

Abstract
Pyrvinium pamoate (PP) is a potent non-competitive inhibitor of the androgen receptor (AR). Using a novel method of target identification, we demonstrate that AR is a direct target of PP in prostate cancer cells. We demonstrate that PP inhibits AR activity via the highly conserved DNA binding domain (DBD), the only AR inhibitor that functions via this domain. Furthermore, computational modeling predicts that pyrvinium binds at the interface of the DBD dimer and the minor groove of the AR response element. Because PP acts through the DBD, PP is able to inhibit the constitutive activity of AR splice variants, which are thought to contribute to the growth of castration resistant prostate cancer (CRPC). PP also inhibits androgen-independent AR activation by HER2 kinase. The anti-androgen activity of pyrvinium manifests in the ability to inhibit the in vivo growth of CRPC xenografts that express AR splice variants. Interestingly, PP was most potent in cells with endogenous AR expression derived from prostate or bone. PP was able to inhibit several other hormone nuclear receptors (NRs), but not structurally unrelated transcription factors. PP inhibition of other NRs was similarly cell-type selective. Using dual-energy X-ray absorptiometry, we demonstrate that the cell-type specificity of PP manifests in tissue-selective inhibition of AR activity in mice, as PP decreases prostate weight and bone mineral density, but does not affect lean body mass. Our results suggest that the non-competitive AR inhibitor pyrvinium has significant potential to treat CRPC, including cancers driven by ligand-independent AR signaling.
Introduction
Despite incredible advances in detection and treatment, prostate cancer remains the second leading cause of cancer death in American men (1). Circulating androgens are essential for normal prostate development as well as the development of prostate cancer through their interactions with the androgen receptor (AR). AR plays a critical role in the development of both primary and advanced prostate cancer, including castration resistant prostate cancer (CRPC), making it the best molecular target for all prostate cancers. In the setting of metastatic cancer, removal of testicular androgens by surgical or chemical castration leads to regression of prostate tumors; however, these cancers universally recur despite very low levels of systemic androgens. Most of these CRPCs remain dependent on AR function (2, 3), and until recently, they have been essentially untreatable. Several molecular mechanisms have been described to account for continued AR signaling in CRPC, including the amplification of AR (2), gain-of-function mutations in AR that confer greater sensitivity to androgens or increased recruitment of AR coactivator proteins (4), LBD-independent N-terminal activation by growth factors, neuropeptides, and inflammatory mediators (5–7), expression of constitutively active AR splice variants (8–11), tumoral conversion of adrenal androgens (12), and intratumoral androgen production (13). These basic discoveries have led to a renewed interest in AR as a target in CRPC and have led to the development of several new drugs that block the AR/androgen signaling axis, including the CYP17 inhibitor abiraterone acetate (14) and the potent competitive antagonist, enzalutamide (15) which have both been approved for use in CRPC by the FDA, as well as other similar agents in clinical trials (16, 17). Because these agents have been so successful, it strongly suggests that the goal of metastatic prostate cancer treatment, until such time as we can prevent or cure it, is to completely inhibit AR activity. However, it appears that recurrence is still a major problem in enzalutamide- and abiraterone-treated patients, and that the acquisition of resistance likely occurs via renewed AR signaling, suggesting the emergence of truly ligand-independent, AR-dependent cancers. Renewed AR signaling, likely via expression of AR splice variants, has recently been shown to be an important mechanism of resistance to abiraterone and enzalutamide in models of CRPC (18, 19). Thus, inhibiting AR via domains that can control activity even in the absence of ligand may be a viable approach to inhibiting the growth of ligand-independent, AR-driven prostate cancers.
We recently reported the discovery of the first non-competitive, ligand-independent AR inhibitor, pyrvinium pamoate (PP) (20). This drug was discovered from a screen for inhibitors of AR conformation change (21). PP inhibits endogenous AR activity with 12nM potency and functions synergistically with competitive antagonists. It does not compete for ligand binding nor does it inhibit AR nuclear accumulation, but it does prevent the recruitment of RNA polymerase II to transcription start sites. PP is tolerated by mice and has potent anti-androgen activity in the mouse prostate. Furthermore, PP inhibits the growth of multiple cultured prostate cancer lines (20). In this work, we significantly advance our mechanistic understanding of PP function, with discoveries that suggest PP will inhibit AR activity in CRPC even when other AR targeted therapies fail.
Results
The androgen receptor is a molecular target of pyrvinium
Pyrvinium, the active component of pyrvinium pamoate (PP), is a potent non-competitive inhibitor of the androgen receptor (AR) (20). Because PP did not disrupt DHT binding and presumably did not interact with the ligand binding pocket, we hypothesized that PP directly interacts with a different region of AR. Therefore, we employed the drug affinity responsive target stability (DARTS) assay to examine whether pyrvinium can directly interact with AR. The DARTS assay is based on the principle that a molecular interaction between a molecule and its target protein(s) confers resistance to proteolysis (22) (Fig. 1A). To validate the assay, LNCaP cells were incubated with increasing concentrations of dihydrotestosterone (DHT) for four hours, at which point, protein was extracted and digested with protease. The digested protein was then resolved with SDS-PAGE and prepared for Western blot (WB) analysis of AR (Fig. 1B). While AR was relatively resistant to low concentrations of protease, binding of DHT to AR reduced the protease susceptibility of the protein even further. This result suggested that DARTS could be used to determine if AR was a target of pyrvinium. LAPC4 prostate cancer cells, which express wild type AR, were incubated with increasing concentrations of the drug then processed as above for WB analysis. AR was protected from proteolysis in cells treated with PP, while GAPDH (Fig. 1C) was not. Casein kinase 1 alpha (CK1α), a potential PP target proposed by Thorne et al. (23) was not protected from proteolysis (Fig. S1). A similar effect was observed in LNCaP prostate cancer cells, which express a mutant AR (Fig. 1D). We also observed that AR was protected from proteolysis when PP was incubated with LNCaP cell extracts following cell lysis (Fig. 1E). Interestingly, we found that PP not only protects AR from proteolysis, but it also decreases the rate of degradation of AR in cyclohexamide treated cells (Fig 1F), similar to DHT (24). This data strongly suggests that AR is a direct binding target of PP in LNCaP and LAPC4 prostate cancer cells.
Fig 1. Pyrvinium protects AR from proteolysis.
