

Abstract
Background
Sepsis-induced inflammation in the gut/peritoneal compartment occurs early in sepsis, and can lead to acute lung injury (ALI). We have suggested that inflammatory ascites drives the pathogenesis of ALI, and that removal of ascites with an abdominal wound vacuum prevents ALI. We hypothesized that the time- and compartment-dependent changes in inflammation that determine this process can be discerned using Principal Component Analysis (PCA) and Dynamic Bayesian Network (DBN) inference.
Methods
To test this hypothesis, data from a previous study were analyzed using PCA and DBN. In that study, two groups of anesthetized, ventilated pigs were subjected to experimental sepsis via intestinal ischemia/reperfusion and placement of a peritoneal fecal clot. The Control Group (n=6) had the abdomen opened at 12 hrs post injury (T12) with attachment of a passive drain. The Peritoneal Suction Treatment (PST) Group (n=6) was treated in an identical fashion except that a vacuum was applied to the peritoneal cavity at T12 to remove ascites and maintained until T48. Multiple inflammatory mediators were measured in ascites and plasma and related to lung function (PaO2/FiO2 ratio [PF] and Oxygen Index [OI]) using PCA and DBN.
Results
PST prevented ALI based on lung histopathology, whereas Control animals developed ALI. Principal Component Analysis revealed that local to the insult (i.e. ascites), primary pro-inflammatory cytokines play a decreased role in the overall response in the treatment group as compared to control. In both groups, multiple, nested positive feedback loops were inferred from DBN, which included interrelated roles for bacterial endotoxin, interleukin-6, transforming growth factor-β1, C-reactive protein, PF, and OI. Von Willebrand Factor was an output in Control, but not PST, ascites.
Conclusions
These combined in vivo and in silico studies suggest that in this clinically realistic paradigm of sepsis, endotoxin drives the inflammatory response in the ascites, interplaying with lung dysfunction in a feed-forward loop that exacerbates inflammation and leads to endothelial dysfunction, systemic spillover, and ALI; PST partially modifies this process.
INTRODUCTION
Sepsis by intestinal ischemia/reperfusion and peritonitis results in massive systemic inflammation with attendant increases in vascular permeability, leading to severe lung injury with pulmonary edema, termed either Acute Lung Injury (ALI) or Adult Respiratory Distress Syndrome (ARDS) (1). In turn, ALI/ARDS are part of the larger process of Multiple Organ Dysfunction Syndrome (MODS) (2), where the first organ to fail in MODS is usually the lung (3). ARDS presents with clinical signs and symptoms of respiratory distress, PaO2/FiO2 ratio below 200, bilateral pulmonary edema, decreased compliance, and increasing oxygen requirements (4). ARDS is a serious clinical problem with over 200,000 cases annually (5) and is resistant to treatment once the syndrome is clinically diagnosed (6). The disease retains disturbingly high mortality (7), costs of care (8), and severe sequelae for survivors (9) despite decades of therapeutic research (10).
The local inflammatory response during gut-associated sepsis is a risk factor for ARDS. Microcirculation in the gut is dramatically impaired in both septic (11, 12) and hemorrhagic shock (13, 14). Impaired microcirculation results in tissue hypoxia and inflammation-induced alteration in both endothelial (15) and epithelial function (16). Increased microvascular permeability in the gut results in intestinal edema and ascites formation (17). The damaged gut is a continual source of inflammation, propagating ARDS and driving other organ damage (16, 18–21).
We have suggested that MODS comes about due to cascading system failure, wherein the positive feedback loop of inflammation → damage → inflammation exceeds compartment-specific thresholds (“tipping points”) (22, 23). We have demonstrated that removal of the inflamed peritoneal ascites using a wound vacuum system would eliminate this “driver” of systemic inflammation, thereby attenuating this positive feedback loops and consequently interrupting the progression of ALI (22, 24). A more complete understanding of the complex relationships between the inflammatory milieu of the ascites and plasma and the mechanism by which ascites removal blocks the development of ALI/ARDS would aid in the translation of this potential therapeutic strategy to the clinical arena.
We hypothesized that the observed prevention of MODS results from a dynamic modification of inflammation after removal of ascites. We have demonstrated previously that we can gain insights into principal drivers and dynamic networks of acute inflammation using Principal Components Analysis (PCA) and Dynamic Bayesian Networks (DBN) (25–28). Accordingly, we used PCA and DBN analyses to determine if removal of ascites was associated with different local (ascites) and systemic (plasma) principal drivers and dynamic networks of inflammatory mediators vs. control. This analysis suggests the presence of complex, time- and compartment-dependent changes in inflammation and lung pathophysiology. Our studies further suggest that these principal drivers and networks could be affected by removal of peritoneal ascites, in essence amounting to modification of this complex response in a manner associated with the reduction or elimination of ALI/ARDS.
MATERIALS AND METHODS
The experimental work forming the basis of the mathematical analysis was previously published (24). The details of those experiments are re-stated below in order to provide a reference point for the subsequent analysis. The experiment was performed in compliance with the National Institutes of Health’s Guidelines on the Use of Laboratory Animals and the CHUA Committee at Upstate University Hospital approved the study protocol.
