

Abstract
Nerve cell metabolic activity is monitored in multiple brain regions, including the hypothalamus and hindbrain dorsal vagal complex (DVC), but it is unclear if individual metabolosensory loci operate autonomously or interact to coordinate central nervous system (CNS) reactivity to energy imbalance. This research addressed the hypothesis that hypoglycemia-associated DVC lactoprivation stimulates hypothalamic AMPK activity and metabolic neurotransmitter expression. As DVC catecholaminergic neurons express biomarkers for metabolic monitoring, we investigated whether these cells are a source of lactate deficit signaling to the hypothalamus. Caudal fourth ventricle (CV4) infusion of the glucose metabolite l-lactate during insulin-induced hypoglycemia reversed changes in DVC A2 noradrenergic, arcuate neuropeptide Y (NPY) and pro-opiomelanocortin (POMC), and lateral hypothalamic orexin-A (ORX) neuronal AMPK activity, coincident with exacerbation of hypoglycemia. Hindbrain lactate repletion also blunted hypoglycemic upregulation of arcuate NPY mRNA and protein. This treatment did not alter hypoglycemic paraventricular oxytocin (OT) and lateral hypothalamic ORX mRNA profiles, but exacerbated or reversed adjustments in OT and ORX neuropeptide synthesis, respectively. CV4 delivery of the monocarboxylate transporter inhibitor, 4-CIN, increased A2 phosphoAMPK (pAMPK), elevated circulating glucose, and stimulated feeding, responses that were attenuated by 6-hydroxydopamine pretreatment. 4-CIN-infused rats exhibited increased (NPY, ORX neurons) or decreased (POMC neurons) pAMPK concurrent with hyperglycemia. These data show that hindbrain lactoprivic signaling regulates hypothalamic AMPK and key effector neurotransmitter responses to hypoglycemia. Evidence that A2 AMPK activity is lactate-dependent, and that DVC catecholamine cells are critical for lactoprivic control of glucose, feeding, and hypothalamic AMPK, implies A2 derivation of this metabolic regulatory stimulus.
Keywords: lactate, A2 noradrenergic neurons, AMPK, laser-catapult microdissection, insulin-induced hypoglycemia, Western blot analysis, qPCR, α-cyano-4-hydroxycinnamate, 6-hydroxydopamine, neuropeptide Y, orexin-A, oxytocin, proopiomelanocortin
cellular metabolic stasis is monitored in discrete brain sites, including the hindbrain dorsal vagal complex (DVC), where specialized neurons at caudal levels of this structure adjust synaptic firing in response to energy imbalance (36, 43). The brain reacts to such signals of energy deficits by activating compensatory physiological and behavioral outflow that increases substrate fuel availability (42). The oxidizable glycolytic end product l-lactate is generated within astrocytes and released into the extracellular space as a vital energy source for central nervous system (CNS) neurons (18). Lactate trafficking between glia and nerve cells is mediated by cell type-specific membrane monocarboxylate transporters, including the neuron-specific transporter MCT2 (9). Neuronal reliance upon lactate is emphasized by recent in vivo evidence that lactate is preferred relative to glucose as an energy substrate when both fuels are available (57). Lactate utilization is a critical monitored variable in hindbrain monitoring of nerve cell metabolic stasis. Our studies show that inhibition of hindbrain monocarboxylate transporter function elevates blood glucose levels and induces Fos expression in hypothalamic metabolic loci, whereas hindbrain lactate repletion during hypoglycemia exacerbates blood glucose decrements (49, 50).
The cellular derivation of hindbrain lactoprivic signaling remains unclear. DVC A2 noradrenergic neurons are a plausible source of this regulatory stimulus, as these cells express the neuronal monocarboxylate transporter variant MCT2 (6), respond electrically to exogenous lactate (24), and undergo transcriptional activation during lactate shortage (51). During hypoglycemia, A2 cells exhibit decreased MCT2, but increased GLUT3 protein expression, suggesting that decreased lactate uptake alone or relative to glucose uptake is a critical manifestation of systemic glucose deficiency to these cell (6). A2 neurons express characterized biomarkers for metabolic sensing, e.g., the low-affinity, high Michaelis constant (Km) hexokinase, glucokinase (6); the inwardly rectifying ATP-dependent potassium channel (KATP) (6); and the cellular energy gauge adenosine 5′-monophosphate-activated protein kinase (AMPK) (13, 14). AMPK is an evolutionarily conserved gauge of cellular energy status that is activated by phosphorylation in response to metabolic stressors that increase the intracellular AMP-to-ATP ratio (22, 27). We recently developed and characterized a novel combinatory approach involving in situ immunocytochemical labeling, laser-catapult microdissection, and high-sensitivity Western blotting of requisite sensitivity for analysis of expression of this and other proteins in small-size pools, e.g., a minimum of 25–50 tyrosine hydroxylase (TH)-immunopositive A2 neurons (13, 14). This technique was applied here to examine the premise that A2 AMPK activation status is regulated by lactate availability. Furthermore, the selective catecholamine neurotoxin 6-hydroxydopamine (6-OHDA) was used as a tool to determine whether DVC catecholaminergic nerve cell integrity is critical for hindbrain lactoprivic augmentation of blood glucose levels and counterregulatory functions.
Our previous work supports a functional connection between hindbrain lactoprivic-senstive neurons and hypothalamic metabolic structures, as pharmacological inhibition of hindbrain monocarboxylate transport induces dose-proportionate manner Fos immunolabeling in those sites (50). A principal aim of the current study was to identify hypothalamic nerve cell populations that respond to hindbrain lactoprivation. Arcuate neuropeptide Y (NPY) neurons are a possible target of such input, because these cells govern energy balance by integrating neural, metabolic hormone, and nutrient signals (17) and are electrically activated by deficits of the latter (19, 39). NPY acts centrally to control circulating insulin, glucagon, and corticosterone secretion (33) and to modulate insulin regulation of hepatic glucose production (55). NPY Y1 receptors function within neural pathways that control hyperphagia and glycemic profiles during insulin-induced hypoglycemia (41). Lateral hypothalamic area (LHA) orexin-A (ORX) neurons exhibit increased synaptic firing, cfos transcription, and preproorexin gene expression during nutrient shortage (10, 11, 20, 37, 56). ORX involvement in glucostasis is implied by evidence for orexin receptor-1-dependent blood glucose and glucagon secretory responses to acute and recurring hypoglycemia (46). The hypothalamic paraventricular nucleus (PVH) serves as the primary conduit for motor control of autonomic and neuroendocrine outflow. The PVH neuropeptide oxytocin (OT) regulates feeding (48) and blood glucose (4). Exogenous OT elevates circulating glucose, glucagon, and corticosterone, in part by central actions, whereas OT receptor antagonism elicits hypoglycemia (3, 4). Here, we used the hypoglycemia plus hindbrain lactate repletion model established in our laboratory (46) to address the hypothesis that hindbrain lactoprivation controls NPY, ORX, and OT gene and/or neuropeptide transmitter expression during hypoglycemia.
Hypothalamic AMPK has garnered broad attention regarding its role in CNS control of systemic energy balance (29, 32, 35). In that brain region, AMPK is expressed in characterized metabolic structures, including the arcuate nucleus of the hypothalamus (ARH), PVH, and ventromedial hypothalamic nucleus (1, 21). In the ARH, this sensor is localized to NPY and pro-opiomelanocortin (POMC) neurons (2, 12, 16, 38). Recent studies suggest that AMPK is also expressed by ORX neurons (28). It remains controversial whether hypothalamic and hindbrain, e.g., A2 metabolosensory neurons function as autonomous monitoring sites or interact to coordinate CNS reactivity to energy imbalance. Here, we adapted the high-sensitivity laser microdissection/Western blot approach described above to investigate the novel premise that AMPK activity in NPY, POMC, and ORX neurons is reliant upon hindbrain lactate status and to determine whether effects of hindbrain lactoprivation on NPY and ORX neurotransmitter gene and/or protein expression correlate with adjustments in AMPK activation.
METHODS AND MATERIALS
Animals
Adult male Sprague-Dawley rats (experiment 1: 360–400 g body wt; experiment 2: 410–440 g body wt) were maintained under a 14-h light:10-h dark lighting schedule (light on at 05.00 h), and fed standard laboratory rat chow and watered ad libitum. All protocols were conducted in accordance with NIH guidelines for care and use of laboratory animals and approved by the ULM Institutional Animal Care and Use Committee. On study day 1, animals were implanted with a 26-gauge stainless-steel cannula guide (product no. C315G/SP; Plastic One, Roanoke, VA) into the caudal fourth ventricle (CV4) (coordinates: 0 mm lateral to midline, 13.3 mm posterior to bregma, and 6.1 mm ventral to the skull surface) under ketamine-xylazine anesthesia (0.1 ml/100 g body wt ip, 90 mg ketamine-10 mg xylazine per ml; Henry Schein, Melville, NY) and transferred to individual cages. All animals used in the present studies exhibited cerebrospinal fluid reflux from the tip of the cannula on day 1 of the study; post mortem histological examination confirmed cannula placements within the CV4, lack of structural damage to underlying hindbrain structures, including the DVC, and lack of necrosis of the hindbrain of rats treated by CV4 delivery of 6-OHDA.