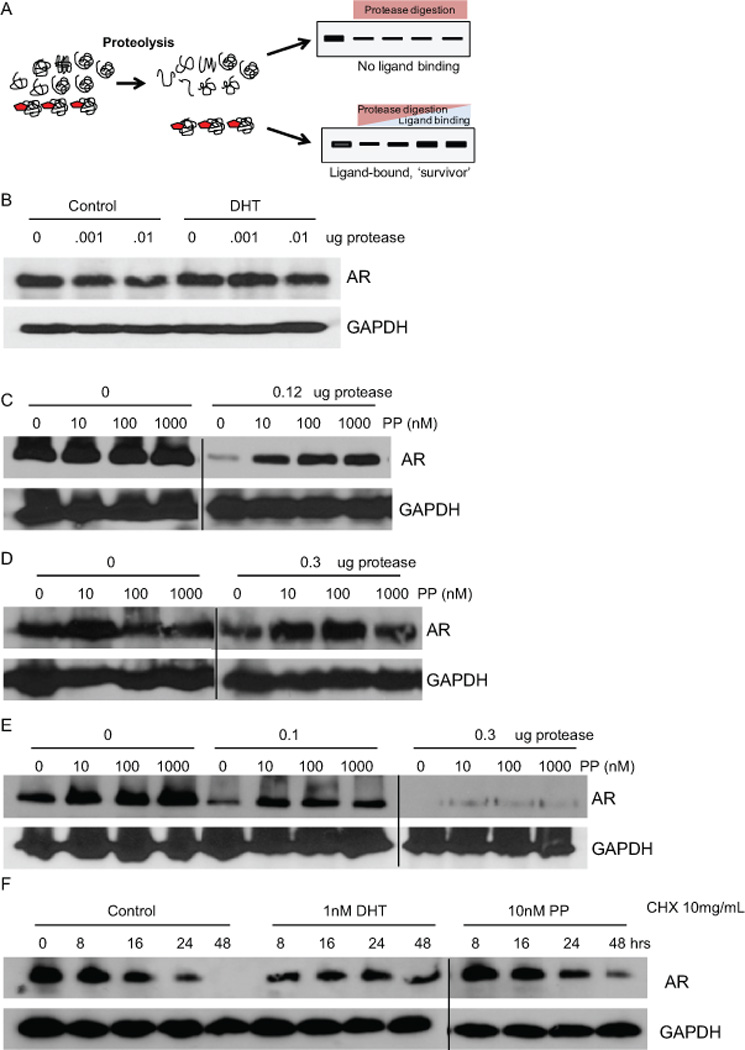
(A) Illustration of DARTS-WB assay where increasing ligand binding enhances the stability of its target protein in the presence of proteases. (B) LNCaP cells were treated with 0.3nM DHT and lysates subjected to digestion with the indicated amount of protease. Digested lysates were resolved by SDS-PAGE and immunoblots were performed to detect AR and GAPDH. DHT, which is known to bind AR, modestly protected AR from proteolysis while GAPDH remained unaffected. LAPC4 (C) or LNCaP (D) cells were treated with 10nM to 1µM of PP for 3 hrs and cell lysates were processed for DARTS-WB as in (B). Similar to DHT, PP protected AR, but not GAPDH, from proteolysis. (E) PP was added to LNCaP lysates for 1hr prior to treatment with protease. DARTS-WB was performed as before and demonstrates that PP can protect AR from proteolysis in lysates as well as intact cells. (F) LNCaP cells were treated with cyclohexamide and vehicle, DHT, or PP for the indicated times and lysates were prepared for WB. PP and DHT decrease the rate of AR degradation.
Pyrvinium acts via the DNA binding domain/hinge region
As it is proposed that PP directly targets AR, we examined what structural and functional domains of the protein participate in the molecular interaction. To define the region, we tested the ability of PP to inhibit the activity of AR truncation mutants (25). We hypothesized that either the N-terminal domain (NTD) or the DNA binding domain (DBD)/hinge region of AR interacted with PP, not the ligand binding domain (LBD), as PP was originally discovered as a non-competitive inhibitor (20). We attempted to examine the activity of AR mutants in prostate cancer cells that do not express endogenous AR; however, we found that PP had much greater potency in prostate cancer cells that endogenously express AR, thus limiting which constructs we were able to test. Thus, we examined PP inhibition of a constitutively active AR mutant that lacks LBD (N644) in LNCaP cells. As expected, PP was able to inhibit its activity (Fig. 2A). When we tested PP’s ability to inhibit an AR in which the DBD was replaced with that of the LexA protein (NLxHC), the domain swapping resulted in loss of drug activity (Fig. 2A). The lack of inhibition was not due to wild type AR activity acting on the LexA luciferase reporter or the lack of nuclear accumulation of the NLxHC construct in the presence of ligand (Fig. S2).
Fig 2. PP acts through the DBD/Hinge region of NRs.
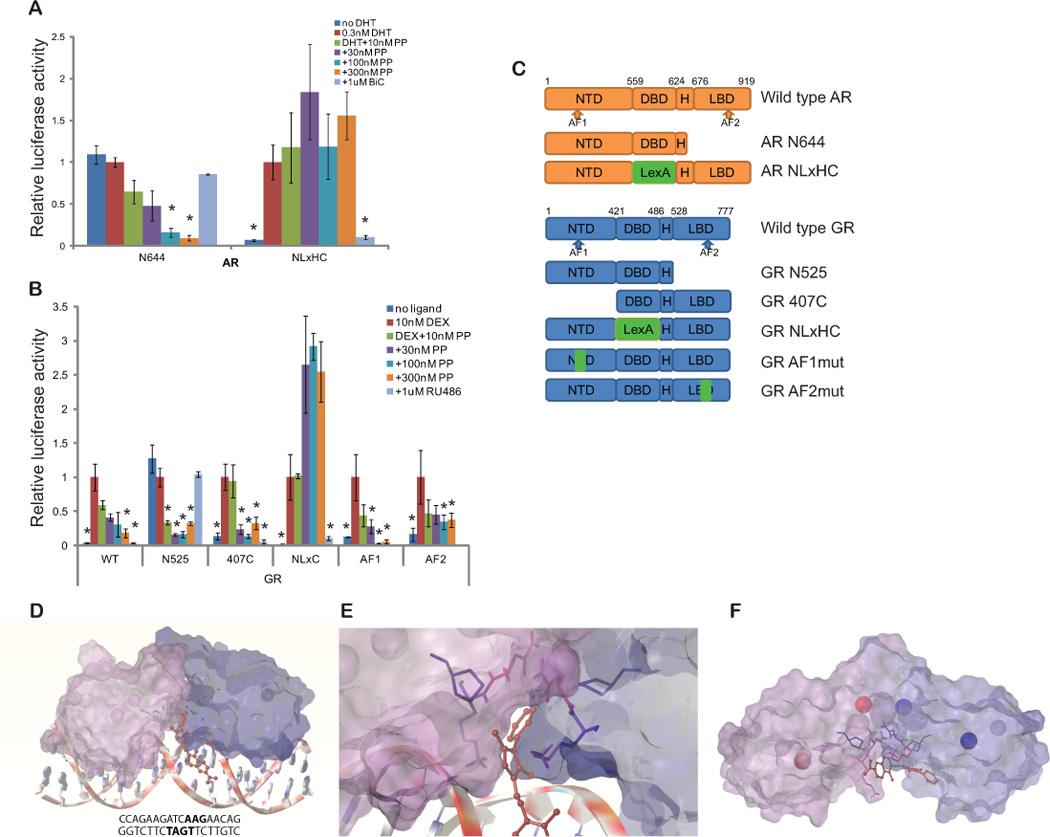
(A) LNCaP cells were transfected with a control plasmid or plasmids encoding the constitutively active AR N644 or AR NLxHC and the appropriate luciferase reporters (PSA-luc or LexA-luc). Cells were treated with the indicated drugs for 24h and luciferase activity was measured, normalizing firefly to renilla luciferase signal. PP inhibits AR N644, which lacks an LBD, while the competitive antagonist bicalutamide (BiC) cannot. However, PP cannot inhibit AR NLxHC, a construct in which the AR DBD is replaced with that of LexA, while BiC can. (B) GR mutants were transfected into LAPC4 cells along the glucocorticoid responsive reporter GilZ-luc and a control renilla reporter. Cells were incubated for 24 hours in 10nM dexamethasone (Dex) along with 30–300nM PP, or the competitive antagonist of GR, RU-486. Both PP and RU-486 inhibited full length GR (WT GR). PP, but not RU-486, inhibited the constitutively active GR N525, which lacks an LBD. PP also inhibited the activity of GR 407C, which lacks an NTD. However, PP was unable to inhibit a construct in which the GR DBD was replaced by the LexA DBD. RU-486 inhibited this construct, as expected. (* p<0.05 compared to DHT (A) or Dex (B) alone by ANOVA. Bars represent the standard error of quadruplicate measurements). (C) Structures of AR and GRα constructs are shown: AF: activator function, DBD: DNA binding domain, H: hinge, and LBD: ligand binding domain. (D) Overview and (E) magnified images are shown of the interaction model with the highest docking score. Potential binding sites were predicted by Surflex-Dock and docking to the site with the best score was simulated by Sybylx1.3. The DNA molecule (ADR3) is depicted as ribbons and coins; pyrvinium is depicted as red ball and stick; AR DBDs are depicted by molecular surface (purple and blue) with Zn ions in Zn finger motifs (red and blue balls); and amino acid residues in the drug interaction in AR DBD were demonstrated with sticks. The phenylpyrrol ring participated in hydrophobic interactions with Lys-609 (pink), Asn-610 (purple), and Pro-612 (blue) of the AR DBD, but no hydrogen bonding was predicted. The quinoline ring of pyrvinium is predicted to interact with residues in the DNA minor groove. (F) Pyrvinium docking with AR was simulated without DNA. The model with the best docking score also predicted an interaction at the interface of the AR DBDs.
