

Abstract
Reduced expression of the p53 family member p63 has been suggested to play a causative role in cancer metastasis. Here we show that ΔNp63α, the predominant p63 isoform, plays a major role in regulation of cell migration, invasion, and cancer metastasis. We identified MAP Kinase Phosphatase 3 (MKP3) as a downstream target of ΔNp63α that is required for mediating these effects. We show that ΔNp63α regulates Extracellular Signal-Regulated Protein Kinase 1 and 2 (Erk1/2) activity via MKP3 in both cancer and non-transformed cells. We further show that exogenous ΔNp63α inhibits cell invasion and is dependent on MKP3 up-regulation for repression. Conversely, endogenous pan-p63 ablation results in increased cell migration and invasion, which can be reverted by reintroducing the ΔNp63α isoform alone, but not by other isoforms. Interestingly, these effects require Erk2, but not Erk1 expression, and can be rescued by enforced MKP3 expression. Moreover, MKP3 expression is reduced in invasive cancers, and reduced p63 expression increases metastatic frequency in vivo. Taken together, these results suggest an important role for ΔNp63α in preventing cancer metastasis by inhibition of Erk2 signaling via MKP3.
Keywords: p63, MKP3, Erk, metastasis, cancer
Introduction
The p63 gene is expressed from two different promoters that generate isoforms either with a p53-homologous transactivation domain (TAp63) or without this domain (ΔNp63). Additionally, alternative splicing gives rise to three different C-termini (α, β, and γ), for a total of six isoforms. The DNA-binding and oligomerization domains are shared by all isoforms and are highly homologous to those of p53. Further, p63α isoforms contain a Sterile Alpha Motif (SAM), important for protein-protein interactions, and a post-inhibitory domain for intramolecular inhibition of gene transactivation (1–3). p63 has been shown to possess pleiotropic functions, including regulation of cell proliferation, survival, apoptosis, differentiation, senescence, and aging (4, 5). Mutations in the p63 gene have been associated with human autosomal dominant developmental diseases affecting primarily the skin and orofacial and limb morphogenesis, including ectrodactyly-ectodermal dysplasia-cleft lip/palate (EEC) syndrome, and ankyloblepharon-ectodermal dysplasia-clefting (AEC) syndrome (6, 7). Mice lacking all p63 isoforms exhibit early post-natal lethality, and severe developmental defects, including a complete lack of all stratified epithelia and ectodermal appendages, severe craniofacial defects, and limb truncation (8, 9). It has been shown that p63 is essential for epithelial stem cell maintenance and epithelial differentiation (10, 11). In addition, p63 deficiency leads to increased cellular senescence and an aging phenotype in mice (12).
A role for p63 in cancer development is demonstrated by a genetic study in which p63 haploinsufficiency in p53+/− mice markedly increases metastatic frequency (13). Further studies show that TAp63+/− and TAp63−/− mice develop spontaneous carcinomas and sarcomas that are highly metastatic (14). Moreover, TAp63 specifically functions in the female germline to protect genomic integrity (15, 16).
ΔNp63α is the predominant p63 isoform expressed in basal epithelial cells and is essential for epithelial development (3, 17). ΔNp63α regulates the expression of several adhesion molecules, including integrins β1, β4 and α6, and the demosome protein PERP (18, 19), which implicates it in cell invasion. Loss of ΔNp63α expression results in cell detachment and anoikis (18), while expression of ΔNp63α inhibits TAp73-mediated apoptosis (20, 21). In line with these observations, ΔNp63α cooperates with Ras to promote skin stem cell proliferation and tumorigenesis (22). Furthermore, ΔNp63α is frequently over-expressed in low-grade squamous cell carcinoma (SCC) (23–25). On the other hand, reduced p63 expression is associated with progression in a variety of cancers, including esophageal SCC, prostate cancer, and melanoma (26–28). Together, these data suggest that ΔNp63α may promote early stages of tumor development, but acts to inhibit cancer metastasis.
However, little is known about the molecular effects by which ΔNp63α, the predominant p63 isoform in cancer cells, affects tumor progression. Here, using breast cancer models, we uncovered an unexpected link to Extracellular signal-Regulated protein Kinases 1 and 2 (Erk1/2). Sustained Erk1/2 activity promotes tumorigenesis by inducing cell growth and proliferation, and it is often up-regulated in advanced cancers (29–31). Moreover, invasive cancer cells have higher Erk1/2 activity (32), which drives cell migration, invasion and metastasis (33–35). Mitogen-Activated Protein (MAP) Kinase Phosphatase 3 (MKP3; also known as DUSP6 or Pyst1) specifically dephosphorylates Erk1/2, thus restraining signal strength and duration, and acting as a putative tumor suppressor (36, 37).
In this study, based on microarray analyses, we uncovered that unlike TAp63 isoforms, ΔNp63α directly activates transcription of MKP3, leading to specific regulation of Erk2 signaling. Thus, we demonstrate that the ΔNp63α isoform plays a critical role in inhibiting cancer cell migration, invasion, and metastasis via a novel signaling mechanism that engages the Erk pathway.
Results
ΔNp63α up-regulates MKP3 expression
To identify ΔNp63α downstream targets through which ΔNp63α executes its biological functions, we profiled gene expression of cells expressing wild type ΔNp63α in comparison to that of cells expressing mutant derivatives or a vector control using Affymetrix gene chip analysis. Accordingly, we stably expressed wild type or mutant ΔNp63α in human breast cancer Hs-578T cells. These cells are highly invasive but not tumorigenic (38), and have no detectable p63 protein expression. The C306R mutation maps to the DNA-binding domain and is associated with EEC syndrome, while the C526W mutation is associated with AEC syndrome and is located in the SAM domain, thus affecting p63α isoforms exclusively (6, 7).
Upon analysis of the microarray, we observed that MAP Kinase Phosphatase 3 (MKP3) gene expression was up-regulated by wild type ΔNp63α to a much greater extent than by the mutant derivatives (Figure 1a). This conclusion was validated by quantitative PCR (Q-PCR) analysis (Figure 1b) and western blotting (Figure 1c).
Figure 1.
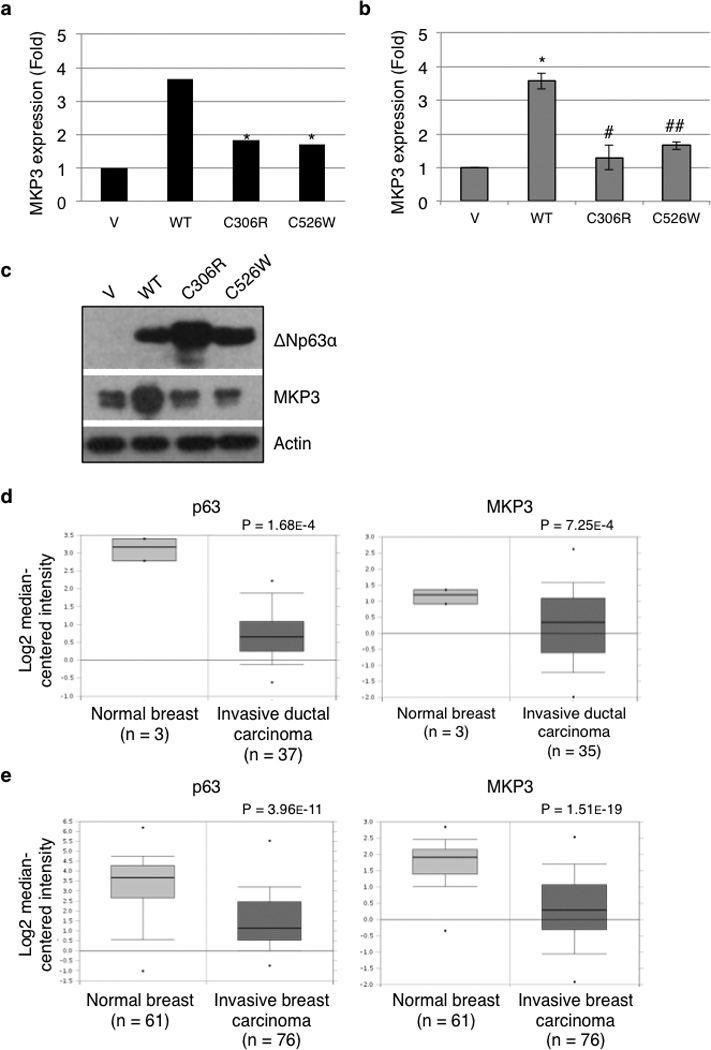
Wild type ΔNp63α, but not disease-associated mutants, up-regulates MKP3 expression. Hs-578T cells were transduced with a recombinant retrovirus encoding wild type murine ΔNp63α (WT), a mutant derivative (C306R or C526W), or a vector control (V). Puromycin-resistant cells were subjected to gene expression profiling by Affymetrix array in two independent experiments performed in triplicate (a), to Q-PCR analysis for MKP3 mRNA levels (b), or to western blotting for MKP3 protein expression (c). The Q-PCR data are presented as means and standard errors (SE) from three independent experiments performed in triplicate. *: P < 0.05 compared to V; **: P < 0.01 compared to V; #: P < 0.05 compared to WT; ##: P < 0.01 compared to WT. (d and e) Box plots representing p63 and MKP3 expression from breast tumor samples at different stages. Oncomine™ (Compendia Bioscience, Ann Arbor, MI) was used for analysis and visualization. (d) Zhao Breast dataset. (e) TCGA Breast dataset.
To explore the clinical relevance of p63-mediated regulation of MKP3 in cancer development, we analyzed the online microarray database Oncomine™, and observed a clear correlation between decreased p63 and MKP3 gene expression and cancer progression in human cancer specimens. Figure 1d showed that expression of both p63 and MKP3 is decreased in invasive ductal breast carcinoma, compared to normal breast tissue (39), and Figure 1e showed similar results using a different dataset. These observations implicate a clear correlation between down-regulation of p63 and MKP3 and breast cancer progression.
