

Abstract
Quantum mechanical (QM) methodology has been employed to study the structure activity relations of DNA and locked nucleic acid (LNA). The QM calculations provide the basis for construction of molecular structure and electrostatic surface potentials from molecular orbitals. The topologies of the electrostatic potentials were compared among model oligonucleotides, and it was observed that small structural modifications induce global changes in the molecular structure and surface potentials. Since ligand structure and electrostatic potential complementarity with a receptor is a determinant for the bonding pattern between molecules, minor chemical modifications may have profound changes in the interaction profiles of oligonucleotides, possibly leading to changes in pharmacological properties. The QM modeling data can be used to understand earlier observations of antisense oligonucleotide properties, that is, the observation that small structural changes in oligonucleotide composition may lead to dramatic shifts in phenotypes. These observations should be taken into account in future oligonucleotide drug discovery, and by focusing more on non RNA target interactions it should be possible to utilize the exhibited property diversity of oligonucleotides to produce improved antisense drugs.
Introduction
The Watson-Crick (W/C) recognition motif, though fundamentally straightforward, leads and directs even the most complex biological processes (Watson and Crick, 1953a, 1953b). It is remarkable how this “digital” bonding motif has been a driver behind numerous applications where native nucleic acids are used as probes (Trapane and Ts'O, 1994; Dangler, 1996; Demidov and Frank-Kamenetskii, 2004). In addition, the recognition motif has served as a design basis for chemically modified nucleic acid analogs employed for highly specific purposes (Uhlmann and Peyman, 1990; Nielsen et al., 1991; Hanvey et al., 1992; Shabarova, 1994; Martin, 1995; Altmann et al., 1996; Corey, 1997; Freier and Altmann, 1997; Cook, 1999; Uhlmann, 2000; Davis et al., 2006; Debart et al., 2007; Eckstein, 2007; Koch and Ørum, 2007; Koch et al., 2008). One of the areas where nucleic acid analogs have extensively been used is for the antisense silencing of gene expression (Zamecnik and Stephenson, 1978). This therapeutic field is based on the inhibition of RNA expression by oligonucleotide analogs designed to specifically recognize, by W/C hydrogen bonding, a complementary target sequence (Zamecnik and Stephenson, 1978).
Over the past 3–4 decades, many nucleic acid analogs have been produced for antisense purposes (Zamecnik and Stephenson, 1978; Uhlmann and Peyman, 1990; Martin, 1995; Freir and Altmann, 1997; Uhlmann, 2000; Wengel, 2001; Koch and Ørum, 2007; Koch et al., 2008; Yamamoto et al., 2011). Despite this extensive work, antisense oligonucleotides have not fully realized their clinical promise. Only a small fraction of the analogs have ever been clinically evaluated, and just two antisense drugs have been approved by the United States Food and Drug Administration for marketing (Kynamro™ and Vitravene™). Enthusiasm for antisense therapeutics was renewed when the nucleic acid analog locked nucleic acid (LNA) was synthesized (Obika et al., 1997; Singh et al., 1998). LNA represents a class of bicyclic nucleic acid analogs, and in the parent LNA nucleoside, the furanose ring is chemically modified by insertion of a 2′-O-CH2-4′ linkage that transforms the furanose to the bicyclic structure. When LNA nucleosides are incorporated into oligonucleotides, the bicyclic modification induces not only substantially increased nuclease stability, but also good mismatch discrimination and high hybridization affinity. LNA nucleosides are equally well combined with native phosphordiester oligonucleotides (PO) or phosphorothioates (PS) (Cohen, 1993; Eckstein, 2007). Driven by its high affinity, LNA has enabled a platform technology for the inhibition of both coding and non-coding RNAs (Wahlestedt et al., 2000; Jepsen and Wengel, 2004; Hansen et al., 2008; Koch et al., 2008; Elmen et al., 2008a, 2008b; Seth et al., 2009; Lanford et al., 2010; Straarup et al., 2010; Lindholm et al., 2011; Obad et al., 2011).
However, in contrast to the simplicity of nucleic acid recognition (W/C), the binding principles are far more complex between oligonucleotides and other biomolecules. For instance, it has been reported that small structural modifications of antisense oligonucleotides (e.g., LNAs) have profound effects on these relations, which are observed as property changes. (Seth et al., 2009; Straarup et al., 2010; Laxton et al., 2011; Stanton et al., 2012). The mechanisms behind this “property diversity” are not understood, and it is clear that a reductionist viewpoint, where the complex properties of a given oligonucleotide (e.g., tissue and cellular uptake, pharmacokinetics, toxicity) are assigned by a simple correlation with the nucleobase sequence alone, neither describes nor predicts these structure/activity changes.
In order to further study the property diversity of oligonucleotides, we have used a methodology employing quantum mechanical calculations (QM). By employing QM calculations, we have established the electronic orbital configuration of selected model oligonucleotides and, based on that, have calculated the molecular electrostatic iso-potential surfaces. These serve as a model for visualizing the three-dimensional changes in molecular structure and electrostatics induced by changes in oligonucleotide composition. Molecular structure and electrostatic iso-potential surfaces serve as models for studying the interaction of oligonucleotides with non-RNA target biomolecules (i.e. proteins), and altered patterns should be reflected in global property changes. The observation of a diversified binding potential of oligonucleotides to non-target molecules can be exploited, and we suggest that a stronger focus on oligonucleotide/non-target interactions could be a targeted approach for making antisense lead candidates with improved pharmacological properties.
Material and Methods
The molecules studied were first constructed ab initio by adding the atoms or whole nucleotides together with a phosphor-sugar backbone using the chemical program SPARTAN ’10 (Wavefunctions, Inc.), or employing the graphics in Gauss-view. After having built the desired molecular structure, the molecule is transferred to a Quantum Mechanical program (e.g. Gaussian program package) (Frisch, 2009) on a supercomputer where the optimization procedure can be carried out.
The electronic properties of the oligonucleotides, both with respect to their molecular orbitals and their electrostatic surfaces, were obtained by QM Hartree-Fock (HF) optimization employing the basis set of Gaussian type functions. In this approach, a large energy functional can be optimized with respect to the electrostatic energy configuration solely on the basis of the electronic density by separating electron and nuclear motions. Once the energy-minimized electronic structure is obtained (i.e., the nuclear coordinates and electron orbitals in the ground state), the electrostatic surfaces and the HOMO (highest occupied molecular orbital) and LUMO (lowest unoccupied molecular orbital) bonding system can be calculated, thus providing information about electron donor/acceptor sites.
More precisely, we employed the Hartree-Fock self-consistent field (HF SCF) QM method with linear combination of atomic orbitals approximation applied to the usual Schrődinger electronic equation (used together with matrix equations of the Roothaan Hall type). We selected HF since it is generally acknowledged that this method provides better optimization results for large biomolecules due to non-local interactions and avoidance of empirical parametrization. The Schrödinger electronic equation can be written as:
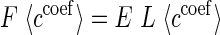 |
where ccoef are wave function orbital coefficients, E are orbital energies, L are overlap terms and F is a so-called Fock matrix given in terms of a core-electron Hamiltonian (H), the Coulomb, and Exchange terms such that:
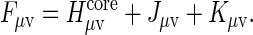 |
The core Hamiltonian is derived from the Schrödinger equation with the one electron wavefunction φμ:
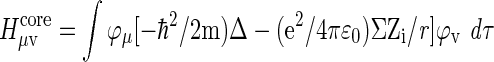 |
where Δ stands for the Laplace operator, m is the electronic mass, e is the electron charge, Zi is the charge of the nucleus i, and the Coulomb and Exchange potential terms are expanded on the basis functions. These are here taken as split-valence basis sets of Gaussian functions denoted as either 6-31G* or 3-21G (less accurate), which describe the number of Gaussian functions in a sum with coefficients. The energies of the molecular parts with wave functions are used to calculate the potential energy surface. This represents the electrostatic potential energy based on wave-function optimization of the orbital structure from where the effective charges can be extracted, or in other words, the electrostatic potential energy a positive test charge is experiencing when approaching the molecule. It was found convenient to describe this potential energy by an iso-surface of defined field strength, which was identical over the entire surface (BOHR, 2013).