Because we could not test the role of the AR NTD in prostatic cell lines that have functional AR signaling, we tested whether PP inhibited the gluticorticoid receptor (GR), which is highly similar to AR in structure (<15, 77, and 50% homologous to NTD, DBD/hinge, and LBD domain, respectively) (26). When GR and the androgen-insensitive GilZ luciferase reporter were expressed in LAPC4 prostate cancer cells, PP suppressed GR activity with potency similar to AR (Fig. 2B). We then tested the ability of PP to inhibit several GR mutants: 407C, which lacks the NTD, N525, which lacks the C-terminal LBD and is the structural equivalent of AR N664, and NLxC, a construct where the DBD was replaced with that of the LexA protein, the structural equivalent of AR NLxHC (27). Deletion of LBD or NTD did not affect drug activity, while the DBD/hinge region was critical for drug inhibition, similar to AR mutants. PP also inhibited the activity of GRs with point mutations in activator function 1 and 2 domains (Fig. 2B) (28, 29).
Computational modeling predicts pyrvinium interaction with the AR DBD
We performed docking and modeling with the aid of unbiased computational analysis. A structure of the AR DBD dimer co-crystallized with the androgen response DNA element ADR3 was downloaded from the Protein Data Bank (PDB ID: 1R4I, (30)) for the docking study. Pyrvinium docking to this structure was accomplished using Surfelx-Dock protomol. By applying soft grid on the dimer interface and comparing the docking score of pyrvinium and inactive pyrvinium derivatives, the small molecule interaction with the DBD and with the DNA minor groove of ADR3 was predicted (Fig. 2D). The phenylpyrrol ring of pyrvinium was located at the DBD dimer interface and interacted with the side chains and backbones of residues Lys-609 and Asn-610, and the side chain of Pro-612, residues located in the second zinc finger module of the DBD (Fig. 2E). No hydrogen bonding was predicted and the docking was driven by hydrophobic interactions, but the docking score of pyrvinium to this site on the DBD was quite reasonable, 6.69 (−log10(Kd)). When docking was simulated without the DNA molecule, pyrvinium binding was still predicted at the interface of DBD dimer with score of 5.48 (Fig. 2F). Residues Phe-582, Ala-586, Tyr-593, Lys-609, Asn-610, Pro-612, and Arg-615 participated in the hydrophobic interaction with pyrvinium.
Pyrvinium inhibits hormone nuclear receptors, not other transcription factors, and only in prostate cells
Having demonstrated GR inhibition by PP, we next examined whether it could inhibit other hormone nuclear receptors (NR) and other transcription factors. PP inhibited the activity of GR, estrogen receptor (ERα), peroxisome proliferator-activated receptor (PPARγ), and thyroid receptor (TR) when transfected into LAPC4 cells with their corresponding luciferase reporters and stimulated with the appropriate ligand (Fig. 3A). However, PP did not inhibit the activity of other transcription factors including TCF/beta-catenin, NF-kappa B, and cAMP response element binding (CREB) protein when LAPC4 cells were transfected with the corresponding luciferase reporters and challenged with the appropriate stimuli. Furthermore, PP did not inhibit endogenous GR activity in A549 lung cancer cells or endogenous ER activity in MCF7 breast cancer cells (Fig. 3B). PP did, however, inhibit the low-level endogenous GR activity in LAPC4 cells (Supplementary Fig. S3). These results suggest that PP can inhibit many NRs, likely because of the high similarity among DBDs of these proteins, but that it can only do so in prostate cells.
Fig 3. Effects of PP on other transcription factors.
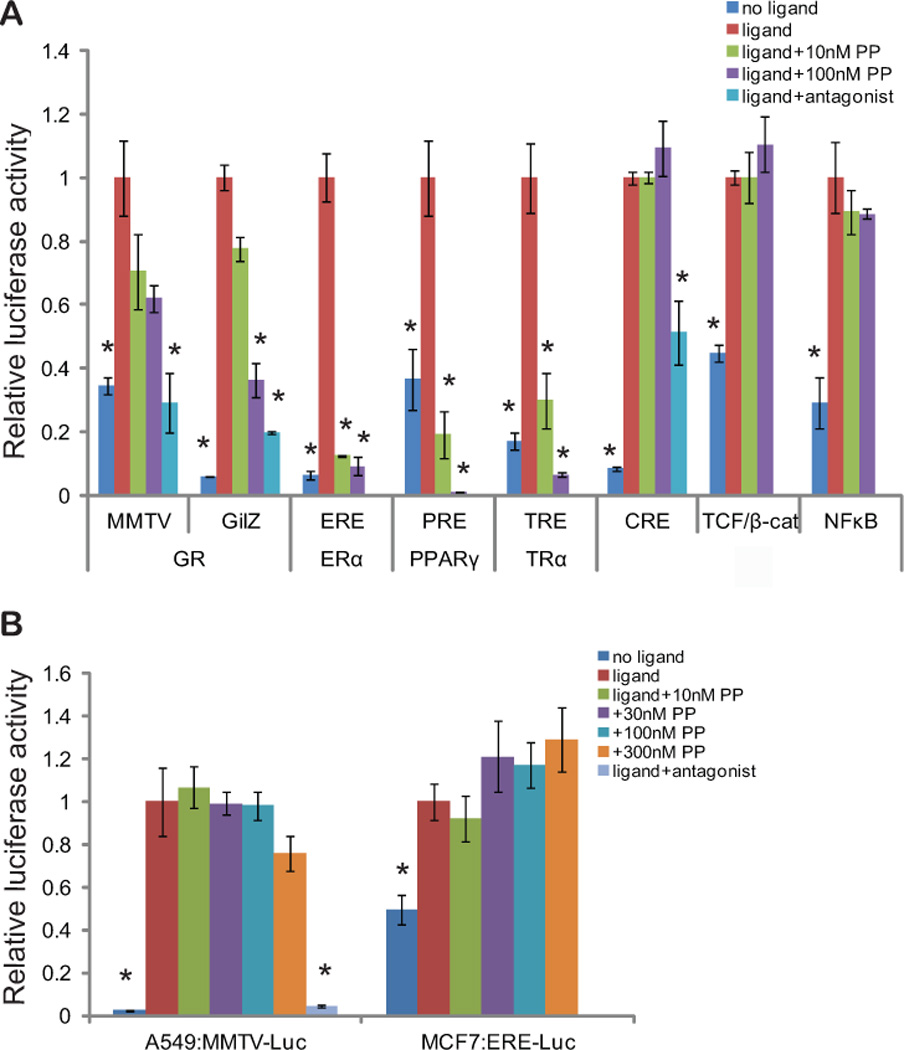
(A) LAPC4 cells were transfected with the indicated transcription factors and/or transcription factor response element driven luciferase plasmids, along with a renilla control plasmid. Two different reporters (MMTV-luc and GilZ-luc) were used to assess PP activity against GR. The following day, quadruplicate wells were treated with the appropriate ligand or stimulus: 10nM dexamethasone (GR), 1nM estradiol (ER-alpha), 100nM GW1929 (PPAR-gamma), 1nM 3’,5,3 triiodo-L-thyronine (TR-alpha), 1uM isoproterenol (CREBP), 32mM LiCl (TCF-Beta catenin), or 1nM TNF-alpha (NF-kappa B). Cells were also treated with increasing concentrations of PP, or, when available, a known competitive antagonist: 100nM RU486 (GR) or 1uM propanolol (CREBP). 24hrs later, luciferase activity was assayed, and the renilla-normalized firefly-luciferase activity is shown relative to ligand treatment for each transcription factor. (B) A549 cells, which endogenously express GR, and MCF7 cells, which endogenously express ER-alpha, were transfected with the appropriate response element luciferase plasmids and a renilla control plasmid. The following day, quadruplicate wells were treated with the appropriate ligand, increasing concentrations of PP, or RU486 (GR). 24hrs later, luciferase activity was assayed, and the renilla-normalized firefly-luciferase activity is shown relative to ligand treatment for each transcription factor. PP inhibits only NRs, and only in prostate cancer cells. (* p<0.05 compared to agonist alone by ANOVA. Bars represent the standard error of quadruplicate measurements).