Experimental Design
Experiment 1: effects of CV4 l-lactate infusion on insulin-induced hypoglycemia and hypoglycemic patterns of feeding, counterregulatory hormone secretion, and hypothalamic phosphoAMPK (pAMPK) and metabolic neurotransmitter gene and protein expression.
On day 10, CV4 infusion of l-lactate (25 μM·2.0 μl−1·h−1) (49, 53) or the vehicle artificial cerebrospinal fluid (aCSF) was initiated 10 min before time 0 (t0; 11.00 h) and continued until +120 min. Infusions were performed using 33-gauge internal injection cannulas (product no. C315I/SP; Plastics One) that projected 0.5 mm beyond the cannula guide into the CV4. At t0, groups of aCSF-infused rats were injected with saline (SAL; n = 5) or neutral protamine Hagedorn insulin (INS; 12.5 U/kg body wt sc; Henry Schein, n = 5) (44), while lactate-infused rats received INS (n = 5). Animals were euthanized by decapitation at +120 min for blood and brain tissue collection. Each hindbrain was snap frozen in cooled isopentane; 10-μm serial frozen tissue sections were cut between −14.36 to −14.86 mm relative to bregma and mounted on P.A.L.M. PEN membrane-coated slides (product no. 911724; Carl Zeiss MicroImaging, Thornwood, NY) for Western blot analysis of AMPK and pAMPK content of laser-microdissected A2 neurons. Each forebrain was frozen and cut into serial 200-μm serial sections in a cryostat. The ARH, LHA, and PVH were each micropunched from separate sides of the hypothalamus with Palkovits-style hollow needles over intervals of −2.00 to −3.00, −1.30 to −3.20, and −1.30 to −1.70 mm posterior to bregma, respectively (53, 54). This approach created, for each animal, two tissue sample pools for each neural structure. ARH, LHA, and PVH sample pools from one hemihypothalamus were used for quantitative real-time RT-PCR (qPCR) measurements of NPY, ORX, and OT mRNAs, respectively; tissue from the remaining half-hypothalamus was evaluated for neuropeptide expression by Western blot. Frozen 10-μm sections were cut over the intervals described above from additional groups of animals (n = 5/group) for analysis of pAMPK expression by NPY, POMC, and ORX neurons. Food intake by each rat was measured between t0 and +120 min.
Experiment 2: effects of DVC catecholaminergic nerve cell destruction on glucose, counterregulatory functions, and hypothalamic AMPK responses to hindbrain lactoprivation.
On days 10 and 12, groups of rats were injected into the CV4 with 6-OHDA [75 μg/day (52); group 1; n = 10] or vehicle, sterile apyrogenic water containing 0.2% ascorbic acid (group 2; n = 10), in a 1.0-μl volume. The additive ascorbic acid attenuates immediate oxidation of 6-OHDA. This 6-OHDA treatment paradigm significantly reduces brain tissue catecholamine levels but does not alter serotonin content (52). On day 14, subsets of each group were treated by CV4 infusion of cyano-4-hydroxycinnamate [4-CIN; 50.0 μg/2.0 μl (49); n = 5] or SAL (n = 5) initiated at t0 (11.00 h) and continued over 30 min. Animals were euthanized at +120 min for blood and brain tissue collection. Each hindbrain was halved. Serial frozen 10-μm sections were collected between −14.36 to −14.86 mm from one hemihindbrain and mounted on PEN membrane slides for Western blot analysis of A2 nerve cell pAMPK expression. The other hemihindbrain was submersion fixed for 12 h in 0.1 M potassium phosphate buffer, pH 7.6, containing 4.0% paraformaldehyde and 0.2% picric acid, sunk in 25% sucrose, and cut into 25-μm serial sections for immunocytochemical confirmation of 6-OHDA-mediated catecholamine nerve cell destruction and verification of lack of neurotoxin effects on DVC neuropeptidergic neurons. Frozen 10-μm sections were also cut from the ARH and LHA over intervals of −2.00 to −3.00 mm and −1.30 to −3.20 mm, respectively, for analysis of pAMPK expression by NPY, POMC, and LHA neurons. For each rat, food intake was measured between t0 and +120 min.
Blood Analyte Measurements
Blood glucose levels were measured using an AccuCheck Advantage glucometer (Roche Diagnostics, Indianapolis, IN) as previously described (26). Plasma glucagon and corticosterone concentrations were determined by radioimmunoassay, as previously reported (7, 44).
Immunocytochemical Assessment of Neurotoxin Destruction of DVC Catecholaminergic Neurons
For each rat, three tissue sections were processed, per level, from the rostral (−13.28 to −13.60 mm), commissural (−13.76 to −14.16 mm), and caudal (−14.36 to −14.86 mm) DVC for TH immunoreactivity (-ir). After being washed with 0.05 M Tris-buffered saline and 0.9% NaCl, pH 7.6 (TBS), sections were preincubated for 60 min with 4.0% normal donkey serum (product no. S30, Millipore, Billerica, MA) and incubated for 48 h at 4°C with mouse monoclonal tyrosine hydroxylase antibodies (1:10,000, product no. 22941, Immunostar, Hudson, WI) diluted in TBS with 0.05% Triton X-100. Tissues were incubated for 2 h with AlexaFluor-488 donkey anti-mouse antibody (1:400, product no. A-21202, Life Technologies, Grand Island, NY) in TBS containing 2.0% normal donkey serum, mounted on glass slides, and coverslipped with Vectashield mounting medium (product no. H-1000, Vector Laboratories, Burlingame, CA). Images were captured with an LSM 5 Pascal confocal scanning laser microscope (Carl Zeiss MicroImaging). Bilateral counts of TH-ir-positive neurons were obtained for the rostral, commissural, and caudal DVC. Additional sections from the commissural and caudal DVC were processed for somatostatin (1:1,000, product no. sc-7819, Santa Cruz Biotechnology, Santa Cruz, CA), corticotropin-releasing hormone (1:500, product no. sc-1759, Santa Cruz Biotechnology), NPY (1:500, product no. NBP1–46535, Novus Biologicals, Littleton, CO), or nitric oxide synthase (1:1,000, product no. 2032307, EMD Millipore, Billerica, MA) immunoreactivity, using AlexaFluor-488 donkey anti-goat (product no. A-11055, Life Technology) or anti-rabbit (1:400, product no. A-21206, Life Technology) secondary antisera and DAPI (product no. D1306, Life Technology).
Laser-Catapult Microdissection of Hindbrain A2 and Hypothalamic NPY, POMC, and ORX Neurons
Mounted frozen tissue sections were fixed for 5 min with cold acetone, blocked with 5% normal serum diluted in 0.1 M sodium phosphate-buffered saline (PBS), 0.9% NaCl, pH 7.2 containing 0.05% Triton X-100, and incubated sequentially with primary antiserum, biotinylated secondary antibodies, and ABC reagent [product no. PK-6102 (mouse), PK-6105 (goat), PK-6101 (rabbit) Vectastain IgG Elite ABC kits; Vector Laboratory] for 20 min each (6). Primary antisera, e.g., mouse monoclonal anti- TH [1:50, product no. 22941, ImmunoStar, Hudson, WI (6)], goat polyclonal anti-NPY [1:50, product no. NBP1–46535; Novus Biol. (8)], rabbit anti-POMC (1:100, product no. sc-20148; Santa Cruz Biotechnology), and goat polyclonal anti-ORX (1:100, product no. sc-8070; Santa Cruz Biotechnology) were diluted in TBS containing 0.1% Tween-20 (product no. P94 16; Sigma Aldrich, St. Louis, MO) and 2% bovine serum albumin (product no. 81003; MP Biomedicals, Solon, OH) (TBS-T-BSA). Antigen-containing neurons were visualized by 3–5 min incubation with Vector DAB Kit reagents (product no. SK-4100; Vector Laboratories), and individually harvested using a Zeiss P.A.L.M. UV-A microlaser (Carl Zeiss MicroImaging). Only neurons exhibiting a visibly intact nucleus and complete labeling of the cytoplasmic compartment were collected. For each protein of interest, pools of n = 50 neurons were established for each treatment group (n = 10 cells per rat).