ΔNp63α modulates Erk1/2 signaling
Since MKP3 is an important negative regulator of Erk1/2, we reasoned that ΔNp63α might regulate Erk1/2 signaling through its modulation of MKP3 expression. To test this hypothesis, we stably expressed ΔNp63α in MDA-MB-231 cells, a highly invasive and tumorigenic human breast cancer cell line with no detectable p63 protein expression. Consistent with our previous results, ΔNp63α significantly up-regulated MKP3 expression at both the mRNA and protein levels (Figures 2a and 2b). Elevated MKP3, however, did not significantly result in down-regulation of Erk1/2 protein phosphorylation (Figure 2b). This was likely due to the presence of hyper-activated Ras(G13D) and Raf(I326T) mutations in MDA-MB-231 cells (40), which overpower the effects of MKP3 on Erk1/2 phosphorylation. Since MKP3 can inhibit Erk1/2 translocation to the nucleus (41), we investigated whether ΔNp63α affects the intracellular distribution of phosphorylated Erk1/2. We immunostained for p63 protein and for phosphorylated Erk1/2 in MDA-MB-231 cells with or without stable expression of ΔNp63α. Strikingly, cells that lacked ΔNp63α expression exhibited strong nuclear phosphorylated Erk1/2 (arrows, Figure 2c), whereas cells with ΔNp63α expression showed a uniform distribution of Erk1/2 phosphorylation between the cytosol and the nucleus. Indeed, approximately forty percent of ΔNp63α-negative cells exhibited strong staining of nuclear Erk1/2 phosphorylation, while less than fifteen percent of ΔNp63α-positive cells showed a similar pattern (Figure 2d). In addition, cellular fractionation experiments showed that ΔNp63α expression resulted in decreased phosphorylated Erk1/2 in the nuclear fraction, as we expected, while no significant changes were observed in the much more abundant cytosolic fraction of phosphorylated Erk1/2 (Figure 2e). Furthermore, the expression of two well-established Erk1/2 transcriptional targets, MMP1 and MMP9 (42, 43), was markedly decreased upon ΔNp63α expression (Figure 2f). Accordingly, MMP1 and MMP9 expression is decreased in human invasive breast cancer biopsies, as assessed by Oncomine™ (Supplementary Figure S1a). Together, these data indicate that ΔNp63α negatively regulates Erk1/2 signaling via MKP3.
Figure 2.
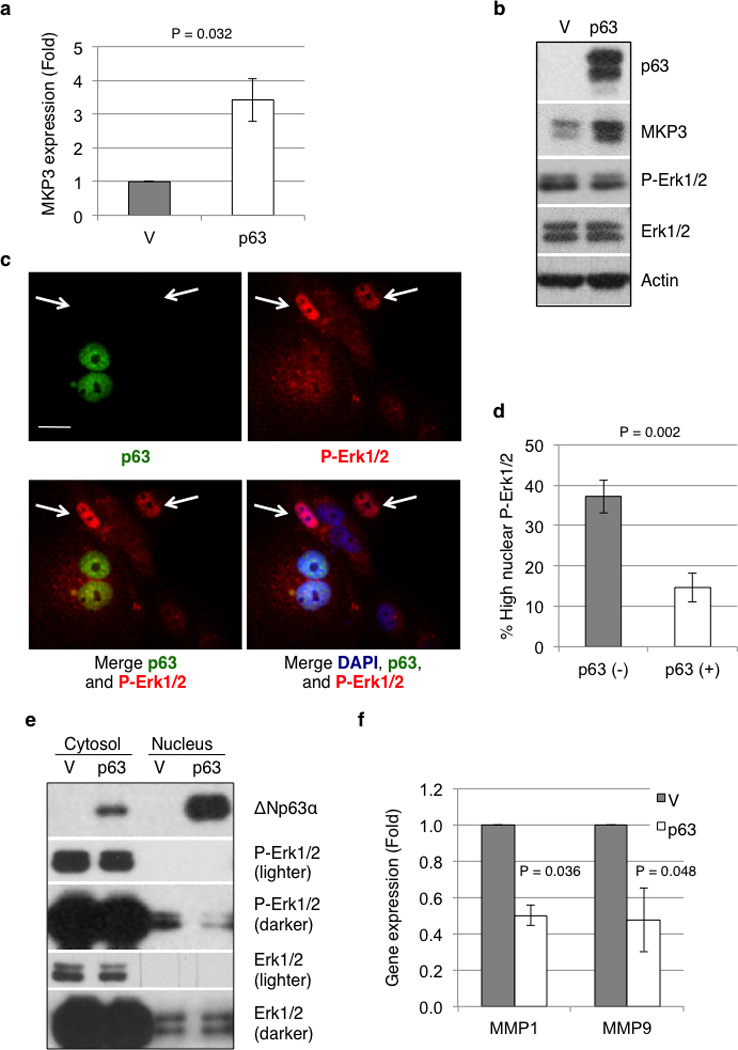
ΔNp63 reduces phosphorylated Erk1/2 in nuclei and attenuates Erk1/2 signaling. MDA-MB-231 cells stably expressing ΔNp63α (p63) or an empty vector control (V) were subjected to Q-PCR analysis for MKP3 mRNA levels (a) and protein expression (b). The Q-PCR data are presented as means and SE from three experiments performed in triplicate (P = 0.032) (c) MDA-MB-231 cells were transduced with retrovirus encoding ΔNp63α. A mixed population of ΔNp63α-positive [p63 (+)] and ΔNp63α-negative [p63 (−)] cells was assessed by immunofluorescence for p63 (green) expression and phosphorylated Erk1/2 (red) levels, and the nuclei were counterstained with DAPI (blue). Arrows denote cells with high levels of nuclear phosphorylated Erk1/2. Scale bar = 20 µm. (d) Cells with high levels of phosphorylated Erk1/2 were scored for both p63 (−) and p63 (+) cells. Data are presented as percentage of cells with high phosphorylated Erk1/2 over total p63 (−) or p63 (+) cells. At least 100 cells in total from five random fields were scored for each of six experiments. Results are presented as means and SE. (e) MDA-MB-231 cells stably expressing ΔNp63α (p63) or vector control (V) were subjected to cellular fractionation to separate the nuclear from the cytosolic fractions, which were then subjected to western blotting as shown. (f) MDA-MB-231 cells stably expressing ΔNp63α (p63) or vector control (V) were subjected to Q-PCR analysis for mRNA levels of MMP1 and MMP9. Results are presented as means and SE from three experiments performed in triplicate.
ΔNp63α inhibits cell invasion in an MKP3-dependent manner
It is well documented that Erk1/2 dysregulation causatively promotes cell migration, invasion, and cancer metastasis (33–35). We therefore asked whether ΔNp63α expression inhibits Erk1/2 and inhibits cancer cell invasion. As shown in Figures 3a and 3b, expression of ΔNp63α in MDA-MB-231 cells significantly reduced cell invasion. We next examined whether ΔNp63α-mediated MKP3 induction is responsible for these effects. Again, ΔNp63α significantly reduced MDA-MB-231 cell invasion, which was significantly reversed by simultaneous MKP3 knockdown using specific shRNAs (Figures 3c, 3d and 3e, and Supplementary Figure S1b). Furthermore, MKP3 ablation restored MMP1 and MMP9 expression in MDA-MB-231 cells expressing ΔNp63α, as assessed by Q-PCR (Figure 3f). Together, these results indicate that ΔNp63α-mediated MKP3 modulation is an important mechanism for regulating cancer cell invasion.
Figure 3.
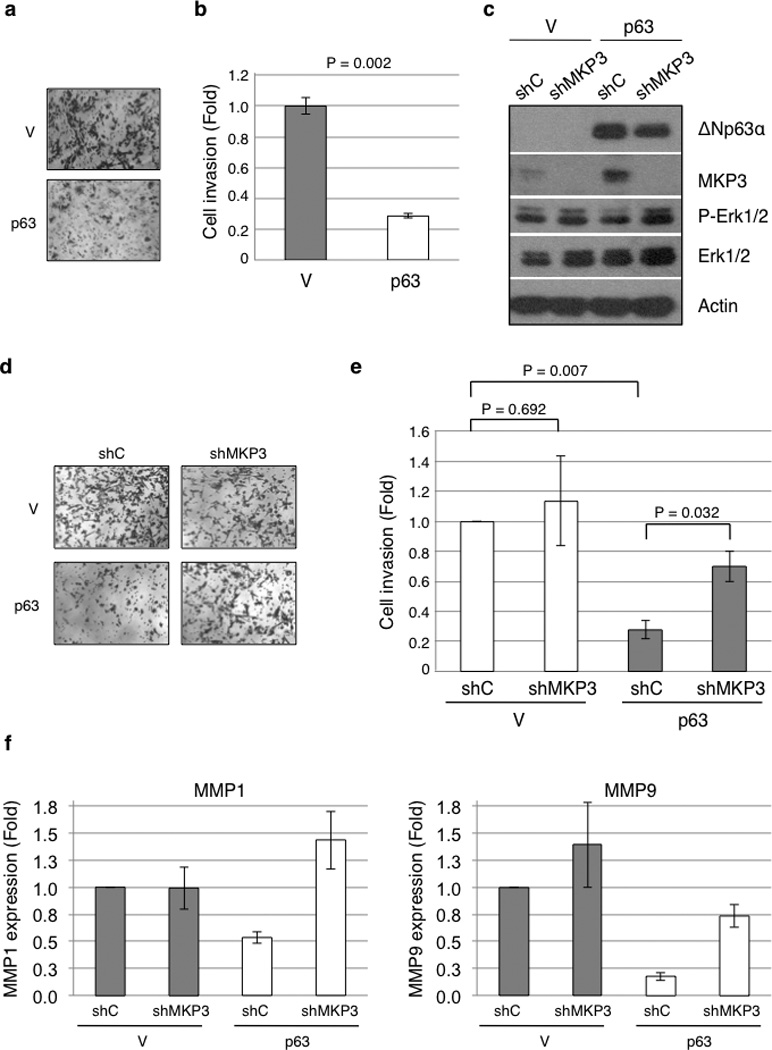
ΔNp63α inhibits cell invasion in an MKP3-dependent manner. (a) MDA-MB-231 cells stably expressing ΔNp63α (p63) or a vector control (V) were subjected to transwell assays for cell invasion. Twenty-four hours after plating, invading cells were fixed and stained with crystal violet and photographed under a light microscope. Representative pictures are shown. (b) Results from invasion assay were quantitated as described in the Materials and Methods section and presented as means and SE from three independent experiments. (c) Stable MDA-MB-231 cells were transduced with recombinant lentivirus encoding an shRNA specific for MKP3 (shMKP3) or for GFP as a control (shC) and subjected to western blotting. (d and e) Stable MDA-MB-231 cells were subjected to transwell assay for invasiveness. Results are presented as means and SE from three independent experiments. (f) Stable MDA-MB-231 cells were subjected to Q-PCR analysis for mRNA levels of MMP1 and MMP9.
Endogenous ΔNp63α regulates MKP3 expression and Erk1/2 activation
Since exogenous ΔNp63α up-regulates MKP3 expression and inhibits cell invasion, we next determined whether endogenous ΔNp63α executes a similar biological function. Human non-transformed mammary epithelial MCF-10A cells and human head and neck squamous cell carcinoma (SCC) FaDu cells express abundant p63, with ΔNp63α as the major protein isoform. When tested, the shRNA against pan-p63 (shp63-1; referred to as shp63 hereafter) effectively ablated endogenous p63 in both MCF-10A and FaDu cells (Figures 4a and 4b). Two other shRNA constructs (shp63-2 and shp63-3) were equally effective in knocking down p63 (Supplementary Figures S2a and S2b). Ablation of p63 by shRNA resulted in a significant decrease in MKP3 expression in both MCF-10A and FaDu cells, as assessed by Q-PCR (Figure 4a and Supplementary Figure S2a), and markedly reduced MKP3 protein levels with a concomitant increase in phosphorylated Erk1/2 (Figure 4b and Supplementary Figure S2b), whereas no significant changes in total Erk1/2 protein or Mek phosphorylation levels were observed (Figure 4b).
