneration by glucose metabolism. Cell Metab 13: 440–449, 2011 [DOI] [PubMed] [Google Scholar]
- 45.Rahier J, Guiot Y, Goebbels RM, Sempoux C, Henquin JC. Pancreatic beta-cell mass in European subjects with type 2 diabetes. Diabetes Obes Metab 10, Suppl 4: 32–42, 2008 [DOI] [PubMed] [Google Scholar]
- 46.Reetz A, Solimena M, Matteoli M, Folli F, Takei K, De Camilli P. GABA and pancreatic beta-cells: colocalization of glutamic acid decarboxylase (GAD) and GABA with synaptic-like microvesicles suggests their role in GABA storage and secretion. EMBO J 10: 1275–1284, 1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Salpeter SJ, Klochendler A, Weinberg-Corem N, Porat S, Granot Z, Shapiro AM, Magnuson MA, Eden A, Grimsby J, Glaser B, Dor Y. Glucose regulates cyclin D2 expression in quiescent and replicating pancreatic beta-cells through glycolysis and calcium channels. Endocrinology 152: 2589–2598, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Samuel VT, Shulman GI. Mechanisms for insulin resistance: common threads and missing links. Cell 148: 852–871, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sone H, Kagawa Y. Pancreatic beta cell senescence contributes to the pathogenesis of type 2 diabetes in high-fat diet-induced diabetic mice. Diabetologia 48: 58–67, 2005 [DOI] [PubMed] [Google Scholar]
- 50.Steil GM, Trivedi N, Jonas JC, Hasenkamp WM, Sharma A, Bonner-Weir S, Weir GC. Adaptation of β-cell mass to substrate oversupply: enhanced function with normal gene expression. Am J Physiol Endocrinol Metab 280: E788–E796, 2001 [DOI] [PubMed] [Google Scholar]
- 51.Suster ML, Kikuta H, Urasaki A, Asakawa K, Kawakami K. Transgenesis in zebrafish with the tol2 transposon system. Methods Mol Biol 561: 41–63, 2009 [DOI] [PubMed] [Google Scholar]
- 52.Terauchi Y, Takamoto I, Kubota N, Matsui J, Suzuki R, Komeda K, Hara A, Toyoda Y, Miwa I, Aizawa S, Tsutsumi S, Tsubamoto Y, Hashimoto S, Eto K, Nakamura A, Noda M, Tobe K, Aburatani H, Nagai R, Kadowaki T. Glucokinase and IRS-2 are required for compensatory beta cell hyperplasia in response to high-fat diet-induced insulin resistance. J Clin Invest 117: 246–257, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Topp BG, McArthur MD, Finegood DT. Metabolic adaptations to chronic glucose infusion in rats. Diabetologia 47: 1602–1610, 2004 [DOI] [PubMed] [Google Scholar]
- 54.Wilson JP, Downs RW, Jr, Feldman JM, Lebovitz HE. β-Cell monoamines: further evidence for their role in modulating insulin secretion. Am J Physiol 227: 305–312, 1974 [DOI] [PubMed] [Google Scholar]
- 55.Yi P, Park JS, Melton DA. Betatrophin: a hormone that controls pancreatic beta cell proliferation. Cell 153: 747–758, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 56.Young SZ, Bordey A. GABA's control of stem and cancer cell proliferation in adult neural and peripheral niches. Physiology (Bethesda) 24: 171–185, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]