qPCR Aalysis of ARH NPY, LHA ORX, and PVH OT mRNA Content of Micropunch-Dissected Hypothalamic Structures
For one hemihypothalamus per rat, total RNA was isolated from pooled ARH (n = 5 punches), LHA (n = 9 punches), and PVH (n = 2 punches) tissue samples using MELT Total RNA isolation kits (product no. AM1983, Ambion, Austin, TX) and evaluated with an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA). cDNA synthesis was performed with RETROscript kit materials (product no. AM1710, Ambion). PCR primers for NPY (forward: 5′-ATGC-TAGGTAACAAACG-3′; reverse: 5′-ATGTAGTGTCGCAGAG-3), prepro-orexin (forward: 5′-CATA-TCCCTGCCCTGGTC-3′; reverse: 5′-GATAGAAGACGGGTTCAGAC-3′), and OT (forward: 5′-GCA-CTGGCTGTTACTTCTTC-3; reverse: 5′-GCTTTGGGCTTTGGGTTAG-3′) were designed with Beacon Designer software (Premier Biosoft International, Palo Alto, CA) and obtained from Genemed Synthesis (San Antonio, TX). PCR cycling parameters were as follows: initial denaturation at 95°C for 3 min, followed by 40 cycles of 1 min each (30 s at 95°C, followed by 30 s at 51°C for NPY, 30 s at 60°C for OT, or 30 s 60°C for ORX, respectively) (6, 8, 40). GAPDH (forward: 5′-ACAGCCGCATCTTCTTGTGC-3′; reverse: 5′-GCCTCACCCCATTTGATGTT-3′) gene expression was measured as a housekeeping control. No-template and -RT controls and melt-curve analyses were performed for each RT-PCR analysis. Amplified PCR products were evaluated by agarose gel electrophoresis. For each animal, ARH NPY, LHA ORX, and PVH OT mRNAs were analyzed by the 2ΔΔCt method.
Western Blot Analysis of NPY, ORX, and OT Protein Levels in Micropunched Hypothalamic Loci and A2, NPY, POMC, and ORX Nerve Cell AMPK and/or pAMPK Content
For each rat, pooled ARH, LHA, and PVH hemihypothalamus tissue punches were collected in microcentrifuge tubes containing 20 μl tissue lysis buffer (2% SDS, 0.05 M DTT, 10% glycerol, 1 mM EDTA, 60 mM Tris·HCl, pH 7.2) (13, 14). For each neuropeptide of interest, 5 immunoblots were run per treatment group. For each group, 3 pools of neurons (50 neurons/pool) of each cell type of interest were collected into separate buffer-containing tubes for AMPK, pAMPK, or α-tubulin immunoblotting. Protein concentrations of heat-denatured tissue or cell lysates were determined with Pierce BCA protein assay reagents (product no. 23225, ThermoScientific, Rockford, IL). Equivalent amounts of sample protein were separated on 4–12% gradient Tris-glycine gels (product no. 412W1.5M10; Jule Biotechnologies, Milford, CT). Membranes were treated with Quentix Western blot signal enhancer (product no. 21050; Pierce, Rockford, IL) and blocked for 2 h in TBS-T-BSA before overnight incubation at 4°C with primary antisera. Pooled micropunch neuropeptide levels were evaluated using goat polyclonal anti-NPY (1:2,000; Novus Biologicals); rabbit polyclonal anti-OT (product no. NBP1-19753, 1:2,000; Novus Biologicals); and goat polyclonal anti-ORX (1:2,000; Santa Cruz Biotechnology) antisera diluted in TBS-T-BSA. Nerve cell pAMPK levels were detected using rabbit polyclonal p-AMPKα1/2 (Thr 172) antibodies (1:1,000, product no. sc-33524, Santa Cruz Biotechnology). The housekeeping protein α-tubulin was probed with a mouse monoclonal anti-α-tubulin antiserum (1:2,000, product no. CP06; Calbiochem, Gibbstown, NJ). Membranes were incubated for 1 h with peroxidase-conjugated goat anti-mouse (1:5,000, product no. NEF822001EA; Perkin Elmer, Boston, MA), goat anti-rabbit (1:5,000, product no. NEF812001EA; Perkin Elmer), or donkey anti-goat (1:5,000, product no. sc-2020; Santa Cruz Biotechnology) secondary antibodies. Membranes incubated with Supersignal West Femto Maximum Sensitivity substrate (product no. 34096; Pierce) were evaluated for chemiluminescence in a Syngene G:box Chemi. Band intensities were quantified by AlphaImager HP V 5.0.1 software (Cell Biosciences, Santa Clara, CA), and protein levels were expressed as ratio of protein band and α-tubulin optical density (O.D.). Protein molecular weight markers were included in each Western blot analysis.
Statistical Analyses
Mean blood glucose levels, food consumption, TH-ir-positive cell counts, neuropeptide mRNA levels, and normalized neuropeptide, AMPK, or pAMPK protein O.D. values were evaluated by one-way ANOVA and Bonferroni's test. Differences of P < 0.05 were deemed significant.
Results
Experiment 1
Figure 1A depicts effects of CV4 l-lactate infusion on blood glucose values in INS-injected rats. The results indicate that at +120 min after INS injection, circulating glucose was significantly decreased compared with SAL-injected euglycemic controls. Lactate infusion to INS-injected rats suppressed glucose levels below that achieved by INS treatment alone. The data in Fig. 1B portray the effects of treatment with INS alone versus the combination of INS + lactate on food intake over the 120-min duration of the study. INS-induced hypoglycemia stimulated food intake over this interval, but this response was completely reversed by hindbrain lactate repletion. Plasma glucagon (Fig. 1C) and corticosterone (Fig. 1D) concentrations were increased at +120 min after induction of hypoglycemia; concurrent CV4 lactate infusion did not modify either hormone profile at this time point. Figure 2 illustrates effects of INS-induced hypoglycemia, with or without CV4 lactate infusion, on AMPK (Fig. 2A) and pAMPK (Fig. 2B) protein levels in laser-microdissected DVC A2 noradrenergic neurons. Total AMPK protein in A2 cells did not differ among treatment groups. A2 cellular pAMPK levels were increased in response to INS but were restored to the baseline range by concomitant CV4 lactate infusion.
Fig. 1.

Effects of caudal fourth ventricular (CV4) infusion of l-lactate on blood glucose levels, hypoglycemic hyperphagia, and glucagon and corticosteorne secretion during insulin-induced hypoglycemia in male rats. A, C, and D depict mean values ± SE (n = 5/group) for blood glucose (mg/dl), plasma glucagon (ng/ml), and plasma corticosterone (ng/ml), respectively, at +2 h after continuous infusion with artificial cerebrospinal fluid (aCSF) or l-lactate (25 μM·2.0 μl−1·h−1) between −10 min and +120 min, and subcutaneous injection of saline (SAL) or neutral protamine Hagedorn insulin (INS; 12.5 U/kg body wt sc) at time 0 (t0). B shows mean grams of consumed food ± SE for n = 5 rats/group between t0 and +120 min after initiation of above treatments. Treatment groups are presented as follows: aCSF + SAL, aCSF + INS, l-lactate + INS. *P < 0.05 vs. aCSF + SAL; **P < 0.05 vs. aCSF + INS. The data show that INS significantly reduced blood glucose and that this decrement was enhanced by concomitant hindbrain lactate infusion in INS-injected rats. INS-induced hypoglycemia stimulated food intake, a response that was abolished by hindbrain lactate administration. Plasma glucagon and corticosterone levels were elevated during hypoglycemia and were not further modified at the time point examined by hindbrain lactate repletion.
Fig. 2.

Effects of CV4 l-lactate infusion on hypoglycemic patterns of adenosine 5′-monophosphate-activated protein kinase (AMPK) and phosphoAMPK (pAMPK) protein expression in dorsal vagal complex (DVC) A2 noradrenergic neurons. A and B depict mean normalized AMPK and pAMPK protein optical density (O.D.) levels ± SE, respectively, in A2 neurons harvested 2 h after treatment with aCSF + SAL, aCSF + INS, or l-lactate + INS. A2 neurons were identified by quick tyrosine hydroxylase (TH)-immunostaining of 10 μm- thick frozen sections through the caudal DVC and harvested by laser-catapult microdissection. Lysates of n = 50 A2 cells per treatment group (n = 10 neurons per rat) were analyzed by Western blot for AMPK, pAMPK, or α-tubulin; protein blots were performed in triplicate. Protein band O.D. measurement were quantified with AlphaImager HP V 5.0.1 software and expressed relative to tubulin. *P < 0.05 vs. aCSF + SAL; **P < 0.05 vs. aCSF + INS. The data show that neither INS nor combinatory INS + lactate treatment altered A2 AMPK expression, whereas A2 pAMPK levels were significantly enhanced during hypoglycemia and normalized by CV4 lactate infusion during hypoglycemia.
