

Abstract
Peroxisome proliferator-activated receptor (PPAR)-δ is a nuclear hormone receptor that is mainly involved in lipid metabolism. Recent studies have suggested that PPAR-δ agonists exert vascular protective effects. The present study was designed to characterize vascular function in mice with genetic inactivation of PPAR-δ in the endothelium. Mice with vascular endothelial cell-specific deletion of the PPAR-δ gene (ePPARδ−/− mice) were generated using loxP/Cre technology. ePPARδ−/− mice were normotensive and did not display any sign of metabolic syndrome. Endothelium-dependent relaxations to ACh and endothelium-independent relaxations to the nitric oxide (NO) donor diethylammonium (Z)-1-(N,N-diethylamino)diazen-1-ium-1,2-diolate were both significantly impaired in the aorta and carotid arteries of ePPARδ−/− mice (P < 0.05). In ePPARδ−/− mouse aortas, phosphorylation of endothelial NO synthase at Ser1177 was significantly decreased (P < 0.05). However, basal levels of cGMP were unexpectedly increased (P < 0.05). Enzymatic activity of GTP-cyclohydrolase I and tetrahydrobiopterin levels were also enhanced in ePPARδ−/− mice (P < 0.05). Most notably, endothelium-specific deletion of the PPAR-δ gene significantly decreased protein expressions of catalase and glutathione peroxidase 1 and resulted in increased levels of H2O2 in the aorta (P < 0.05). In contrast, superoxide anion production was unaltered. Moreover, treatment with catalase prevented the endothelial dysfunction and elevation of cGMP detected in aortas of ePPARδ−/− mice. The findings suggest that increased levels of cGMP caused by H2O2 impair vasodilator reactivity to endogenous and exogenous NO. We speculate that chronic elevation of H2O2 predisposes PPAR-δ-deficient arteries to oxidative stress and vascular dysfunction.
Keywords: endothelial dysfunction, nitric oxide, tetrahydrobiopterin, carotid artery, hydrogen peroxide, peroxisome proliferator-activated receptor-δ, endothelial nitric oxide synthase
peroxisome proliferator-activated receptor (PPAR)-δ, together with PPAR-α and PPAR-γ, constitute the PPAR subfamily of the nuclear receptor superfamily (6). All three PPAR subtypes share structural and functional similarities and control transcription through binding to specific PPAR-responsive elements in the target gene promoters and also repress gene transcription by a mechanism that is independent of DNA binding (19). PPAR-δ is ubiquitously expressed in tissues (35), and activation of PPAR-δ exerts many favorable metabolic effects, including reduced weight gain, increased skeletal muscle metabolic rate, and improved insulin sensitivity (30, 38, 44, 51). In addition, it has been previously demonstrated that treatment with a PPAR-δ agonist suppresses vascular inflammation and prevents atherosclerosis in apolipoprotein E-deficient mice (5, 29), indicating that stimulation of PPAR-δ is vasoprotective (49).
The vascular endothelium plays an essential role in the regulation of vascular tone (31). Endothelial dysfunction characterized by reduced bioavailability of nitric oxide (NO) is an early event in the pathogenesis of atherosclerosis (40). NO is mainly formed in endothelial cells from l-arginine by the enzymatic activity of endothelial NO synthase (eNOS) (34). Activation of smooth muscle guanylate cyclase by NO causes increased formation of cGMP and vasodilation (37). Relevant to our study, PPAR-δ is expressed in endothelial cells (17, 36), and activation of PPAR-δ results in increased NO bioavailability and preserved endothelial function (14, 47, 55). Moreover, both in vitro and in vivo studies (2, 14, 17, 41, 50) have indicated that increased antioxidant capacity is one of the important molecular mechanisms involved in the protective effects of PPAR-δ activation.
While endothelium-specific deletion of PPAR-γ impairs endothelium-dependent relaxations to ACh in mice aortas (27), the effects of PPAR-δ deficiency on endothelial function are unknown. PPAR-δ knockout mice have been developed; however, global deletion of PPAR-δ leads to embryonic lethality and growth retardation in surviving mice (4, 35). Tissue-restricted gene-targeting technology provides an alternative approach to address the tissue-specific role of PPAR-δ (9). To date, there have been no reported studies regarding the vascular phenotype of endothelium-specific PPAR-δ-deficient mice (ePPARδ−/− mice). Therefore, the objective of the present study was to characterize vascular endothelial function in conduit arteries of ePPARδ−/− mice.
MATERIALS AND METHODS
Generation of ePPARδ−/− mice.
Male ePPARδ−/− (PPARδflox/flox; Cre+) mice and their wild-type (WT) littermates (PPARδflox/flox; Cre−) were kindly provided by R. M. Evans (The Salk Institute, La Jolla, CA). Originally, mice harboring loxP sites on either side of exon 4 of the PPAR-δ gene were generated and backcrossed to C57BL/6 mice as previously described (4). Floxed PPAR-δ mice were crossed with transgenic mice expressing Cre recombinase (Tie2-Cre, stock no. 004128, Jackson Laboratory, Bar Harbor, ME). Selected descendants were bred to generate both floxed ePPARδ−/− mice and their littermate controls.
Male ePPARδ−/− (PPARδflox/flox; Cre+) mice were crossed with female control (PPARδflox/flox; Cre−) mice to generate offspring in our laboratory. At 4–5 wk of age, mice were anesthetized with 1% isoflurane, and tail snips were analyzed by PCR. Genotyping was performed using Cre primers according to the protocol from the Jackson Laboratory (stock no. 004128). To confirm endothelium-specific deletion of PPAR-δ, dissected aortas were rinsed in Krebs solution, and endothelial cells were flushed out of the aorta with lysis buffer (PureLink RNA Mini Kit, Ambion, Carlsbad, CA). The isolation and purification of total mRNA were performed according to the manufacturer's instructions (PureLink RNA Mini Kit, Ambion). Thereafter, cDNA was synthetized using SuperScript III (Invitrogen, Carlsbad, CA) and was subjected to PCR analysis using primers spanning the deletion site (exon 4) in the PPAR-δ gene (GenBank Accession No. NM_011145, forward: 5′-TTCCTCCCCTTCCTCCCTGC-3′ and reverse: 5′-GTGGACCCCGTAGTGGAAGC-3′). As a reference control, GAPDH (GenBank Accession No. NM_008084, forward: 5′-TGCCAAGGCTGTGGGCAAGG-3′ and reverse: 5′-TGGGCCCTCAGATGCCTGCT-3′) was used. The following conditions were used for PCR: 95°C for 30 s, 58°C for 30 s, and 72°C for 1 min (29 cycles). To confirm endothelial integrity, eNOS primers (forward: 5′-CCTGCCCCCATGACTTTG-3′ and reverse: 5′-TCCCGGTAGAGATGGTCCAG-3′) were used (25). The resulting PCR products were separated using agarose gel electrophoresis, and the bands were visualized.