Pyrvinium exhibits a cell and tissue-type selective activity
In our initial small molecule screen, we observed that PP had a much greater ability to inhibit AR activity in LAPC4 prostate cancer cells compared to HEK293 kidney cells (21). Cell type-specific action of PP was also suggested by the fact that PP was able to inhibit NRs only in prostate cancer cells but not in A549 or MCF7 cells (Fig. 3). Hence, we further determined the activity of PP in a broad range of cell lines, most derived from clinically-relevant, AR-expressing tissues (Fig. 4). Pyrvinium effectively inhibited AR activity (IC50 ~8–30nM) in prostate-derived cells that had endogenous and functional AR signaling: normal mouse prostate epithelial cells (PrEC) and human prostate cancer cell lines CWR22Rv1, LNCaP, LNCaP-C4-2, and LAPC4 (Fig. 4A). Interestingly, PP could not inhibit AR activity in most other cell lines examined. When prostate cancer cells that did not endogenously express AR (PC3, DU145) and immortalized non-cancerous prostate cells (RWPE1 and BPH1) were transfected with AR and luciferase reporters, they responded to DHT but were not inhibited by PP treatment (Fig. 4B). Transfection of exogenous AR into endogenous AR-expressing prostate cancer cells did not affect PP potency, suggesting that the potency of PP is dependent on cell type, not the expression of exogenous vs. endogenous AR (Fig. S3). PP also had no effect in cell lines isolated from various tissues including embryonic kidney (HEK293), adipocytes (3T3-L1), testis (TM4), muscle (RD, HMC-tert, and C2C12), neurons (C17 and GT1-7), uterine endometrium (T-HESC), mammary (MCF7 and MCF10a), ovarian granulosa (HGL5), lung cancer (A549), and cervical cancer (HeLa) (Fig. 4D). However, two cell lines derived from bone, pre-osteoblasts (hFOB1.19) and osteosarcoma (U2OS), responded to DHT and were inhibited by PP when AR and a reporter were transfected (Fig. 4C). The cell type selectivity of PP extended to the regulation of endogenous genes as well, as Q-PCR revealed that PP inhibited androgen-regulated gene expression in LAPC4 (20), LNCaP, and hFOB1.19 bone cells, but not RWPE1 or HMC-tert cells expressing exogenous AR (Fig. S4). Cell type selectivity of PP was not the result of differential distribution of PP within cells (Fig. S5).
Fig 4. Cell and tissue-selective activity of PP.
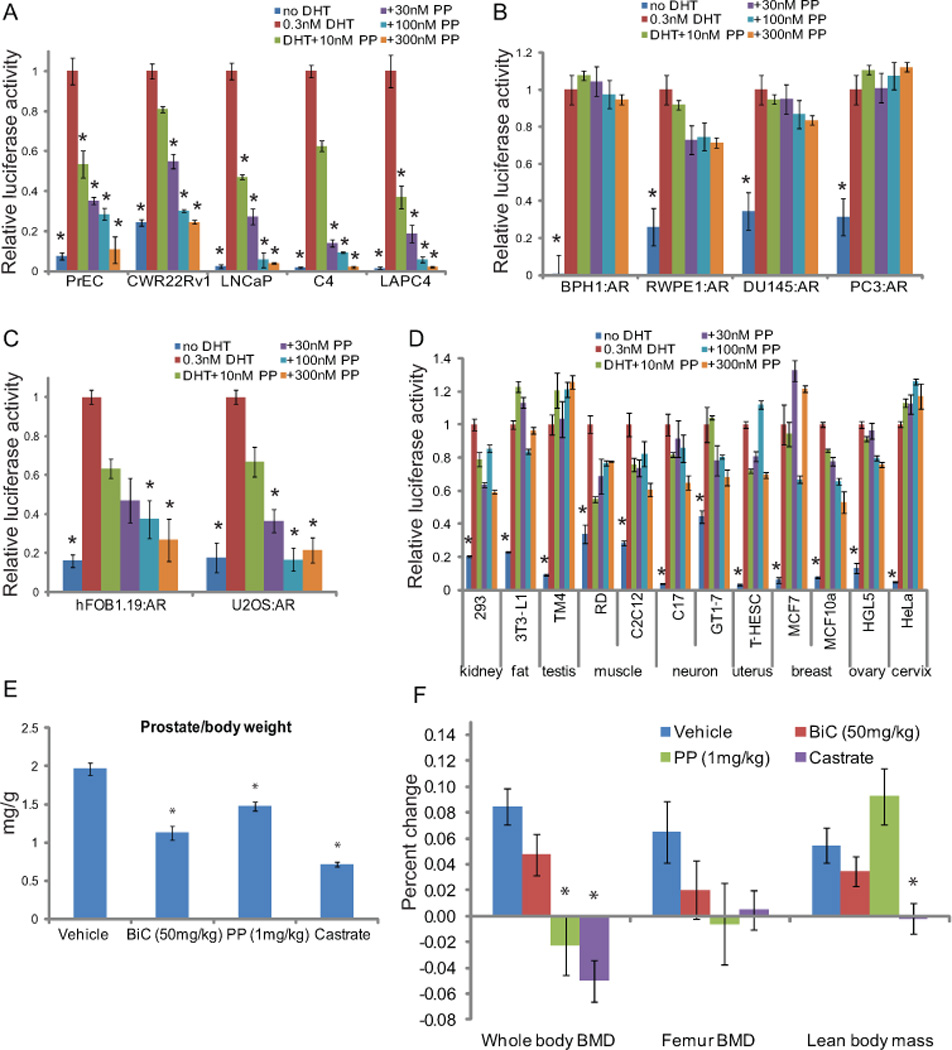
(A) Normal prostate epithelial cells (PrEC), the androgen-dependent LNCaP and LAPC4, and the androgen-independent LNCaP-C4 and CWR22Rv1 prostate cancer cells, all of which endogenously express AR, were transfected with PSA-luc and a renilla control plasmid. The following day, quadruplicate wells were treated with 0.3nM DHT and increasing concentrations of PP, or vehicle. 24hrs later, luciferase activity was assayed, and the renilla-normalized PSA-luciferase activity is shown relative to DHT treatment for each cell. (B,C,D) The immortalized normal prostate cells, BPH-1 and RWPE-1, and AR-negative prostate cancer cells, PC3 and DU145 (B), bone-derived cells, hFOB1.19 and U2OS (C), or cells derived from various tissues (D) were transfected with AR and luciferase plasmids, and assayed as in (A). PP failed to inhibit AR activity in AR-negative prostate cell lines and cells of many different types, but did inhibit AR activity in bone-derived cells. (E,F) Mice (n=9/cohort) were treated once daily with vehicle, PP, or bicalutamide (BiC), or were castrated as a control. Whole body and femur bone mineral density, as well as lean body mass were assessed by DEXA prior to treatment and at the end of the experiment. The percent change in each of these metrics is shown (E). Prostate weight was also measured. Castration, and to a lesser degree BiC treatment, resulted in decreased BMD, LBM, and prostate weight. PP treatment decreased BMD and prostate weight, but not lean body mass, demonstrating that the cell type selectivity of PP is reflected in vivo. (* p<0.05 compared to agonist alone (A–D) or vehicle (E,F) by ANOVA. Bars represent the standard error of quadruplicate measurements).