Animals and preparations
Complete and detailed surgical methods can be found in the original analysis (24); more succinct methods are included here. Female Yorkshire pigs (21–38 kg) were anesthetized with ketamine/xylazine to maintain a surgical plane of anesthesia. An open tracheotomy was performed and the animal connected to a G5 ventilator (Hamilton Medical, Reno, NV) with initial settings during the surgical preparation as follows: tidal volume (Vt) of 12 mL/kg, respiratory rate (RR) of 15 breaths/min, titrated to maintain PaCO2 within the normal range (35–45 cmH2O), FiO2 of 21%, and positive end-expiratory pressure (PEEP) of 3 cmH2O. Lung volume history was standardized by recruiting the lung using the PV TOOL (Hamilton Medical). The lung was inflated to a peak pressure of 30 cmH2O, held for 5 s, and deflated back to 5 cmH2O of PEEP. Under sterile conditions, a left carotid artery catheter was placed for blood chemistry and gas content measurements (Roche Cobras b211; Roche Diagnostics, Indianapolis, IN), and systemic arterial pressure monitoring. A veinotomy was performed on the left external jugular vein for placement of a triple lumen catheter, allowing anesthesia, fluid, and antibiotic administration. A right external jugular Swan-Ganz catheter (7 French) was placed for measurement of pulmonary artery (PAP) and wedge pressures (PAW), sampling of mixed venous blood gases, and cardiac output (CO; Agilent CMS-2001, Boebingen, Germany). A Foley catheter was inserted into the bladder for measurement of urine output (UOP), collection of urine samples, and was connected to a pressure transducer leveled at midline to measure intra-abdominal pressure (IAP). The health of the animal was determined by normal hemodynamic and lung function parameters following instrumentation and normal blood gases and chemistries.
Study protocol
A midline laparotomy was performed, and the superior mesenteric artery (SMA) was isolated and clamped for 30 min to induce intestinal ischemia. After 30 min, the clamp was removed, and reperfusion was confirmed by the reappearance of the mesenteric pulse and the return of normal color to the bowel. At this point an enterotomy of 2 cm was performed to harvest feces (0.5 mL/kg) in the cecum and combined with 2 mL/kg of the pig’s blood to create a fecal-blood clot. A catheter was placed in Morrison pouch between the liver and right kidney and brought out through the skin for collection of peritoneal ascites. Collected ascites was flash-frozen for measurement of inflammatory mediators. The abdomen was then closed with sutures and the time recorded as T0 (i.e. 0 h after injury). For the first 12 h of the protocol, the animals were treated in an identical fashion. All animals received a regimen of intravenous fluids and antibiotics at a dose and quantity established in our initial experiments (Fluids and antibiotics).
The entire abdomen was reopened at T12, and the V.A.C. Abdominal Dressing System (KCI, Inc., San Antonio, Tex) was applied to the open wound as per packet instructions. At this point, animals were randomly assigned to treatment groups as follows:
Control group (Passively Drained - PD; n = 6) had the dressing placed, but the vacuum was not activated (i.e. negative pressure was not applied); however, the drain line was left open to allow passive drainage of ascites.
Treatment group (PST; n = 6) had the dressing placed and the vacuum activated so that negative pressure (−125 mmHg) was applied continuously for the remainder of the experiment.
Assessment of inflammatory mediators
Inflammatory mediators were measured in the plasma and peritoneal ascites fluid (Pfluid). Ascites fluid was collected from an abdominal drain in the posterior gutters independent of the vacuum drainage tubing. Ascites and plasma fluid were collected at Baseline, T6, T9, T12, T15, T18, T21, T24, T27, T36, T42, T48. All samples were spun at 3,000 RPM at 4°C for 10 min, snap-frozen in liquid nitrogen, and stored at −80°C.
TNF-α, IL-1β, IL-6, IL-8, IL-12, and transforming growth factor-β1 (TGF-β1; R&D Systems, Minneapolis, MN), IL-10, C5a (Immunobiological Laboratories, Minneapolis, MN), von Willebrand factor (vWf; American Diagnostics Inc. Stamford, CT), and C-reactive protein (CRP) (Immunology Consultant’s Laboratories, Inc., New Burg, OR) were measured using pig-specific enzyme-linked immunosorbent assays according to the manufacturer’s assigned specifications. Prostaglandin E2 (PGE2; Oxford Biomedical Research, Oxford, MI) and antioxidants (Cayman Chemical Company, Ann Arbor, MI) were tested using commercially available ELISA kits. Endotoxin was tested using an end-point chromogenic Limulus amebocyte lysate assay (Lonza Group Ltd., Basel, Switzerland). Total protein was measured in plasma and Pfluid using a bicinchoninic colorimetric assay (BCA; Pierce Biotechnology, Rockford, IL). Urine proteins were first precipitated using technical resource 0049.0 (Pierce Biotechnology). Blood cultures (aerobic and anaerobic) and Gram staining were performed by the Upstate Medical University Clinical Pathology Department using standard techniques.
Dynamic Bayesian Network (DBN) inference
Time courses of cytokine measurements from each experiment were used as input for a Dynamic Bayesian Network (DBN) inference algorithm. Given time-series data, DBN provides a way of inferring causal relationships among variables (e.g. inflammatory mediators and physiological parameters) based on probabilistic measure. Unlike standard correlative approaches, DBNs consider the joint distribution of the entire dataset when making inferences about the dependencies among variables or nodes in the network. In a DBN, variables are shown as nodes and the interconnections are shown as edges. The values of each node are assumed to be distributed according to a chosen model (e.g. Gaussian) and the relationships among nodes are defined by the structure of the directed network and the corresponding conditional probability distributions of the interacting nodes. Network structure is inferred by a sampling technique that iteratively proposes candidate structures and evaluates them based on how well they explain the observed data using a specified scoring criterion, until reaching convergence on a network structure with the highest score.