Figure 4.
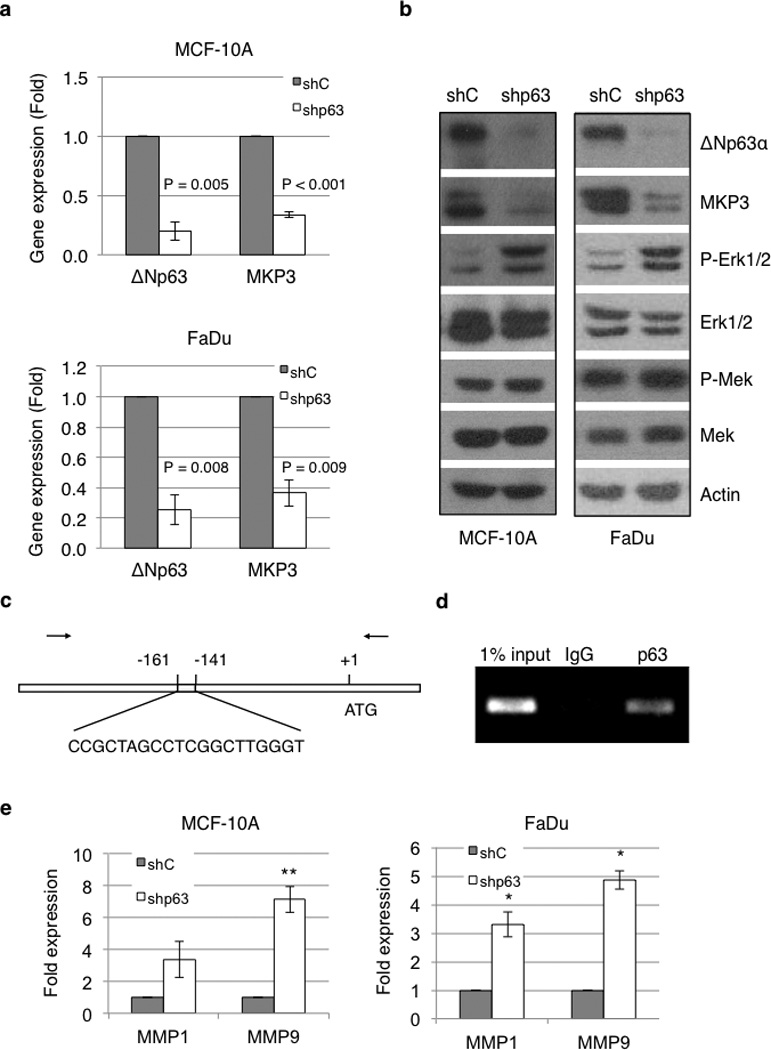
ΔNp63α regulates MKP3 expression and Erk1/2 activity. (a) MCF-10A and FaDu cells were infected with recombinant lentivirus encoding shRNA specific for pan-p63 (shp63) or for GFP as a control (shC). ΔNp63 and MKP3 mRNA levels were analyzed by Q-PCR. Results are presented as means and SE from three independent experiments performed in triplicate. (b) Whole-cell lysates were subjected to western blotting as indicated. (c) Diagram of the MKP3 gene and promoter locus highlighting a putative p63 binding site, and the primers (arrows) used for PCR amplification of the immunoprecipitated DNA. (d) Binding of p63 to this putative binding site was assessed in FaDu cells by chromatin immunoprecipitation (ChIP) using a specific p63 antibody (4A4) or a control mouse IgG, followed by PCR amplification. (e) MCF-10A cells infected with recombinant lentivirus encoding shRNA specific for p63 (shp63) or GFP as a control (shC) were subjected to Q-PCR analyses for MMP1 and MMP9 mRNA levels. Results are presented as means and SE from three experiments performed in triplicate. *: P < 0.05; **: P < 0.01.
In order to further investigate the molecular mechanism by which ΔNp63α regulates MKP3 expression, we asked whether ΔNp63α directly binds to the promoter of the MKP3 gene. Computational analysis of the human MKP3 gene and promoter revealed several potential p63/p53 binding sites, including a putative p63 binding site located 141 base pairs upstream from the translational start site (Figure 4c). Chromatin immunoprecipitation (ChIP) using a specific p63 antibody showed direct binding of p63 to this site (Figure 4d), suggesting that ΔNp63α exerts direct transcriptional regulation on MKP3. Consistently, knockdown of p63 resulted in marked increases in MMP1 and MMP9 expression in both MCF-10A and FaDu cells (Figure 4e). Taken together, these findings indicate that MKP3 is a direct ΔNp63α transcriptional target, and that ΔNp63α negatively regulates Erk1/2 activity via MKP3.
p63 ablation enhances cell migration and invasion
Next, we investigated the biological consequences of p63-mediated modulation of Erk1/2 activity, focusing on cell migration and invasion. Knockdown of all p63 isoforms in MCF-10A and FaDu cells led to a dramatic increase in cell migration and invasion (Figures 5a and 5b). In addition, p63 knockdown in MCF-10A cells resulted in a morphological change from cuboidal and cobblestone-like to elongated and spindle-like morphology (Supplementary Figure S2d). To study the underlying mechanism by which p63 modulates cell migration, we examined the distribution of focal adhesions by immunostaining for phospho-paxillin, a component of the focal adhesion complex. It has been shown that large focal adhesions orderly assembled around the periphery of the cells are indicative of low cell motility due to poor turnover of the focal adhesion complex (44, 45). As shown in Figure 5c, while control MCF-10A cells exhibited a normal staining pattern of large focal adhesions around the periphery, knockdown of p63 in these cells led to a punctuated and disorganized pattern of small focal adhesions, indicative of increased turnover and enhanced motility.
Figure 5.
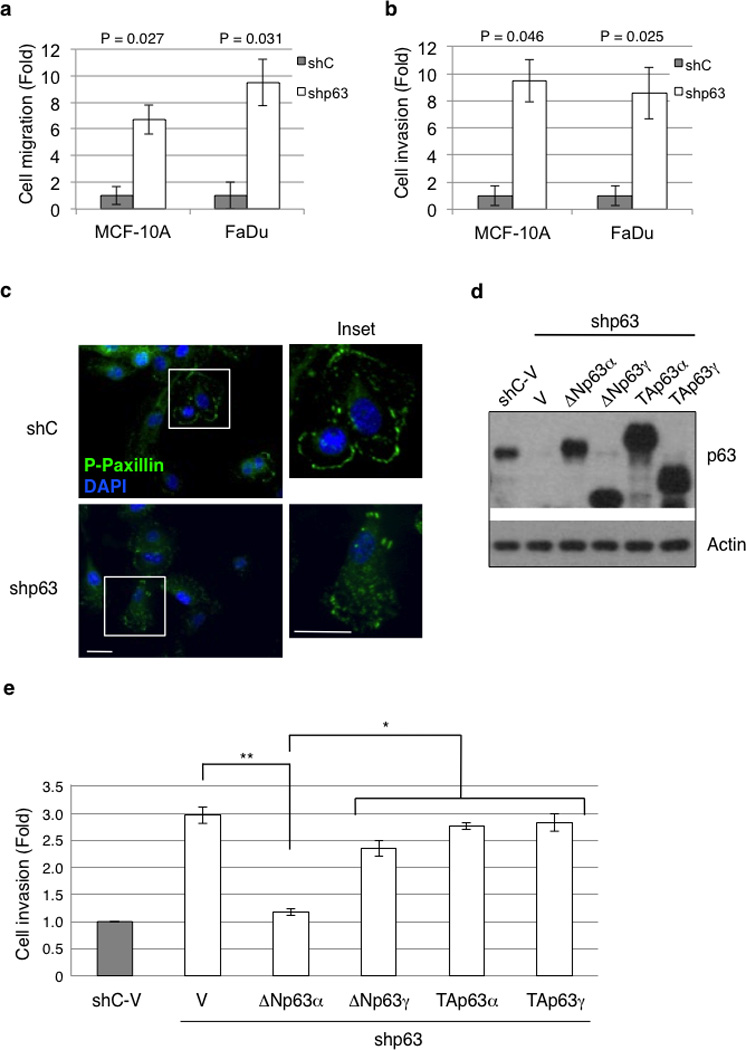
ΔNp63α, but not other p63 isoforms, inhibits cell migration and invasion. (a and b) MCF-10A and FaDu cells stably expressing shRNA specific for pan-p63 (shp63) or GFP (shC) were subjected to cell migration (a) or invasion assays (b), using transwell systems. Migratory and invasive cells were fixed and stained with crystal violet and quantitated as described in the Materials and Methods section. Results are presented as means and SE from three experiments. (c) MCF-10A cells infected with recombinant lentivirus encoding shRNA specific for p63 (shp63) or GFP as a control (shC) were grown in fresh normal growth media (see Materials and Methods) for two hours prior to fixation. Cells were then immunostained for phospho-paxillin (pTyr118; green) and counterstained with DAPI (blue). A representative figure from two independent experiments performed in triplicate is shown. Note the punctuated staining of focal adhesion complexes in p63-ablated cells in contrast to the organized peripheral staining in control cells (insets). Scale bars = 20 µm. (d and e) MCF-10A cells infected with recombinant lentivirus encoding shRNA specific for p63 (shp63) or GFP as a control (shC) were transiently transfected with mouse Myc-tagged ΔNp63α, ΔNp63γ, TAp63α, TAp63γ, or a vector control (V) as shown. Cells were subjected to western blotting (d) or to invasion assays using transwell systems (e). Results are presented as means and SE from two independent experiments. *: P < 0.05; **: P < 0.01.
To investigate whether a particular p63 isoform(s) specifically mediates these effects, we introduced Myc-tagged murine ΔNp63α, ΔNp63γ, TAp63α, or TAp63γ into MCF-10A cells that were ablated for endogenous p63, followed by western blot analysis (Figure 5d), and cell invasion assays (Figure 5e and Supplementary Figure S3a). Reintroduction of ΔNp63α fully restored the non-invasive phenotype, similar to that of parental cells, while expression of ΔNp63γ exhibited only a marginal effect in cell invasion. In sharp contrast, reintroduction of TAp63α or TAp63γ exhibited hardly any effect, despite abundant expression of these proteins (Figure 5d and 5e). These results demonstrate that ΔNp63α is the primary p63 isoform that regulates cell invasion in this system.
p63 ablation-induced cell motility is dependent on Erk2, but not Erk1, and can be rescued by enforced MKP3 expression
Our results indicate that loss of ΔNp63α leads to both higher Erk1/2 activity and increased cell migration/invasion. We then investigated whether up-regulation of Erk1/2 activity is responsible for the observed increases in cell invasion upon p63 ablation. Indeed, inhibition of Erk1/2 phosphorylation with a pharmacological Mek inhibitor, U0126 (Supplementary Figure S3b), completely restored the non-invasive phenotype (Figure 6a). Accordingly, U0126 treatment clearly inhibited MMP1 and MMP9 up-regulation elicited by p63 knockdown in MCF-10A cells (Supplementary Figure S3c).