To determine if this cell type selectivity translated into tissue selectivity in vivo, we treated mice with 1mg/kg PP by IP injection daily for 6 weeks and monitored changes in bone, muscle and fat tissue by dual X-ray absorption (DEXA). DEXA is commonly used to assess androgen-dependent changes in these tissues (31, 32). We found that PP, as well as castration and bicalutamide, decreased prostate weight (Fig. 4E) and bone mineral density (Fig. 4F). However, unlike castration and bicalutamide, PP did not affect the lean body mass, suggesting that the cell-type selectivity of PP manifests as tissue selectivity in vivo.
Pyrvinium inhibits ligand-independent activity of AR splicing variants and N-terminal AR activation
Because PP non-competitively inhibits AR by interacting with the DBD, we hypothesized that it could inhibit the ligand-independent activation of AR. This includes the constitutive activity of AR splice variants (ARvs) as well as activation of AR via N-terminal cross-talk. ARvs have been shown to be expressed during the emergence of CRPC and are proposed to drive the proliferation of prostate cancers in the absence of ligand, as all ARvs lack an LBD and are constitutively active (9–11). Five different clinically relevant ARvs or a control plasmid were transfected into LNCaP cells and each demonstrated the ability to stimulate reporter gene activity in the absence of ligand. PP inhibited these ARvs as potently as it inhibited full-length AR, both in the presence and absence of DHT, while bicalutamide was not effective (Fig. 5A and B). Similar results were observed when expression of full-length AR was eliminated by transfection of a specific siRNA (Fig. 5C), suggesting that the activity of PP is not dependent on the presence of full-length AR.
Fig 5. PP inhibits AR splice variants and N-terminal AR activation.
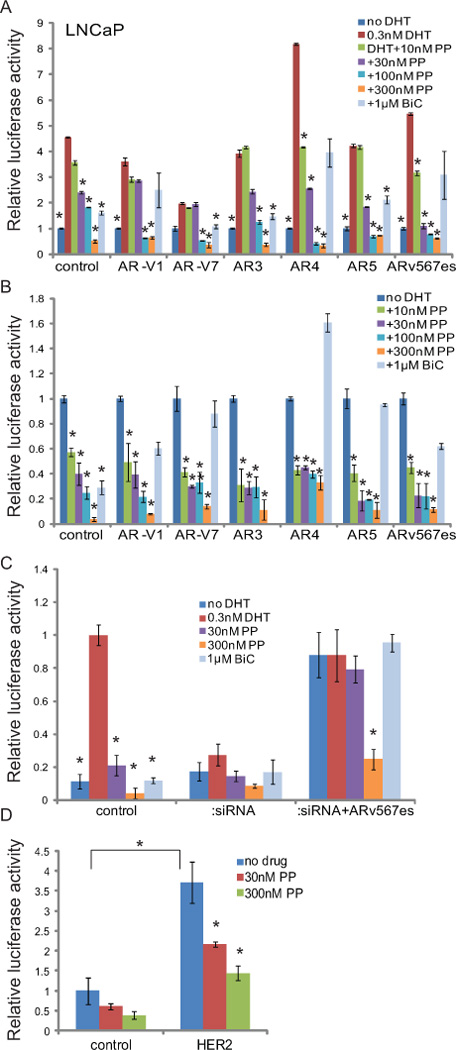
(A,B) LNCaP cells were transfected with PSA-luc, a control renilla reporter, and the indicated AR variants, all of which are constitutively active and lack an LBD. PP, but not the competitive antagonist bicalutamide, was able to inhibit these variants, both in the presence (A) and absence (B) of DHT. (C) LNCaP cells were transfected with control siRNA or siRNA targeting exon 7 of AR. Cells were also transfected with the indicated AR variant expression vectors and treated with the indicated drugs. AR activity was assessed as in (A). (D) LNCaP cells were transfected with control or HER2 expression vectors, which increases AR activity via N-terminal activation in the absence of ligand. PP is able to inhibit this activation, suggesting that it can inhibit ligand-independent AR activity. (* p<0.05 compared to DHT alone (A,C), no DHT (B), or HER2 alone (D) by ANOVA. Bars represent the standard error of quadruplicate measurements)
Several growth factors and other pathways have been found to activate AR in the absence of ligand and are associated with occurrence of castration resistant prostate cancer (33). Addition of a ligand for the HER2 kinase, heregulin, or over-expression of HER2 itself can increase transcriptional activity of AR in the absence of ligand via N-terminal activation (7, 34). When AR activity was induced in LNCaP cells by over-expression of HER2, PP was also able to potently inhibit the N-terminal activation of AR in the absence of ligand (Fig. 5D).
Pyrvinium inhibits the castration-resistant growth of ARv-expressing 22Rv1 xenografts
Because PP inhibited the activity of all ARv’s examined, we hypothesized that it could inhibit the castration-resistant growth of 22Rv1 cells, which express ARv’s (35). To test this, we castrated nude mice, injected 22Rv1 cells subcutaneously, and when palpable tumors arose, separated them into control (n=9), 1mg/kg PP (n=8), and 3mg/kg PP (n=4) cohorts. PP was administered continuously for four weeks by implantation of osmotic pump, over which time tumor growth was measured by caliper. Mice were euthanized at the end of the four week life of the pump or when tumors reached 15mm in diameter. PP-treated mice showed a dose-dependent inhibitory effect on the growth of 22Rv1 xenografts, decreasing both the relative growth rate (Fig 6A) and the time it took tumors to reach the 15mm end-point (Fig 6B). The difference in growth of 3mg/kg PP-treated mice and control mice at final tumor measurement was significant (p<.05). One of the tumors in the 3mg/kg PP-treated mice regressed substantially and one of the tumors in the 1mg/kg PP-treated mice was cured. Furthermore, PP inhibited AR-dependent gene expression in the tumors in a dose-dependent fashion (Fig 6C). These results suggest that the anti-AR activity of PP can inhibit the growth of CRPCs driven by ARv expression.