Male mice used in experiments were maintained on standard chow with free access to drinking water. At 12–16 wk of age, WT and ePPARδ−/− mice were anesthetized with an overdose of pentobarbital (200–250 mg/kg body wt ip) followed by exsanguination via cardiac puncture for blood collection. In some experiments that required only WT animals, male C57BL/6 mice were obtained from the Jackson Laboratory. Aortas and common carotid arteries were carefully removed and dissected free from connective tissue in cold (4°C) modified Krebs-Ringer solution [containing (in mmol/l) 118.6 NaCl, 4.7 KCl, 2.5 CaCl2, 1.2 MgSO4, 1.2 KH2PO4, 25.1 NaHCO3, 10.1 glucose, and 0.026 EDTA]. Animal housing facilities and experimental protocols were approved by the Institutional Animal Care and Use Committee of the Mayo Clinic and complied with the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals.
Systolic blood pressure.
Systolic blood pressure was recorded in quiescent mice by the tail-cuff method (Harvard Apparatus, Kent, UK) as previously described (13).
Glucose and cholesterol profile.
Blood samples obtained through right ventricular puncture were immediately transferred to a tube containing EDTA. After centrifugation at 2,000 rpm (4°C, 10 min), supernatants were stored at −80°C until assayed. Cholesterol and high-density lipoprotein levels were measured using a Hitachi 912 chemistry analyzer (Roche Diagnostics, Indianapolis, IN) as previously described (14). Glucose levels were measured in whole blood with Accu Check (Roche Diagnostics, Indianapolis, IN).
Leptin levels.
Circulating leptin levels were quantified in plasma using sandwich enzyme immunoassay techniques (R&D Systems, Minneapolis, MN).
Vascular reactivity experiments with carotid arteries.
Four-millimeter-long segments of common carotid arteries were studied under pressurized conditions in small vessel chambers (Living Systems Instrumentation, Burlington, VT) as previously described (11). Carotid arteries were equilibrated for 60 min, after which their resting lumen diameter and media thickness were recorded. Concentration-dependent relaxations to ACh (10−9–10−5 mol/l) were obtained during submaximal contractions to the thromboxane analog 9,11-dideoxy-11α,9α-epoxymethano-PGF2α (U-46619). Concentrations of U-46619 (3 × 10−8–10−7 mol/l) were selected to obtain the same submaximal contractions in WT and ePPARδ−/− mice (59 ± 5% and 54 ± 5%, respectively, n = 6, P > 0.05). After washout and equilibration, responses to the NO donor diethylammonium (Z)-1-(N,N-diethylamino)diazen-1-ium-1,2-diolate (DEA-NONOate; 10−9–10−5 mol/l) were obtained.
Vascular reactivity experiments with aortic rings.
Isolated aortic rings were mounted horizontally between a fixed and a movable stainless steel hook and placed in organ chambers for recording isometric tension (PowerLab, AD Instruments, Colorado Springs, CO) as previously described (10). Data were acquired using LabChart Pro software (AD Instruments). In some rings, endothelial cells were mechanically removed and studied in parallel. Aortic rings were preincubated without or with catalase (1,400 U/ml for 20 min), and endothelium-dependent relaxations to ACh (10−9–10−5 mol/l) were obtained during submaximal contractions to phenylephrine. Concentrations of phenylephrine (3 × 10−8–3 × 10−7 mol/l) were selected to achieve the same submaximal contractions in WT and ePPARδ−/− mouse aortas without or with catalase. After washout and equilibration, endothelium-independent relaxations to DEA-NONOate (10−10–10−5 mol/l) were studied after preincubation without or with catalase (1,400 U/ml for 20 min).
Measurements of GTP-cyclohydrolase I enzyme activity and biopterin levels.
Aortas were homogenized in buffer containing 50 mmol/l Tris (pH 7.4), 1 mmol/l DTT, and 1 mmol/l EDTA at 4°C and were centrifuged at 10,000 rpm. Enzymatic activities of GTP-cyclohydrolase I (GTPCH I) were obtained by the amount of neopterin formation (13). Biopterin levels were determined after differential oxidation under acid [which converts both tetrahydrobiopterin (BH4) and 7,8-dihydrobiopterin (BH2) to biopterin] and base (which converts only BH2 to biopterin) conditions by reverse-phase HPLC (Beckman Coulter) as previously described. BH4 content was calculated from the difference in biopterin levels after acid and base oxidations (12).
Detection of superoxide anion.
Intracellular superoxide anion production was quantified using a HPLC/fluorescence assay that uses dihydroethidium as a probe as previously described (13).
Measurement of H2O2.
An Amplex red H2O2 assay kit (Life Technologies, Grand Island, NY) was used to perform measurements of H2O2 content in the mouse aorta (13). Briefly, aortas were homogenized in buffer solution provided by the kit and centrifuged at top speed. H2O2 measurements were performed in supernatants according the manufacturer's instructions and normalized against protein levels.
Measurement of cGMP levels.
A cGMP colorimetric ELISA kit (Cell Biolabs, San Diego, CA) was used. In brief, aortas (10–15 mm in length) were incubated for 60 min in minimal essential medium (containing 0.1% BSA, 100 U/ml penicillin, and 100 μg/ml streptomycin, GIBCO) with 3-isobutyl-1-methylxanthine (IBMX; 100 μmol/l) to inhibit the degradation of cyclic nucleotides by phosphodiesterases. Aortas were then removed and quickly frozen in liquid N2 until assayed. Some of the aortas were incubated with catalase (1,400 U/ml) for 60 min before IBMX. Aortas were homogenized in lysis buffer provided by the kit and centrifuged at top speed. Supernatants were used for cGMP measurements according the manufacturer's instructions and normalized against protein levels.
In separate experiments, WT (C57BL/6) mouse aortas with or without endothelium were used to study the effect of H2O2 on cGMP production. In brief, aortas were incubated for 60 min in IBMX-containing minimal essential medium, and the soluble guanylyl cyclase inhibitor 1H-[1,2,4]oxadiazolo-[4,3-a]quinoxalin-1-one (ODQ; 10 μmol/l; 30 min) was then added to some of the rings. Afterward, aortic rings were incubated for 30 min with 100 μM H2O2. This concentration was selected based on a previous study (46).
Western blot analysis.
Whole aortas were homogenized in lysis buffer (14). Equal amounts of protein (50 μg) were separated by SDS-PAGE and transferred to nitrocellulose membranes (Amersham), after which the membranes were probed using primary antibodies against eNOS and Ser1177-phosphorylated eNOS (BD Biosciences); CuZnSOD, MnSOD, extracellular (ec)SOD (Enzo), catalase, and β-actin (Sigma); and glutathione peroxidase 1 (GPx-1; AbFrontier). Bands were visualized using a commercially available ECL kit (Amersham), and densitometry was carried out using NIH Image (Scion-Image, Scion, Frederick, MD).
To evaluate the contribution of the endothelium, aortas without and with endothelium were studied in parallel. Aortas were dissected free from surrounding fat and were opened lengthwise. The endothelial surface was incubated with 0.1% collagenase in PBS for 1 min at room temperature. Endothelial cells were removed carefully and softly using a surgical blade, and the aorta was washed with Krebs buffer and homogenized as described above.
Calculations and statistical analysis.
All results are expressed as means ± SE; n is the number of animals from which tissues were harvested. Relaxations (expressed as a percentage of maximal relaxations induced by papaverine) were determined for each individual concentration-response curve by nonlinear regression analysis. Concentration-response curves of the different groups were compared by ANOVA for repeated measurements followed by Bonferroni's correction. WT and ePPARδ−/− mice without and with endothelium were compared by two-way ANOVA for multiple comparisons. For simple comparisons between two groups of WT and ePPARδ−/− mice, an unpaired Student's t-test was used where appropriate. P values of <0.05 were considered significant.