In the HF calculation, the lowest energy states are found by optimization under the constraints of self-consistent (SCF) field procedures that obey the laws of electrodynamics. The HF calculations are based on independent particle models where the electrons experience a mean field from the electrons and the nuclei. In the Density Functional Theory approach, the exchange potential is based on electron densities, while the HF method uses wave functions also describing non-local exchange potentials.
The HF calculations are stepwise processes: Initially, single stranded molecules were constructed. DNA or LNA oligonucleotide (nucleobase and sugar-phosphate backbone) consists of hundreds of atoms. Next, the molecule was subjected to a basic electrostatic optimization to determine if the molecular structure is stable. Finally, the coordinate file was transferred to a high performing supercomputer that optimizes the proposed electronic structure using variational methods, and constructs the electronic orbitals that are consistent with the electrostatic functionals.
The process of proposing a system of wave functions and checking their consistency according to their electronic densities is then carried out for many iterations (ca. 1,000) and is terminated when a convergent state is reached. In a complete quantum mechanical calculation of a molecular structure, it is important to include solvent effects on the naked (gas phase) molecule. Water molecules are here included indirectly by altering the dielectric constant (relative permittivity 75) in a cavity. From the electrostatic iso-surfaces, in addition to charge densities, differential geometric properties such as topology can also be obtained. These geometric properties appear in the connectedness of the surface and the curvature values (see Figs. 1–6).
Fig. 1.
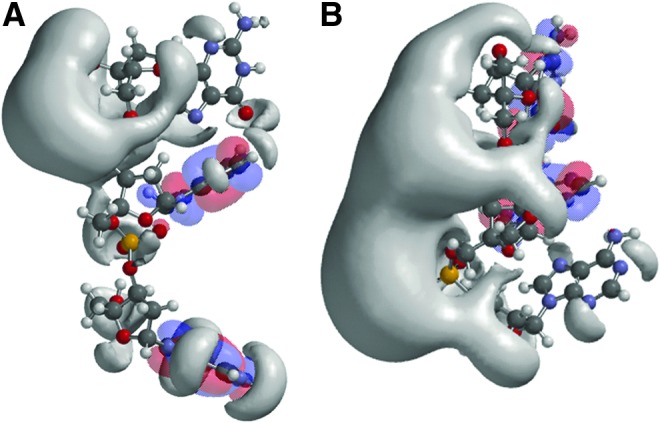
Structures and frontier orbitals (red/blue) for (A) DNA and (B) LNA with the sequence 5′-AAG-3′ (5′ end at bottom of structure). The electrostatic potential (negative) is shown in gray and represented by an Iso-potential value of −83 kJ/mol.
Fig. 2.
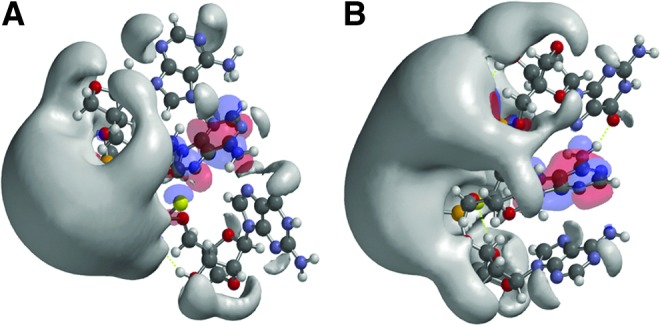
Structures and frontier orbitals (red/blue) for LNA trinucleotides with sequence 5′-AAG-3′, chemically modified by phosphorothioate internucleoside linkages (PS). In (A), the configuration of PS is RS and in (B) the configuration is SR. The 5′ end is at the top in A and at the bottom in B. The electrostatic potential (negative) is shown in gray and represented by an Iso-potential value of −83 kJ/mol.
Fig. 3.
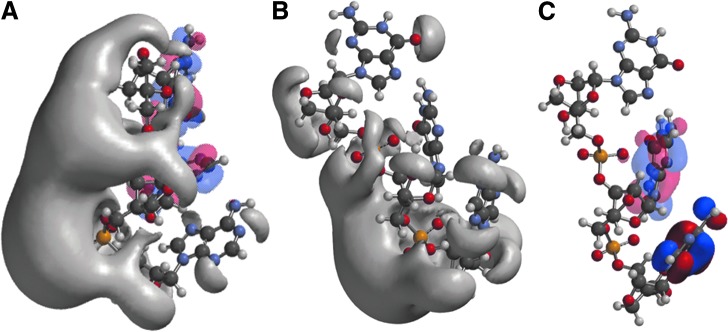
Structures and frontier orbitals (red/blue) for LNA tri-nucleotides with sequence 5′-AAG-3′ (A) and 5′-AGG-3′ (B). The 5′ end is at the bottom of the figure. In (C) the HOMO-LUMO orbitals are explicitly shown for LNA 5′-AGG-3′. The electrostatic potential (negative) is shown in gray and represented by an Iso-potential value of −83 kJ/mol.
Fig. 4.
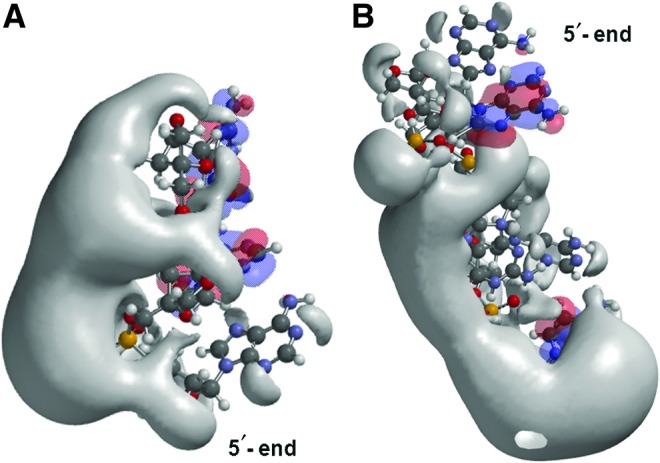
Structures and frontier orbitals (red/blue) for (A) LNA tri-nucleotide with sequence 5′-AAG-3′, and (B) LNA penta-nucleotide with sequence 5′-AAGAC-3′. The 5′ end is at the bottom in (A) and is at the top in (B). The electrostatic potential (negative) is shown in gray and represented by an Iso-potential value of −83 kJ/mol.
Fig. 5.