Figure 6.
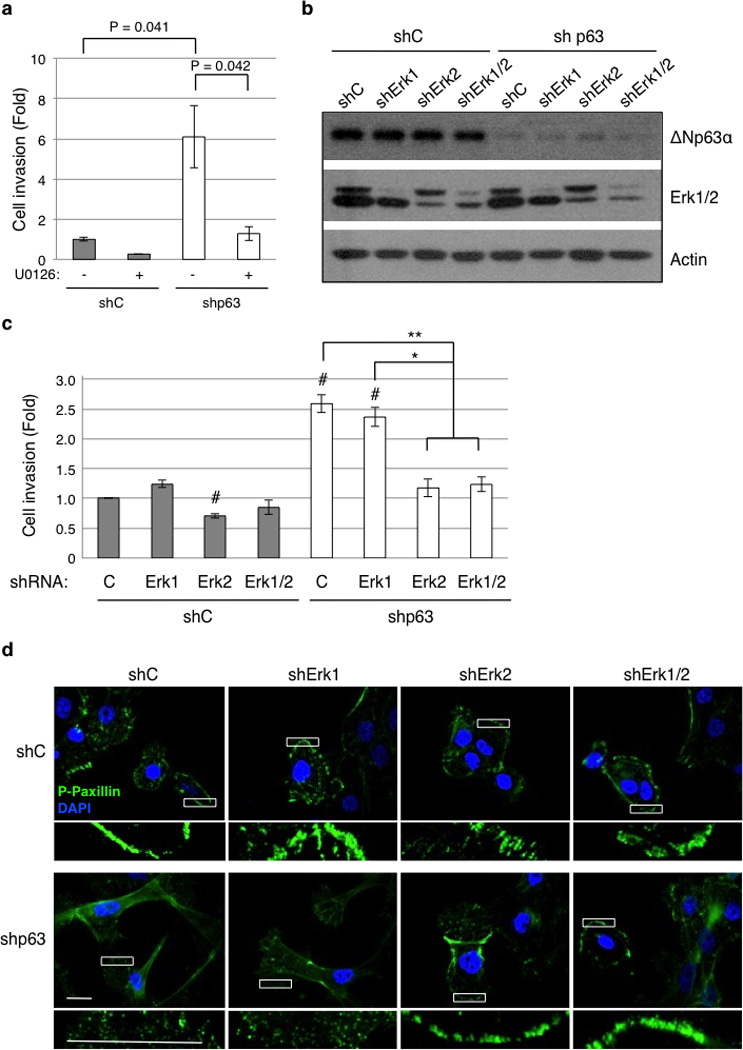
p63 ablation-induced cell motility and invasion requires Erk2, but not Erk1. (a) MCF-10A cells infected with lentivirus encoding an shRNA specific for pan-p63 (shp63) or GFP as a control (shC) were subjected to cell invasion assays using transwell systems in the presence of 10 µM U0126 (+) or DMSO as a vehicle control (−). Results are presented as means and SE from three independent experiments. (b) MCF-10A cells expressing shRNA against p63 (shp63) or GFP as a control (shC) were infected with a lentivirus encoding shRNA against Erk1 and/or Erk2, or a control shRNA against GFP (shC). Cells were subjected to western blotting (b) or to cell invasion assays (c). Results from invasion assays are presented as means and SE from three independent experiments. *: P < 0.05; **: P < 0.01; #: P < 0.05 compared to shC-C (first column). (d) MCF-10A cells expressing shRNAs as indicated above were grown in fresh normal growth media for two hours prior to fixation. Cells were then immunostained for phospho-paxillin (pTyr118; green) and counterstained with DAPI (blue). Representative figures from two independent experiments performed in triplicate are shown. A magnified view of the respective boxed area is shown below each image. Note the organized peripheral staining of focal adhesion complexes in p63-ablated cells with reduced Erk2 expression. Scale bar = 20 µm.
Since Erk1 and Erk2 possess similar biochemical activities, we studied the effect of Erk1 or Erk2 specific ablation upon p63 knockdown in MCF-10A cells (Figure 6b). Surprisingly, knockdown of Erk1 had little effect on cell invasion. In sharp contrast, knockdown of Erk2 alone significantly reduced invasion of p63-deficient cells (Figure 6c and Supplementary Figure S3d). Notably, simultaneous knockdown of Erk1 and Erk2 did not reduce cell invasion further, compared to Erk2 knockdown alone.
We then examined focal adhesions by immunostaining for phospho-paxillin. The p63-deficient cells again exhibited a punctuated, disorganized pattern of focal adhesions. Knockdown of Erk2 in these cells led to formation of large focal adhesions organized around the periphery of the cell, similar to those of the control cells (Figure 6d). On the other hand, knockdown of Erk1 did not affect focal adhesion patterns observed in either the control cells or in cells defective of p63 (Figure 6d). Together, these results demonstrate that ΔNp63α-mediated modulation of focal adhesions and cell migration/invasion is dependent on Erk2, but not on Erk1.
Next, we studied the role of MKP3 in p63-mediated regulation of cell motility. In line with our previous results, ablation of MKP3 in MCF-10A cells led to an increase in MMP1 expression and to a much higher level of MMP9 expression (Supplementary Figure S4a). Knockdown of p63 in MCF-10A cells reproducibly led to increased cell migration and invasion, concomitant with a reduction in MKP3 protein expression and increased Erk1/2 phosphorylation (Figure 7). Importantly, exogenous expression of MKP3 in these cells (Figure 7a) effectively reduced cell migration and invasion (Figures 7b and 7c, and Supplementary Figure S4b). In addition, MKP3 over-expression in p63-ablated MCF-10A cells also restored focal adhesion organization back to large complexes orderly arranged around the periphery of the cells (Figure 7d). These data strongly support an important role for MKP3 in negative regulation of cell migration and invasion.
Figure 7.
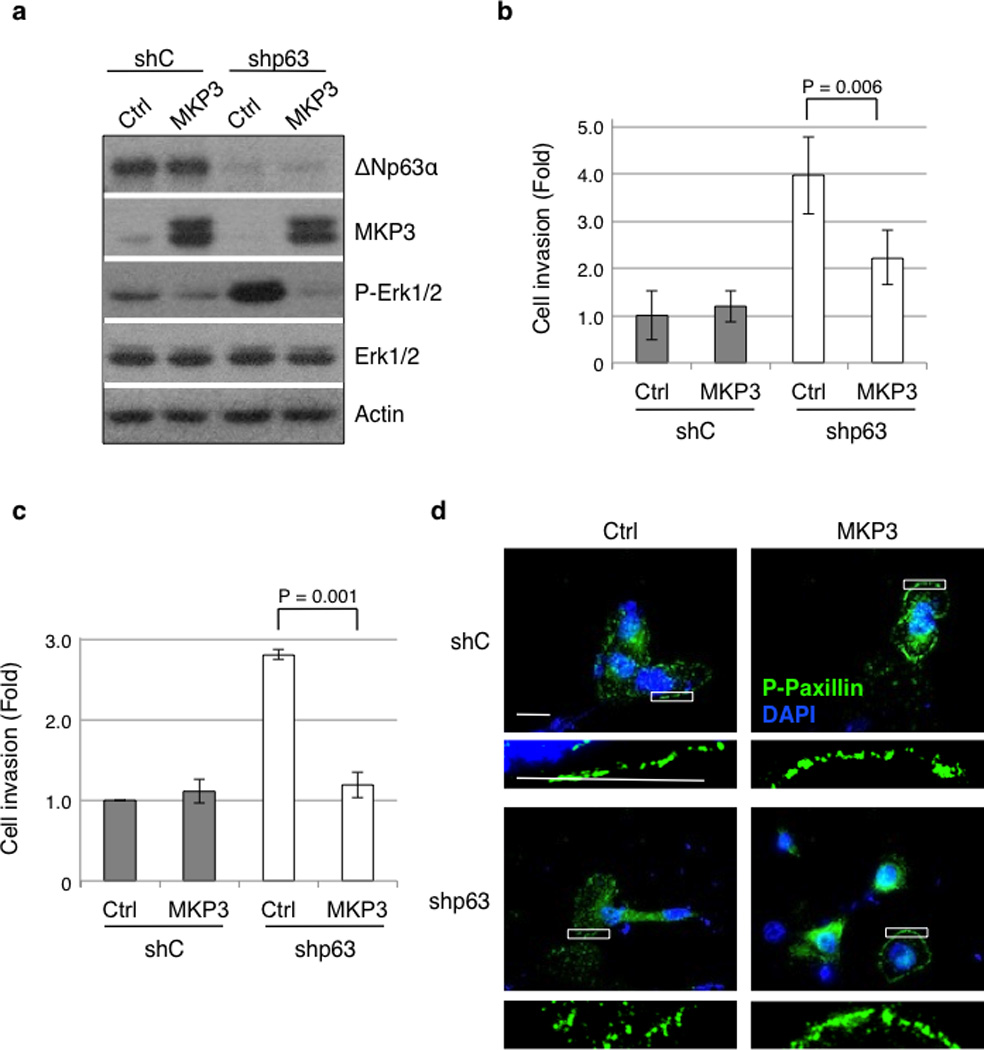
MKP3 expression overcomes p63 ablation-induced cell migration and invasion. MCF-10A cells expressing shRNA against p63 (shp63) or a control shRNA against GFP (shC) were transiently transfected with MKP3 or a vector control (Ctrl). Cells were subjected to western blotting (a), or cell migration (b) and invasion assays (c), using transwell systems. Migratory and invasive cells were fixed and stained with crystal violet and quantitated as described in the Materials and Methods section. Results are presented as means and SE from three independent experiments. (d) MCF-10A cells expressing shRNAs and expression vectors as indicated were grown in fresh normal growth media for two hours prior to fixation. Cells were then immunostained for phospho-paxillin (pTyr118; green) and counterstained with DAPI (blue). Two independent experiments were performed in triplicate. Representative pictures are shown. A magnified view of the respective boxed area is shown below each image. Scale bar = 20 µm.