RESULTS
Characterization of ePPARδ−/− mice.
All mouse offspring were viable and grew and developed normally. Body weight did not differ between WT littermates and ePPARδ−/− mice (Table 1). Measurements of systolic blood pressure indicated that ePPARδ−/− mice were normotensive (Table 1). In addition, plasma lipid profiles, leptin, and blood glucose levels were unaltered in ePPARδ−/− mice (Table 1). PCR analysis of isolated endothelial cells from WT and ePPARδ−/− mouse aortas provided evidence showing that the PPAR-δ gene is present in endothelial cells, and some expression was found in vascular smooth muscle cells and adventitia (Fig. 1). The use of primers of exon 4 of the PPAR-δ gene showed that exon 4 was absent in isolated endothelial cells of ePPARδ−/− mice (Fig. 1).
Table 1.
Characteristics of WT and ePPARδ−/− mice
| Parameters | WT Mice | ePPARδ−/− Mice |
|---|---|---|
| Systolic blood pressure, mmHg | 112 ± 1 | 114 ± 2 |
| Body weight, g | 24 ± 2.0 | 25 ± 1 |
| Glucose, mg/dl | 140 ± 11 | 130 ± 12 |
| Cholesterol, mmol/l | 1.59 ± 0.08 | 1.61 ± 0.14 |
| High-density lipoprotein, mmol/l | 1.29 ± 0.05 | 1.26 ± 0.08 |
| Leptin, ng/ml | 3.88 ± 1.59 | 5.60 ± 1.55 |
Data are means ± SE; n = 6–8. WT mice, wild-type mice; ePPARδ−/− mice, endothelium-specific peroxisome proliferator-activated receptor-δ-deficient mice. P > 0.05.
Fig. 1.

A: PCR analysis of peroxisome proliferator-activated receptor (PPAR)-δ mRNA in the aorta of wild-type (WT) and endothelium-specific PPAR-δ-deficient mice (ePPARδ−/− mice). Endothelial cells (ECs) were isolated from mouse aortas and subjected to PCR analysis as described in materials and methods. The remaining aortas without endothelium were also analyzed. The PCR product was 124 bp for the exon 4-deleted PPAR-δ allele. GAPDH was used as a loading control (168 bp). B: endothelial nitric oxide (NO) synthase (eNOS; 62 bp) was used to verify endothelial integrity. Cre−, WT littermates; Cre+, ePPARδ−/− mice.
Structure and function of common carotid arteries in ePPARδ−/− mice.
The lumen diameter of common carotid arteries did not differ between WT and ePPARδ−/− mice (344 ± 8 and 335 ± 6 μm, respectively, n = 6). In addition, media thickness was unaltered in ePPARδ−/− mice (41 ± 4 μm) compared with WT littermates (44 ± 2 μm, n = 6). However, endothelium-dependent relaxations to ACh were significantly impaired in common carotid arteries of ePPARδ−/− mice (P < 0.05 vs. WT littermates; Fig. 2A). Moreover, endothelium-independent relaxations to the NO donor DEA-NONOate were also significantly reduced in ePPARδ−/− mice (P < 0.05 vs. WT mice; Fig. 2B).
Fig. 2.

Endothelium-dependent relaxations to ACh (A) and endothelium-independent relaxations to the NO donor diethylammonium (Z)-1-(N,N-diethylamino)diazen-1-ium-1,2-diolate (DEA-NONOate; B) of right common carotid arteries in WT littermates and ePPARδ−/− mice. Please note that relaxations to ACh and DEA-NONOate were significantly reduced in ePPARδ−/− mice compared with WT mice. All data are expressed as percent relaxation of the submaximal contraction to U-46619; n = 6. *P < 0.05 (by ANOVA with Bonferroni's correction).
Endothelial function of aortas in ePPARδ−/− mice.
Endothelium-dependent relaxations to ACh were also significantly impaired in ePPARδ−/− aortas (P < 0.05 vs. WT aortas; Fig. 3A). Moreover, increased contractions were observed in ePPARδ−/− aortas at higher concentrations of ACh (10−6–10−5 mol/l). Incubation with catalase significantly improved relaxations to ACh in ePPARδ−/− aortas (P < 0.05; Fig. 3C), whereas it had no effect in WT aortas (Fig. 3B).
Fig. 3.

A: endothelium-dependent relaxations to ACh in isolated aortic rings of WT littermates and ePPARδ−/− mice. B: effect of catalase (1,400 U/ml) on relaxations to ACh in WT mouse aortas. C: effect of catalase (1,400 U/ml) incubation on relaxations to ACh in aortas of ePPARδ−/− mice. All data are expressed as percent relaxation of the submaximal contraction to phenylephrine; n = 6. *P < 0.05 WT mice; †P < 0.05 vs. ePPARδ−/− mice (by ANOVA with Bonferroni's correction).
Endothelium-independent relaxations to DEA-NONOate were impaired, and the sensitivity (pD2) to DEA-NONOate was significantly shifted to the right in aortas of ePPARδ−/− mice with intact endothelium (P < 0.05; Fig. 4A and Table 2). With removal of endothelial cells, sensitivity was generally shifted to the left in all groups of mice (Table 2). However, relaxations to the NO donor were significantly improved in ePPARδ−/− arteries without endothelium compared with aortas with endothelium (P < 0.05; Table 2), and the difference between WT and ePPARδ−/− mice was abolished (Fig. 4B). Interestingly, catalase also significantly improved endothelium-independent relaxations in ePPARδ−/− aortas with intact endothelium, whereas it had no effect in aortas without endothelium (Table 2).
Fig. 4.

Relaxations to the NO donor DEA-NONOate in isolated aortic rings of WT littermates and ePPARδ−/− mice. A: relaxations to DEA-NONOate were significantly reduced in ePPARδ−/− mice compared with WT mice (n = 6). *P < 0.05 (by ANOVA with Bonferroni's correction). B: removal of ECs (E−) abolished the difference (n = 6). All data are expressed as percent relaxation of the submaximal contraction to phenylephrine.
Table 2.
Effects of catalase on reactivity to DEA-NONOate in WT and ePPARδ−/− mouse aortas
| WT Mice | WT Mice + Catalase | ePPARδ−/− Mice | ePPARδ−/− Mice + Catalase | |
|---|---|---|---|---|
| With endothelium | 7.60 ± 0.09 | 7.49 ± 0.07 | 7.37 ± 0.05* | 7.66 ± 0.09† |
| Without endothelium | 7.75 ± 0.07 | 8.02 ± 0.06‡ | 7.86 ± 0.11‡ | 8.04 ± 0.09‡ |
Data are means ± SE; n = 6. All values represent −log EC50 (in mol/l). DEA-NONOate, diethylammonium (Z)-1-(N,N-diethylamino)diazen-1-ium-1,2-diolate.
P < 0.05 vs. WT mice;
P < 0.05 vs. ePPARδ−/− mice;
P < 0.05 vs. aortas with endothelium (by two-way ANOVA).