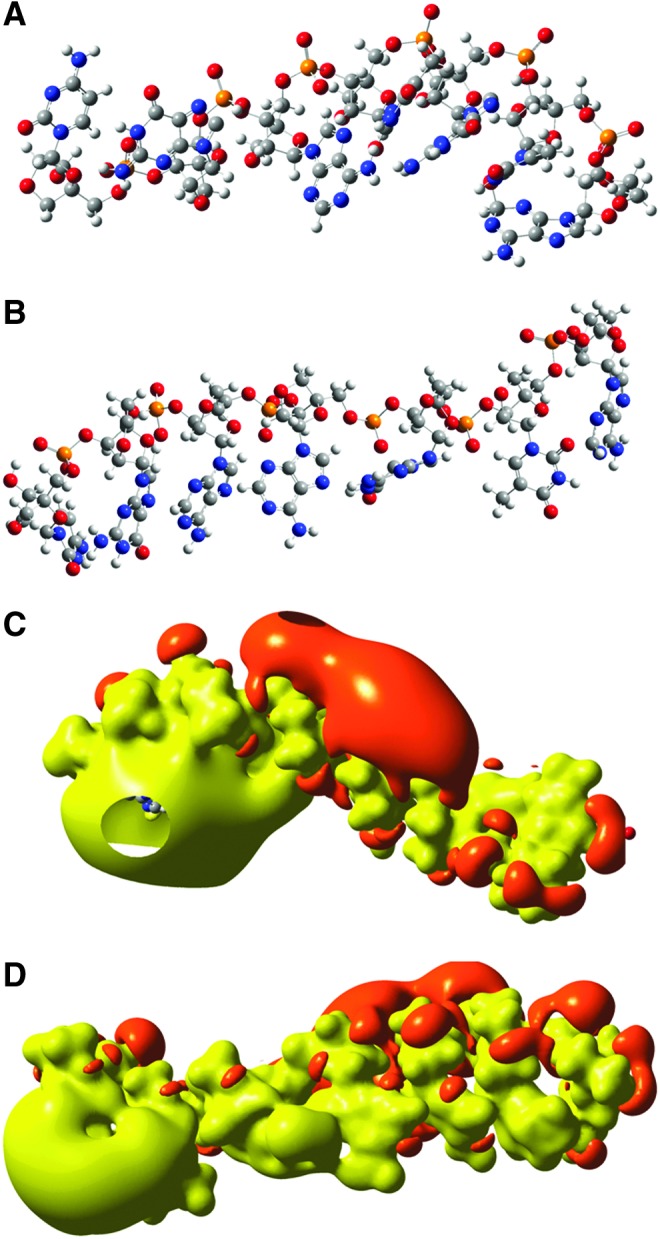
Three-dimensional chemical structures of the LNA heptanucleotides (A) 5′-ATGTAGC-3′ and (B) 5′-ATGAAGC-3′. The 5′ end is placed to the right. The topology of the electrostatic potentials of the two heptanucleotides is shown in (C) 5′-ATGTAGC-3′ and (D) 5′-ATGAAGC-3′. The energy of the electrostatic iso-potential surfaces is set to 1.63 eV (157 kJ/mol). The negative potential is marked with red and the positive with yellow. The 5′ end is to the right.
Fig. 6.
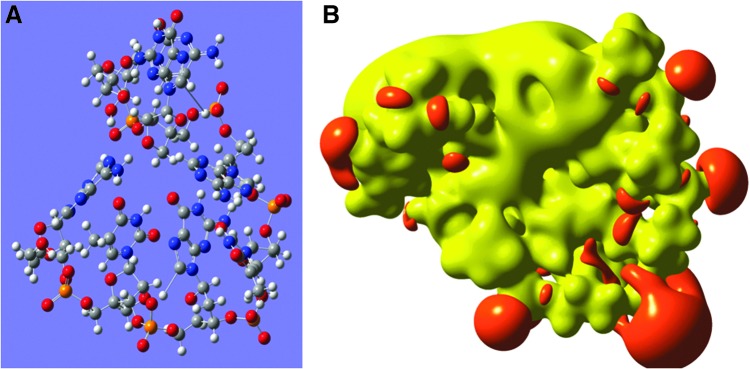
Three-dimensional chemical structure of the LNA-DNA PO gap-mer 5′-ATGTAGC-3′ (LNA nts underlined) (A), and in (B) the electrostatic potential represented by an iso-surface value of iso=0.06H=1.62eV=156 kJ/mol. The positive electrostatic potential is shown in yellow and the negative in red.
An important question is which role the initial conditions play on the quantum electronic structure calculation and optimization and if changed initial conditions will lead to different conformational structures. These could be purely technical initial conditions such as the choice of basis set for the quantum mechanical optimization. In order to assess the dependence of the initial conditions of the calculated optimized structures of a given molecule, we chose a DNA oligomer as a test case. Hence, we calculated optimized electronic structures of the DNA-7mer of ATGTAGC using the basis set 3-21G and 6-31G*, where the latter is the double size of the former: The measure for the difference of the electronic structures can be the size of the HOMO/LUMO gap (quantum mechanically) and more classically, the end-to-end distance of the molecular structure. For the smaller basis set 3-21G, the HOMO-state is −0.30787 H and the LUMO-state is −0.04727; and thus, the H/L gap is g=0.26060. The end-to-end distance is l=15.75 Å, measured between specific atoms separated with long distance (the exocyclic nitrogen on the 5′ end adenosine and the 3′ carbon of the deoxyribose of the 3′ end cytosine). For the larger basis set 6-31G*, the HOMO-state is −0.31649 and the LUMO-state −0.04342, and the gap becomes g=0.27307. The end-to-end distance was in this case l=16.95 Å. Thus, we see only little change in the resulting quantum electronic structures, making the optimized structures more unique. It should be noted that uniqueness of the quantum electronic structures are related to calculated minima. In particular for larger molecules (>3nts) more minimized structures exist, but importantly such other structures and their related MEPs are specific for the given compound. Altogether, the QM calculations presented here produce optimized electronic structures and molecular electrostatic potentials and importantly, the outcome of QM calculations appear less dependent on initial conditions than for example, classical molecular dynamics.
The QM calculation method employed here has been experimentally verified by spectroscopy studies of other biomolecules (peptides). The theoretically derived spectra required electronic wave functions, and hence QM, in order to get the quantum states, or wave functions for the matrix element, of the atomic tensors describing vibrational states. It was reported in this study that these calculations were in accordance with experimentally determined vibrational spectra of bio-molecules obtained by infrared absorption and RAMAN scattering (Jalkanen et al., 2001; Abdali et al., 2002).
In this study we have also included calculations on oligonucleotides containing phosphorothioate (PS) internucleoside linkage. Sulfur is introduced during chemical synthesis and replaces one of the non-bridging oxygens. Since synthesis of PS modifications creates a chiral phosphorous, and since these are normally not resolved, antisense oligonucleotides are diastereomeric mixtures. The number of diastereoisomers introduced is equal to 2n, where n is the number of PSs made in the oligonucleotide. PSs play a central role in the activity of antisense oligonucleotides (AONs). PSs are more nucleases resistant than native POs and secure that antisense oligonucleotides designed with DNA nucleotide segments are nucleolytic stable. Compared with POs, PS modified oligonucleotides are also better taken up by cells, and systemically PSs exhibit better tissue distribution and general pharmacokinetic properties. Due to these properties PSs are frequently included in AONs (Lebedeva et al., 2000; Uhlmann, 2000; Krutzfeldt et al., 2005; Koch et al., 2008; Lanford et al., 2010; Stein et al., 2010; Straarup et al., 2010; Koller et al., 2011; Moschos et al., 2011; Obad et al., 2011; Stanton et al., 2012).
Results
The QM calculated molecular iso-potentials were compared for either native phosphordiester oligonucleotides (PO) or chemically modified phosphorothioates (PS) (Cohen, 1993; Eckstein, 2007). The value of the iso-potentials was chosen to be an energy ranging from 10 - 20 kcal/mol. This value corresponds to the energy of 2–5 H-bonds and characterizes a potential binding to a receptor.