Figure 3 depicts effects of hindbrain lactate repletion on hypoglycemic patterns of ARH NPY (Fig. 3A), PVH OT (Fig. 3B), and LHA ORX (Fig. 3C) mRNA and protein expression. ARH NPY mRNA and protein levels were each elevated after INS injection; these responses were correspondingly attenuated or completely reversed by exogenous lactate delivery to the hindbrain. PVH OT mRNA and protein expression were inhibited by INS; lactate did not modify the latter response but exacerbated the decline in OT neuropeptide content. LHA ORX mRNA and protein content were respectively decreased and increased during INS-induced hypoglycemia. Lactate infusion had no impact on this gene response but reversed hypoglycemic stimulation of ORX neurotransmitter levels. Hypoglycemia increased pAMPK levels in ARH NPY (Fig. 4A) and LHA ORX (Fig. 4C) neurons but suppressed this protein profile in ARH POMC neurons (Fig. 4B). Hindbrain lactate infusion during hypoglycemia normalized AMPK activity in NPY and ORX neurons and reversed the decline in pAMPK in POMC cells to increase AMPK activation beyond that measured in controls.
Fig. 3.
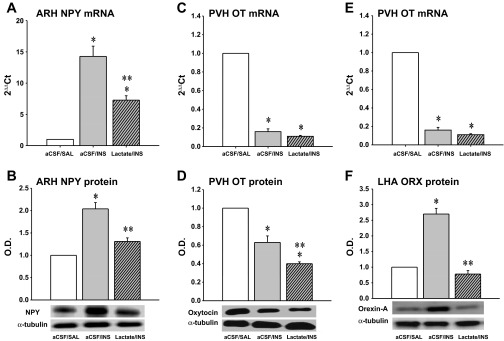
Effects of CV4 l-lactate infusion on hypoglycemia-associated patterns of arcuate nucleus (ARH) neuropeptide Y (NPY), paraventricular (PVH) oxytocin (OT), and lateral hypothalamic area (LHA) orexin (ORX) mRNA and protein expression. The data depict mean ARH NPY, PVH OT, and LHA ORX mRNA (A, B, and C) and normalized protein O.D. levels (C, D, and E) ± SE (n = 5 rats/group) 2 h after treatment with aCSF + SAL, aCSF + INS, or l-lactate + INS. For each animal, micropunches of the ARH, PVN, and LHA were obtained from one side of the hypothalamus in serial 200-μm-thick frozen sections over coordinates intervals of −2.00 to −3.00, −1.30 to −3.20, and −1.30 to −1.70 mm posterior to bregma, respectively, for qPCR analysis of NPY, OT, and ORX mRNA, respectively. Micropunches of these structures collected over identical intervals from the other hemihypothalamus were utilized for neuropeptide and α-tubulin Western blotting. *P < 0.05 vs. aCSF + SAL; **P < 0.05 vs. aCSF + INS. The data show that hypoglycemia increased ARH NPY gene and protein expression; concomitant CV4 lactate infusion only partially reversed this transcriptional response but normalized NPY protein profiles. PVH OT mRNA and neuropeptide levels were both diminished during hypoglycemia; hindbrain lactate repletion did not modify this gene profile but further suppressed OT protein levels. LHA ORX gene and protein expression were decreased or increased, respectively, in response to hypoglycema; lactate infusion to hypoglycemic animals did not alter this transcriptional response but normalized ORX protein production.
Fig. 4.
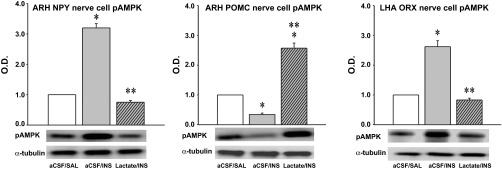
Effects of CV4 l-lactate infusion on ARH NPY, ARH pro-opiomelanocortin (POMC), and LHA ORX nerve cell pAMPK protein expression. The data depict mean normalized pAMPK levels ± SE in NPY (A), POMC (B), and ORX (C) neurons harvested 2 h after treatment with aCSF + SAL, aCSF + INS, or l-lactate + INS. Ten-micrometer frozen sections of the ARH or LHA were labeled for NPY or POMC versus ORX, respectively, using avidin-biotin-peroxidase immunocytochemical methods; antigen-containing neurons were subsequently harvested by laser-catapult microdissection. Lysates of n = 50 NPY-, POMC-, or ORX-ir cells per treatment group (n = 10 neurons per rat) were analyzed by Western blotting for pAMPK or α-tubulin; protein blots were performed in triplicate. *P < 0.05 vs. aCSF + SAL; **P < 0.05 vs. aCSF + INS. The data show that hypoglycemic augmentation of NPY and ORX and suppression of POMC pAMPK expression was reversed by hindbrain lactate delivery.
Experiment 2
Results presented in Fig. 5 show that CV4 4-CIN administration resulted in a significant increase in DVC A2 nerve cell pAMPK. The data in Fig. 6 show that mean numbers of TH-ir-positive neurons were significantly decreased at rostral, commissural, and caudal levels of the DVC in 6-OHDA- versus vehicle-pretreated rats. Figures 7A and 8B illustrate the stimulatory effects of this treatment paradigm on glucose levels and food intake, respectively. Pretreatment with the catecholamine neurotoxin 6-OHDA did not alter basal glucose or feeding (aCSF + SAL vs. 6-OHDA + SAL) but effectively blunted both hyperglycemic and hyperphagic responses to 4-CIN. 4-CIN increased glucagon (Fig. 7C) and corticosterone (Fig. 7D) secretion. Augmented glucagon and corticosterone output in 6-OHDA-pretreated animals was respectively unchanged or decreased, respectively, by 4-CIN.
Fig. 5.

Effects of CV4 administration of the monocarboxylate transporter inhibitor α-cyano-4-hydroxycinnamate (4-CIN) on pAMPK protein expression in A2 neurons. The data depict mean normalized pAMPK levels ± SE in A2 neurons harvested 2 h after CV4 infusion of SAL, or 4-CIN (50 μg·2.0 μl−1·30 min−1). TH-ir-positive A2 neurons were laser microdissected from 10-μm-frozen sections of the caudal DVC. Lysates of n = 50 A2 cells per treatment group (n = 10 neurons per rat) were analyzed by Western blotting for pAMPK or α-tubulin; protein blots were performed in triplicate. O.D. measures for pAMPK were normalized to α-tubulin. *P < 0.05 vs. SAL. The data show that A2 pAMPK expression was significantly increased by inhibition of hindbrain monocarboxylate transporter function.
Fig. 6.

Effects of CV4 6-hydroxydopamine (6-OHDA) administration on numbers of DVC tyrosine hydroxylase (TH)-immunoreactive neurons. In experiment 2, 25-μm serial sections were cut through one hemihindbrain (obtained at +2 h on the day of the study) of animals pretreated with 6-OHDA (75 μg/day; days −12 and −10) versus vehicle before continuous CV4 infusion of 4-CIN (50.0 μg/2.0 μl) between t0 and +30 min. Sections through the rostral, commissural, and caudal DVC were processed for immunofluorescence detection of TH-ir; images were captured with a Zeiss LSM 5 Pascal confocal microscope. Top: data presented in the chart show that neurotoxin administration significantly reduced mean bilateral counts of TH-ir-positive neurons at each level of the DVC. Bottom: representative photomicrographs depict immunostaining for TH in the caudal DVC of vehicle- (A) versus 6-OHDA (B)-treated animals.
Fig. 7.

Effects of CV4 administration of 4-CIN on blood glucose levels, food intake, and glucagon and corticosterone secretion in male rats. After pretreatment by intra-CV4 administration of 6-OHDA on days −12 and −10, adult male rats were infused with SAL or 4-CIN into the CV4. In A, C, and D, bars depict mean values ± SE (n = 5 rats/group) for blood glucose levels (mg/dl), plasma glucagon (pg/ml), and plasma corticsterone (ng/ml) 2 h after treatment with vehicle + SAL, vehicle + 4-CIN, 6-OHDA + SAL, or 6-OHDA + 4-CIN. In B, bars depict mean grams of food consumed ± SE (n = 5 rats/group) between t0 and +120 min after initiation of above treatments. *P < 0.05 vs. matched SAL control; **P < 0.05 vs. vehicle + 4-CIN; #P < 0.05 vs. vehicle + SAL. The data show that 4-CIN elicited hyperglycemia and hyperphagia and that these responses were either blunted or prevented by 6-OHDA pretreatment. 4-CIN-induced increases in glucagon and corticosterone secretion were unaffected or reversed, respectively, by 6-OHDA.