Protein expression of eNOS in ePPARδ−/− mice.
Western blot analysis showed that the protein expression of phosphorylated eNOS at Ser1177 was significantly decreased in aortas of ePPARδ−/− mice, whereas the protein expression of eNOS was unchanged (P < 0.05 vs. WT mice; Fig. 5). Conversely, the protein expression of inducible NO synthase in WT and ePPARδ−/− blood vessels was weak and did not differ between both groups (data not shown).
Fig. 5.

Top: representative Western blot analysis of protein expressions of phosphorylated (p-)eNOS at Ser1177 in aortas of WT and ePPARδ−/− mice. Bottom: bar graph showing the results of the relative optical densitometry (OD) compared with eNOS protein (n = 8). *P < 0.05 vs. WT mice.
Biopterin metabolism in ePPARδ−/− mice.
BH4 levels were significantly increased in ePPARδ−/− aortas (P < 0.05 vs. WT aortas; Fig. 6A), whereas levels of BH2, the oxidative product of BH4, were unchanged (Fig. 6B). The resulting BH4-to-BH2 ratio did not differ between WT and ePPARδ−/− mice (Fig. 6C). To investigate the mechanism of increased BH4 levels, we examined the enzymatic activity of GTPCH I. Interestingly, GTPCH I activity was significantly increased in ePPARδ−/− aortas (P < 0.05 vs. WT aortas; Fig. 6D), whereas the protein expression of GTPCH I was unaltered (Fig. 6, E and F).
Fig. 6.
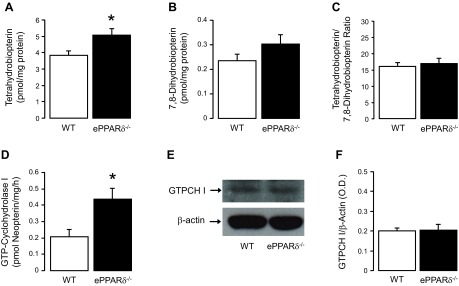
Quantitative HPLC analysis of vascular biopterin metabolism in ePPARδ−/− mice. A–D: bar graphs showing tetrahydrobiopterin levels (A; n = 6), 7,8-diydrobiopterin levels (B; n = 6), the ratio of tetrahydrobiopterin to 7,8-diydrobiopterin (C; n = 6), and the enzymatic activity of GTP-cyclohydrolase I (GTPCH I; D; n = 5). E: GTPCH I protein expression (n = 4) in aortas of WT littermates and ePPARδ−/− mice. Please note that the bar graph of GTPCH I protein indicates the results of the relative densitometry compared with β-actin protein (F). All results are shown as means ± SE. *P < 0.05 vs. WT mice.
Measurements of superoxide anion and H2O2 in ePPARδ−/− mice.
Levels of the stable fluorescent product 2-hydroxyethidium, which is formed from the reaction between dihydroethidium and superoxide anion, were similar between WT and ePPARδ−/− aortas (Fig. 7A). In contrast, levels of H2O2 were significantly increased in aortas of ePPARδ−/− mice (P < 0.05; Fig. 7B).
Fig. 7.

Quantitative analysis of superoxide anion (A; n = 5) and H2O2 levels (B; n = 8) in aortas of WT littermates and ePPARδ−/− mice. All results were normalized against tissue protein levels. *P < 0.05 vs. WT mice.
cGMP levels in ePPARδ−/− mice.
cGMP levels were significantly enhanced in aortas from ePPARδ−/− mice (P < 0.05 vs. WT mice; Fig. 8A). In vitro incubation of ePPARδ−/− aortas with catalase (1,400 U/ml) significantly decreased cGMP levels (P < 0.05 vs. control ePPARδ−/− aortas; Fig. 8B). Furthermore, incubation of WT aortas with H2O2 resulted a significant increase in cGMP levels, which was blocked by treatment with ODQ (P < 0.05 vs. WT aortas; Fig. 8C). Removal of endothelial cells abolished the stimulatory effects of H2O2 on cGMP levels (Fig. 8D).
Fig. 8.

A: basal cGMP levels in aortas of WT littermates and ePPARδ−/− mice (n = 9–11). B: effects of in vitro incubation with catalase (1,400 U/ml) on cGMP levels in ePPARδ−/− mouse aortas (n = 6). C: effects of H2O2 (100 μmol/l) on cGMP levels in WT mouse aortas in the absence or presence of the soluble guanylyl cyclase inhibitor 1H-[1,2,4]oxadiazolo-[4,3-a]quinoxalin-1-one (ODQ; 10 μmol/l, n = 5). D: effects of endothelium removal (E−) on cGMP levels in WT mouse aortas in the presence or absence of H2O2 (100 μmol/l; n = 7). All results were normalized against tissue protein levels. *P < 0.05 vs. WT mice; † P < 0.05 vs. control ePPARδ−/− mice; #P < 0.05 vs. H2O2.
Protein expressions of antioxidants in ePPARδ−/− mice.
Western blot analyses indicated that expressions of SOD isoforms CuZnSOD, MnSOD, and ecSOD were unaltered in aortas of ePPARδ−/− mice (Fig. 9). Interestingly, protein expressions of the H2O2 detoxifying enzymes catalase and GPx-1 were both significantly decreased in ePPARδ−/− mice (P < 0.05 vs. WT mice; Fig. 10). Removal of the endothelium abolished the differences between WT and ePPARδ−/− mice (Fig. 10).
Fig. 9.
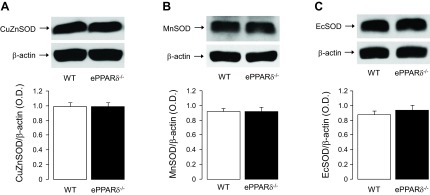
Top: Representative Western blot analysis of CuZnSOD (A), MnSOD (B), and extracellular (ec)SOD (C) protein expression in aortas of WT littermates and ePPARδ−/− mice. Bottom: bar graphs showing the results of the relative densitometry compared with β-actin protein (n = 8–9).
Fig. 10.
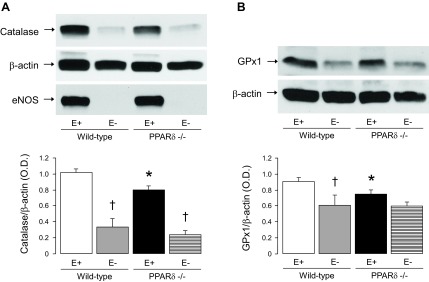
Top: representative Western blot analysis of catalase (A) and glutathione peroxidase 1 (GPx-1; B) protein expression in WT littermates and ePPARδ−/− mouse aortas in the presence (E+) or absence (E−) of ECs. Please note that removal of the endothelium eliminated the differences between WT and ePPARδ−/− mice. Successful removal of ECs was confirmed by the absence of eNOS. Bottom: bar graphs showing the results of the relative densitometry compared with β-actin protein. Results are means ± SE; n = 10 for E+ and 4 for E−. *P < 0.05 vs. WT mice; †P < 0.05 vs. E+ (by two-way ANOVA).