Reduced p63 expression enhances metastasis in vivo
We further investigated the pathological role of reduced ΔNp63α expression in breast cancer metastasis. We chose to use MCF-10A cells because they express abundant endogenous ΔNp63α protein, yet they are non-tumorigenic. However, MCF-10A cells expressing hyperactive murine Her2/Neu (V664E; referred to as NeuT) are tumorigenic in a nude mouse model, as we have shown previously (46). Thus, we transformed MCF-10A cells by stable transfection with NeuT. These cells exhibited neoplastic phenotypes, including loss of contact inhibition and formation of foci when grown in monolayer (Supplementary Figure S5a). Notably, p63 knockdown in NeuT-expressing cells resulted in increased cell invasion, compared to NeuT cells expressing a control shRNA (Supplementary Figures S4b and S4c). We then injected MCF-10A-NeuT cells with or without p63 ablation into the lateral tail vein of recipient nude mice, an established lung metastasis model (44). Forty-five days after injection, mice were euthanized and their lungs dissected and inspected for metastatic nodules (Figure 8a). Three out of five mice injected with NeuT-shp63 cells exhibited three, four, and five large metastatic nodules each, visible on the surface of the lungs, whereas only one out of six mice injected with NeuT-shC cells formed just one observable metastatic nodule (Figure 8b). Furthermore, histological analysis demonstrated normal lung architecture in the majority of the control mice, while lungs from mice injected with NeuT-shp63 cells showed frequent infiltration of metastatic cells (Figure 8c). Taken together, these results highlight the importance of ΔNp63α reduction as an important factor in promoting metastasis in vivo.
Figure 8.
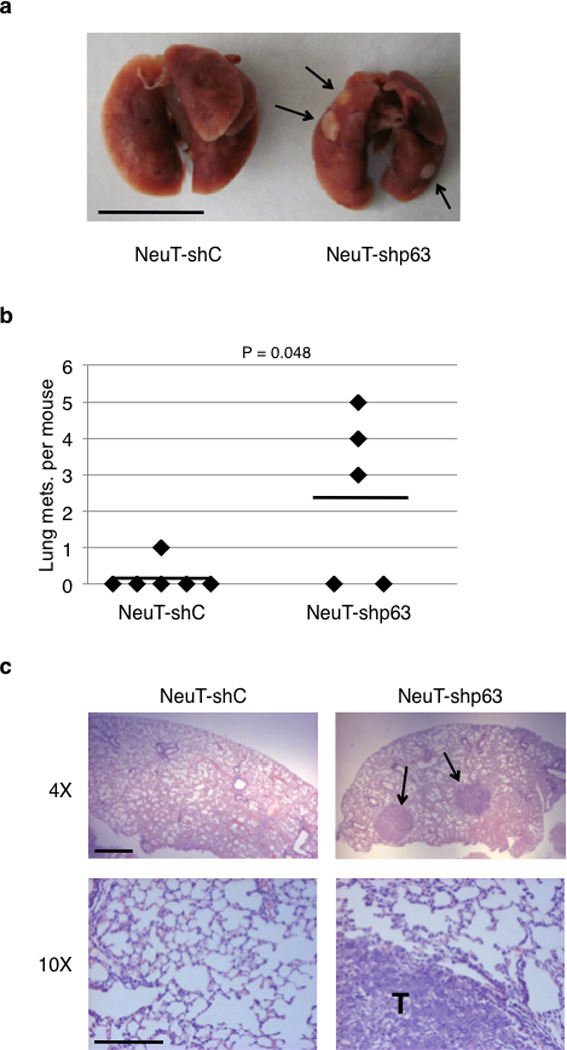
Reduced p63 expression enhances metastasis in vivo. MCF-10A cells transformed with NeuT were infected with lentivirus expressing shRNA against p63 (NeuT-shp63) or GFP (NeuT-shC). NCr nude mice were injected intravenously with 1.0×106 NeuT-shC or NeuT-shp63 cells. Mice were observed daily and euthanized after 45 days. (a) Lungs were dissected, fixed, and inspected for metastatic nodules on their surface. Arrows point to metastatic nodules. Scale bar = 1.0 cm. (b) Graph represents the number of metastatic nodules in the lungs per mouse. A horizontal line indicates the mean for each group. (c) Lungs were fixed, embedded in paraffin, sectioned, and stained by H&E for histological analysis. Metastatic nodules are indicated by arrows on the top panel and by a T on the bottom panel. Scale bars = 200 µm.
Discussion
In this study, we demonstrated that the major protein isoform of p63, ΔNp63α, plays a critical role in modulation of cell migration and invasion, as well as cancer metastasis, in an Erk2-dependent pathway. First, exogenous expression of wild type ΔNp63α, but not of the disease-associated mutant derivatives, inhibits cancer cell invasion. Second, ΔNp63α transcriptionally up-regulates MKP3, which in turn inhibits Erk1/2 signaling and reduces cell motility. Third, knockdown of p63 promotes cell migration and invasion, which is effectively rescued by expression of MKP3 or ΔNp63α, but not by ΔNp63γ or TAp63 isoforms. Fourth, knockdown of p63 facilitates turnover of focal adhesions and cell invasion, which is overcome by ablation of Erk2, but not Erk1. Furthermore, reduction of p63 markedly enhances metastasis in a lung metastasis model, which is reminiscent of clinical observations in which expression of both p63 and MKP3 are inversely correlated with cancer progression.
Erk1/2 signaling plays a pivotal role in regulating cell migration via multiple mechanisms, including induction of membrane protrusion and regulation of focal adhesion turnover (47). Here, we reproducibly observe punctuated, small focal adhesions upon p63 ablation, concomitant with increased Erk1/2 activity. These data are consistent with the notion that reduction of ΔNp63α stimulates Erk1/2 activity and promotes cell migration. Interestingly, it has been shown that Erk2 is responsible for Ras-induced epithelial-to-mesenchymal transition (EMT) and increased cell migration and invasion (48). Our data show that reduction of only Erk2, but not Erk1, effectively reverses p63 ablation-induced cell migration. These studies suggest that Erk2 is a major effector that integrates a variety of different signals in regulation of cell migration. It is plausible that Erk2 specifically phosphorylates components of the focal adhesion complex to impact its turnover.
In addition to migration, Erk1/2 can also promote cell invasion by transcriptional up-regulation of genes involved in cell invasion, such as MMP1, MMP2, and MMP9 (42, 43, 49). Our gain- and loss-of-function experiments demonstrate that ΔNp63α modulates Erk1/2 activity via MKP3, which in turn regulates MMP1 and MMP9 expression. Hence, reduced ΔNp63α expression leads to up-regulation of MMP1 and MMP9, thereby promoting cell invasion. Interestingly, it has been shown that ΔNp63α can induce transcription of Id-3, which then inhibits Ets-1-mediated MMP2 gene expression and cell invasion (50). Notably, Erk1/2 can phosphorylate and activate Ets-1 to stimulate MMP1 and MMP9 gene expression and cell invasion (42, 43). Therefore, it is likely that ΔNp63α regulates cell invasion via direct modulation of Erk1/2 activity. It has been reported, however, that p53 can indirectly impact transcription of MMP1 and MMP9 via various transcriptional factors such as AP-1 and NF-kB (51, 52). Although our data strongly suggest that ΔNp63α modulates MMP1 and MMP9 gene expression via Erk1/2, we cannot rule out the possibility that ΔNp63α may also modulate MMP1 and MMP9 expression via different mechanisms. Furthermore, Fukushima and colleagues (53) reported that loss of ΔNp63α in certain urothelial carcinoma cell lines results in increased N-Cadherin expression, leading to increased Erk1/2 signaling. In this study using mammary epithelial or head and neck SCC systems, we did not detect any significant changes in N-Cadherin expression. It remains possible that ΔNp63α modulates Erk1/2 activity by different mechanisms in a cell context-dependent manner.
Erk1/2 activity is tightly controlled spatially and temporally. Aberrant Erk1/2 activity in advanced cancers has often been linked to up-regulated Ras and/or Raf signaling or increased receptor tyrosine kinase activity (54). However, accumulating evidence indicate that dephosphorylation plays an equally important role for regulation of Erk1/2 activity (55–57). MKP3 specifically dephosphorylates both the threonine and tyrosine residues in the activation loop of Erk1/2, an important mechanisms for restraining Erk1/2 activity (58). In this study, we demonstrate that ΔNp63α regulates Erk1/2 activity in an MKP3-dependent manner. We show that ΔNp63α up-regulates MKP3 expression leading to inhibition of cell invasion, which is markedly rescued by ablation of MKP3. Notably, knockdown of p63 induces cell invasion, which is fully reverted by ectopic expression of MKP3, yet p63 ablation-induced cell migration is only partially reverted by MKP3. These results suggest that MKP3 is a critical player in p63-mediated modulation of cell invasion, while additional factors are important for p63-mediated regulation of cell migration.
Activation of Erk1/2 due to p63 ablation could arise from activation of upstream kinases in the MAP kinase cascade. However, despite repeated efforts, we did not detect significant changes in Mek phosphorylation, suggesting that p63 modulates Erk1/2 activity via MKP3, rather than by enhancing upstream kinase activity.
The two major p63 isoforms, TAp63 and ΔNp63, have been shown to execute overlapping as well as distinct biological functions. Recently, Flores and colleagues (14) showed that TAp63 suppresses metastasis through coordinate regulation of Dicer and miR-130b (14). Our study demonstrates that ΔNp63α impacts the Erk1/2 pathway, leading to inhibition of cell migration and invasion, in accordance with the observations that reduced ΔNp63α expression up-regulates genes involved in cell motility and induces cell invasion in SCC and keratinocyte cell lines (59–61). Thus, both TAp63 and ΔNp63 function to inhibit metastasis likely through different mechanisms.
In this study, we show that two human disease-associated mutations in ΔNp63α, C304R in the DNA binding domain and C526W in the SAM domain, do not up-regulate MKP3, and can not affect cell migration and invasion (unpublished data). These observations highlight the importance of DNA-binding activity and protein-protein interaction via the SAM domain for the function of ΔNp63α in regulation of cell motility. Our data also show that, unlike ΔNp63α, ΔNp63γ is unable to rescue MCF-10A cell invasion elicited by p63 ablation. Notably, it was reported that ablation of p63α and p63β, but leaving p63γ expression intact, resulted in much increased cell invasion, compared to pan-p63 knockdown (62), suggesting that ΔNp63γ might promote cell invasion. Although we have not observed these effects of ΔNp63γ in our system, it might be possible that the level of expression and the ratio between different p63 isoforms affect the results observed.
More recently, it has been shown that mutant p53 (mtp53) promotes metastasis through inhibition of p63. Piccolo and colleagues (63) show that mtp53 inhibits the anti-metastatic activity of p63 by forming a trimeric complex between mtp53, Smad2 and p63 upon TGFβ stimulation. In addition, Vousden and colleagues (64) show that expression of mtp53 (R175H or R273H) in p53 null H1299 cells results in increased invasion, which can be reversed by TAp63α expression, suggesting that mtp53 suppresses endogenous TAp63 to promote cell invasion. In this study, we observe that inhibition of ΔNp63α in MCF-10A and FaDu cells increases migration and invasion. Importantly, MCF-10A cells express wild type p53, while FaDu cells express truncated p53 from one allele and DNA binding mutant (p53-R248L) from the other allele (65). Thus, the ability of ΔNp63α to suppress migration and invasion is likely independent of p53. However, it remains possible that reduced p63 may cooperate with mtp53 to promote cell migration/invasion. Clearly, the interplay between different p63 isoforms, mutant p53 proteins, and p53 family members can largely determine the overall anti-metastatic activity.