Fig. 8.
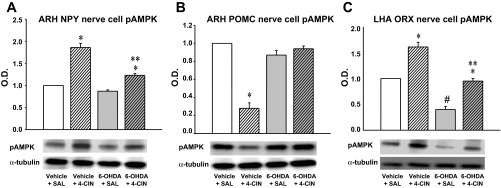
Effects of 6-OHDA on ARH NPY, ARH POMC, and LHA ORX nerve cell pAMPK expression in CV4 4-CIN-treated rats. Bars depict mean normalized pAMPK levels ± SE in NPY (A), POMC (B), and ORX (C) neurons harvested at 2 h after treatment with vehicle + SAL, vehicle + 4-CIN, 6-OHDA + SAL, or 6-OHDA + 4-CIN. Lysates of n = 50 NPY-, POMC-, or ORX-ir cells per treatment group (n = 10 neurons per rat) were analyzed by Western blot for pAMPK and α-tubulin expression; each protein blot were performed in triplicate. *P < 0.05 vs. matched SAL control; **P < 0.05 vs. vehicle + 4-CIN; #P < 0.05 vs. vehicle + SAL. Intra-CV4 delivery of 4-CIN significantly elevated NPY and decreased POMC nerve cell pAMPK levels, responses that were respectively blunted or abolished by 6-OHDA. 4-CIN also elevated pAMPK expression in ORX neurons; 6-OHDA pretreatment inhibited baseline ORX neuropeptide profiles and normalized pAMPK expression hypoglycemia to levels measured in vehicle + SAL controls.
As shown in Fig. 8A, levels of pAMPK in laser-microdissected ARH NPY neurons were dramatically augmented in response to hindbrain 4-CIN delivery. This protein profile was significantly diminished in NPY neurons from 6-OHDA + 4-CIN versus vehicle + 4-CIN treatment groups. Figure 8B shows that ARH POMC neurons from 4-CIN-treated animals had significantly diminished pAMPK content compared with SAL controls. This negative sensor response to hindbrain monocarboxylate transporter inhibition was abolished in rats pretreated with 6-OHDA, despite drug-induced augmentation of blood glucose levels. As shown in Fig. 8C, 4-CIN elevated pAMPK levels in LHA ORX neurons. Animals pretreated with 6-OHDA exhibited a significant reduction in baseline pAMPK in this cell population. ORX neurons from vehicle + SAL and 6-OHDA + 4-CIN rats had similar pAMPK levels, despite elevated blood glucose levels in the latter group.
Figures 9 and 10 indicate that 6-OHDA pretreatment did not significantly alter numbers of DVC neurons expressing immunoreactivity for corticotropin-releasing hormone or neuronal nitric oxide synthase, respectively. Patterns of immunolabeling with antisera for somatostatin and neuropeptide Y revealed immunopositive nerve cell processes, but not cell bodies in the DVC.
Fig. 9.

Effects of CV4 6-OHDA administration on DVC corticotropin-releasing hormone (CRH)-immunoreactive nerve cell counts. Sections through the commissural and caudal DVC of SAL-injected vehicle- versus 6-OHDA-pretreated rats were processed for confocal microscopic analysis of CRH-ir. Top: data in the chart shown indicate that 6-OHDA did not alter mean bilateral counts of CRH-ir-positive neurons in either DVC region. Representative photomicrographs depict immunostaining for cytoplasmic CRH-ir alone (A and B) or merged CRH-ir and nuclear NAPI labeling (C and D) after vehicle (left-hand column) versus 6-OHDA (right-hand column) treatment.
Fig. 10.

Effects of CV4 6-OHDA administration on DVC neuronal nitric oxide synthase (nNOS)-immunoreactive nerve cell counts. Sections through the commissural and caudal DVC of SAL-injected vehicle- versus 6-OHDA-pretreated rats were processed for confocal microscopic analysis of nNOS-ir. Top: data in the chart show that 6-OHDA had no effect on nNOS-immunoreactive cell numbers at either DVC level. Representative photomicrographs depict immunostaining for nNOS-ir alone (A and B) or merged nNOS-ir and DAPI labeling (C and D) following vehicle (left-hand column) versus 6-OHDA (right-hand column) treatment.
DISCUSSION
The present research provides novel evidence that availability of the glucose metabolite lactate, within the caudal hindbrain, regulates local and extra-hindbrain AMPK activity, key hypothalamic metabolic neuropeptide transmitter synthesis, and glucostasis. We previously reported that DVC A2 noradrenergic neurons express AMPK and that this enzyme is activated in response to hypoglycemia in male rats (13). Current results identify lactoprivation as a stimulus for A2 nerve cell AMPK phosphorylation during systemic glucose dyshomeostasis and implicate this cell type as a source of origin of lactoprivic signaling to the hypothalamus. Data show that caudal hindbrain monocarboxylate transporter inhibition increases or decreases pAMPK levels in hypothalamic anabolic (ARH NPY, LHA ORX) versus catabolic (ARH POMC) neurons, respectively, coincident with hyperglycemia, hyperphagia, and elevated glucagon and corticosterone secretion. Hypoglycemia elicited analogous changes in pAMPK profiles in these hypothalamic cell groups, responses that were reversed by caudal hindbrain lactate repletion. Hypoglycemic augmentation of ARH NPY and LHA ORX protein expression and food intake were also normalized by lactate provision to the caudal hindbrain. These present studies show that hindbrain lactoprivation potently stimulates endocrine and behavioral outflow that elevates blood glucose through mechanisms that may involve one or more metabolic effector cell groups examined here. Moreover, the ability of hindbrain substrate fuel status to alter hypothalamic assessment of energy status despite local and peripheral signals of metabolic disturbance supports the novel concept that hindbrain energy state supplants that of the hypothalamus in regulating CNS responses to metabolic compensation.
The ubiquitous cellular energy sensor AMPK is expressed in multiple brain regions, including the hypothalamus and hindbrain. Initial research on central AMPK regulation of body-wide metabolic stasis focused on the hypothalamus, but recent studies implicate hindbrain DVC AMPK in neural control of food intake (23) and glucostasis (25). Our work was the first to localize this sensor to a specific neurochemical phenotype in the DVC, e.g., A2 noradrenergic neurons, where it is revealed to be hypoglycemia sensitive (13). Here, we found that hypoglycemia-induced A2 AMPK activation is reversed by CV4 lactate infusion, whereas 4-CIN delivery to the caudal hindbrain increases pAMPK expression in these cells. These findings demonstrate a correlation between lactate deficiency and AMPK activation in A2 neurons; our presumption that increased sensor activity is mediated, in part, by decreased energy production awaits experimental confirmation. Current efforts by our group aim to identify the molecular mechanisms whereby A2 AMPK activity regulates noradrenergic input to central metabolic structures. Evidence for concurrent down- and upregulation of MCT2 and GLUT3 proteins, respectively, in A2 neurons during hypoglycemia (6) implies that enhanced AMPK activity reflects, in part, diminished lactate utilization. This notion is supported by current proof that A2 pAMPK levels are augmented by pharmacological suppression of hindbrain monocarboxylate transport. Taken together, these results support the view that reduced decreased lactate uptake, alone or relative to glucose, may be a critical manifestation to A2 cells during hypoglycemia. The current studies do not exclude the possibility that A2 neurons may utilize both AMPK-dependent and -independent mechanisms to monitor cellular lactate status.