Our analysis was carried out in MATLAB™ (The MathWorks, Inc., Natick, MA), using an algorithm adapted from Grzegorczyk & Husmeier (29) and revised recently by our group (30, 31). Briefly, the algorithm uses an inhomogeneous dynamic changepoint model, with a Bayesian Gaussian with score equivalence (BGe) scoring criterion. For each node, a new set of parent nodes was sampled directly from the posterior distribution and the local scores computed using the BGe scoring model. Each node was subject to a fan-in restriction of three parent (i.e. effector) nodes. The marginal posterior edge probability, the likelihood of observing each edge (interaction) in the network, was estimated by calculating the average occurrence of each edge in the highest scoring networks that were sampled. The inference procedure was run individually for each pig and the marginal edge probabilities averaged across all runs. The thickness of edges was weighted by this number, and only edges with an averaged marginal edge probability greater than 0.5 were included in the final consensus network for each condition (Fig 1). We note that the program is probabilistic and thus applying this hard cutoff can lead to minor differences in results from different runs.
Figure 1. Process flow for calculating consensus Dynamic Bayesian Networks.

DBN inference is performed on time course data from each individual pig. The estimated probability of occurrence of each edge is averaged over all individual pigs within the Control or PST group, and only edges with a value greater than 0.5 are included in the final consensus network structure.
Principal Component Analysis (PCA)
The goal of this analysis was to identify the subsets of variables (in the form of orthogonal normalized linear combinations of the original variables, called principal components) that are most strongly correlated with a given experimental compartment and procedure (Ascites and plasma, control and PST), and thereby might be considered principal drivers of each response. PCA is a non-parametric statistical method of reducing the dimensionality of a dataset to a few principal components, i.e. those that could potentially account for the most variability in the dataset (32, 33). This method is based on the hypothesis that a variable that changes during a process is important to that process. Recently, in a mouse model of trauma/hemorrhagic shock, we have demonstrated the utility of PCA for suggesting key inflammatory influences based on levels of circulating mediators in blood serum (25). We have also shown that PCA can shed insights into the acute inflammatory response in endotoxemic swine (26, 27) and in septic rats (26).
To perform PCA, the data were first normalized for each variable (i.e. a given value divided by the maximum value for that variable), so that all variable levels were converted into the same scale (from 0 to 1). In this way, any artifactual effects on variance due to the different ranges of concentration observed for different variables were eliminated. Only sufficient components to capture at least 95% of the variance in the data were considered. From these leading principal components, the coefficient (weight) associated with each variable was multiplied by the eigenvalue associated with that principal component. This product represented the contribution of a given variable to the variance accounted for in that principal component. The overall score given to each variable is the sum of its scores in each component. This gives a measure of a variable’s contribution to the overall variance of the system. The variables with the largest scores are the ones who contributed most to the variance of the process being studied. More specifically, the overall PCA score was calculated in the following way: , where i is the index of component and j is the index of variable. Wi,j is the amount that the jth mediator contributes to the ith component. The complete Matlab® code for this analysis can be found as supplementary material to our previously published work (25).
Previously Reported Results
Removal of ascites with PST prevented ARDS based on PF ratio, compliance, and histology (Fig 2). Histology also showed attenuation of MODS based on intestine, lung, liver, and kidney histopathology, whereas Control animals developed MODS (24). Our previously published work also includes beneficial effects of PST on hemodynamics, lung function, and blood chemistry (24). Also, we did observe a reduction in plasma cytokine concentration (TNFa, IL-12, IL-6 and IL-1β) following suction removal of ascites and a reduction in ascites cytokine concentration (IL-8 and IL-8) (24). Other inflammatory mediators remained at the same concentration in the ascites and both groups of animals had positive blood cultures for aerobic and anaerobic bacteria (24).
Figure 2. Lung histopathology shows reduced damage in PST animals.
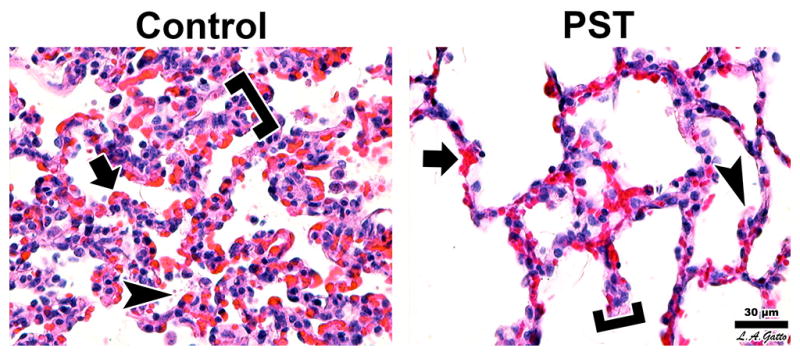
Among the No Suction animals the lesions were more pronounced than in the Suction group. Alveolar walls appeared thicker (brackets) due to folding and collapse, alveolar capillaries were congested (arrow), and the air compartment exhibited edematous fibrin deposits (arrowhead). Suction animals had the same lesions but to a lesser extent (H&E, 40x objective, bar = 30μm).