Initially, the effects of modifying the ribose backbone were examined. DNA-PO-AAG was compared with all LNA-PO-AAG (Fig. 1). From the structural representation, DNA is generally more bent while LNA is more stretched. This is consistent with the more rigid bicyclic structure of LNA (Obika et al., 1997; Nielsen et al., 2000; Petersen et al., 2000, 2002; Nielsen and Spielmann, 2005; Hughesmann et al., 2011). When the iso-potential surfaces are added, the surface of LNA-PO-AAG is constricted and concentrated on the phosphate backbone face of the molecule. The HOMO orbital was shifted from the 5′ adenine in DNA to the 3′ guanine of the LNA, while the LUMO orbital resides on the central adenine in both. Interestingly, the electrostatic potential of the 3′ PO in the DNA is weak, much weaker in fact than for the other phosphate in the trimer, and exhibits a potential comparable to the lone pairs on the nucleobases—particularly the 5′ adenine.
Next, the effects of changed internucleoside linkages and PS configuration were examined. LNA-PO-AAG (Fig. 1) was compared with LNA-PS-AAG (with the PSs in the RS configuration) (Fig. 2A). The PS configuration was determined by assigning the hydrogen to sulfur (S–P single bond). The LNA-PS-AAG (RS) modification results in a large potential change and exhibits a more scattered topology compared to LNA-PO-AAG, with a shift of the electrostatic potential towards the 5′ end (Figs. 1, 2A). Interestingly, with the PS in the SR configuration the topology reverts into a coherent structure resembling the compact form of the PO congener (Figs. 1B, 2B). PS modification also induces changes in the localization of the frontier orbitals. The HOMO-LUMO orbitals are split between adenine and guanine for LNA-PO-AAG but the LUMO resides on the central adenines for both PSs, irrespective of configuration. Interestingly, the HOMO resides on sulfur on the PSs in the S configuration.
The effects of changing/mutating a nucleobase were examined. LNA-PO-AAG was compared with the mutant LNA-PO-AGG, (Fig. 3). The single nucleotide mutation causes a shift in the iso-potential from a compact surface for the AAG sequence (Fig. 3A) to a more scattered surface for the mutant, AGG sequence, placing a stronger field on 5′ adenine (Fig. 3B). In addition, the potential for the 3′ PO is weakened. The HOMO shifts from the 3′ guanine to the 5′-adenine, while the LUMO is located on the central guanine (Fig. 3A, C).
The effects of increasing the nucleotide sequence from trimer to pentamer were also examined. The potential of the LNA-PO-AAG is concentrated around the central/and 5′-adenosines. Adding two nucleotides (A and C) changes the iso-potential concentration towards the 3′ end and leaves the two 5′-adenines with much less electrostatic density (Fig. 4). The frontier orbitals also shift from the central adenine and 3′ guanine in the trimer to the penultimate 5′ adenine and the 3′ adenine in the pentamer. Electrostatically, the potential of the pentamer is unique.
Sequence/topology relations were examined for larger oligonucleotides. The biologically active LNA-PO-7-mer (5′-ATGTAGC-3′) (Fig. 5A, C) was compared with the single base pair mutant (5′-ATGAAGC-3′) (Fig. 5B, D) and with the DNA-PO-ATGTAGC (not shown). The LNA-PO-7-mer (5′-ATGTAGC-3′) (Fig. 5A, C) was originally designed by Obad et al. (as fully PS modified) to target the seed sequence of microRNA (miR)-221 and miR-222 and accordingly, was able to inhibit both microRNAs due to seed sequence homology (Obad et al., 2011).
The interchange of the central thymine with adenine does not cause a change in the position of the frontier orbitals. In both cases, the HOMO orbital is found on the adenine closest to the 3′ end and the LUMO is found on the 5′ adenine. However, changing the backbone to DNA causes a shift in both the position and energy of the frontier orbitals. Thus, in the DNA oligonucleotide, the HOMO orbital is on the 3′ cytosine (E=−8.38 eV), and the LUMO orbital is on the 5′ adenine (E=−1.29 eV) (data not shown). The LNA (5′-ATGTAGC-3′) has the HOMO orbital on the adenine closest to the 3′ end (E=−8.04 eV) and the LUMO orbital on 5′ adenine (E=−4.55 eV). Thus, the gap is reduced for the LNA oligonucleotide (ΔE=−3.49 eV) compared with the iso-sequential DNA oligonucleotide (ΔE=−7.09 eV). The molecular structure of the DNA-PO and LNA-PO oligonucleotide with same sequence (5′-ATGTAGC-3′) are very different, with the DNA oligonucleotide being bent (ca. 90°), and the LNA oligonucleotide with a more straight structure. Correspondingly, the topologies of the electrostatic potentials are also very different and each unique. Interestingly, the central thymidine/adenosine mutation causes also a global change in topologies of the two LNA oligonucleotides (Fig. 5C, D). The topology of the electrostatic potential of the mutant is more evenly distributed over the molecule with a weaker negative potential (red). The central mutation also causes a 3′ end weakening of the positive potential (yellow) (Fig. 5C, D).
Finally, a study was also carried out on a LNA-DNA PO 7-gapmer. The compound has the same nucleotide sequence as in the parent LNA compound mentioned above (5′-ATGTAGC-3′), but the middle three nucleotides were substituted with DNA PO nucleotides. The resulting optimized structure (Fig. 6A) was more compact and bend—almost globular—and the increased flexibility converged by the DNA nucleotides brought the two LNA ends close to each other. The HOMO-LUMO gap was ΔE=−4.05 eV, and the established iso-surface had a disrupted or scattered potential surface very different from the potential of the iso-sequential full LNA 7-mer (Fig. 6 B). The electronic data on the 7-mers confirm that there are no particular boundary effects in this study, although it could be anticipated that special boundary effects on the 3-mers were dominating.
Discussion
The QM data illustrate that small modifications can profoundly change the structure, polarity and topology of the molecular electrostatic potential (MEP) of oligonucleotides. Introduction of a single base mutation (Figs. 3, 5) induces global structural and MEP topology changes, as do oligonucleotide design changes for iso-sequential compounds (Figs. 5, 6). Changing the sugar backbone from DNA to LNA can lead to strong polarity changes in the backbone phosphates (Fig. 1). The molecular structure and MEP may also be different among diastereoisomers of the same sequence (Fig. 2). As further illustrated in Fig. 4, structure and MEP topology for any given compound is unique; structural modifications lead to topologies that cannot be viewed simply as additions to a precursor structure. The wave-function representation illustrates that chemical modifications influence both structure and MEP, not only at the site of incorporation, but throughout the entire molecule.
Molecular structure, MEP strength, and topology are determinants for the interaction with and the binding pattern between molecules. Generally, ligand-receptor binding processes are very complex, the net binding energy is a balance between molecular structure and MEP on one side and counteracting desolvation effects on the other (Chong et al., 1998; Kangas and Tidor, 1999; Sheinerman et al., 2000; Muzet et al., 2003; Sulea and Purisima, 2003; Burgoyne and Jackson, 2006; Kraut et al., 2006; Dhimoy et al., 2008, 2013; Kenny, 2009). Thus, binding cannot be automatically assumed to take place even if perfect structure and MEP complementarity exists between ligand and receptor. However, in some systems, ligand-receptor MEP complementarity appears to play a more determinant role for binding to take place. These receptor-ligand systems are characterized by ligand binding to concave pockets of a receptor where binding is driven thermodynamically by hydrogen bonding (Kangas and Tidor, 1999; Muzet et al., 2003). This has importance for oligonucleotide–protein binding. Oligonucleotide binding proteins usually share a common concave structure called the OB (oligonucleotide binding) fold (Sun and Shamoo, 2003; Harris et al., 2012). Oligonucleotide binding to the fold is driven by hydrogen (H)-bonding which is further stabilized by hydrophobic interactions and stacking between nucleobases and aromatic amino acids. Interestingly, strong binding is also observed to negatively charged proteins, and the charged backbone may also interact either via H-bonding or salt bridge formation. Notably, however, the phosphates are predominately exposed to the highly dielectric solvent (Max et al., 2006; Zeeb et al., 2006). The fact that any energy strength can be selected as an iso-value for the electrostatic potentials (e.g., fixed to H-bonding energies) means that the MEPs are informative about how well H-bonding may occur locally. Altogether, it can be assumed that electrostatic complementarity plays a more determinant role for single stranded oligonucleotide–protein interactions.