Prior research on the role of hypothalamic NPY, ORX, and OT in regulation of food intake and glucostasis relied chiefly on neuropeptide gene expression as an indicator of probable changes in neurotransmitter signaling during dyshomeostasis. The present studies show that NPY gene and protein profiles were both elevated, whereas OT mRNA and protein expression were each reduced during hypoglycemia. On the contrary, ORX gene and protein profiles deviated in response to this challenge, with only the latter undergoing augmentation. For each neurotransmitter investigated here, hindbrain lactate repletion exerted dissimilar effects on hypoglycemic patterns of mRNA versus protein expression. For instance, this treatment paradigm only partially reversed increases in NPY mRNA but effectively normalized NPY protein expression. Furthermore, this treatment had no effect on OT or ORX gene profiles, but either exacerbated (OT) or completely reversed (ORX) these protein responses to hypoglycemia. Taken together, these data imply that input on hindbrain lactate status may differentially regulate NPY, OT, and ORX transcription versus translation/posttranslational processing during hypoglycemia, with the latter being a critical target level of control. Additional work is needed to determine if effects of hindbrain lactate signaling on neuropeptide production correlate with adjustments in synaptic neurotransmitter release. Normalization of NPY and ORX neurotransmitter production in INS + lactate animals, despite exacerbated hypoglycemia, supports the prospect that these hypothalamic cell groups may exhibit relatively greater sensitivity to hindbrain lactate status versus adjustments in blood glucose decrements. It should be noted that the extent of hindbrain lactate repletion effects on hypoglycemia-associated patterns of hypothalamic neuropeptide yield likely reflects a balance of opposing signals, namely hindbrain energy sufficiency, signified by normalized A2 pAMPK profiles, versus intensified blood glucose decrements. Further effort is required to ascertain if lactoprivic regulation of NPY, ORX, and OT involves direct innervation of these neuropeptidergic cell groups by A2 neurons, or alternatively, is mediated by polysynaptic pathways that originate with these cells. It would also be beneficial to learn if NPY, ORX, and OT protein responses to hindbrain lactate repletion are causally related to concurrent normalization of food intake during hypoglycemia. We previously reported that LHA ORX and PVH OT mRNA levels were increased or unchanged, respectively, in response to acute hypoglycemia (5, 45), while the current qPCR data demonstrate a reduction in both gene profiles in response to this condition. Differences between this and earlier studies include utilization of greater rostrocaudal sample sizes and micropunch volume here; thus, prior findings may reflect, in part, distinctive reactivity of regional populations of ORX and OT neurons to hypoglycemia.
Combinatory laser-catapult microdissection and high-sensitivity Western blotting was applied here, for the first time, to investigate AMPK activity state in hypothalamic metabolosensory neurons in vivo during insulin-induced hypoglycemia. Our data show that ARH NPY and POMC neurons exhibit increased or decreased pAMPK, respectively, in response to this metabolic stress. These results concur with earlier in vitro observations that adjustments in media glucose provision alter sensor activity in immortalized hypothalamic cell lines (12, 38). The current research provides novel evidence for hypoglycemic enhancement of LHA ORX nerve cell pAMPK, data that extend recent immunocytochemical reports of AMPK-ir in this cell type (28). Inhibition of hindbrain monocarboxylate transport elicited similar adjustments in pAMPK profiles in these cell groups, concurrent with hyperglycemia, as this protein profile was increased in NPY and ORX neurons but decreased in POMC nerve cells. These results, together with evidence that CV4 lactate infusion during hypoglycemia normalizes NPY, POMC, and ORX nerve cell AMPK activity despite heightened hypoglycemia, demonstrate that hindbrain lactostasis is a critical determinant of hypothalamic AMPK function, independent of secondary sequelae such as changes in blood glucose levels. Parallel adjustments in NPY and ORX nerve cell AMPK content versus NPY and ORX protein, but not mRNA expression in both INS- and lactate + INS-treated rats, suggest that translation/posttranslational processing of these neurotransmitters may be more sensitive, relative to NPY and ORX gene transcription, to AMPK control. Taken together, results provide novel evidence that hindbrain lactoprivic signaling is a critical controller of phosphorylation of hypothalamic AMPK and manufacture of key hypothalamic effector neurotransmitters during hypoglycemia.
6-OHDA pretreatment blunted 4-CIN-induced stimulation of NPY and LHA pAMPK and prevented inhibition of POMC nerve cell pAMPK content by 4-CIN, results that indicate catecholamine involvement in hindbrain lactoprivic regulation of these hypothalamic sensors. The 6-OHDA treatment paradigm used here destroyed TH-ir neurons throughout the rostrocaudal extent of the DVC. Since this manipulation did not differentiate between C2 adrenergic neurons and A2 noradrenergic neurons [which reside at rostral and caudal levels of the DVC, respectively, and coexist in the commissural DVC], our results do not shed light on individual roles of these cell types in lactoprivic signaling to the hypothalamus. Since A2 neurons express metabolosensory biomarkers, including AMPK (6, 13, 14), it is quite likely that attenuating effects of 6-OHDA on hindbrain lactoprivic regulation of hypothalamic AMPK activity reflect, in part, loss of these cells; nonetheless, this conclusion requires experimental verification. Original electrophysiological mapping studies of the DVC showed that substrate fuel-sensitive neurons reside primarily at caudal levels of this structure (36), findings that were recently confirmed by reports that AMPK regulation of feeding occurs within the caudal, not rostral DVC (23). While it is doubtful that 6-OHDA ablation of C2 neurons had a substantial impact on DVC lactoprivic signaling, the possibility that C2 nerve cells function downstream of A2 neurons or metabolic monitoring cells located external to the DVC cannot be discounted. This prospect is supported by preliminary data from our laboratory that C2 neurons do not express pAMPK (Shrestha and Briski, unpublished observations).
The current studies confirm earlier reports that hindbrain lactoprivation elevates circulating glucose levels (49) and demonstrate this manipulation also increases food intake and glucagon and corticosterone release. The data also show that hindbrain lactate repletion during hypoglycemia normalizes net food intake but not glucagon or corticosterone secretion. Parallel normalization of food intake and NPY and ORX neuropeptide synthesis in lactate + INS-treated rats supports the likely involvement of one or both neurotransmitters in this behavioral response. While OT is reported to inhibit feeding (30) and to stimulate glucagon and corticosterone secretion (3, 4, 15), this neurotransmitter may not regulate feeding or hormone profiles during lactate repletion, as this manipulation exacerbated hypoglycemic suppression of PVH OT neuropeptide levels. Since glucagon and corticosterone were evaluated at a single time point, e.g., +120 min after induction of hypoglycemia with or without lactate infusion, we do not know whether lactate repletion reduced or otherwise altered the release of one or both hormones before or after that singular time point. Current results do not exclude a role for the hypothalamic neuropeptides evaluated here in patterns of hypoglycemic hyperglucagonemia and hypercorticosteronemia but rather support the notion that regulation of these counterregulatory hormones reflects a balance between input (of peripheral origin) on blood glucose levels versus (central signals of) nerve cell metabolic status. The ability versus inability of lactate repletion to normalize feeding versus glucagon and corticosterone secretion may indicate that the feeding circuitry is more sensitive to hindbrain lactate status than absolute blood glucose levels. Conversely, glucagon and corticosterone secretion was increased coincident with hyperglycemia, following 4-CIN treatment. Taken together, these results suggest that signals of privation, either systemic or cellular, supersede stimuli that signal sufficiency. Baseline glucagon and corticosterone output were both augmented by CV4 6-OHDA administration, whereas blood glucose and food intake were not disturbed by this manipulation. These results may reflect development of “supersensitivity” to diminished catecholamine input by receptors involved in regulation of the former but not the latter functions (34). Current data show that neurotoxin-treated animals exhibited blunted or absence of 4-CIN-induced hyperglycemia, hyperphagia, and hyperglucagonemia, respectively, data that implicate catecholamine neurons, including AMPK-expressing A2 neurons, as a principal source of hindbrain lactoprivic control of these functions. On the other hand, 6-OHDA reversed the direction of 4-CIN effect on corticosterone secretion from stimulation to inhibition; this outcome suggests that the strength of catecholamine signaling may dictate the direction of lactoprivic influence on this particular hormone. Demonstrable residual effects of 4-CIN treatment in 6-OHDA-treated animals suggest that A2 neurons may not be the sole source of lactoprivic regulatory signaling; further studies are needed to determine potential involvement of populations observed here to escape neurotoxin destruction, e.g., corticotropin-releasing hormone and nitrergic neurons.
Other investigators observed that systemic injection of the glucose antimetabolite 2-deoxy-d-glucose does not alter dorsomedial hindbrain pAMPK levels (31). The current studies differ significantly from that work in that pharmacological induction of glucopenia deviates from the physiological model of insulin-induced hypoglycemia in that it elicits elevated, rather than decreased, blood glucose. We have observed that fourth ventricular administration of 5-thio-glucose inhibits A2 nerve cell pAMPK, coincident with elevated glucose (Ibrahim and Briski, unpublished observations). Another difference between these studies concerns sample size and neuroanatomical resolution. Our samples consisted of immuno-identified A2 cells whereas Li et al. (31) analyzed pAMPK in 2.0-mm thick tissue punches, an approach that would preclude neural structure, let alone neuron population discrimination. Our results also diverge from reports that manipulation of DVC AMPK activity does not impact whole hypothalamic pAMPK (23). We surmise that averaged pAMPK protein measures could likely obscure concomitant adjustments in this protein profile in different locations within the hypothalamus, including reverse changes in direction of NPY and POMC pAMPK levels.