RESULTS
We have previously suggested that PST attenuates ALI/ARDS based on clinical and physiologic data in our original publication of this study (24). Herein, we sought to gain further insights into this complex response through the use of data-driven computational modeling. Principal Component Analysis revealed that local to the insult (i.e. ascites), primary pro-inflammatory cytokines play a decreased role in the overall response in the treatment group compared to control. In ascites, IL-6, TNF-α, and IL-1β were ranked 2nd, 4th, and 5th, respectively, by PCA for the control group. These mediators decreased to 6th, 7th, and 8th in ascites of animals treated with PST (Fig 3A-B). A similar but less pronounced effect was seen systemically, with these cytokines decreasing from 1st, 3rd, and 4th in plasma readings to 1st, 4th and 7th (Fig 3C-D). Interestingly, the oxygen index (OI = FiO2 * Pm / PaO2) was markedly lower in rank in PST compared to control for both ascites and plasma (Fig 3A-D). OI is the only routine measure that takes into account both oxygenation and lung pressure (34), and has been found to be predictive of acute hypoxic respiratory failure in children (35). Additionally, in our previous work, we have shown that OI correlates with predicted overall “damage”, a simulated index of whole-animal health status in a two-compartment ordinary differential equation model of porcine endotoxemia (27).
Figure 3. Principal Component Analysis suggests decreased importance of primary pro-inflammatory mediators in treatment response.

Ascites = Inflammatory mediator found in the ascites. Plasma = Inflammatory mediator found in the plasma. Note that the PaO2/FiO2 ratio (PF) and oxygen index (OI) are global measures included in both ascites and plasma analyses. Control: The Control Group (n=6) did not have the ascites suctioned from the peritoneal cavity. PST: The Peritoneal Suction Treatment Group (n=6) was treated in an identical fashion except that a vacuum was applied to the peritoneal cavity to remove ascites and maintained until T48 hrs. Variables are ordered by the sum of their contribution to all components, with contributions to individual components represented by different colored sections of the bars. In ascites the rankings of IL-6, TNF-α and IL-1β decrease from control to PST, a similar effect is seen to a lesser extent in plasma. OI decreases markedly in rank from control to PST in both ascites and plasma.
Dynamic Bayesian Network inference suggested that endotoxin drives the inflammatory response in the ascites, interplaying with PF ratio (i.e. lung dysfunction), in an apparent feed-forward loop that exacerbates inflammation characterized by IL-6 and IL-1β (in the Control group) and IL-1β only (in the PST group). The implication of this DBN analysis that IL-6 plays a reduced role in the PST group compared to Control is in agreement with the PCA results discussed above (Fig 4). In the plasma, and in agreement with prior studies (36), DBN inferred the induction of TGF-β1 and CRP by IL-6 in both Control and PST groups. TGF-β1, CRP and IL-6 formed the central nodes of the networks in both Control and PST, with cross-regulation and self-feedback among all three nodes.
Figure 4. Dynamic Bayesian Network analysis suggest conserved peritoneal and systemic inflammatory network in porcine MODS model, but with altered primary outputs.

Ascites = Inflammatory mediator found in the ascites. Plasma = Inflammatory mediator found in the plasma. Panel A: The Control Group (n=6) did not have the ascites suctioned from the peritoneal cavity. Panel B: The Peritoneal Suction Treatment Group (n=6) was treated in an identical fashion except that a vacuum was applied to the peritoneal cavity to remove ascites and maintained until T48 hrs. Multiple inflammatory mediators in ascites and plasma, PaO2/FiO2 ratio (PF), and Oxygen Index (OI) were interrelated using DBN inference. The weight of the arrows denotes strength of interaction. While overall network structure was maintained, Peritoneal Suction led to a notably different output in the Ascites (Loss of IL-6 as an output).
DISCUSSION
Prior studies of trauma and sepsis in both animals and humans have suggested that an appropriately robust inflammatory response is necessary for appropriate resolution of the insult, with dysregulated inflammation being the hallmark of morbidity and mortality (37, 38). The adaptive responsiveness to stress can be observed both at the physiological and inflammatory levels, which reinforces the concept that these processes are interlinked (39). These interconnections, in turn, can drive a multi-compartment, feed-forward cycle of inflammation → organ dysfunction → inflammation, which we hypothesize underlies the phenomenon of MODS. The responses to Gram-negative bacterial infection involves events initially driven by pathogen-associated molecular pattern (PAMP) molecules such as endotoxin, which in turn stimulate the production of chemokines and pro-inflammatory cytokines; this process is kept in check by anti-inflammatory mechanisms (predominantly anti-inflammatory cytokines). Subsequent to pro-inflammatory activation is the production of damage-associated molecular pattern molecules (DAMP’s), which are also released from tissues undergoing physiologic stress such as ischemia/reperfusion injury. In turn, DAMP’s cause the further release of pro-inflammatory chemokines and cytokines, and this overall process in essence represents a multi-compartment, feed forward loop of inflammation → damage/dysfunction → inflammation (22, 40, 41). Given this pathophysiological process, it follows that therapy for MODS may need to target the multi-compartment nature of acute inflammation in settings such as sepsis (22, 23, 42). This hypothesis is supported by our recent findings of the effect of hemoadsorption in a rat model of Gram-negative sepsis that, based on PCA identification of primary mediators present in both groups, hemoadsorption appears to re-compartmentalize inflammation while reducing organ dysfunction and improving clearance of bacteria (26). We have also used Dynamic Network Analysis, a technique based on undirected correlations among significantly altered mediators, in concert with PCA to define dynamic inflammatory interactions in mouse trauma/hemorrhage (25). More recently, we used DBN to define dynamic inflammation networks in human pediatric acute liver failure, an analysis that distinguished spontaneous survivors from non-survivors in this disease setting (28).