Here, we uncovered that the ΔNp63α-MKP3-Erk2 pathway is an important mechanism in controlling cell migration and invasion. Whether this axis represents a true physiological pathway in vivo and whether dysregulation of this pathway is pathologically relevant are critical questions. It is well documented that activation of Erk1/2 is a causative factor for cancer progression and metastasis (32). Accumulating evidence clearly indicate a clinical correlation between reduction of p63 and/or MKP3 expression and cancer progression, including SCC, breast, lung, and pancreatic cancers (26–28, 39, 56, 57, 66). Interestingly, in human head and neck SCC where ΔNp63α is often over-expressed, expression of MKP3 is also increased (67, 68). Furthermore, using an established lung metastasis model, we show that reduction of p63 significantly increases metastasis in mice. Notably, Her2/Neu can transform MCF-10A cells and promotes rapid tumor formation in nude mice, yet these tumors exhibit limited metastatic potential (46). Interestingly, we showed that knockdown of p63 significantly enhances the metastatic potential of these cells, indicating that reduction of ΔNp63α greatly potentiates Her2-mediated malignant activity.
Altogether, this study demonstrates that dysregulation of the ΔNp63α-MKP3-Erk2 pathway is important for cancer metastasis. Further studies will be necessary to decipher the specific contribution from each p63 isoform and their interactions with other p53 family members so as to develop strategies for more efficacious treatments and preventions against cancer metastasis.
Materials and Methods
Cell culture
Human non-transformed mammary epithelial MCF-10A cells were maintained in DMEM:F12 media [1:1 mixture of Dulbecco's modified Eagle's medium (DMEM) and Ham's F12 medium with reduced Ca2+ (0.04 mM; Invitrogen Inc., Carlsbad, CA)], supplemented with 20 ng/mL epidermal growth factor (Invitrogen Inc., Carlsbad, CA), 100 ng/mL cholera toxin (Sigma, St. Louis, MO), 10 g/mL insulin (Sigma, St. Louis, MO), 500 ng/mL (95%) hydrocortisone (Sigma, St. Louis, MO), and 5% of Chelex-treated horse serum (Invitrogen Inc., Carlsbad, CA). Human head and neck squamous cell carcinoma FaDu cells, human breast cancer MDA-MB-231 cells, and HEK-293T cells were maintained in DMEM supplemented with 10% fetal bovine serum (FBS; Invitrogen Inc., Carlsbad, CA). Human breast cancer Hs-578T cells were cultured in DMEM supplemented with 10% FBS and 10 g/mL of insulin. All cells were grown in media supplemented with 1% l-glutamine and 1% penicillin-G/streptomycin sulfate at 37 °C in a humidified incubator under 5% CO2.
Plasmid transfections, viral infections and RNA interference
Transient transfections were carried out using Lipofectamine 2000 (Invitrogen Inc., Carlsbad, CA). Expression plasmids include myc-tagged murine pcDNA3-ΔNp63α, pcDNA3-ΔNp63γ, pcDNA3-TAp63α and pcDNA3-TAp63γ, murine pMSCVpuro-ΔNp63α, and human myc-tagged pSG5-MKP3. Retrovirus were amplified by transfecting HEK-293T cells with 3µg each of Gag/Pol and Env packaging plasmids and the retroviral expression plasmid using Lipofectamine 2000. Lentivirus were amplified by transfecting HEK-293T cells with 1.2 µg each of Gag/Pol, Tat and Env, 2.4 µg Vsv-g packaging plasmids, and 24 µg of the lentiviral expression plasmid using TransIT (Mirus, Madison, WI). Lentiviral-based shRNA against GFP, human p63, Erk1 and Erk2 in pLKO.1puro backbones were obtained from Open Biosystems. The shRNA against human MKP3 is in a pSICOR(puro) plasmid.
Western blot analysis and immunofluorescence
Cells were lysed in EBC250 lysis buffer (69). Nuclear extraction was performed with cytoskeletal (CSK) buffer (10 mM PIPES (pH 6.8), 100 mM NaCl, 300 mM sucrose, 3 mM MgCl2, 1 mM EGTA, 1 mM DTT, 10 mM NaF, 0.1% Triton X-100, 1 mM ATP). Proteins were resolved by SDS-PAGE, transferred to polyvinylidene difluoride membranes (NEN LifeSciences, Waltham, Massachusetts) and hybridized to an appropriate primary antibody and HRP-conjugated secondary antibody for subsequent detection by enhanced chemiluminescence (Thermo Fisher Scientific Inc., Rockford, IL). For immunostaining, cells grown on coverslips were fixed with 3.7% formaldehyde in PBS, permeabilized with 0.1% Triton X-100 in PBS, blocked with 1% bovine serum albumin (BSA) in PBS, and sequentially incubated with primary and secondary antibodies prepared in 4% BSA. Coverslips were mounted with ProLong Gold antifade reagent with DAPI (Invitrogen Inc., Carlsbad, CA). Using an Axiovert 200M microscope (Carl Zeiss, Germany) under 40% or 100% objective lenses, fluorescent images were acquired using an AxioCam MRm (Carl Zeiss, Germany) and processed with AxioVision 4 software. Monoclonal antibody specific for p63 (4A4) and goat anti-actin antibody (C-11) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Rabbit anti-MKP3 (3058), rabbit anti-phospho-Erk1/2 (9109), rabbit anti-Erk1/2 (9102), rabbit anti-phospho-Mek (9121), rabbit anti-Mek (9122), and rabbit anti-phospho-paxillin (pTyr118; 2541) antibodies were purchased from Cell Signaling (Danvers, MA). Goat anti-mouse IgG-HRP (sc-2005), goat anti-rabbit IgG-HRP (sc-2004), and donkey anti-goat IgG-HRP (sc-2020) secondary antibodies for western blotting were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Secondary antibodies used for immunostaining, including goat anti-mouse Alexa Fluor 488 (A11001), goat anti-rabbit Alexa Fluor 488 (A11008) and goat anti-rabbit Alexa Fluor 594 (A11012), were purchased from Invitrogen (Carlsbad, CA).
Affymetrix array and Q-PCR analysis
Total RNA was isolated using TRIzol (Gibco Life Technologies, Rockville, MD) or RNeasy Mini Kit (Qiagen, Chatsworth, CA), followed by reverse transcription using SuperScript III First-Strand Synthesis System (Invitrogen Inc., Carlsbad, CA). Array analysis was performed using U133Av2.0 human gene chip (Affymetrix, Santa Clara, CA) in two independent experiments. Q-PCR was performed in a 7300 Real-Time PCR System (Applied Biosystems, Foster City, CA) using QuantiTect SYBR Green PCR Kit (Qiagen, Chatsworth, CA) according to manufacturer’s instructions. Primers used for Q-PCR are listed in Table S1 in the Supplementary Material.
Assays for cell migration and invasion
Cell migration was measured using 6.5 mm, 8.0 µm-pore polycarbonate membrane transwell inserts (BD Biosciences, San Jose, CA). Cell invasion was measured using matrigel-coated inserts (BD Biosciences, San Jose, CA). Cells (1.0 × 105 to 2.5 × 105) were suspended in serum-free DMEM (for MDA-MB-231 and FaDu cells) or DMEM:F12 (for MCF-10A cells) media containing 0.5% gelatin, and seeded into the inner chamber. The outer chamber was filled with normal growth media (as described previously for each cell type) containing 0.5% gelatin. Cells were incubated for 16 to 24 hours. Non-migrating cells were carefully removed with a cotton swab. Migrating cells were stained with 0.5% crystal violet in 70% ethanol for ten minutes at room temperature, and photographed under a Zeiss light microscope. At least 100 cells in total from five random fields were counted. Alternatively, the stained cells were lyzed with 2% SDS in PBS and subjected to spectrophotometric analysis at 570 nm.
In vivo metastasis assays
MCF-10A cell were infected with pBABE-NeuT or pBABE. Puromycin-resistant cells were then infected with lentivirus expressing shRNA against p63 (NeuT-shp63) or GFP (NeuT-shC). 1.0×106 NeuT-shC or NeuT-shp63 cells in 200 µL PBS were injected into the lateral tail vein of six NCr nude six-week old female mice (CrTac:NCr-Foxn1nu; Taconic). Mice were observed daily and euthanized after 45 days. During the course of the experiment, one NeuT-shp63 mouse died for reasons unclear. The lungs were dissected and inspected for metastatic nodules. Lungs were then fixed overnight in 3.7% formaldehyde, embedded in paraffin, and sectioned onto microscope slides for haematoxylin and eosin (H&E) staining and histological analysis. All animal studies were approved by the Institutional Animal Care and Use Committee.
Statistical analysis
Quantitative data was analyzed statistically using student’s t-test to assess significance. Data are presented as means ± standard error (SE), as noted on figure legends.
Bioinformatic analysis of gene expression
Oncomine™ (Compendia Bioscience, Ann Arbor, MI) was used for analysis and visualization.
Supplementary Material
Acknowledgements
We thank Dr. Frank McKeon (Harvard Medical School, Boston, MA) for providing the myc-tagged murine pcDNA3-ΔNp63α, pcDNA3-ΔNp63γ, pcDNA3-TAp63α and pcDNA3-TAp63γ expression plasmids, and Dr. Tyler Jacks (Massachusetts Institute of Technology, Cambridge, MA) for providing a plasmid encoding shRNA against human MKP3. We also thank Dr. Xixi Cao (Baylor College of Medicine) for help with Oncomine™ bioinformatics analysis. This work was supported by NIH grants (CA79804 and GM70017) and by the National Key Basic Research Program (973 Program) of China (2012CB910700) to Z-X. X., and United States Department of Defense Congressionally Directed Medical Research Programs grant (W81XWH-10-1-0161) to J.B.
Footnotes
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc).
Conflict of interest
The authors declare no conflict of interest.