Perspectives and Significance
The present studies provide novel evidence for functional connectivity between hindbrain and hypothalamus sensors, with input from the former regulating the latter, as opposed to autonomous operation of these neuroanatomically distinct populations. This work justifies the need for systems-level perspective in research on CNS reactivity to glucose dyshomeostasis. Future work is likely to focus on anatomical and neurochemical foundations of communication among individual metabolosensory cell groups as well as downstream signaling from these cells to integrative and motor elements of the central glucoregulatory circuitry. Moreover, effort is also needed to determine whether individual sensors detect and differentially prioritize a unique set of nutrient, hormone, and neurochemical signals.
GRANTS
This research was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grant DK-085122.
REFERENCES
- 1.Alquier T, Kawashima J, Tsuji Y, Kahn BB. Role of hypothalamic adenosine 5′-monophosphate-activated protein kinase in the impaired counterregulatory response induced by repetitive neuroglucopenia. Endocrinology 148: 1367–1375, 2007 [DOI] [PubMed] [Google Scholar]
- 2.Andrews ZB, Liu ZW, Wallingford N, Erion DM, Borok E, Friedman JM, Tschöp MH, Shanabrough M, Cline G, Shulman GI, Coppola A, Gao XB, Horvath TL, Diano S. UCP2 mediates ghrelin's action on NPY/AgRP neurons by lowering free radicals. Nature 454: 846–851, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bjorkstrand E, Eriksson M, Uvnas-Moberg K. Evidence of a peripheral and a central effect of oxytocin on pancreatic hormone release in rats. Neuroendocrinology 63: 337–383, 1996 [DOI] [PubMed] [Google Scholar]
- 4.Bjorkstrand E, Eriksson M, Uvnas-Moberg K. Plasma levels of oxytocin after food deprivation and hypoglycaemia, and effects of 1-deamino-2-d-tyr-(OEt)-4-Thr-8-Orn-oxytocin on blood glucose levels in rats. Acta Physiol Scand 144: 355–359, 1992 [DOI] [PubMed] [Google Scholar]
- 5.Briski KP, Kale AY, Vavaiya KV. Impact of recurring intermediate insulin-induced hypoglycemia on hypothalamic paraventricular corticotropin-releasing hormone, oxytocin, vasopressin, and glucokinase gene profiles: role of type II glucocorticoid receptors. Exp Brain Res 195: 499–507, 2009 [DOI] [PubMed] [Google Scholar]
- 6.Briski KP, Koshy Cherian A, Genabai NK, Vavaiya KV. In situ coexpression of glucose and monocarboxylate transporter mRNAs in metabolic-sensitive dorsal vagal complex catecholaminergic neurons: transcriptional reactivity to insulin-induced hypoglycemia and caudal hindbrain glucose or lactate repletion during insulin-induced hypoglycemia. Neuroscience 164: 1152–1160, 2009 [DOI] [PubMed] [Google Scholar]
- 7.Briski KP, Nedungadi TP. Adaptation of feeding and counter-regulatory hormone responses to intermediate insulin-induced aa in the ovariectomised female rat: effects of oestradiol. J Neuroendocrinol 21: 578–585, 2009 [DOI] [PubMed] [Google Scholar]
- 8.Briski KP, Nedungadi TP, Koshy Cherian A. Effects of hypoglycaemia on neurotransmitter and receptor gene expression in arcuate neuropeptide Y/agouti-related peptide neurones. J Neuroendocrinol 22: 599–607, 2010 [DOI] [PubMed] [Google Scholar]
- 9.Broer S, Rahman B, Pellegri G, Pellerin L, Martin JL, Verleysdonk S, Hamprecht B, Magistretti PJ. Comparison of lactate transport in astroglial cells and monocarboxylate transporter (MCT 1) expressing Xenopus laevis oocytes. Expression of two different monocarboxylate transporters in astroglial cells and neurons. J Biol Chem 272: 30096–30102, 1997 [DOI] [PubMed] [Google Scholar]
- 10.Burdakov D, Gerasimenko O, Verkhratsky A. Physiological changes in glucose differentially modulate the excitability of hypothalamic melanin-concentrating hormone and orexin neurons in situ. J Neurosci 25: 2429–2433, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cai XJ, Evans ML, Lister CA, Leslie RA, Arch JR, Wilson S, Williams G. Hypoglycemia activates orexin neurons and selectively increases hypothalamic orexin-B levels: responses inhibited by feeding and possibly mediated by the nucleus of the solitary tract. Diabetes 50: 105–112, 2001 [DOI] [PubMed] [Google Scholar]
- 12.Cai F, Gyulkhandanyan AV, Wheeler MB, Belsham DD. Glucose regulates AMP-activated protein kinase activity and gene expression in clonal, hypothalamic neurons expressing proopiomelanocortin: additive effects of leptin or insulin. J Endocrinol 192: 605–614, 2007 [DOI] [PubMed] [Google Scholar]
- 13.Cherian AK, Briski KP. Quantitative RT PCR and immunoblot analyses reveal acclimated A2 noradrenergic neuron substrate fuel transporter, glucokinase, phospho-AMPK, and dopamine-beta-hydroxylase responses to hypoglycemia. J Neurosci Res 89: 1114–1124, 2011 [DOI] [PubMed] [Google Scholar]
- 14.Cherian AK, Briski KP. A2 noradrenergic nerve cell metabolic transducer and nutrient transporter adaptation to hypoglycemia: impact of estrogen. J Neurosci Res 90: 1347–1358, 2012 [DOI] [PubMed] [Google Scholar]
- 15.Chiodera P, Volpi R, Capretti L, Speroni G, Marcato A, Rossi G, Coiro R. Hypoglycemia-induced arginine vasopressin and oxytocin release is mediated by glucoreceptors located inside the blood-brain barrier. Neuroendocrinology 55: 655–659, 1992 [DOI] [PubMed] [Google Scholar]
- 16.Claret M, Smith MA, Batterham RL, Selman C, Choudhury AI, Fryer LG, Clements M, Al-Qassab H, Heffron H, Xu AW, Speakman JR, Barsh GS, Viollet B, Vaulont S, Ashford ML, Carling D, Withers DJ. AMPK is essential for energy homeostasis regulation and glucose sensing by POMC and AgRP neurons. J Clin Invest 117: 2325–2336, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cone RD, Cowley MA, Butler AA, Fan W, Marks DL, Low MJ. The arcuate nucleus as a conduit for diverse signals relevant to energy homeostasis. Intl J Obes Relat Metab Disord 25: S63–S67, 2001 [DOI] [PubMed] [Google Scholar]
- 18.Dringen R, Gebhardt R, Hamprecht B. Glycogen in astrocytes: possible function as lactate supply for neighboring cell. Brain Res 623: 208–214, 1993 [DOI] [PubMed] [Google Scholar]
- 19.Fioramonti X, Contié S, Song Z, Routh VH, Lorsignol A, Pénicaud L. Characterization of glucosensing neuron subpopulations in the arcuate nucleus: integration in neuropeptide Y and pro-opio melanocortin networks? Diabetes 56: 1219–1227, 2007 [DOI] [PubMed] [Google Scholar]
- 20.Griffond B, Risold PY, Jacquemard C, Colard C, Fellmann D. Insulin-induced hypoglycemia increases preprohypocretin (orexin) mRNA in the rat lateral hypothalamic area. Neurosci Lett 262: 77–80, 1999 [DOI] [PubMed] [Google Scholar]
- 21.Han SM, Namkoong C, Jang PG, Park IS, Hong SW, Katakami H, Chun S, Kim SW, Park JY, Lee KU, Kim MS. Hypothalamic AMP-activated protein kinase mediates counter-regulatory responses to hypoglycaemia in rats. Diabetologia 48: 2170–2178, 2005 [DOI] [PubMed] [Google Scholar]
- 22.Hardie DG. Minireview: The AMP-activated protein kinase cascade: the key sensor of cellular energy status. Endocrinology 144: 5179–5183, 2003 [DOI] [PubMed] [Google Scholar]
- 23.Hayes MR, Skibicka KP, Bence KK, Grill HJ. Dorsal hindbrain 5′-adenosine monophosphate-activated protein kinase as an intracellular mediator of energy balance. Endocrinology 150: 2175–2182, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Himmi T, Perrin J, Dallaporta M, Orsini JC. Effects of lactate on glucose-sensing neurons in the solitary tract nucleus. Physiol Behav 74: 391–397, 2001 [DOI] [PubMed] [Google Scholar]