We sought to utilize similar methodology to gain insights into how PST ameliorates MODS and, particularly, ALI/ARDS. We have previously suggested that PST attenuates ALI/ARDS based on clinical and physiologic data in our original publication of this study (24). The initial study utilized the clinically applicable porcine model of multiple organ injury, and showed that application of PST 12 hrs following injury significantly reduced lung, kidney, liver, and intestinal pathology and improved pulmonary mechanics. Lung compliance was significantly improved and PST animals had significantly lower mean airway pressures, peak inspiratory pressure (PIP), and plateau pressure (Pplat). Quantitative histologic analysis of all lung fields showed a significant decrease in atelectasis, fibrinous deposits, and leukocyte infiltration in animals treated with PST vs. control. Also, PST significantly decreased IL-6 and IL-8 levels in ascites fluid and plasma levels of TNF-α, IL-12, IL-6, and IL-1β were significantly reduced in PST animals (24). It is evident that PST removed toxic ascites; however, only the concentrations of IL-8 and IL-6 were lowered significantly. All other inflammatory mediators remained at the same concentrations between groups; there was no difference in endotoxin concentration, and all animals had positive cultures for both aerobic and anaerobic bacteria (24). Thus, simple removal of bacteria does not seem to be the mechanism underlying the protective effect of PST.
These data were then used in our current study for in silico analyses, which in turn suggest that principal inflammatory drivers, and their interconnections, 1) differ between local vs. systemic compartments in the control setting (sepsis/ALI/ARDS), and 2) are altered by PST. Equally importantly, we infer from our analyses that inflammation and lung function affect each other iteratively, in essence forming a feed-forward loop, associated with the development of ALI/ARDS. To our knowledge, this is the first study to demonstrate this feed-forward loop explicitly in the context of dynamic networks.
Our studies suggest that, in the context of our animal model, sepsis/ALI/ARDS are propagated locally (in the peritoneum, as assessed in the ascites fluid) in a feed-forward manner involving bacterial endotoxin and lung dysfunction in both control and PST animals. In both control and PST, this core motif leads to the production of chemokines (IL-8) and cytokines (IL-1β). This process impacts downstream physiological process that, ultimately, affects the OI (again, in both control and PST animals). A crucial difference between the two experimental groups, however, is IL-6: this key inflammation biomarker is an outcome of the core inflammation/organ dysfunction process in control animals, but is absent in PST animals. Given that IL-6 is the biomarker that typically distinguishes adverse outcomes in numerous inflammatory conditions including sepsis/ALI/ARDS (36, 43, 44), we take this result to suggest that PST modifies inflammation in a favorable fashion so as to reduce the production of IL-6. This hypothesis is supported also by PCA, in which IL-6 appears to be a less-important driver of inflammation in PST vs. control animals. Future studies will be aimed at modulating IL-6 experimentally in our animal model, though genetic and/or pharmacologic manipulations are not as straightforward in the pig model as in mice or rats.
It is generally assumed that the failure of local inflammation to contain an infection is a key feature of the progression to sepsis (44–46). At the systemic level, our in silico analyses suggest that both control and PST animals exhibit a feed-forward process driven in some way by IL-6. We infer this process because the core motif inferred in the plasma of control animals involves IL-6 driving the cytokine TGF-β1 along with CRP, and both TGF-β1 and CRP are known to be stimulated by IL-6 (36). The key difference between control and PST animals with regard to systemic inflammation is that, in control animals, IL-10 is inferred to be produced as a consequence of this core IL-6/TGF-β1/CRP motif; this key anti-inflammatory cytokine is absent in the PST animals, perhaps suggesting a more robust ability to control infection.
There are several limitations to our study. First, it is also possible that differential intra-vs. extra-corporeal environment conditions may account for some of the changes we infer, but that while this can’t be ruled out, we believe otherwise. Also, a relatively small number of animals and mediators were studied. These limitations are inherent in large-animal studies, especially in swine where the number of mediators that can be assessed is smaller than that of mice or humans. However, this limitation is mitigated by the clinical realism of the animal model, and hence the likelihood of translation to the human setting of the key results obtained herein. Another key limitation is that the data-driven modeling techniques used herein are, at best, quasi-mechanistic and should be interpreted with care. Future studies in which specific mediators are modulated in order to test predictions of these in silico analyses, combined with studies involving explicitly mechanistic computational models, are needed in order to validate the hypotheses raised by the current study.
CONCLUSION
In conclusion, we suggest that combined in vivo and in silico studies imply that dynamic modification of networks controlling inflammatory and physiologic (dys)function during the process of ascites removal is a possible mechanism of prevention of ALI/ARDS. These studies further highlight the utility of data-driven computational tools in the study of complex processes such as sepsis.