The interaction/binding of oligonucleotides to proteins is an important parameter influencing toxicity, plasma half-life, tissue accumulation, cellular uptake, and activity of antisense oligonucleotides (Beltinger et al., 1995; Prakash et al., 2002; Watanabe et al., 2006; Lai et al., 2007a, 2007b; Stein et al., 2009). Since H-bonding to folded protein structures (e.g., OB fold) is important for oligonucleotide–protein binding we can assume that structural and MEP complementarity are determinants for binding/interaction to take place. Consequently, oligonucleotides with marked differences in MEP are likely to exhibit differences in protein binding. Assisted by the QM data, it is evident that small structural modifications may lead to profound changes in the binding/interaction profile (i.e., the phenotypic profile). As an example, a single nucleotide modification (e.g., perfect match vs. one base pair mismatch) may lead to major, or “all or none,” phenotypic changes. The QM modeling data explains and illustrates reasons underlying some of the unexpected observations relating to major phenotypic changes observed for otherwise “closely related” molecules (vide infra) (Seth et al., 2009; Straarup et al., 2010; Laxton et al., 2011; Stanton et al., 2012; Hagedorn et al., 2013).
The data also underlines that caution should be taken in making broad predictions about the effects of particular chemical modifications. For instance, in nearly all cases, incorporation of LNA nucleotides will lead to higher affinity and nuclease stability. However, how a single LNA modification, or other structural units for that matter, will influence the global effects, on a cellular or systemic level is more complex. As an example of how the global bioproperties of LNA gapmer phosphorothioate oligonucleotides can be affected by seemingly minimal changes, a series of these 16-mer oligonucleotides, targeted to Bcl-2, were synthesized that differed only by a single base in the gap. After gymnotic delivery of the oligos to 518A2 cells (Soifer et al., 2012; Stein et al., 2010), the phosphorylation patterns of the PI3K/Akt/mTor pathway were examined. Each oligo silenced Bcl-2 protein expression to the same extent. However, the levels of phosphorylation of PI3K, Akt, and mTOR were different for every oligo (Cy A. Stein, personal communication). Another example was recently reported where a specific LNA 13-mer was hepatotoxic. However, adding just a single additional LNA nt rendered the resulting 14-mer non-hepatotoxic (Hagedorn et al., 2013). This demonstrates that single-nucleotide substitutions may induce major phenotypic changes—changes that are not “inherited” from the single nucleotide per se, but result more likely from the nucleotide insertion/substitution in a specific sequence/compound context. At this point in time, we can predict some effects of single-nucleotide substitutions, but not how single-nucleotide modifications will alter the global interactions. In practice, this also means that caution should be taken in over-generalizing about data arising from comparative studies using single base pair mismatch control oligonucleotides. The potential to produce very different phenotype, induced by the single-nucleotide modification compared to the “full match” oligonucleotide, may be so great that any observed differences in effects may be caused by the altered global interaction profile, and only minimally resulting from the mismatch hybridization effect with the RNA target.
Antisense oligonucleotides are composed of many discrete structural units each bringing unique properties (e.g., nucleobases, PO/PS internucleotide linkages, DNA/RNA/LNA backbones, base sequence motifs, etc.). These units collectively engender, in a sequence context, the properties of the individual molecule. The QM data shown here illustrate that each structural element, and potentially their diastereoisomers (Fig. 3), may induce property changes if substituted by another structural element. This large property diversity points to the existence of a wide “property space” of oligonucleotides (Koch, 2013). It should be possible to exploit this understanding in antisense drug discovery by testing many combinations of a structural units, with the aim to explore the property space, to be able to select better candidates with unique/extreme properties (Fig. 7). For instance, optimized lead candidates should exhibit an averaged optimal interaction profile for the global interaction determinant for effective cellular uptake, efficient intracellular trafficking, low toxicity, and for potent inhibition of the target RNA.
Fig. 7.

Illustration of “extreme properties” of drug candidates. Most oligonucleotides tend to cluster around a central value for each variable (phenotype) in search. The dashed circles illustrate the atypical and extreme properties that are selected for in drug discovery: high pharmacological activity combined with low toxicity.
The question is then how large should the oligonucleotide pool be and how many structural units—pharmacophores—should be combined? We are not able to answer that question now, and numbers becomes quickly a limiting factor. For example, the number of unique 14-mers constructed from 4 different nucleotide building blocks is 414 (2.7×108). If the number of building blocks is increased by adding 4 extra nucleotides (e.g., four LNA and four DNA nucleotides) an expanded property space will be formed. However, in order to explore all combinations 814=4.4×1012 compounds would have to be tested. Numbers like these are known issues from classical small molecule drug discovery and cannot be managed in practice. Nevertheless, in contrast to small molecule drug discovery, antisense offers a great advantage: The RNA-target binding motif is known! Due to the requirement of complementary base pairing to the target, the relevant compound number is immediately reduced by ∼6 orders of magnitude (depending on the length and number of modifications considered). This reduces but still leaves too many compounds to handle, further reduction required. In this case, property prediction algorithms are the preferred tools, and although the QM data illustrate a more intricate structure/activity relationship of oligonucleotides, it is far from chaotic. However, new and improved prediction algorithms must be constructed from larger training data sets, where the property of interest has been measured experimentally, and correlated with several structural entities. These include nucleobase sequence, backbone, inter-nucleotide linkages, or any modification of these in combination and related to a specific molecular context. We are currently establishing such large databases of thousands of oligonucleotides tested both in vitro and in vivo. A simplified decomposition of oligonucleotides has been developed, combined with a random forest classification that with ∼80% significance can predict the hepatotoxic potential of LNA/DNA-gapmer oligonucleotides (Hagedorn et al., 2013). Predicted experimental outcome by in silico algorithms is linked with uncertainty. Nevertheless, they offer a knowledge based strategy to reduce the relevant compound number, by zooming in on the selection of oligonucleotides where the probability is highest for finding unique/extreme properties (Fig. 7). It is therefore not necessary to test all combinations of a structural unit selection. Testing much smaller numbers (e.g., 102–104), may be suffice for a thorough investigation of the property space.
The data presented here are based on QM calculations for 3-, 5-, and 7-mer oligonucleotides; these lengths are significantly shorter than the ones used for antisense purposes (12- to 16-mers). It is therefore relevant to ask if the structural and MEP sensitivity to the chemical modifications observed here also is the case for the 12- to 16-mers. We have not yet been able to complete QM calculations on longer oligonucleotides because such calculations are very CPU demanding. However, experimental observations of large property diversity and the “all or none” phenotypes of closely related antisense compounds (vide supra) could indicate that single modifications may affect not only the site of substitution, but can affect the entire molecule as well. In that case, a wave function representation could be useful, and accordingly large variations in structure and MEP would also be expected for longer oligonucleotides.