References
- 1.Serber Z, Lai HC, Yang A, Ou HD, Sigal MS, Kelly AE, et al. A C-terminal inhibitory domain controls the activity of p63 by an intramolecular mechanism. Mol Cell Biol. 2002 Dec 1;22(24):8601–8611. doi: 10.1128/MCB.22.24.8601-8611.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thanos CD, Bowie JU. p53 Family members p63 and p73 are SAM domain-containing proteins. Protein Sci. 1999 Aug 1;8(8):1708–1710. doi: 10.1110/ps.8.8.1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang A, Kaghad M, Wang Y, Gillett E, Fleming MD, Dötsch V, et al. p63, a p53 homolog at 3q27-29, encodes multiple products with transactivating, death-inducing, and dominant-negative activities. Mol Cell. 1998 Sep 1;2(3):305–316. doi: 10.1016/s1097-2765(00)80275-0. [DOI] [PubMed] [Google Scholar]
- 4.Candi E, Cipollone R, Rivetti di Val Cervo P, Gonfloni S, Melino G, Knight R. p63 in epithelial development. Cell Mol Life Sci. 2008 Oct 1;65(20):3126–3133. doi: 10.1007/s00018-008-8119-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Melino G. p63 is a suppressor of tumorigenesis and metastasis interacting with mutant p53. Cell Death Differ. 2011 Jul 15; doi: 10.1038/cdd.2011.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Celli J, Duijf P, Hamel BC, Bamshad M, Kramer B, Smits AP, et al. Heterozygous germline mutations in the p53 homolog p63 are the cause of EEC syndrome. Cell. 1999 Oct 15;99(2):143–153. doi: 10.1016/s0092-8674(00)81646-3. [DOI] [PubMed] [Google Scholar]
- 7.McGrath JA, Duijf PH, Doetsch V, Irvine AD, de Waal R, Vanmolkot KR, et al. Hay-Wells syndrome is caused by heterozygous missense mutations in the SAM domain of p63. Hum Mol Genet. 2001 Feb 1;10(3):221–229. doi: 10.1093/hmg/10.3.221. [DOI] [PubMed] [Google Scholar]
- 8.Yang A, Schweitzer R, Sun D, Kaghad M, Walker N, Bronson RT, et al. p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature. 1999 Apr 22;398(6729):714–718. doi: 10.1038/19539. [DOI] [PubMed] [Google Scholar]
- 9.Mills AA, Zheng B, Wang XJ, Vogel H, Roop DR, Bradley A. p63 is a p53 homologue required for limb and epidermal morphogenesis. Nature. 1999 Apr 22;398(6729):708–713. doi: 10.1038/19531. [DOI] [PubMed] [Google Scholar]
- 10.Senoo M, Pinto F, Crum CP, McKeon F. p63 Is essential for the proliferative potential of stem cells in stratified epithelia. Cell. 2007 May 4;129(3):523–536. doi: 10.1016/j.cell.2007.02.045. [DOI] [PubMed] [Google Scholar]
- 11.Shalom-Feuerstein R, Lena AM, Zhou H, De La Forest Divonne S, Van Bokhoven H, Candi E, et al. ΔNp63 is an ectodermal gatekeeper of epidermal morphogenesis. Cell Death Differ. 2010 Dec 3; doi: 10.1038/cdd.2010.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Keyes WM, Wu Y, Vogel H, Guo X, Lowe SW, Mills AA. p63 deficiency activates a program of cellular senescence and leads to accelerated aging. Genes Dev. 2005 Sep 1;19(17):1986–1999. doi: 10.1101/gad.342305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Flores ER, Sengupta S, Miller JB, Newman JJ, Bronson R, Crowley D, et al. Tumor predisposition in mice mutant for p63 and p73: evidence for broader tumor suppressor functions for the p53 family. Cancer Cell. 2005 Apr 1;7(4):363–373. doi: 10.1016/j.ccr.2005.02.019. [DOI] [PubMed] [Google Scholar]
- 14.Su X, Chakravarti D, Cho MS, Liu L, Gi YJ, Lin Y-L, et al. TAp63 suppresses metastasis through coordinate regulation of Dicer and miRNAs. Nature. 2010 Oct 21;467(7318):986–990. doi: 10.1038/nature09459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gonfloni S, Di Tella L, Caldarola S, Cannata SM, Klinger FG, Di Bartolomeo C, et al. Inhibition of the c-Abl-TAp63 pathway protects mouse oocytes from chemotherapy-induced death. Nat Med. 2009 Oct 1;15(10):1179–1185. doi: 10.1038/nm.2033. [DOI] [PubMed] [Google Scholar]
- 16.Suh E-K, Yang A, Kettenbach A, Bamberger C, Michaelis AH, Zhu Z, et al. p63 protects the female germ line during meiotic arrest. Nature. 2006 Nov 30;444(7119):624–628. doi: 10.1038/nature05337. [DOI] [PubMed] [Google Scholar]
- 17.Candi E, Rufini A, Terrinoni A, Dinsdale D, Ranalli M, Paradisi A, et al. Differential roles of p63 isoforms in epidermal development: selective genetic complementation in p63 null mice. Cell Death Differ. 2006 Jun 1;13(6):1037–1047. doi: 10.1038/sj.cdd.4401926. [DOI] [PubMed] [Google Scholar]
- 18.Carroll DK, Carroll JS, Leong C-O, Cheng F, Brown M, Mills AA, et al. p63 regulates an adhesion programme and cell survival in epithelial cells. Nat Cell Biol. 2006 Jun 1;8(6):551–561. doi: 10.1038/ncb1420. [DOI] [PubMed] [Google Scholar]
- 19.Ihrie RA, Marques MR, Nguyen BT, Horner JS, Papazoglu C, Bronson RT, et al. Perp is a p63-regulated gene essential for epithelial integrity. Cell. 2005 Mar 25;120(6):843–856. doi: 10.1016/j.cell.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 20.Rocco JW, Leong C-O, Kuperwasser N, DeYoung MP, Ellisen LW. p63 mediates survival in squamous cell carcinoma by suppression of p73-dependent apoptosis. Cancer Cell. 2006 Jan 1;9(1):45–56. doi: 10.1016/j.ccr.2005.12.013. [DOI] [PubMed] [Google Scholar]
- 21.DeYoung MP, Johannessen CM, Leong C-O, Faquin W, Rocco JW, Ellisen LW. Tumor-specific p73 up-regulation mediates p63 dependence in squamous cell carcinoma. Cancer Res. 2006 Oct 1;66(19):9362–9368. doi: 10.1158/0008-5472.CAN-06-1619. [DOI] [PubMed] [Google Scholar]
- 22.Keyes WM, Pecoraro M, Aranda V, Vernersson-Lindahl E, Li W, Vogel H, et al. ΔNp63α is an oncogene that targets chromatin remodeler Lsh to drive skin stem cell proliferation and tumorigenesis. Cell Stem Cell. 2011 Feb 4;8(2):164–176. doi: 10.1016/j.stem.2010.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hibi K, Trink B, Patturajan M, Westra WH, Caballero OL, Hill DE, et al. AIS is an oncogene amplified in squamous cell carcinoma. Proc Natl Acad Sci USA. 2000 May 9;97(10):5462–5467. doi: 10.1073/pnas.97.10.5462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Massion PP, Taflan PM, Jamshedur Rahman SM, Yildiz P, Shyr Y, Edgerton ME, et al. Significance of p63 amplification and overexpression in lung cancer development and prognosis. Cancer Res. 2003 Nov 1;63(21):7113–7121. [PubMed] [Google Scholar]
- 25.Weber A, Bellmann U, Bootz F, Wittekind C, Tannapfel A. Expression of p53 and its homologues in primary and recurrent squamous cell carcinomas of the head and neck. Int J Cancer. 2002 May 1;99(1):22–28. doi: 10.1002/ijc.10296. [DOI] [PubMed] [Google Scholar]
- 26.Haqq C, Nosrati M, Sudilovsky D, Crothers J, Khodabakhsh D, Pulliam BL, et al. The gene expression signatures of melanoma progression. Proc Natl Acad Sci USA. 2005 Apr 26;102(17):6092–6097. doi: 10.1073/pnas.0501564102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Su H, Hu N, Shih J, Hu Y, Wang Q-H, Chuang EY, et al. Gene expression analysis of esophageal squamous cell carcinoma reveals consistent molecular profiles related to a family history of upper gastrointestinal cancer. Cancer Res. 2003 Jul 15;63(14):3872–3876. [PubMed] [Google Scholar]
- 28.Vanaja DK, Cheville JC, Iturria SJ, Young CYF. Transcriptional silencing of zinc finger protein 185 identified by expression profiling is associated with prostate cancer progression. Cancer Res. 2003 Jul 15;63(14):3877–3882. [PubMed] [Google Scholar]
- 29.Dumesic PA, Scholl FA, Barragan DI, Khavari PA. Erk1/2 MAP kinases are required for epidermal G2/M progression. J Cell Biol. 2009 May 4;185(3):409–422. doi: 10.1083/jcb.200804038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yamamoto T, Ebisuya M, Ashida F, Okamoto K, Yonehara S, Nishida E. Continuous ERK activation downregulates antiproliferative genes throughout G1 phase to allow cell-cycle progression. Curr Biol. 2006 Jun 20;16(12):1171–1182. doi: 10.1016/j.cub.2006.04.044. [DOI] [PubMed] [Google Scholar]
- 31.Vicent S, López-Picazo JM, Toledo G, Lozano MD, Torre W, Garcia-Corchón C, et al. ERK1/2 is activated in non-small-cell lung cancer and associated with advanced tumours. Br J Cancer. 2004 Mar 8;90(5):1047–1052. doi: 10.1038/sj.bjc.6601644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Adeyinka A, Nui Y, Cherlet T, Snell L, Watson PH, Murphy LC. Activated mitogen-activated protein kinase expression during human breast tumorigenesis and breast cancer progression. Clin Cancer Res. 2002 Jun 1;8(6):1747–1753. [PubMed] [Google Scholar]
- 33.Krueger JS, Keshamouni VG, Atanaskova N, Reddy KB. Temporal and quantitative regulation of mitogen-activated protein kinase (MAPK) modulates cell motility and invasion. Oncogene. 2001 Jul 12;20(31):4209–4218. doi: 10.1038/sj.onc.1204541. [DOI] [PubMed] [Google Scholar]
- 34.McCawley LJ, Li S, Wattenberg EV, Hudson LG. Sustained activation of the mitogen-activated protein kinase pathway. A mechanism underlying receptor tyrosine kinase specificity for matrix metalloproteinase-9 induction and cell migration. J Biol Chem. 1999 Feb 12;274(7):4347–4353. doi: 10.1074/jbc.274.7.4347. [DOI] [PubMed] [Google Scholar]