Figure 5. Summary of complex compartment and treatment-specific inflammatory networks.

Local inflammation in the ascites, arising from the introduction of Endotoxin, causes a rise in both pro- and anti-inflammatory mediators, leading to lung dysfunction. Once established, lung dysfunction perpetuates through a feedback loop with pro-inflammatory mediators. Translocation of pro-inflammatory mediators from ascites to plasma leads to widespread systemic inflammation. Differences between PST and Control are noted.
Acknowledgments
Funding: NIH R33-HL-089082, NIH P50-GM-53789, and KCI Inc.
ABBREVIATIONS
- ACS
Abdominal Compartment Syndrome
- BL
Baseline
- CHUA
Committee for the Humane Use of Animals
- CO
Cardiac Output
- DBN
Dynamic Bayesian Network
- IAH
Intra-abdominal Hypertension
- IAP
Intra-abdominal Pressure
- MODS
Multiple Organ Dysfunction Syndrome
- OI
Oxygen Index
- Pm
Mean Airway Pressure
- PAP
Pulmonary Artery Pressure
- PAW
Pulmonary Capillary Wedge Pressure
- PCA
Principle Component Analysis
- PD
Passive Drain
- PEEP
Positive End Expiratory Pressure
- PF
PaO2/FiO2 ratio
- PST
Peritoneal Suction Treatment
- RR
Respiratory Rate
- SIRS
systemic inflammatory response syndrome
- SMA
Superior Mesenteric Artery
- UOP
Urine Output
- Vt
Tidal Volume
References
- 1.Ware LB. Pathophysiology of acute lung injury and the acute respiratory distress syndrome. Seminars in respiratory and critical care medicine. 2006;27:337–349. doi: 10.1055/s-2006-948288. [DOI] [PubMed] [Google Scholar]
- 2.Jean-Baptiste E. Cellular mechanisms in sepsis. Journal of intensive care medicine. 2007;22:63–72. doi: 10.1177/0885066606297123. [DOI] [PubMed] [Google Scholar]
- 3.Regel G, Grotz M, Weltner T, Sturm JA, Tscherne H. Pattern of organ failure following severe trauma. World journal of surgery. 1996;20:422–429. doi: 10.1007/s002689900067. [DOI] [PubMed] [Google Scholar]
- 4.Ware LB, Matthay MA. The acute respiratory distress syndrome. The New England journal of medicine. 2000;342:1334–1349. doi: 10.1056/NEJM200005043421806. [DOI] [PubMed] [Google Scholar]
- 5.Rubenfeld GD, Herridge MS. Epidemiology and outcomes of acute lung injury. Chest. 2007;131:554–562. doi: 10.1378/chest.06-1976. [DOI] [PubMed] [Google Scholar]
- 6.Brower RG, Fessler HE. Another "negative" trial of surfactant. Time to bury this idea? American journal of respiratory and critical care medicine. 2011;183:966–968. doi: 10.1164/rccm.201101-0018ED. [DOI] [PubMed] [Google Scholar]
- 7.Villar J, Blanco J, Anon JM, Santos-Bouza A, Blanch L, Ambros A, Gandia F, Carriedo D, Mosteiro F, Basaldua S, Fernandez RL, Kacmarek RM, Network A. The ALIEN study: incidence and outcome of acute respiratory distress syndrome in the era of lung protective ventilation. Intensive care medicine. 2011;37:1932–1941. doi: 10.1007/s00134-011-2380-4. [DOI] [PubMed] [Google Scholar]
- 8.Cheung AM, Tansey CM, Tomlinson G, Diaz-Granados N, Matte A, Barr A, Mehta S, Mazer CD, Guest CB, Stewart TE, Al-Saidi F, Cooper AB, Cook D, Slutsky AS, Herridge MS. Two-year outcomes, health care use, and costs of survivors of acute respiratory distress syndrome. American journal of respiratory and critical care medicine. 2006;174:538–544. doi: 10.1164/rccm.200505-693OC. [DOI] [PubMed] [Google Scholar]
- 9.Herridge MS, Tansey CM, Matte A, Tomlinson G, Diaz-Granados N, Cooper A, Guest CB, Mazer CD, Mehta S, Stewart TE, Kudlow P, Cook D, Slutsky AS, Cheung AM Canadian Critical Care Trials G. Functional disability 5 years after acute respiratory distress syndrome. The New England journal of medicine. 2011;364:1293–1304. doi: 10.1056/NEJMoa1011802. [DOI] [PubMed] [Google Scholar]
- 10.McIntyre RC, Jr, Pulido EJ, Bensard DD, Shames BD, Abraham E. Thirty years of clinical trials in acute respiratory distress syndrome. Critical care medicine. 2000;28:3314–3331. doi: 10.1097/00003246-200009000-00034. [DOI] [PubMed] [Google Scholar]
- 11.Ince C. The microcirculation is the motor of sepsis. Critical care. 2005;9 (Suppl 4):S13–19. doi: 10.1186/cc3753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van Haren FM, Sleigh JW, Pickkers P, Van der Hoeven JG. Gastrointestinal perfusion in septic shock. Anaesthesia and intensive care. 2007;35:679–694. doi: 10.1177/0310057X0703500505. [DOI] [PubMed] [Google Scholar]
- 13.Matheson PJ, Wilson MA, Garrison RN. Regulation of intestinal blood flow. The Journal of surgical research. 2000;93:182–196. doi: 10.1006/jsre.2000.5862. [DOI] [PubMed] [Google Scholar]