Conclusion
The QM oligonucleotide modeling illustrates that structure activity relations are complex interrelations of individual chemical components and their specific structural position. Property diversity is mechanistically driven by the molecular structure and the derived molecular electrostatic potentials. Nucleobase sequence is not the only determinant for oligonucleotide properties; the global properties are derived from each structural unit in concert with the molecular composition. The diversified recognition potential between oligonucleotides and non-target biomolecules should be exploited. This could be done by testing more combinations of selected structural units. In a targeted approach, the binding/interactions between lead compounds and non-target biomolecules should be optimized together with target RNA inhibition.
Acknowledgments
The author wishes to thank Professor Cy A. Stein, City of Hope Medical Center, and Senior Director Christoph Rosenbohm, Santaris Pharma A/S, for fruitful discussions and for commenting in this article.
Author Disclosure Statement
This work is made by authors employed full-time by Santaris Pharma A/S (TK, ML, and HØ) and by authors (HGB and IS) that are employed by the Technical University of Denmark (DTU).
References
- ABDALI S., JALKANEN K.J., BOHR H., SUHAI S., and NIEMINEN R.M. (2002). The VA and VCD spectra of various isotopes of L-alanine in aqueous solution. Chem. Phys. 282,219–235 [Google Scholar]
- ALTMANN K.-H., DEAN N.M., FABBRO D., FREIER S.M., GEIGER T., HÄNER R., HÜSKEN D., MARTIN P., MONIA B.P., MÜLLER M., et al. (1996). Second generation OD antisense oligonucleotides: from nuclease resistance to biological efficacy in animals. Chimia 50,168–176 [Google Scholar]
- BELTINGER C., SARAGOVI H.U., SMITH R.M., LESAUTEUR L., SHAH N., DEDIONISIO L., CHRISTENSEN L., RAIBLE A., JARETT L., and GEWIRTZ A.M. (1995). Binding, uptake, and intracellular trafficking of phosphorothioate-modified oligodeoxynucleotides. J. Clin. Invest 95,1814–1823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- BOHR H.G. (2013). Perspectives in quantum nanobiology and biophysical chemistry. Current Phys. Chem. 3,4–8 [Google Scholar]
- BURGOYNE N.J., and JACKSON R.M. (2006). Predicting protein interaction sites: binding hot-spots in protein-protein and protein-ligand interfaces. Bioinformatics 22,1335–1342 [DOI] [PubMed] [Google Scholar]
- CHONG L.T., DEMPSTER S.E., HENDSCH Z.S., LEE L.P., and TIDOR B. (1998). Computation of electrostatic complements to proteins: a case of charge stabilized binding. Protein Sci. 7,206–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- COHEN J.S. (1993). Phosphorothioate oligonucleotides. In: Antisense Research and Applications. Crooke S.T. and Lebleu B., eds. (CRC Press: Boca Raton, FL: ), pp. 205–223 [Google Scholar]
- COOK P.D. (1999). Making drugs out of oligonucleotides: a brief review and perspective. Nucleosides Nucleotides 18,1141–1162 [DOI] [PubMed] [Google Scholar]
- COREY D.R. (1997). Peptide nucleic acids: expanding the scope of recognition. TIBTECH 15,224–229 [DOI] [PubMed] [Google Scholar]
- DANGLER C.A. (1996). Nucleic Acid Analysis: Principles and Bioapplications. (Wiley-Liss: New York: ), pp. 1–475 [Google Scholar]
- DAVIS S., LOLLO B., FREIER S., and ESAU C. (2006). Improved targeting of miRNA with antisense oligonucleotides. Nucleic Acids Res. 34,2294–2304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DEBART F., ABES S., DEGLANE G., MOULTON H.M., CLAIR P., GAIT M.J., VASSEUR J.J., and LEBLEU B. (2007). Chemical modifications to improve the cellular uptake of oligonucleotides. Curr. Top. Med. Chem. 7,727–737 [DOI] [PubMed] [Google Scholar]
- DEMIDOV V.V., and FRANK-KAMENETSKII M.D. (2004). Two sides of the coin: affinity and specificity of nucleic acid interaction. Trends Biochem. Sci. 29,62–71 [DOI] [PubMed] [Google Scholar]
- DHIMOY R.K., BALANARAYAN P., ANDSHRIDHAR G.R. (2008). An appraisal of Poincaré-Hopf relation and application to topography of molecular electrostatic potentials. J. Chem. Phys. 129, 174103. [DOI] [PubMed] [Google Scholar]
- DHIMOY R.K., BALANARAYAN P., and SHRIDHAR G.R. (2013). Signatures of molecular recognition from topography of electrostatic potential. J. Chem. Sci. 121,815–821 [Google Scholar]
- ECKSTEIN F. (2007). The versatility of oligonucleotides as potential therapeutics. Expert. Opin. Biol. Ther. 7,1021–1034 [DOI] [PubMed] [Google Scholar]
- ELMEN J., LINDOW M., SCHUTZ S., LAWRENCE M., PETRI A., OBAD S., LINDHOLM M., HEDTJARN M., HANSEN H.F., BERGER U., GULLANS S., KEARNEY P., SARNOW P., STRAARUP E.M., and KAUPPINEN S. (2008a). LNA-mediated microRNA silencing in non-human primates. Nature 452,896–899 [DOI] [PubMed] [Google Scholar]
- ELMEN J., LINDOW M., SILAHTAROGLU A., BAK M., CHRISTENSEN M., LIND-THOMSEN A., HEDTJARN M., HANSEN J.B., HANSEN H.F., STRAARUP E.M., MCCULLAGH K., KEARNEY P., and KAUPPINEN S. (2008b). Antagonism of microRNA-122 in mice by systemically administered LNA-antimiR leads to up-regulation of a large set of predicted target mRNAs in the liver. Nucleic Acids Res. 36,1153–1162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- FREIER S.M., and ALTMANN K.-H. (1997). The ups and downs of nucleic acid duplex stability: structure-stability studies on chemically-modified DNA:RNA duplexes. Nucleic Acid Res. 25,4429–4443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- FRISCH M.J., TRUCKS G.W.; SCHLEGEL H.B.; SCUSERIA G.E.; ROBB M.A.; CHEESEMAN J.R.; SCALMANI G.; BARONE V.; MENNUCCI B.; PETERSSON G.A., et al. (2009). Gaussian 09, (Gaussian, Inc., Wallingford CT: ) [Google Scholar]
- HAGEDORN P.H., YAKIMOV V., OTTOSEN S., KAMMLER S., NIELSEN N.F., HOG A.M., HEDTJARN M., MELDGAARD M., MOLLER M.R., ORUM H., et al. (2013). Hepatotoxic potential of therapeutic oligonucleotides can be predicted from their sequence and modification pattern. Nucleic Acid Ther. 23,302–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- HANSEN J.B., FISKER N., WESTERGAARD M., KJAERULFF L.S., HANSEN H.F., THRUE C.A., ROSENBOHM C., WISSENBACH M., ORUM H., and KOCH T. (2008). SPC3042: a proapoptotic survivin inhibitor. Mol. Cancer Ther. 7,2736–2745 [DOI] [PubMed] [Google Scholar]