- 35.Webb CP, Van Aelst L, Wigler MH, Woude GF. Signaling pathways in Ras-mediated tumorigenicity and metastasis. Proc Natl Acad Sci USA. 1998 Jul 21;95(15):8773–8778. doi: 10.1073/pnas.95.15.8773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ekerot M, Stavridis M, Delavaine L, Mitchell M, Staples C, Owens D, et al. Negative feedback regulation of FGF signalling by DUSP6/MKP-3 is driven by ERK1/2 and mediated by Ets factor binding to a conserved site within the DUSP6/MKP-3 gene promoter. Biochem J. 2008 Mar 6; doi: 10.1042/BJ20071512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li C, Scott DA, Hatch E, Tian X, Mansour SL. Dusp6 (Mkp3) is a negative feedback regulator of FGF-stimulated ERK signaling during mouse development. Development. 2007 Jan 1;134(1):167–176. doi: 10.1242/dev.02701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fu SW, Schwartz A, Stevenson H, Pinzone JJ, Davenport GJ, Orenstein JM, et al. Correlation of expression of BP1, a homeobox gene, with estrogen receptor status in breast cancer. Breast Cancer Res. 2003 Jan 1;5(4):R82–R87. doi: 10.1186/bcr602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhao H, Langerød A, Ji Y, Nowels KW, Nesland JM, Tibshirani R, et al. Different gene expression patterns in invasive lobular and ductal carcinomas of the breast. Mol Biol Cell. 2004 Jun 1;15(6):2523–2536. doi: 10.1091/mbc.E03-11-0786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hollestelle A, Elstrodt F, Nagel JHA, Kallemeijn WW, Schutte M. Phosphatidylinositol-3-OH kinase or RAS pathway mutations in human breast cancer cell lines. Mol Cancer Res. 2007 Feb 1;5(2):195–201. doi: 10.1158/1541-7786.MCR-06-0263. [DOI] [PubMed] [Google Scholar]
- 41.Karlsson M, Mathers J, Dickinson RJ, Mandl M, Keyse SM. Both nuclear-cytoplasmic shuttling of the dual specificity phosphatase MKP-3 and its ability to anchor MAP kinase in the cytoplasm are mediated by a conserved nuclear export signal. J Biol Chem. 2004 Oct 1;279(40):41882–41891. doi: 10.1074/jbc.M406720200. [DOI] [PubMed] [Google Scholar]
- 42.Jinnin M, Ihn H, Mimura Y, Asano Y, Yamane K, Tamaki K. Matrix metalloproteinase-1 up-regulation by hepatocyte growth factor in human dermal fibroblasts via ERK signaling pathway involves Ets1 and Fli1. Nucleic Acids Res. 2005 Jan 1;33(11):3540–3549. doi: 10.1093/nar/gki648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu S, Liang Y, Huang H, Wang L, Li Y, Li J, et al. ERK-dependent signaling pathway and transcriptional factor Ets-1 regulate matrix metalloproteinase-9 production in transforming growth factor-beta1 stimulated glomerular podocytes. Cell Physiol Biochem. 2005 Jan 1;16(4–6):207–216. doi: 10.1159/000089846. [DOI] [PubMed] [Google Scholar]
- 44.Yang S, Zhang JJ, Huang X-Y. Orai1 and STIM1 are critical for breast tumor cell migration and metastasis. Cancer Cell. 2009 Feb 3;15(2):124–134. doi: 10.1016/j.ccr.2008.12.019. [DOI] [PubMed] [Google Scholar]
- 45.Webb DJ, Donais K, Whitmore LA, Thomas SM, Turner CE, Parsons JT, et al. FAK-Src signalling through paxillin, ERK and MLCK regulates adhesion disassembly. Nat Cell Biol. 2004 Feb 1;6(2):154–161. doi: 10.1038/ncb1094. [DOI] [PubMed] [Google Scholar]
- 46.Meng L, Gabai VL, Sherman MY. Heat-shock transcription factor HSF1 has a critical role in human epidermal growth factor receptor-2-induced cellular transformation and tumorigenesis. Oncogene. 2010 Sep 16;29(37):5204–5213. doi: 10.1038/onc.2010.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huang C, Jacobson K, Schaller MD. MAP kinases and cell migration. J Cell Sci. 2004 Sep 15;117(Pt 20):4619–4628. doi: 10.1242/jcs.01481. [DOI] [PubMed] [Google Scholar]
- 48.Shin S, Dimitri CA, Yoon S-O, Dowdle W, Blenis J. ERK2 but not ERK1 induces epithelial-to-mesenchymal transformation via DEF motif-dependent signaling events. Mol Cell. 2010 Apr 9;38(1):114–127. doi: 10.1016/j.molcel.2010.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kuo L, Chang H-C, Leu T-H, Maa M-C, Hung W-C. Src oncogene activates MMP-2 expression via the ERK/Sp1 pathway. J Cell Physiol. 2006 Jun 1;207(3):729–734. doi: 10.1002/jcp.20616. [DOI] [PubMed] [Google Scholar]
- 50.Higashikawa K, Yoneda S, Tobiume K, Saitoh M, Taki M, Mitani Y, et al. DeltaNp63alpha-dependent expression of Id-3 distinctively suppresses the invasiveness of human squamous cell carcinoma. Int J Cancer. 2009 Jun 15;124(12):2837–2844. doi: 10.1002/ijc.24280. [DOI] [PubMed] [Google Scholar]
- 51.Liu J, Zhan M, Hannay JA, Das P, Bolshakov SV, Kotilingam D, et al. Wild-type p53 inhibits nuclear factor-kappaB-induced matrix metalloproteinase-9 promoter activation: implications for soft tissue sarcoma growth and metastasis. Molecular cancer research : MCR. 2006 Nov;4(11):803–810. doi: 10.1158/1541-7786.MCR-06-0201. [DOI] [PubMed] [Google Scholar]
- 52.Sun Y, Wenger L, Rutter JL, Brinckerhoff CE, Cheung HS. p53 down-regulates human matrix metalloproteinase-1 (Collagenase-1) gene expression. The Journal of biological chemistry. 1999 Apr 23;274(17):11535–11540. doi: 10.1074/jbc.274.17.11535. [DOI] [PubMed] [Google Scholar]
- 53.Fukushima H, Koga F, Kawakami S, Fujii Y, Yoshida S, Ratovitski E, et al. Loss of DeltaNp63alpha promotes invasion of urothelial carcinomas via N-cadherin/Src homology and collagen/extracellular signal-regulated kinase pathway. Cancer Res. 2009 Dec 15;69(24):9263–9270. doi: 10.1158/0008-5472.CAN-09-1188. [DOI] [PubMed] [Google Scholar]
- 54.Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer. 2003 Jan 1;3(1):11–22. doi: 10.1038/nrc969. [DOI] [PubMed] [Google Scholar]
- 55.Zhang Z, Kobayashi S, Borczuk AC, Leidner RS, Laframboise T, Levine AD, et al. Dual specificity phosphatase 6 (DUSP6) is an ETS-regulated negative feedback mediator of oncogenic ERK-signaling in lung cancer cells. Carcinogenesis. 2010 Jan 22; doi: 10.1093/carcin/bgq020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Furukawa T, Sunamura M, Motoi F, Matsuno S, Horii A. Potential tumor suppressive pathway involving DUSP6/MKP-3 in pancreatic cancer. Am J Pathol. 2003 Jun 1;162(6):1807–1815. doi: 10.1016/S0002-9440(10)64315-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Okudela K, Yazawa T, Woo T, Sakaeda M, Ishii J, Mitsui H, et al. Down-regulation of DUSP6 expression in lung cancer: its mechanism and potential role in carcinogenesis. Am J Pathol. 2009 Aug 1;175(2):867–881. doi: 10.2353/ajpath.2009.080489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Maillet M, Purcell N, Sargent M, York A, Bueno O, Molkentin J. Dusp6 (MKP3) null mice show enhanced ERK1/2 phosphorylation at baseline and increased myocyte proliferation in the heart affecting disease susceptibility. J Biol Chem. 2008 Aug 27; doi: 10.1074/jbc.M806085200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Barbieri CE, Tang LJ, Brown KA, Pietenpol JA. Loss of p63 leads to increased cell migration and up-regulation of genes involved in invasion and metastasis. Cancer Res. 2006 Aug 1;66(15):7589–7597. doi: 10.1158/0008-5472.CAN-06-2020. [DOI] [PubMed] [Google Scholar]
- 60.Higashikawa K, Yoneda S, Tobiume K, Taki M, Shigeishi H, Kamata N. Snail-induced down-regulation of DeltaNp63alpha acquires invasive phenotype of human squamous cell carcinoma. Cancer Res. 2007 Oct 1;67(19):9207–9213. doi: 10.1158/0008-5472.CAN-07-0932. [DOI] [PubMed] [Google Scholar]
- 61.Kommagani R, Leonard M, Lewis S, Romano R, Sinha S, Kadakia M. Regulation of VDR by {Delta}Np63{alpha} is associated with inhibition of cell invasion. J Cell Sci. 2009 Jul 21; doi: 10.1242/jcs.049619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lindsay J, McDade SS, Pickard A, McCloskey KD, McCance DJ. The role of {delta}Np63{gamma} in epithelial to mesenchymal transition. J Biol Chem. 2010 Dec 2; doi: 10.1074/jbc.M110.162511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Adorno M, Cordenonsi M, Montagner M, Dupont S, Wong C, Hann B, et al. A Mutant-p53/Smad complex opposes p63 to empower TGFbeta-induced metastasis. Cell. 2009 Apr 3;137(1):87–98. doi: 10.1016/j.cell.2009.01.039. [DOI] [PubMed] [Google Scholar]
- 64.Muller PAJ, Caswell PT, Doyle B, Iwanicki MP, Tan EH, Karim S, et al. Mutant p53 Drives Invasion by Promoting Integrin Recycling. Cell. 2009 Dec 1;139(7):1327–1341. doi: 10.1016/j.cell.2009.11.026. [DOI] [PubMed] [Google Scholar]
- 65.Eicheler W, Zips D, Dörfler A, Grénman R, Baumann M. Splicing mutations in TP53 in human squamous cell carcinoma lines influence immunohistochemical detection. J Histochem Cytochem. 2002 Feb 1;50(2):197–204. doi: 10.1177/002215540205000207. [DOI] [PubMed] [Google Scholar]
- 66.Xu S, Furukawa T, Kanai N, Sunamura M, Horii A. Abrogation of DUSP6 by hypermethylation in human pancreatic cancer. J Hum Genet. 2005 Jan 1;50(4):159–167. doi: 10.1007/s10038-005-0235-y. [DOI] [PubMed] [Google Scholar]
- 67.Ginos MA, Page GP, Michalowicz BS, Patel KJ, Volker SE, Pambuccian SE, et al. Identification of a gene expression signature associated with recurrent disease in squamous cell carcinoma of the head and neck. Cancer Res. 2004 Jan 1;64(1):55–63. doi: 10.1158/0008-5472.can-03-2144. [DOI] [PubMed] [Google Scholar]
- 68.Talbot SG, Estilo C, Maghami E, Sarkaria IS, Pham DK, O-charoenrat P, et al. Gene expression profiling allows distinction between primary and metastatic squamous cell carcinomas in the lung. Cancer Res. 2005 Apr 15;65(8):3063–3071. doi: 10.1158/0008-5472.CAN-04-1985. [DOI] [PubMed] [Google Scholar]
- 69.Ying H, Chang DLF, Zheng H, McKeon F, Xiao Z-XJ. DNA-binding and transactivation activities are essential for TAp63 protein degradation. Mol Cell Biol. 2005 Jul 1;25(14):6154–6164. doi: 10.1128/MCB.25.14.6154-6164.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.