- 14.Zakaria el R, Li N, Garrison RN. Mechanisms of direct peritoneal resuscitation-mediated splanchnic hyperperfusion following hemorrhagic shock. Shock. 2007;27:436–442. doi: 10.1097/01.shk.0000245017.86117.4e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Farand P, Hamel M, Lauzier F, Plante GE, Lesur O. Review article: organ perfusion/permeability-related effects of norepinephrine and vasopressin in sepsis. Canadian journal of anaesthesia = Journal canadien d'anesthesie. 2006;53:934–946. doi: 10.1007/BF03022837. [DOI] [PubMed] [Google Scholar]
- 16.Fink MP, Delude RL. Epithelial barrier dysfunction: a unifying theme to explain the pathogenesis of multiple organ dysfunction at the cellular level. Critical care clinics. 2005;21:177–196. doi: 10.1016/j.ccc.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 17.Mayberry JC, Welker KJ, Goldman RK, Mullins RJ. Mechanism of acute ascites formation after trauma resuscitation. Archives of surgery. 2003;138:773–776. doi: 10.1001/archsurg.138.7.773. [DOI] [PubMed] [Google Scholar]
- 18.Marshall JC. Inflammation, coagulopathy, and the pathogenesis of multiple organ dysfunction syndrome. Critical care medicine. 2001;29:S99–106. doi: 10.1097/00003246-200107001-00032. [DOI] [PubMed] [Google Scholar]
- 19.Johnson D, Mayers I. Multiple organ dysfunction syndrome: a narrative review. Canadian journal of anaesthesia = Journal canadien d'anesthesie. 2001;48:502–509. doi: 10.1007/BF03028318. [DOI] [PubMed] [Google Scholar]
- 20.Suliburk J, Helmer K, Moore F, Mercer D. The gut in systemic inflammatory response syndrome and sepsis. Enzyme systems fighting multiple organ failure. European surgical research Europaische chirurgische Forschung Recherches chirurgicales europeennes. 2008;40:184–189. doi: 10.1159/000110859. [DOI] [PubMed] [Google Scholar]
- 21.Deitch EA. Role of the gut lymphatic system in multiple organ failure. Current opinion in critical care. 2001;7:92–98. doi: 10.1097/00075198-200104000-00007. [DOI] [PubMed] [Google Scholar]
- 22.An G, Nieman G, Vodovotz Y. Computational and systems biology in trauma and sepsis: current state and future perspectives. International journal of burns and trauma. 2012;2:1–10. [PMC free article] [PubMed] [Google Scholar]
- 23.An G, Nieman G, Vodovotz Y. Toward computational identification of multiscale "tipping points" in acute inflammation and multiple organ failure. Annals of biomedical engineering. 2012;40:2414–2424. doi: 10.1007/s10439-012-0565-9. [DOI] [PubMed] [Google Scholar]
- 24.Kubiak BD, Albert SP, Gatto LA, Snyder KP, Maier KG, Vieau CJ, Roy S, Nieman GF. Peritoneal negative pressure therapy prevents multiple organ injury in a chronic porcine sepsis and ischemia/reperfusion model. Shock. 2010;34:525–534. doi: 10.1097/SHK.0b013e3181e14cd2. [DOI] [PubMed] [Google Scholar]
- 25.Mi Q, Constantine G, Ziraldo C, Solovyev A, Torres A, Namas R, Bentley T, Billiar TR, Zamora R, Puyana JC, Vodovotz Y. A dynamic view of trauma/hemorrhage-induced inflammation in mice: principal drivers and networks. PloS one. 2011;6:e19424. doi: 10.1371/journal.pone.0019424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Namas RA, Namas R, Lagoa C, Barclay D, Mi Q, Zamora R, Peng Z, Wen X, Fedorchak MV, Valenti IE, Federspiel WJ, Kellum JA, Vodovotz Y. Hemoadsorption reprograms inflammation in experimental gram-negative septic peritonitis: insights from in vivo and in silico studies. Molecular medicine. 2012;18:1366–1374. doi: 10.2119/molmed.2012.00106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nieman G, Brown D, Sarkar J, Kubiak B, Ziraldo C, Dutta-Moscato J, Vieau C, Barclay D, Gatto L, Maier K, Constantine G, Billiar TR, Zamora R, Mi Q, Chang S, Vodovotz Y. A two-compartment mathematical model of endotoxin-induced inflammatory and physiologic alterations in swine. Critical care medicine. 2012;40:1052–1063. doi: 10.1097/CCM.0b013e31823e986a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Azhar N, Ziraldo C, Barclay D, Rudnick DA, Squires RH, Vodovotz Y Pediatric Acute Liver Failure Study G. Analysis of serum inflammatory mediators identifies unique dynamic networks associated with death and spontaneous survival in pediatric acute liver failure. PloS one. 2013;8:e78202. doi: 10.1371/journal.pone.0078202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grzegorczyk M, Husmeier D. Improvements in the reconstruction of time-varying gene regulatory networks: dynamic programming and regularization by information sharing among genes. Bioinformatics. 2011;27:693–699. doi: 10.1093/bioinformatics/btq711. [DOI] [PubMed] [Google Scholar]