- HANVEY J.C., PEFFER N.J., BISI J.E., THOMSON S.A., CADILLA R., JOSEY J.A., RICCA D.J., HASSMAN C.F., BONHAM M.A., AU K.G., et al. (1992). Antisense and antigene properties of peptide nucleic acid. Science 258,1481–1485 [DOI] [PubMed] [Google Scholar]
- HARRIS C.R., MACKOY T., MACHADO C.D.ANA XU D., ROHS R., and FENLEY O.M. (2012). Opposite attract: shape and electrostatic complementarity in Protein-DNA Complexes. In: Innovations in Biomolecular Modeling and Simulations, Volume 2 Schlick T., ed. (Royal Society of Chemistry; ), pp. 53–80 [Google Scholar]
- HUGHESMANN C.B., TURNER R.F.B., and HAYNES C.A. (2011). Role of heat capacity change in understanding and modeling melting thermodynamics of complementary duplexes containing standard and nucleobase-modified LNA. Biochemistry 50,5354–5368 [DOI] [PubMed] [Google Scholar]
- JALKANEN J.K, NIEMINEN M.R., FRIMAND K., BOHR J., BOHR H., WADE C.R., TAJKHORSHID E., and SUHAI S. (2001). A comparison of aqueous solvent models used in the calculation of the Raman and ROA spectra od L-alanine. Chem. Phys. 265,125–151 [Google Scholar]
- JEPSEN J.S., and WENGEL J. (2004). LNA-Antisense rivals siRNA for gene silencing. Curr. Opin. Drug Discov. Devel. 7:188–194 [PubMed] [Google Scholar]
- KANGAS E., and TIDOR B. (1999). Charge optimization leads to favorable electrostatic binding free energy. Phys. Rev. E. Stat. Phys. Plasmas. Fluids Relat. Interdiscip. Topics 59,5958–5961 [DOI] [PubMed] [Google Scholar]
- KENNY P.W. (2009). Hydrogen bonding, electrostatic potential, and molecular design. J. Chem. Inf. Model. 49,1234–1244 [DOI] [PubMed] [Google Scholar]
- KOCH T. (2013). LNA antisense: a review. Curr. Phys. Chem. 3,55–68 [Google Scholar]
- KOCH T., and ØRUM H. (2007). Locked nucleic acid. In: Antisense Drug Discovery. Crooke S.T., ed. (CRC Press: Boca Raton, FL: ), pp. 519–565 [Google Scholar]
- KOCH T., ROSENBOHM C., HANSEN H.F.F., HANSEN B., STRAARUP E.M., and KAUPPINEN S. (2008). Locked nucleic acid: properties and therapeutic aspects. In: Therapeutic Oligonucleotides. Jens Kurreck, ed. (RSC Publishing: Cambridge, UK: ), pp. 103–141 [Google Scholar]
- KOLLER E., VINCENT T.M., CHAPPELL A., DE S., MANOHARAN M., and BENNETT C.F. (2011). Mechanisms of single-stranded phosphorothioate modified antisense oligonucleotide accumulation in hepatocytes. Nucleic Acids Res. 39,4795–4807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- KRAUT D.A., SIGALA P.A., PYBUS B., LIU C.W., RINGE D., PETSKO G.A., and HERSCHLAG D. (2006). Testing electrostatic complementarity in enzyme catalysis: hydrogen bonding in the ketosteroid isomerase oxyanion hole. PLoS Biol. 4, e99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KRUTZFELDT J., RAJEWSKY N., BRAICH R., RAJEEV K.G., TUSCHL T., MANOHARAN M., and STOFFEL M. (2005). Silencing of microRNAs in vivo with ‘antagomirs'. Nature 438,685–689 [DOI] [PubMed] [Google Scholar]
- LAI J.C., BROWN B.D., VOSKRESENSKIY A.M., VONHOFF S., KLUSSMAN S., TAN W., COLOMBINI M., WEERATNA R., MILLER P., BENIMETSKAYA L., and STEIN C.A. (2007A). Comparison of d-g3139 and its enantiomer L-g3139 in melanoma cells demonstrates minimal in vitro but dramatic in vivo chiral dependency. Mol. Ther. 15,270–278 [DOI] [PubMed] [Google Scholar]
- LANFORD R., HILDEBRANDT-ERIKSEN E.S., PETRI A., PERSSON R.,LINDOW M., MUNK M.E., KAUPPINEN S., and ØRUM H. (2010). Therapeutic silencing of microRNA-122 in Primates with chronic hepatitis C virus infection. Science 327,198–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LAXTON C., BRADY K., MOSCHOS S., TURNPENNY P., RAWAL J., PRYDE D.C., SIDDERS B., CORBAU R., PICKFORD C., and MURRAY E.J. (2011). Selection, optimization, and pharmacokinetic properties of a novel, potent antiviral locked nucleic acid-based antisense oligomer targeting hepatitis C virus internal ribosome entry site. Antimicrob. Agents Chemother. 55,3105–3114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEBEDEVA I., BENIMETSKAYA L., STEIN C.A., and VILENCHIK M. (2000). Cellular delivery of antisense oligonucleotides. Eur. J. Pharm. Biopharm. 50,101–119 [DOI] [PubMed] [Google Scholar]
- LINDHOLM M.W., ELMEN J., FISKER N., HANSEN H.F., PERSSON R., MOLLER M.R., ROSENBOHM C., ORUM H., STRAARUP E.M., and KOCH T. (2011). PCSK9 LNA Antisense Oligonucleotides Induce Sustained Reduction of LDL Cholesterol in Nonhuman Primates. Mol. Ther. 20,376–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MARTIN P. (1995). Ein neuer zugang zu 2′-O-alkylribonucleosiden und eigenschaften deren oligonucleotiden. Helvetica Chimica Acta 78,486–504 [Google Scholar]
- MAX K.E., ZEEB M., BIENERT R., BALBACH J., and HEINEMANN U. (2006). T-rich DNA single strands bind to a preformed site on the bacterial cold shock protein Bs-CspB. J. Mol. Biol. 360,702–714 [DOI] [PubMed] [Google Scholar]
- MOSCHOS S.A., FRICK M., TAYLOR B., TURNPENNY P., GRAVES H., SPINK K.G., BRADY K., LAMB D., COLLINS D., ROCKEL T.D., et al. (2011). Uptake, efficacy, and systemic distribution of naked, inhaled short interfering RNA (siRNA) and locked nucleic acid (LNA) antisense. Mol. Ther. 19,2163–2168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MUZET N., GUILLOT B., JELSCH C., HOWARD E., and LECOMTE C. (2003). Electrostatic complementarity in an aldose reductase complex from ultra-high-resolution crystallography and first-principles calculations. Proc. Natl. Acad. Sci. U. S. A. 100,8742–8747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- NIELSEN K.E., SINGH S.K., WENGEL J., and JACOBSEN J.P. (2000). Solution structure of an LNA hybridized to DNA: NMR study of the d(CT(L)GCT(L)T(L)CT(L)GC):d(GCAGAAGCAG) duplex containing four locked nucleotides. Bioconjug. Chem. 11,228–238 [DOI] [PubMed] [Google Scholar]
- NIELSEN K.E., and SPIELMANN H.P. (2005). The structure of a mixed LNA/DNA:RNA duplex is driven by conformational coupling between LNA and deoxyribose residues as determined from (13)C relaxation measurements. J. Am. Chem. Soc. 127,15273–15282 [DOI] [PubMed] [Google Scholar]
- NIELSEN P.E., EGHOLM M., BERG R.H., and BUCHARDT O. (1991). Sequence-selective recognition of DNA by strand displacement with a thymine-substituted polyamide. Science 254,1497–1500 [DOI] [PubMed] [Google Scholar]