DISCUSSION
The present study was designed to determine how genetic inactivation of PPAR-δ in the endothelium affects the vascular function of large conduit arteries. Here, we present several novel findings. First, in aortas and carotid arteries of endothelium-specific PPAR-δ-deficient mice, endothelium-dependent and -independent relaxations were significantly impaired. Second, phosphorylation of eNOS at Ser1177 was also reduced in ePPARδ−/− mouse aortas. Third, deletion of the endothelium-specific PPAR-δ gene significantly increased the biosynthesis of BH4 in aortic walls. Fourth, protein expressions of catalase and GPx-1 were downregulated, which resulted in significantly increased H2O2 content in the aortas of ePPARδ−/− mice. Finally, increased levels of cGMP were detected in aortas of ePPARδ−/− mice, and this increase in cGMP was inhibited in aortas treated with catalase.
Emerging evidence supports the notion that activation of PPAR-δ exerts beneficial effects on vascular endothelial function (49). Indeed, treatment with PPAR-δ ligands improved endothelium-dependent relaxations in peripheral and cerebral arteries by increasing antioxidant capacity and a subsequent increase in the bioavailability of NO (14, 41, 47). These observations are important because normal NO signaling is vasoprotective. In the present study, deletion of PPAR-δ in the endothelium caused an impairment of endothelium-dependent relaxations to ACh in aortas and carotid arteries. This impaired relaxation may be caused by a decreased enzymatic activity of eNOS because in ePPARδ−/− mouse aortas, we also detected a downregulation of protein expression of phosphorylated eNOS at Ser1177. Phosphorylation of eNOS at serine has been reported to increase the enzymatic activity of eNOS and NO production, thereby accounting, in part, for the protection of vascular function (3). The most likely molecular mechanism for decreased eNOS phosphorylation at Ser1177 may be explained by the high levels of H2O2 detected in the aorta, as in previous studies reporting that long-term exposure of H2O2 can attenuate eNOS phosphorylation (22), increase the expression of arginase I (45), and impair endothelium-dependent relaxations (46). Likewise, reductions in NO-mediated signaling have been reported in GPx-1-deficient mice aortas under basal conditions (15). This is consistent with our observation that catalase was able to improve endothelium-dependent relaxations to ACh in aortas of ePPARδ−/− mice. We also wish to point out that this impairment of endothelial function in ePPARδ−/− mice was not caused by the increase in blood pressure or by an elevation of cholesterol or glucose levels.
We did not investigate the involvement of the cyclooxygenase pathway, and, therefore, we cannot rule out whether impaired endothelium-dependent relaxations in ePPARδ−/− mice were caused by increased generation of prostaglandins. We (10) have previously shown that endothelium-dependent relaxations to ACh in WT mouse aortas are entirely dependent on NO. Furthermore, treatment with catalase completely prevented increased contractions to higher concentrations of ACh (10−6–10−5 mol/l), thus suggesting that the impairment of endothelium-dependent relaxations in ePPARδ−/− mice is mediated by H2O2. In this regard, it has been previously reported that H2O2 is involved in the increased synthesis of prostaglandins causing endothelial dysfunction (16). Whether cyclooxygenase is activated in ePPARδ−/− mice remains to be determined.
Alteration of the responsiveness of vascular smooth muscle cells to NO may also explain to the impairment of endothelium-dependent relaxations. Indeed, we observed that endothelium-independent relaxations to the NO donor DEA-NONOate were reduced in aortas and carotid arteries of ePPARδ−/− mice, thereby indicating that reduced reactivity of NO in vascular smooth muscle cells may be responsible for the impaired endothelium-dependent relaxations to ACh. Moreover, previous studies (33, 52) have shown that an elevation of cGMP reduced the vasodilator effect to NO. It is important to note that exogenous catalase administration improved endothelium-independent relaxations of aortas in ePPARδ−/− mice. Likewise, removal of the endothelium also abolished the difference in DEA-NONOate responses between WT and ePPARδ−/− mouse aortas, suggesting that increased H2O2 levels in the endothelium are responsible for the impairment of relaxations to DEA-NONOate. Furthermore, catalase did not affect endothelium-independent relaxations to the NO donor in ePPARδ−/− mouse aortas without endothelium, ruling out the possibility that H2O2 production is increased in vascular smooth muscle cells. This is in line with our observations that protein expressions of catalase and GPx-1 were not different between WT and ePPARδ−/− mouse aortas without endothelium.
The impaired endothelial function in ePPARδ−/− mice may be due to suboptimal production of BH4 because BH4 is an essential cofactor for optimal enzymatic activity of eNOS in endothelial cells (24). In the present study, however, we found that endothelium-specific deletion of the PPAR-δ gene significantly increased BH4 levels in aortas, whereas levels of BH2 remained unchanged. Furthermore, the enzymatic activity of GTPCH I was enhanced in ePPARδ−/− mouse aortas, indicating that the increase in BH4 levels was caused by increased de novo biosynthesis of BH4 via GTPCH I. The elevation of BH4 again indicates that the impaired endothelial function was not due to BH4 deficiency, which would cause eNOS uncoupling and generation of superoxide anion (24).
Several potential mechanisms may explain the observed increase in BH4 production. It is well established that proinflammatory cytokines upregulate the expression and activity of GTPCH I, thereby increasing levels of BH4 (39). Based on established anti-inflammatory effects of PPAR-δ activation, it is conceivable that genetic inactivation of PPAR-δ in the endothelium may mimic a proinflammatory environment (5). However, this explanation is unlikely because a proinflammatory environment would decrease expression of eNOS (56). In the present study, eNOS protein expression was not affected by deletion of the PPAR-δ gene in the vascular endothelium. It is possible that the levels of BH4 are dependent on levels of H2O2 because H2O2 is also known as a stimulator of GTPCH I enzymatic activity and production of BH4 (43).
The elevated H2O2 levels in ePPARδ−/− mice may be caused by decreased expression and function of H2O2 detoxifying enzymes. Indeed, loss of PPAR-δ in the endothelium downregulated protein expressions of both catalase and GPx-1, and this, in turn, elevated production of H2O2 in mouse aortas. More importantly, removal of endothelial cells abolished the differences between WT and ePPARδ−/− mice, suggesting that the decreased catalase and GPx-1 protein expressions are endothelium specific. Both catalase and GPx-1 are antioxidant enzymes that catalyze the dismutation of H2O2 to oxygen and water and thus protect cells against this potentially toxic molecule (1). In addition, a functional PPAR response element is located in the promoter region of the catalase gene, indicating that catalase expression is directly regulated by PPAR (20). In this regard, recent evidence indicates that treatment of mice with the PPAR-δ ligand GW-501516 increased protein expression of catalase, thereby protecting vascular function by reducing the concentration of H2O2 (41). Moreover, overexpression of catalase can inhibit NF-κB activation and protect cells and tissues from inflammation and the development of atherosclerosis (32, 53).