- 25.Ibrahim B, Tamrakar P, Gujar A, Koshy Cherian A, Brisk KP. Caudal fourth ventricular administration of the AMPK activator 5-aminoimiazole-4-carboxamide-riboside regulates glucose and counterregulatory hormone profiles, dorsal vagal complex metabolosensory neuron function, and hypothalamic Fos expression. J Neurosci Res 91: 1226–1238, 2013 [DOI] [PubMed] [Google Scholar]
- 26.Kale AY, Paranjape SA, Briski KP. I.c.v administration of the nonsteroidal glucocorticoid receptor antagonist, CP4–72555, prevents exacerbated hypoglycemia during repeated insulin administration. Neuroscience 140: 555–565, 2006 [DOI] [PubMed] [Google Scholar]
- 27.Kahn BB, Alquier T, Carling D, Hardie DG. AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab 1: 15–25, 2005 [DOI] [PubMed] [Google Scholar]
- 28.Kuhla B, Görs S, Metges CC. Hypothalamic orexin A expression and the involvement of AMPK and PPAR-gamma signalling in energy restricted dairy cows. Arch Tierz 54: 567–575, 2011 [Google Scholar]
- 29.Lage R, Diéguez C, Vidal-Puig A, López M. AMPK: a metabolic gauge regulating whole-body energy homeostasis. Trends Mol Med 14: 539–549, 2008 [DOI] [PubMed] [Google Scholar]
- 30.Leng G, Onaka T, Caquineau C, Sabatier N, Tobin VA, Takayanagi Y. Oxytocin and appetite. Prog Brain Res 170: 137–51, 2008 [DOI] [PubMed] [Google Scholar]
- 31.Li AJ, Wang Q, Ritter S. Participation of hindbrain AMP-activated protein kinase in glucoprivic feeding. Diabetes 60: 436–442, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lim CT, Kola B, Korbonits M. AMPK as a mediator of hormonal signalling. J Mol Endocrinol 44: 87–97, 2010 [DOI] [PubMed] [Google Scholar]
- 33.Marks JL, Waite K. Intracerebroventricular neuropeptide Y acutely influences glucose metabolism and insulin sensitivity in the rat. J Neuroendocrinol 9: 99–103, 1997 [DOI] [PubMed] [Google Scholar]
- 34.Mileson BE, Lewis MH, Mailman RB. Dopamine receptor “supersensitivity” occurring without receptor up-regulation. Brain Res 561: 1–10, 1991 [DOI] [PubMed] [Google Scholar]
- 35.Minokoshi M, Alquier T, Furukawa N, Kim YB, Lee A, Xue B, Mu J, Foufelle F, Ferré P, Birnbaum MJ, Stuck BJ, Kahn BB. AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature 428: 569–574, 2004 [DOI] [PubMed] [Google Scholar]
- 36.Mizuno Y, Oomura Y. Glucose responding neurons in the nucleus tractus solitarius of the rat: in vitro study. Brain Res 307: 109–116, 1984 [DOI] [PubMed] [Google Scholar]
- 37.Moriguchi T, Sakurai T, Nambu T, Yanagisawa M, Goto K. Neurons containing orexin in the lateral hypothalamic area of the adult rat brain are activated by insulin-induced acute hypoglycemia. Neurosci Lett 264: 101–104, 1999 [DOI] [PubMed] [Google Scholar]
- 38.Mountjoy PD, Bailey SJ, Rutter GA. Inhibition by glucose or leptin of hypothalamic neurons expressing neuropeptide Y requires changes in AMP-activated protein kinase activity. Diabetologia 50: 168–177, 2007 [DOI] [PubMed] [Google Scholar]
- 39.Murphy BA, Fioramonti X, Jochnowitz N, Fakira K, Gagen K, Contie G, Lorsignol A, Penicaud L, Martin WJ, Routh VH. Fasting enhances the response of arcuate neuropeptide Y-glucose-inhibited neurons to decreased extracellular glucose. Am J Physiol Cell Physiol 296: C746–C756, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nedungadi TP, Briski KP. Effects of estradiol on acute and recurrent insulin-induced hypoglycemia-associated patterns of arcuate neuropeptide Y, proopiomelanocortin, and cocaine- and amphetamine related-transcript gene expression in the ovariectomized rat. Neuroendocrinology 86: 270–276, 2007 [DOI] [PubMed] [Google Scholar]
- 41.Nedungadi TP, Briski KP. Effects of intracerebroventricular administration of the NPY-Y1 receptor antagonist, 1229U91, on hyperphagic and glycemic responses to acute and chronic intermediate insulin-induced hypoglycemia in female rats. Regul Pept 159: 14–18, 2010 [DOI] [PubMed] [Google Scholar]
- 42.Nijima A. Nervous regulation of metabolism. Prog Neurobiol 33:135–147; 1988 [DOI] [PubMed] [Google Scholar]
- 43.Oomura Y, Yoshimatsu H. Neural network of glucose monitoring system. J Auton Nerv Syst 10: 359–372, 1984 [DOI] [PubMed] [Google Scholar]
- 44.Paranjape SA, Briski KP. Recurrent insulin-induced hypoglycemia causes site-specific patterns of habituation or amplification of CNS neuronal genomic activation. Neuroscience 130: 957–970, 2005 [DOI] [PubMed] [Google Scholar]
- 45.Paranjape SA, Vavaiya KV, Kale AY, Briski KP. Habituation of insulin-induced hypoglycemic transcription activation of lateral hypothalamic orexin-A-containing neurons to recurring exposure. Regul Pept 135: 1–6, 2006 [DOI] [PubMed] [Google Scholar]
- 46.Paranjape SA, Vavaiya KV, Kale AY, Briski KP. Role of dorsal vagal motor nucleus orexin- receptor-1 in glycemic responses to acute versus repeated insulin administration. Neuropeptides 41: 111–116, 2007 [DOI] [PubMed] [Google Scholar]
- 47.Parikh R, Marks JL. Metabolic and orexigenic effects of intracerebroventricular neuropeptide Y are attenuated by food deprivation. J Neuroendocrinol 9: 789–795, 1997 [DOI] [PubMed] [Google Scholar]
- 48.Parker JA, Bloom SR. Hypothalamic neuropeptides and the regulation of appetite. Neuropharmacology 63: 18–30, 2012 [DOI] [PubMed] [Google Scholar]
- 49.Patil GD, Briski KP. Lactate is a critical “sensed” variable in caudal hindbrain monitoring of CNS metabolic stasis. Am J Physiol Regul Integr Comp Physiol 289: R1777–R1786, 2005 [DOI] [PubMed] [Google Scholar]
- 50.Patil GD, Briski KP. Induction of Fos immunoreactivity labeling in forebrain metabolic loci by caudal fourth ventricular administration of the monocarboxylate transporter inhibitor, α-cyano-4-hydroxycinnamic acid. Neuroendocrinology 82: 49–57, 2005 [DOI] [PubMed] [Google Scholar]
- 51.Patil GD, Briski KP. Transcriptional activation of nucleus tractus solitarii/area postrema catecholaminergic neurons by pharmacological inhibition of caudal hindbrain monocarboxylate transporter function. Neuroendocrinology 81: 96–102, 2005 [DOI] [PubMed] [Google Scholar]
- 52.Selvage DJ, Lee SY, Parsons LH, Seo DO, Rivier CL. A hypothalamic-testicular neural pathway is influenced by brain catecholamines, but not testicular blood flow. Endocrinology 145: 1750–1759, 2004 [DOI] [PubMed] [Google Scholar]
- 53.Vavaiya KV, Briski KP. Caudal hindbrain lactate administration alters glucokinase, SUR1, and neuronal substrate fuel transporter gene expression in the dorsal vagal complex, lateral hypothalamic area, and ventromedial nucleus hypothalamus of hypoglycemic male rats. Brain Res 1176: 62–70, 2007 [DOI] [PubMed] [Google Scholar]
- 54.Vavaiya KV, Briski KP. Effects of caudal hindbrain lactate infusion on insulin-induced hypoglycemia and neuronal substrate transporter, glucokinase, and sulfonylurea receptor-1 gene expression in the ovariectomized female rat dorsal vagal complex: impact of estradiol. J Neurosci Res 86: 694–701, 2008 [DOI] [PubMed] [Google Scholar]
- 55.van den Hoek AM, Voshol PJ, Karnekamp BN, Buijs RM, Romijn JA, Havekes LM, Pijl H. Intracerebroventricular neuropeptide Y infusion precludes inhibition of glucose and VLDL production by insulin. Diabetes 53: 2529–2534, 2004 [DOI] [PubMed] [Google Scholar]
- 56.Williams RH, Alexopoulos H, Jensen LT, Fugger L, Burdakov D. Adaptive sugar sensors in hypothalamic feeding circuits. Proc Natl Acad Sci USA 105: 11975–11980, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wyss MT, Jolivet R, Buck A, Magistretti PJ, Weber B. In vivo evidence for lactate as a neuronal energy source. J Neurosci 31: 7477–7485, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]