- OBAD S., DOS SANTOS C.O., PETRI A., HEIDENBLAD M., BROOM O., RUSE C., FU C., LINDOW M., STENVANG J., STRAARUP E.M., et al. (2011). Silencing of microRNA families by seed-targeting tiny LNAs. Nat. Genet. 43,371–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- OBIKA S., NANBU D., HARI Y., MORIO J.A.K., IN Y., ISHIDA T., and IMANISHI T. (1997). Synthesis of 2′-0,4′-C-methyleneuridine and -cytidine. Novel bicyclic nucleosides having a fixed c3-endo sugar puckering. Tetrahedron Lett. 38,8735–8738 [Google Scholar]
- PETERSEN M., BONDENSGAARD K., WENGEL J., and JACOBSEN J.P. (2002). Locked nucleic acid (LNA) recognition of RNA: NMR solution structures of LNA:RNA hybrids. J. Am. Chem. Soc. 124,5974–5982 [DOI] [PubMed] [Google Scholar]
- PETERSEN M., NIELSEN C.B., NIELSEN K.E., JENSEN G.A., BONDENSGAARD K., SINGH S.K., RAJWANSHI V.K., KOSHKIN A.A., DAHL B.M., WENGEL J., and JACOBSEN J.P. (2000). The conformations of locked nucleic acids (LNA). J. Mol. Recognit. 13,44–53 [DOI] [PubMed] [Google Scholar]
- PRAKASH T.P., MANOHARAN M., KAWASAKI A.M., FRASER A.S., LESNIK E.A., SIOUFI N., LEEDS J.M., TEPLOVA M., and EGLI M. (2002). 2′-O-[2-(methylthio)ethyl]-modified oligonucleotide: an analogue of 2′-O-[2-(methoxy)-ethyl]-modified oligonucleotide with improved protein binding properties and high binding affinity to target RNA. Biochemistry 41,11642–11648 [DOI] [PubMed] [Google Scholar]
- SETH P.P., SIWKOWSKI A., ALLERSON C.R., VASQUEZ G., LEE S., PRAKASH T.P., WANCEWICZ E.V., WITCHELL D., and SWAYZE E.E. (2009). Short antisense oligonucleotides with novel 2′-4′ conformationaly restricted nucleoside analogues show improved potency without increased toxicity in animals. J. Med. Chem. 52,10–13 [DOI] [PubMed] [Google Scholar]
- SHABAROVA Z.A., Bogdanov A.A. (1994). Advanced organic chemistry of nucleic acids, (VCH: Weinheim, Germany: ), pp. 389–471 [Google Scholar]
- SHEINERMAN F.B., NOREL R., and HONIG B. (2000). Electrostatic aspects of protein-protein interactions. Curr. Opin. Struct. Biol. 10,153–159 [DOI] [PubMed] [Google Scholar]
- SINGH S.K., NIELSEN P., KOSHKIN A., and WENGEL J. (1998). LNA (locked nucleic acids): synthesis and high-affinity nucleic acid recognition. Chem. Commun. 455–456 [Google Scholar]
- SOIFER H.S., KOCH T., LAI J., HANSEN B., HOEG A., OERUM H., and STEIN C.A. (2012). Silencing of gene expression by gymnotic delivery of antisense oligonucleotides. Methods Mol. Biol. 815,333–346 [DOI] [PubMed] [Google Scholar]
- STANTON R., SCIABOLA S., SALATTO C., WENG Y., MOSHINSKY D., LITTLE J., WALTERS E., KREEGER J., DIMATTIA D., CHEN T., et al. (2012). Chemical modification study of antisense gapmers. Nucleic Acid Ther. 22,344–359 [DOI] [PubMed] [Google Scholar]
- STEIN C.A., HANSEN J.B., LAI J., WU S., VOSKRESENSKIY A., HOG A., WORM J., HEDTJARN M., SOULEIMANIAN N., MILLER P., SOIFER H.S., et al. (2010). Efficient gene silencing by delivery of locked nucleic acid antisense oligonucleotides, unassisted by transfection reagents. Nucleic Acids Res. 38, e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STEIN C.A., WU S., VOSKRESENSKIY A.M., ZHOU J.F., SHIN J., MILLER P., SOULEIMANIAN N., and BENIMETSKAYA L. (2009). G3139, an anti-Bcl-2 antisense oligomer that binds heparin-binding growth factors and collagen I, alters in vitro endothelial cell growth and tubular morphogenesis. Clin. Cancer Res. 15,2797–2807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- STRAARUP E.M., FISKER N., HEDTJARN M., LINDHOLM M.W., ROSENBOHM C., AARUP V., HANSEN H.F., ORUM H., HANSEN J.B., and KOCH T. (2010). Short locked nucleic acid antisense oligonucleotides potently reduce apolipoprotein B mRNA and serum cholesterol in mice and non-human primates. Nucleic Acids Res. 38,7100–7111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- SULEA T., and PURISIMA E.O. (2003). Profiling charge complementarity and selectivity for binding at the protein surface. Biophys. J. 84,2883–2896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- SUN S., and SHAMOO Y. (2003). Biochemical characterization of interactions between DNA polymerase and single-stranded DNA-binding protein in bacteriophage RB69. J. Biol. Chem. 278,3876–3881 [DOI] [PubMed] [Google Scholar]
- TRAPANE T., and TS'O P. (1994). Triplex formation at single-stranded nucleic acid target sites of unrestricted sequences by two added strands of oligonucleotides: a proposed model. J. Am. Chem. Soc. 116,10437–10449 [Google Scholar]
- UHLMANN E. (2000). Recent advances in the medicinal chemistry of antisense oligonucleotides. Curr. Opin. Drug Discov. Devel. 3,203–213 [PubMed] [Google Scholar]
- UHLMANN E., and PEYMAN A. (1990). Antisense oligonucleotides: a new therapeutic principle. Chem. Rev. 90,544–579 [Google Scholar]
- WAHLESTEDT C., SALMI P., GOOD L., KELA J., JOHNSSON T., HOKFELT T., BROBERGER C., PORRECA F., LAI J., REN K., et al. (2000). Potent and nontoxic antisense oligonucleotides containing locked nucleic acids. Proc. Natl. Acad. Sci. U. S. A 97,5633–5638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- WATANABE T.A., GEARY R.S., and LEVIN A.A. (2006). Plasma protein binding of an antisense oligonucleotide targeting human ICAM-1 (ISIS 2302). Oligonucleotides 16,169–180 [DOI] [PubMed] [Google Scholar]
- WATSON J.D., and CRICK F.H.C. (1953a). A structure of deoxyribose nucleic acid. Nature 171,737–738 [DOI] [PubMed] [Google Scholar]
- WATSON J.D., and CRICK F.H.C. (1953b). Genetical implications of the structure of deoxyribonucleic acid. Nature 171,964–967 [DOI] [PubMed] [Google Scholar]
- WENGEL J. (2001). LNA (locked nucleic acid). In: Antisense Drug Technology; Principles, Strategies, and Applications. Crooke S.T., ed. (Marcel Dekker: New York: ), pp. 339–357 [Google Scholar]
- YAMAMOTO T., NAKATANI M., NARUKAWA K., and OBIKA S. (2011). Antisense drug discovery and development. Future Med. Chem. 3,339–365 [DOI] [PubMed] [Google Scholar]
- ZAMECNIK P.C., and STEPHENSON M.L. (1978). Inhibition of Rous sarcoma virus replication and cell transformation by a specific oligonucleotide. Proc. Natl. Acad. Sci. U. S. A. 75,280–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZEEB M., MAX K.E., WEININGER U., LOW C., STICHT H., and BALBACH J. (2006). Recognition of T-rich single-stranded DNA by the cold shock protein Bs-CspB in solution. Nucleic Acids Res. 34,4561–4571 [DOI] [PMC free article] [PubMed] [Google Scholar]