Fig 6. PP inhibits CRPC xenograft growth.
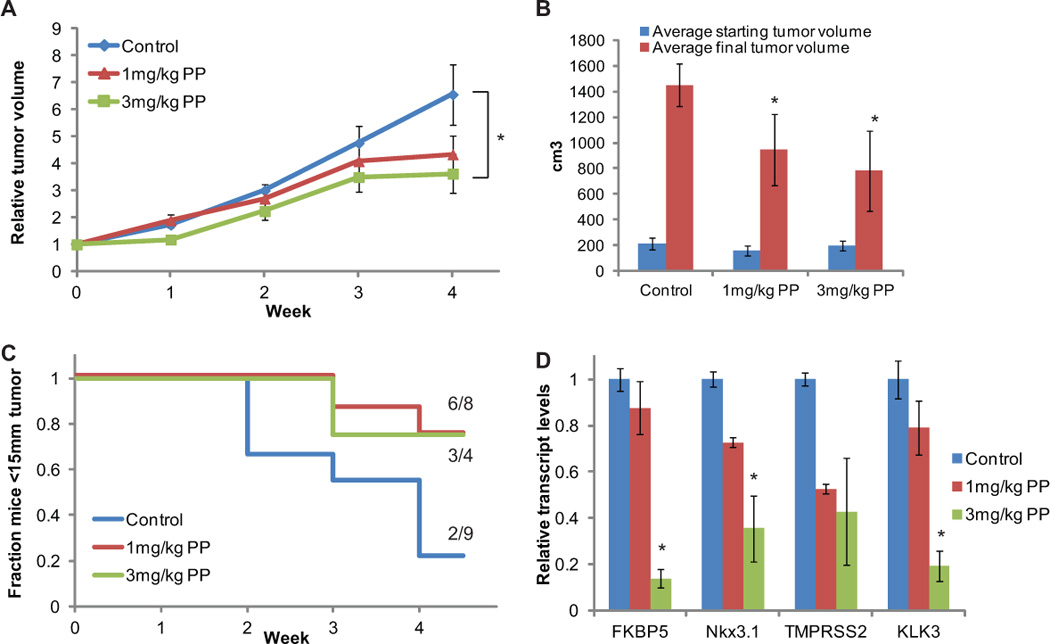
(A,B) Nude mice were castrated, injected with 22Rv1 cells subcutaneously, and when palpable tumors arose, separated them into control (n=9), 1mg/kg PP (n=8), and 3mg/kg PP (n=4) cohorts. PP was administered continuously for four weeks by implantation of osmotic pump, over which time tumor growth was measured by caliper. Mice were euthanized at the end of the four week life of the pump or when tumors reached 15mm in diameter. (A) Growth relative to tumor volume at start of treatment. (B) Weekly fraction of animals whose tumors remained <15mm in diameter. (C) RNA was extracted from tumors and reverse-transcribed, and the expression of the indicated AR-regulated genes was quantified relative to housekeeping gene RPL19. PP slowed the growth of tumors and inhibited AR activity in tumors in a dose-dependent fashion. (Bars represent standard error. * p<0.05 compared to control using ANOVA)
Discussion
Pyrvinium was the first reported non-competitive small molecule inhibitor of AR (20). The 12nM potency of PP in cell culture studies rivals the best competitive antagonists, but its detailed mechanism of action was not clear. Here, we make several important advances in our understanding of the mechanism of PP, which have significant ramifications for treatment of prostate cancer. First, we demonstrate that AR is a direct molecular target of PP. Using the DARTS assay, we demonstrated that PP protected AR from proteolysis in intact LAPC4 and LNCaP cells, and in LNCaP cell extracts. While it is possible that PP binding simply enhanced the stability of a different protein that forms a complex with AR, thereby indirectly stabilizing AR, the observations that PP decreases the rate of AR degradation and that it protected AR in LNCaP extracts argue against this. We chose to use the DARTS assay because no modification of the small molecule that may compromise its molecular interactions or activity is required and the method does not rely on physiological activity of drug, as are often required in traditional methods of target identification (22, 36, 37). Furthermore, many traditional methods are incompatible with pyrvinium because it has a charged nitrogen residue that non-specifically interacts with proteins and other molecules in many in vitro systems.
While it may seem obvious that AR would be a direct target of PP, this was not necessarily true given that the cell-based assay of AR conformation change that was used to identify PP could very well have identified indirect regulators of AR. For instance, it was possible that PP targeted a kinase that provided an essential phosphorylation on AR. In fact, PP has been reported to target several different proteins, including CK1α, which PP activated in vitro with a dose range similar to its inhibition of AR (23). Because CK1α controls the activity of β catenin, a known AR coactivator, we thought that PP may indirectly inhibit AR activity via its effects on the CK1α - β catenin pathway. However, DARTS experiments failed to demonstrate protection of CK1α by PP in prostate cancer cells and extensive experiments clearly demonstrated that CK1α plays no role in the inhibition of AR activity by PP in prostate cancer cells (Fig. S1). Furthermore, β catenin is known to interact with the AR LBD (38), so it is not likely to be involved in the inhibition of AR by PP, since PP targets the DBD. We did not rule out the possibility that PP targets additional proteins in the prostate cancer cells and are performing unbiased DARTS experiments to identify additional potential PP targets.
Demonstrating that PP inhibits AR by interacting with the DBD was the second important advance in our understanding of its mechanism of action. We used two complementary approaches to demonstrate that PP functions via the DBD. First, AR and GR constructs demonstrated the importance of the DBD for the inhibitory activity of PP. Second, computational modeling predicted an interaction of PP with the DBD domains of the AR dimer in simulations performed with or without a DNA molecule present. Interaction between PP and AR DBD is predicted at three residues of the second zinc-finger domain. These residues have no known specific function other than Pro-612 in the recognition helix (30). The region predicted for drug binding, KNCPS (Cys-611 in interaction with Zn) is conserved in GRα (progesterone receptor and mineralcorticoid receptor as well), but in other hormone NRs which were inhibited by PP (Fig. 3A), the sequence was less conserved (KSCQA in ERα, NKCQY in PPARγ, and NQCQL in TRα. Considering that PP only inhibits NRs in cells with a functional endogenous AR, it is possible that the inhibitory effect may be due to formation of heterodimers with AR (39, 40). Alternatively, PP could interrupt the association between a co-regulatory protein and the DBD of AR and other NRs.