- 30.Azhar N, Ziraldo C, Barclay D, Rudnicka D, Squires R, Vodovotz Y. Analysis of serum inflammatory mediators identifies unique dynamic networks associated with death and spontaneous survival in pediatric acute liver failure. PloS one. 2013 doi: 10.1371/journal.pone.0078202. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zaaqoq AM, Namas R, Almahmoud K, Krishnan S, Azhar N, Mi Q, Zamora R, Brienza DM, Billiar TR, Vodovotz Y. IP-10, a potential driver of neurally-controlled IL-10 and morbidity in human blunt trauma. Critical care medicine. 2014 doi: 10.1097/CCM.0000000000000248. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Janes KA, Yaffe MB. Data-driven modelling of signal-transduction networks. Nature reviews Molecular cell biology. 2006;7:820–828. doi: 10.1038/nrm2041. [DOI] [PubMed] [Google Scholar]
- 33.Vodovotz Y, Billiar TR. In silico modeling: methods and applications to trauma and sepsis. Critical care medicine. 2013;41:2008–2014. doi: 10.1097/CCM.0b013e31829a6eb4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Funk DJ, Lujan E, Moretti EW, Davies J, Young CC, Patel MB, Vaslef SN. A brief report: the use of high-frequency oscillatory ventilation for severe pulmonary contusion. The Journal of trauma. 2008;65:390–395. doi: 10.1097/TA.0b013e31817f283f. [DOI] [PubMed] [Google Scholar]
- 35.Trachsel D, McCrindle BW, Nakagawa S, Bohn D. Oxygenation index predicts outcome in children with acute hypoxemic respiratory failure. American journal of respiratory and critical care medicine. 2005;172:206–211. doi: 10.1164/rccm.200405-625OC. [DOI] [PubMed] [Google Scholar]
- 36.Jawa RS, Anillo S, Huntoon K, Baumann H, Kulaylat M. Analytic review: Interleukin-6 in surgery, trauma, and critical care: part I: basic science. Journal of intensive care medicine. 2011;26:3–12. doi: 10.1177/0885066610395678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pinsky MR. Sepsis: a pro- and anti-inflammatory disequilibrium syndrome. Contributions to nephrology. 2001:354–366. doi: 10.1159/000060100. [DOI] [PubMed] [Google Scholar]
- 38.Namas R, Ghuma A, Torres A, Polanco P, Gomez H, Barclay D, Gordon L, Zenker S, Kim HK, Hermus L, Zamora R, Rosengart MR, Clermont G, Peitzman A, Billiar TR, Ochoa J, Pinsky MR, Puyana JC, Vodovotz Y. An adequately robust early TNF-alpha response is a hallmark of survival following trauma/hemorrhage. PloS one. 2009;4:e8406. doi: 10.1371/journal.pone.0008406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gomez H, Mesquida J, Hermus L, Polanco P, Kim HK, Zenker S, Torres A, Namas R, Vodovotz Y, Clermont G, Puyana JC, Pinsky MR. Physiologic responses to severe hemorrhagic shock and the genesis of cardiovascular collapse: can irreversibility be anticipated? The Journal of surgical research. 2012;178:358–369. doi: 10.1016/j.jss.2011.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vodovotz Y, Csete M, Bartels J, Chang S, An G. Translational systems biology of inflammation. PLoS computational biology. 2008;4:e1000014. doi: 10.1371/journal.pcbi.1000014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vodovotz Y, Constantine G, Rubin J, Csete M, Voit EO, An G. Mechanistic simulations of inflammation: current state and future prospects. Mathematical biosciences. 2009;217:1–10. doi: 10.1016/j.mbs.2008.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dick TE, Molkov YI, Nieman G, Hsieh YH, Jacono FJ, Doyle J, Scheff JD, Calvano SE, Androulakis IP, An G, Vodovotz Y. Linking Inflammation, Cardiorespiratory Variability, and Neural Control in Acute Inflammation via Computational Modeling. Frontiers in physiology. 2012;3:222. doi: 10.3389/fphys.2012.00222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ivady B, Beres BJ, Szabo D. Recent advances in sepsis research: novel biomarkers and therapeutic targets. Current medicinal chemistry. 2011;18:3211–3225. doi: 10.2174/092986711796391598. [DOI] [PubMed] [Google Scholar]
- 44.Dellinger RP, Carlet JM, Masur H, Gerlach H, Calandra T, Cohen J, Gea-Banacloche J, Keh D, Marshall JC, Parker MM, Ramsay G, Zimmerman JL, Vincent JL, Levy MM Surviving Sepsis Campaign Management Guidelines C. Surviving Sepsis Campaign guidelines for management of severe sepsis and septic shock. Critical care medicine. 2004;32:858–873. doi: 10.1097/01.ccm.0000117317.18092.e4. [DOI] [PubMed] [Google Scholar]
- 45.Marshall JC, Maier RV, Jimenez M, Dellinger EP. Source control in the management of severe sepsis and septic shock: an evidence-based review. Critical care medicine. 2004;32:S513–526. doi: 10.1097/01.ccm.0000143119.41916.5d. [DOI] [PubMed] [Google Scholar]
- 46.Schein M, Marshall J. Source control for surgical infections. World journal of surgery. 2004;28:638–645. doi: 10.1007/s00268-004-7505-2. [DOI] [PubMed] [Google Scholar]