A significant increase of cGMP levels was also detected in aortas of ePPARδ−/− mice compared with WT mice. H2O2 is the most likely candidate responsible for activation of the cGMP pathway because similar observations have been reported in several studies (7, 18, 26, 42). Moreover, incubation of ePPARδ−/− mouse aortas with catalase significantly reduced levels of cGMP, thereby indicating that H2O2 causes increased production of cGMP in ePPARδ−/− aortas. Consistent with this observation, catalase normalized relaxations to ACh and relaxations to DEA-NONOate. Notably, exogenous H2O2 increased cGMP levels only in aortas with intact endothelium. This observation suggests that the H2O2-induced elevation of cGMP levels in ePPARδ−/− aortas might be dependent on chronically increased formation of NO in endothelial cells (46). A previous study (21) has demonstrated that H2O2 can increase Ca2+ levels in the endothelium and activate eNOS. Consistent with this interpretation, H2O2 was not able to increase cGMP levels in endothelium-denuded arteries. In agreement with our observations, previous studies have reported that in vascular smooth muscle cells (54) or in endothelium-denuded arteries (23), H2O2 did not affect the production of cGMP, again suggesting that the presence of an intact endothelium is required for H2O2 to increase cGMP.
It has been previously reported that genetic inactivation of PPAR-δ reduces adipogenesis in mice (4) and that PPAR-δ-deficient mice are more prone to weight gain on a high-fat diet (51). In contrast, activation of PPAR-δ is protective against obesity and lipid accumulation (44, 51). Moreover, prior studies (8, 48) have reported a potential inhibitory function of PPAR-δ expression and function of leptin. Conversely, circulating leptin levels were increased in neuron-specific PPAR-δ-deficient mice, indicating that leptin was regulated by PPAR-δ (28, 48). However, in our study, we did not observe any changes in body weight and circulating levels of leptin in ePPARδ−/− mice, suggesting that the vascular alterations observed in these mice are independent of leptin.
A previous study by Kleinhenz and colleagues (27) reported that endothelium-specific deletion of the PPAR-γ gene increased ROS (although the exact nature of the ROS was not identified), impaired endothelium-dependent relaxations to ACh in aortas, and increased arterial blood pressure. Moreover, levels of endothelial NO were significantly reduced (27). In the present study, however, we found that in ePPARδ−/− mice, endothelial dysfunction is caused by selective reduction of the antioxidant capacity responsible for the inactivation of H2O2 and subsequent increase in H2O2 levels. Thus, it is becoming apparent that in the endothelium, each PPAR subserves an isoform-specific set of functions (6). Of note, there are no reported studies regarding endothelium-specific PPAR-α-deficient mice.
In conclusion, we demonstrate that endothelium-specific deletion of the PPAR-δ gene causes an impairment of the vascular responsiveness to NO. Increased cGMP levels caused by elevation of H2O2 appear to be an important mechanism underlying the impairment of vasodilator function. Thus, functional endothelial PPAR-δ maintains normal expression of catalase and GPx-1, thereby protecting the vascular wall from the effects of excessive production of H2O2. In addition, we speculate that the loss of PPAR-δ in endothelial cells, and the consequent increase in the local concentration of H2O2, predisposes the blood vessel wall to oxidative stress.
GRANTS
This work was supported by National Institutes of Health (NIH) Grants HL-91867 and HL-111062 and the Mayo Foundation (to Z. S. Katusic) and by NIH Grants HL-088093, DK-057978, DK-090962, HL-105278, and ES-010337, the Glenn Foundation for Medical Research, the Leona M. and Harry B. Helmsley Charitable Trust, Ipsen/Biomeasure, the California Institute for Regenerative Medicine, The Ellison Medical Foundation, and the Howard Hughes Medical Institute (to R. M. Evans). R. M. Evans is an Investigator of the Howard Hughes Medical Institute at the Salk Institute and March of Dimes Chair in Molecular and Developmental Biology.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: L.V.d., T.H., and L.-J.T. performed experiments; L.V.d., T.H., and A.V.R.S. analyzed data; L.V.d., T.H., A.V.R.S., L.-J.T., R.M.E., and Z.S.K. interpreted results of experiments; L.V.d., T.H., and A.V.R.S. prepared figures; L.V.d. and Z.S.K. drafted manuscript; L.V.d., T.H., A.V.R.S., and Z.S.K. edited and revised manuscript; L.V.d., T.H., A.V.R.S., L.-J.T., R.M.E., and Z.S.K. approved final version of manuscript; R.M.E. and Z.S.K. conception and design of research.
ACKNOWLEDGMENTS
The authors thank Leslie Smith for technical assistance.
REFERENCES
- 1.Aebi H. Catalase in vitro. Methods Enzymol 105: 121–126, 1984 [DOI] [PubMed] [Google Scholar]
- 2.Ali F, Ali NS, Bauer A, Boyle JJ, Hamdulay SS, Haskard DO, Randi AM, Mason JC. PPARδ and PGC1α act cooperatively to induce haem oxygenase-1 and enhance vascular endothelial cell resistance to stress. Cardiovasc Res 85: 701–710, 2010 [DOI] [PubMed] [Google Scholar]
- 3.Atochin DN, Wang A, Liu VW, Critchlow JD, Dantas AP, Looft-Wilson R, Murata T, Salomone S, Shin HK, Ayata C, Moskowitz MA, Michel T, Sessa WC, Huang PL. The phosphorylation state of eNOS modulates vascular reactivity and outcome of cerebral ischemia in vivo. J Clin Invest 117: 1961–1967, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barak Y, Liao D, He W, Ong ES, Nelson MC, Olefsky JM, Boland R, Evans RM. Effe cts of peroxisome proliferator-activated receptor δ on placentation, adiposity, and colorectal cancer. Proc Natl Acad Sci USA 99: 303–308, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barish GD, Atkins AR, Downes M, Olson P, Chong LW, Nelson M, Zou Y, Hwang H, Kang H, Curtiss L, Evans RM, Lee CH. PPARδ regulates multiple proinflammatory pathways to suppress atherosclerosis. Proc Natl Acad Sci USA 105: 4271–4276, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brown JD, Plutzky J. Peroxisome proliferator-activated receptors as transcriptional nodal points and therapeutic targets. Circulation 115: 518–533, 2007 [DOI] [PubMed] [Google Scholar]
- 7.Burke-Wolin T, Abate CJ, Wolin MS, Gurtner GH. Hydrogen peroxide-induced pulmonary vasodilation: role of guanosine 3′,5′-cyclic monophosphate. Am J Physiol Lung Cell Mol Physiol 261: L393–L398, 1991 [DOI] [PubMed] [Google Scholar]