The quinoline ring of pyrvinium is predicted to interact with the bases and side chains of residues A11, A12, G13, T26, G27, A28, and T29 in the ADR3 sequence, most of which are in the minor groove (5’-CCAGAACATCAAGAACAG-3’/5’-CTGTTCTTGATGTTCTGG-3’). Interestingly, binding of pyrvinium to this interface does not disrupt AR binding to AREs but prevents recruitment of RNA polymerase II, as shown from chromatin immunoprecipitation experiments in our previous study (20). Our current and previous results suggest a model in which PP allows AR to bind to DNA but interferes with a protein or proteins that interact with AR and facilitate recruitment of RNA pol II. This protein is likely expressed in a tissue-selective fashion as PP exhibits tissue-selective AR inhibition. This protein also likely interacts with the DBD, although molecular interactions at the regions other than DBD could be affected as PP alters the overall conformation of AR (21). We have initiated co-crystallization studies to confirm the proposed binding model of PP and experiments are underway to identify tissue-specific proteins whose interactions with AR are altered by PP. An alternative hypothesis is that PP ‘locks’ the AR onto the DNA, preventing the normal dynamic interaction of the AR with DNA and/or the normal protein turnover, thus decreasing transcriptional activity. There is intense interest in the development of small molecules that inhibit protein-DNA or protein-protein interactions (41, 42). PP may inhibit transcription initiation by stabilizing rather than obstructing DNA-protein-protein interactions. We are now working to determine if the dynamic interaction of AR with DNA is altered by PP.
PP is the only AR inhibitor shown to act via the DBD, the most highly conserved domain in AR. Allosteric inhibitors of the LBD have been reported (43), and recently EPI-001 has been reported to act via the AF1 of the NTD (44), but both the LBD and AF1 are hotspots for mutation in prostate cancer (45). Although many different mutations are found in the Ar gene in PC patients, few have been found in the DBD, likely because mutations in the DBD would render the AR non-functional. In fact, a recent publication suggested that the second alpha helix in the DBD, which pyrvinium is predicted to bind, is an ideal drug development target because it is essential for the structure of the DBD and AR function (46). Therefore, in contrast to LBD-targeted agents and the new NTD-targeted agent EPI-001, mutations that would render cancers PP-resistant are highly unlikely.
The major obstacle faced by agents targeting the AR/androgen axis is resistance due to continued AR signaling. This occurs following hormone therapy and now appears to occur following treatment with novel agents such as abiraterone and enzalutamide. The expression of constitutively active ARvs and N-terminal activation of AR can drive ligand-independent growth of recurrent cancers. For instance, the potent AR antagonist cannot inhibit the growth of 22Rv1 cells (18). PP potently inhibits the activity of all ARvs and suppresses androgen-independent N-terminal AR activation, which manifests in its ability to inhibit the castration-resistant growth of 22Rv1 xenografts in vivo. This was reflected by slowing tumor volume increase, which is most pronounced in the time it took tumors to reach the 15mm endpoint (Fig 6B). Because mice that were euthanized due to reaching 15mm tumors prior to the end of the four week study were removed from analysis of tumor volume increase (Fig 6A), the difference among groups does not appear as dramatic as it actually is. If we were to estimate the size of tumors in control animals at four weeks based on prior growth rate, the size of tumors in control animals would be more than five times that of tumors in PP-treated animals. Though PP treatment only caused tumor regression in a few mice, this is likely the result of the poor pharmicodynamics of PP. PP is highly insoluble in aqueous solution and preliminary studies show that it has poor distribution when injected IP. We attempted to overcome these limitations by solubilizing PP in PEG-400 and administering it continuously by osmotic pump implanted IP; however, tumor penetration is likely less than ideal. Indeed, we observed that PP was more effective in tumors with a smaller volume at onset of treatment. Optimized derivatives, compounds with greater solubility and oral absorption would likely improve the efficacy of PP in in vivo models. Studies are underway to synthesize and examine PP derivative for lead compound development. It has also been postulated that GR signaling may substitute AR signaling in some prostate cancers facing AR blockade (47). Our results indicate that PP also inhibits GR activity in prostate cancer cells, suggesting it would be effective in cancers that are driven by GR signaling as well.
PP has shown exquisite tissue-selective activity. Hormone therapy and other AR inhibitors are fraught with side-effects due to inhibition of AR activity in non-diseased tissues, such as skeletal muscle, bone, and the brain (48). Our cell culture and animal data suggest that PP will have reduced side-effects compared to traditional AR/androgen axis targeted therapies as it only inhibits AR activity in the prostate and bone. Thus, PP may lead to an effective, less debilitating drug for CRPC.
Materials and Methods
Reagents
Steroid hormones, pyrvinium pamoate, and other chemicals were purchased from Steraloids, MP Biomedicals, and SigmaAldrich. Expression plasmids for GR mutants were a gift of Dr. Michael J. Garabedian. AR variant expression constructs were kindly provided by the authors of references (9–11, 25). TCF/LEF reporters were a gift of Dr. Randall Moon via Dr. Ravi Bhatia.
Cell lines and culture conditions
Cells were generously provided by Stephen Plymate (LNCaP-C4), Victor Beshay (HGL5, immortalized human granulose-lutein cells), Pamela Mellon (GT1-7, immortalized gonadotropin-releasing hormone neurons), Robert Reiter (LAPC4, prostate adenocarcinoma), Kenneth Parkinson (HMC-tert), Eric Bolton (mouse primary epithelial cells), and Carlotta Glackin (MCF10a). Other cells were originally purchased from the ATCC. LNCaP, CWR22Rv1, and LNCaP-C4-2 cells were maintained in phenol-red free RPMI supplemented with 10% (LNCaP) or 5% charcoal-stripped (22Rv1 and C4-2) FBS and antibiotics (Mediatech/Cellgro). PC3, DU145, BPH-1, HEK293, 3T3-L1, C17, HeLa, A549, MCF7, U2OS, C2C12, HMC-tert, and RD cells were maintained in DMEM supplemented with 10% FBS and antibiotics (Mediatech/Cellgro). RWPE-1 cells were maintained in keratinocyte serum free media supplemented with EGF and bovine pituitary extract (Gibco). Primary mouse epithelial cells were maintained in PrEGM media (Gibco). hFOB1.19 cells were maintained in phenol red-free DMEM:Ham’s F12 with 2.5 mM L-glutamine 0.3 mg/ml G418, and 10% FBS. T-HESC cells were maintained in phenol red-free DMEM:Ham’s F12 with 10% C/S FBS, 3.1g/L glucose, 1mM sodium pyruvate, 1.5g/L sodium bicarbonate, 1% ITS premix, and antibiotics. TM4 cells were maintained in phenol red-free DMEM:Ham’s F12 with 1.2g/L sodium bicarbonate, 5% horse serum, 2.5% FBS, and antibiotics. MCF10a cells were maintained in phenol red-free DMEM:Ham’s F12 with 100ng/mL cholera enterotoxin, 10ug/ml insulin, 0.5ug/ml hydrocortisone, 20ng/ml EGF, 1% L-glutamine, 5% heat-inactivated horse serum, and antibiotics. HGL5 cells were maintained in phenol red-free DMEM:Ham’s F12 with 10% FBS, 1% ITS premix, 1ug/ml gentamicin, and antibiotics. GT1-7 cells were maintained in DMEM with 10% FBS, 110mg/L sodium pyruvate, and antibiotics.
Immunoblotting and immunoprecipitation
293-AR or LNCaP cells were treated with drugs for 24–72hrs and then lysed in TBS with 0.1% Tween with protease inhibitors. Lysates were applied for immunoblot assays or immunoprecipitation with anti-AR antibody (Santa Cruz Biotechnology, cat no. 7305). Western blot was used to detect AR, casein kinase 1 alpha (EnCor, CPCA-CK1a), β-catenin or phosphor-β-catenin (Cell Signaling, cat no.9562 and 9564), or GAPDH (Santa Cruz, cat no. 47724) as a loading control.