- 8.Chen W, Wang LL, Liu HY, Long L, Li S. Peroxisome proliferator-activated receptor delta-agonist, GW501516, ameliorates insulin resistance, improves dyslipidaemia in monosodium l-glutamate metabolic syndrome mice. Basic Clin Pharmacol Toxicol 103: 240–246, 2008 [DOI] [PubMed] [Google Scholar]
- 9.Cheng L, Ding G, Qin Q, Huang Y, Lewis W, He N, Evans RM, Schneider MD, Brako FA, Xiao Y, Chen YE, Yang Q. Cardiomyocyte-restricted peroxisome proliferator-activated receptor-δ deletion perturbs myocardial fatty acid oxidation and leads to cardiomyopathy. Nat Med 10: 1245–1250, 2004 [DOI] [PubMed] [Google Scholar]
- 10.d'Uscio LV, Baker TA, Mantilla CB, Smith L, Weiler D, Sieck GC, Katusic ZS. Mechanism of endothelial dysfunction in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol 21: 1017–1022, 2001 [DOI] [PubMed] [Google Scholar]
- 11.d'Uscio LV, Smith LA, Katusic ZS. Hypercholesterolemia impairs endothelium-dependent relaxations in common carotid arteries of apolipoprotein E-deficient mice. Stroke 32: 2658–2664, 2001 [DOI] [PubMed] [Google Scholar]
- 12.d'Uscio LV, Milstien S, Richardson D, Smith L, Katusic ZS. Long-term vitamin C treatment increases vascular tetrahydrobiopterin levels and nitric oxide synthase activity. Circ Res 92: 88–95, 2003 [DOI] [PubMed] [Google Scholar]
- 13.d'Uscio LV, Smith LA, Katusic ZS. Differential effects of eNOS uncoupling on conduit and small arteries in GTP-cyclohydrolase I-deficient hph-1 mice. Am J Physiol Heart Circ Physiol 301: H2227–H2234, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.d'Uscio LV, Das P, Santhanam AV, He T, Younkin SG, Katusic ZS. Activation of PPARδ prevents endothelial dysfunction induced by overexpression of amyloid-β precursor protein. Cardiovasc Res 96: 504–512, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dayal S, Brown KL, Weydert CJ, Oberley LW, Arning E, Bottiglieri T, Faraci FM, Lentz SR. Deficiency of glutathione peroxidase-1 sensitizes hyperhomocysteinemic mice to endothelial dysfunction. Arterioscler Thromb Vasc Biol 22: 1996–2002, 2002 [DOI] [PubMed] [Google Scholar]
- 16.Erdei N, Bagi Z, Edes I, Kaley G, Koller A. H2O2 increases production of constrictor prostaglandins in smooth muscle leading to enhanced arteriolar tone in type 2 diabetic mice. Am J Physiol Heart Circ Physiol 292: H649–H656, 2007 [DOI] [PubMed] [Google Scholar]
- 17.Fan Y, Wang Y, Tang Z, Zhang H, Qin X, Zhu Y, Guan Y, Wang X, Staels B, Chien S, Wang N. Suppression of pro-inflammatory adhesion molecules by PPAR-δ in human vascular endothelial cells. Arterioscler Thromb Vasc Biol 28: 315–321, 2008 [DOI] [PubMed] [Google Scholar]
- 18.Fujimoto S, Mori M, Tsushima H. Mechanisms underlying the hydrogen peroxide-induced, endothelium-independent relaxation of the norepinephrine-contraction in guinea-pig aorta. Eur J Pharmacol 459: 65–73, 2003 [DOI] [PubMed] [Google Scholar]
- 19.Gervois P, Fruchart JC, Staels B. Drug Insight: mechanisms of action and therapeutic applications for agonists of peroxisome proliferator-activated receptors. Nat Clin Pract Endocrinol Metab 3: 145–156, 2007 [DOI] [PubMed] [Google Scholar]
- 20.Girnun GD, Domann FE, Moore SA, Robbins ME. Identification of a functional peroxisome proliferator-activated receptor response element in the rat catalase promoter. Mol Endocrinol 16: 2793–2801, 2002 [DOI] [PubMed] [Google Scholar]
- 21.Hu Q, Corda S, Zweier JL, Capogrossi MC, Ziegelstein RC. Hydrogen peroxide induces intracellular calcium oscillations in human aortic endothelial cells. Circulation 97: 268–275, 1998 [DOI] [PubMed] [Google Scholar]
- 22.Hu Z, Chen J, Wei Q, Xia Y. Bidirectional actions of hydrogen peroxide on endothelial nitric-oxide synthase phosphorylation and function: co-commitment and interplay of Akt and AMPK. J Biol Chem 283: 25256–25263, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Iida Y, Katusic ZS. Mechanisms of cerebral arterial relaxations to hydrogen peroxide. Stroke 31: 2224–2230, 2000 [DOI] [PubMed] [Google Scholar]
- 24.Katusic ZS, d'Uscio LV, Nath KA. Vascular protection by tetrahydrobiopterin: progress and therapeutic prospects. Trends Pharmacol Sci 30: 48–54, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kerkela R, Karsikas S, Szabo Z, Serpi R, Magga J, Gao E, Alitalo K, Anisimov A, Sormunen R, Pietila I, Vainio L, Koch WJ, Kivirikko KI, Myllyharju J, Koivunen P. Activation of hypoxia response in endothelial cells contributes to ischemic cardioprotection. Mol Cell Biol 33: 3321–3329, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim SM, Byun JS, Jung YD, Kang IC, Choi SY, Lee KY. The effects of oxygen radicals on the activity of nitric oxide synthase and guanylate cyclase. Exp Mol Med 30: 221–226, 1998 [DOI] [PubMed] [Google Scholar]
- 27.Kleinhenz JM, Kleinhenz DJ, You S, Ritzenthaler JD, Hansen JM, Archer DR, Sutliff RL, Hart CM. Disruption of endothelial peroxisome proliferator-activated receptor-γ reduces vascular nitric oxide production. Am J Physiol Heart Circ Physiol 297: H1647–H1654, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kocalis HE, Turney MK, Printz RL, Laryea GN, Muglia LJ, Davies SS, Stanwood GD, McGuinness OP, Niswender KD. Neuron-specific deletion of peroxisome proliferator-activated receptor delta (PPARδ) in mice leads to increased susceptibility to diet-induced obesity. PLOS ONE 7: e42981, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee CH, Chawla A, Urbiztondo N, Liao D, Boisvert WA, Evans RM, Curtiss LK. Transcriptional repression of atherogenic inflammation: modulation by PPARδ. Science 302: 453–457, 2003 [DOI] [PubMed] [Google Scholar]
- 30.Leibowitz MD, Fievet C, Hennuyer N, Peinado-Onsurbe J, Duez H, Bergera J, Cullinan CA, Sparrow CP, Baffic J, Berger GD, Santini C, Marquis RW, Tolman RL, Smith RG, Moller DE, Auwerx J. Activation of PPARδ alters lipid metabolism in db/db mice. FEBS Lett 473: 333–336, 2000 [DOI] [PubMed] [Google Scholar]
- 31.Lüscher TF, Vanhoutte PM. The Endothelium: Modulator of Cardiovascular Function. Boca Raton, FL: CRC, 1990, p. 1–215 [Google Scholar]
- 32.Nilakantan V, Spear BT, Glauert HP. Liver-specific catalase expression in transgenic mice inhibits NF-κB activation and DNA synthesis induced by the peroxisome proliferator ciprofibrate. Carcinogenesis 19: 631–637, 1998 [DOI] [PubMed] [Google Scholar]
- 33.Olmos L, Mombouli JV, Illiano S, Vanhoutte PM. cGMP mediates the desensitization to bradykinin in isolated canine coronary arteries. Am J Physiol Heart Circ Physiol 268: H865–H870, 1995 [DOI] [PubMed] [Google Scholar]
- 34.Palmer RMJ, Ashton DS, Moncada S. Vascular endothelial cells synthesize nitric oxide from l-arginine. Nature 333: 664–666, 1988 [DOI] [PubMed] [Google Scholar]
- 35.Peters JM, Lee SS, Li W, Ward JM, Gavrilova O, Everett C, Reitman ML, Hudson LD, Gonzalez FJ. Growth, adipose, brain, and skin alterations resulting from targeted disruption of the mouse peroxisome proliferator-activated receptor β(δ). Mol Cell Biol 20: 5119–5128, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Piqueras L, Reynolds AR, Hodivala-Dilke KM, Alfranca A, Redondo JM, Hatae T, Tanabe T, Warner TD, Bishop-Bailey D. Activation of PPARβ/δ induces endothelial cell proliferation and angiogenesis. Arterioscler Thromb Vasc Biol 27: 63–69, 2007 [DOI] [PubMed] [Google Scholar]
- 37.Rapoport RM, Draznin MB, Murad F. Endothelium-dependent relaxation in rat aorta may be mediated through cyclic GMP-dependent protein phosphorylation. Nature 306: 174–176, 1983 [DOI] [PubMed] [Google Scholar]
- 38.Riserus U, Sprecher D, Johnson T, Olson E, Hirschberg S, Liu A, Fang Z, Hegde P, Richards D, Sarov-Blat L, Strum JC, Basu S, Cheeseman J, Fielding BA, Humphreys SM, Danoff T, Moore NR, Murgatroyd P, O'Rahilly S, Sutton P, Willson T, Hassall D, Frayn KN, Karpe F. Activation of peroxisome proliferator-activated receptor (PPAR) δ promotes reversal of multiple metabolic abnormalities, reduces oxidative stress, and increases fatty acid oxidation in moderately obese men. Diabetes 57: 332–339, 2008 [DOI] [PubMed] [Google Scholar]
- 39.Rosenkranz-Weiss P, Sessa WC, Milstien S, Kaufman S, Watson CA, Pober JS. Regulation of nitric oxide synthesis by proinflammatory cytokines in human umbilical vein endothelial cells. Elevations in tetrahydrobiopterin levels enhance endothelial nitric oxide synthase specific activity. J Clin Invest 93: 2236–2243, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ross R. Cell biology of atherosclerosis. Annu Rev Physiol 57: 791–804, 1995 [DOI] [PubMed] [Google Scholar]
- 41.Santhanam AV, d'Uscio LV, He T, Katusic ZS. PPARδ agonist GW501516 prevents uncoupling of endothelial nitric oxide synthase in cerebral microvessels of hph-1 mice. Brain Res 1483: 89–95, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sato A, Sakuma I, Gutterman DD. Mechanism of dilation to reactive oxygen species in human coronary arterioles. Am J Physiol Heart Circ Physiol 285: H2345–H2354, 2003 [DOI] [PubMed] [Google Scholar]
- 43.Shimizu S, Shiota K, Yamamoto S, Miyasaka Y, Ishii M, Watabe T, Nishida M, Mori Y, Yamamoto T, Kiuchi Y. Hydrogen peroxide stimulates tetrahydrobiopterin synthesis through the induction of GTP-cyclohydrolase I and increases nitric oxide synthase activity in vascular endothelial cells. Free Radic Biol Med 34: 1343–1352, 2003 [DOI] [PubMed] [Google Scholar]
- 44.Tanaka T, Yamamoto J, Iwasaki S, Asaba H, Hamura H, Ikeda Y, Watanabe M, Magoori K, Ioka RX, Tachibana K, Watanabe Y, Uchiyama Y, Sumi K, Iguchi H, Ito S, Doi T, Hamakubo T, Naito M, Auwerx J, Yanagisawa M, Kodama T, Sakai J. Activation of peroxisome proliferator-activated receptor δ induces fatty acid β-oxidation in skeletal muscle and attenuates metabolic syndrome. Proc Natl Acad Sci USA 100: 15924–15929, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thengchaisri N, Hein TW, Wang W, Xu X, Li Z, Fossum TW, Kuo L. Upregulation of arginase by H2O2 impairs endothelium-dependent nitric oxide-mediated dilation of coronary arterioles. Arterioscler Thromb Vasc Biol 26: 2035–2042, 2006 [DOI] [PubMed] [Google Scholar]
- 46.Thomas SR, Schulz E, Keaney JF., Jr Hydrogen peroxide restrains endothelium-derived nitric oxide bioactivity–role for iron-dependent oxidative stress. Free Radic Biol Med 41: 681–688, 2006 [DOI] [PubMed] [Google Scholar]
- 47.Tian XY, Wong WT, Wang N, Lu Y, Cheang WS, Liu J, Liu L, Liu Y, Lee SS, Chen ZY, Cooke JP, Yao X, Huang Y. PPARδ activation protects endothelial function in diabetic mice. Diabetes 61: 3285–3293, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wagner KD, Benchetrit M, Bianchini L, Michiels JF, Wagner N. Peroxisome proliferator-activated receptor β/δ (PPARβ/δ) is highly expressed in liposarcoma and promotes migration and proliferation. J Pathol 224: 575–588, 2011 [DOI] [PubMed] [Google Scholar]
- 49.Wang N. PPAR-δ in vascular pathophysiology. PPAR Res 2008: 164163, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang P, Liu J, Li Y, Wu S, Luo J, Yang H, Subbiah R, Chatham J, Zhelyabovska O, Yang Q. Peroxisome proliferator-activated receptor δ is an essential transcriptional regulator for mitochondrial protection and biogenesis in adult heart. Circ Res 106: 911–919, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang YX, Lee CH, Tiep S, Yu RT, Ham J, Kang H, Evans RM. Peroxisome-proliferator-activated receptor δ activates fat metabolism to prevent obesity. Cell 113: 159–170, 2003 [DOI] [PubMed] [Google Scholar]
- 52.Yamashita T, Kawashima S, Ohashi Y, Ozaki M, Rikitake Y, Inoue N, Hirata K, Akita H, Yokoyama M. Mechanisms of reduced nitric oxide/cGMP-mediated vasorelaxation in transgenic mice overexpressing endothelial nitric oxide synthase. Hypertension 36: 97–102, 2000 [DOI] [PubMed] [Google Scholar]
- 53.Yang H, Roberts LJ, Shi MJ, Zhou LC, Ballard BR, Richardson A, Guo ZM. Retardation of atherosclerosis by overexpression of catalase or both Cu/Zn-superoxide dismutase and catalase in mice lacking apolipoprotein E. Circ Res 95: 1075–1081, 2004 [DOI] [PubMed] [Google Scholar]
- 54.Yang M, Yang Y, Zhang S, Kahn AM. Insulin-stimulated hydrogen peroxide increases guanylate cyclase activity in vascular smooth muscle. Hypertension 42: 569–573, 2003 [DOI] [PubMed] [Google Scholar]
- 55.Zarzuelo MJ, Jimenez R, Galindo P, Sanchez M, Nieto A, Romero M, Quintela AM, Lopez-Sepulveda R, Gomez-Guzman M, Bailon E, Rodriguez-Gomez I, Zarzuelo A, Galvez J, Tamargo J, Perez-Vizcaino F, Duarte J. Antihypertensive effects of peroxisome proliferator-activated receptor-β activation in spontaneously hypertensive rats. Hypertension 58: 733–743, 2011 [DOI] [PubMed] [Google Scholar]
- 56.Zemse SM, Chiao CW, Hilgers RH, Webb RC. Interleukin-10 inhibits the in vivo and in vitro adverse effects of TNF-α on the endothelium of murine aorta. Am J Physiol Heart Circ Physiol 299: H1160–H1167, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]