Cloning of AR expression constructs, AR NLxHC
The LexA DBD was amplified from the GR NLxC plasmid (27) and ligated into the BglII/BamHI site of the p6R ARN556 plasmid (25). The region representing the C-terminal 297aa of AR (hinge region and CTD) was amplified and ligated into the BamHI/XbaI site. Thus ARNLxHC lacks aa557-622.
Drug affinity responsive target stability (DARTS) assay and stability assay
Whole cell extract was prepared as previously described (49) and protein concentration was measured using Bio-Rad Protein Assay kit, then applied for DARTS assay (22). Experimental conditions were optimized for amount of protein used in reaction, concentration of small molecule drug, drug treatment condition, protease, and duration of protease digestion. Cancer cells were treated with pyrvinium prior to protein extraction or the drug was added to cell extract. 40 to 100µg of protein was used for each reaction. Mixed protease Pronase (Roche) was selected among thermolysine (Sigma-Aldrich) and trpsin (EMD Chemicals) tested. Assay conditions are described in detail in the figure legend. Samples processed by DARTS reaction were separated in 9% SDS-PAGE and transfer to PVDF membrane for immunoblot assays. Anti-AR and -GAPDH and HRP-conjugated secondary antibodies were purchased from SantaCruz Biotechnology. For stability assays, cells were incubated with or without DHT and PP in the presence of cyclohexamide (100mg/ml). Cells were lysed at various times up to 48hrs post-treatment and prepared for Western blot analysis as described above.
Transfection and luciferase assays
1–2 days prior to transfection, cells were placed in media containing charcoal-stripped serum. For all transfections, pools of cells were transfected using Lipofectamine Plus (Invitrogen) with PSA-luciferase (50), MMTV-luciferase, or appropriate plasmids containing response elements for different transcription factors driving luciferase expression, and pRL-SV40 (Promega) as a control. In some cases, 50ng/6 well (0.2 ~ 1×106 cells, depending on cell type) of expression plasmids for HER2, AR or other transcription factors, or AR and GR mutants, or control plasmids were transfected along with the luciferase reporter plasmids. In one experiment, siRNA targeting full-length AR (Qiagen AR6) was transfected with plasmids. The following day, the cells were plated in quadruplicate with drugs in 96 well plates. 24hrs later luciferase production was measured (Dual luciferase assay kit, Promega). ANOVA methods were used to determine statistically significant differences among the groups using a Tukey test for planned comparisons.
qPCR analysis
Total RNA was isolated from cells or homogenized tumor tissue using an RNAeasy kit (Qiagen). RNA was reverse-transcribed (MMLV-RT; Invitrogen), and the expression of androgen-regulated genes was assessed by qPCR using a StepOne Real Time PCR System (Applied Biosystems), using SYBR green (Invitrogen) as the detecting dye and Rox (Invitrogen) as the reference dye. Primer sequences can be found in the supplementary methods. Differences between treated (x) and no DHT control (y) samples were normalized to RPL19 transcript levels (i.e., androgen-unresponsive) and determined with the following calculation: (2[Ctxgene1 − Ctygene1])/(2[CtxRPL19 − CtyRPL19]). ANOVA methods were used to determine statistically significant differences among the groups using a Tukey test for planned comparisons.
Docking study
Structural information for the AR DNA-binding domain (DBD) was downloaded from the Protein Data Bank (PDB ID: 1R4I) (30) for the docking study. The structure was built on a co-crystal structure of rat AR DBD dimer complex with an 18-mer oligonucleotide of an AR direct repeat element. The amino acid sequence is 100% homologous to human AR DBD. The receptor structure was prepared by following a protein preparation protocol for the docking analysis by using Sybylx1.3 (Tripos Inc). Pyrvinium and derivatives were prepared by ISISdraw and Sybyl ligand preparation tool and compiled into a ligand library. The ligand binding site was identified by Surflex-Dock protomol. The novel AR ligand binding site locates at the dimer and DNA interface. Surflex-Dock was used to dock the pyrvinium compound library to the AR DBD complex. Ligand and ring flexibility was allowed when generating the molecular fragments and soft grid treatment was used in docking. The ligands were optimized before docking experiments by a BFGS quasi-Newton method and an internal Dreiding force field. The ligand docking poses are ranked with an empirically derived scoring function based on the binding affinities of protein-ligand complexes and on their X-ray structures. The docked ligand pose was minimized in the context of the receptor. The protein hydrogen atoms at the binding site were allowed to move in the optimization process.
Xenograft studies
All animal experiments were approved by the City of Hope IACUC. Male nude mice were castrated and five days later injected with 2×106 22Rv1 cells mixed with matrigel (1:1) subcutaneously into the dorsal flank. At the onset of palpable tumor (2–3 weeks), an osmotic pump (Alzet 1004) containing 1mg/kg or 3mg/kg PP in 20% DMSO/80% PEG-400 was implanted in the intraperitoneal cavity. Tumor growth was measured weekly by caliper until the tumor reached 15mm in diameter (IACUC endpoint) or the pump expired (four weeks), at which point animals were euthanized and organs and tumors harvested for downstream analysis. Tumor volume was estimated by the formula: V = π/6*f (l*w)3/2 (51). Growth rate was determined by the percent increase over initial tumor volume over time. ANOVA methods were used to determine statistically significant differences among the groups using the final growth rate measurement for each animal.
DEXA experiments
All animal experiments were approved by the UCSF IACUC. 10 adult male FVB mice were treated by IP injection once daily (M-F) for six weeks with 1mg/kg PP, 100mg/kg, or vehicle (DMSO/PEG-400). Another cohort of mice was castrated at the onset of the study as a positive control. Animals were weighed once a week throughout the study. 24 hours after the final dosing, animals were weighed, sacrificed, and the prostates were dissected and weighed. Prior to the first and last drug administration, fat and lean body mass, whole-body and femur areal bone mineral density and bone mineral content were determined by dual-energy X-ray absorptiometry (DXA), as instructed by the manufacturer (Lunar PIXImus mouse densitometer). To test for differences in treatment groups, analysis of variance methods were used for planned comparisons between treatment groups that were defined by linear contrast statements.
Supplementary Material
Acknowledgements
This work was supported by NIH K99/R00 CA138711 to J.O. Jones. The authors would like to thank S. Plymate, Y. Qiu, and J. Luo for contributing ARv plasmids, R. Moon and R. Bhatia for contributing beta-catenin reporters, and M.J. Garabedian for contributing GR plasmids. We would also like to thank those who contributed cell lines, as indicated in the methods. We would like to thank Y. Yuan and the Bioinformatics core facility for help with computer modeling. We would like to thank B. Hann and the pre-clinical therapeutics core facility at UCSF for help with mouse experiments. Finally, we would like to thank M. Diamond, E. Bolton, K. Yamamoto and the rest of the Yamamoto lab for helpful discussions of this project. The project described was supported by Grant Number P30 CA033572 from the National Cancer Institute. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Cancer Institute or NIH.
Support: This work was supported by NIH K99/R00 CA138711 to J.O. Jones
Footnotes
Supporting information associated with this manuscript is available and can be found online at http://pubs.acs.org/journal/acbcct.
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63:11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 2.Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, Rosenfeld MG, Sawyers CL. Molecular determinants of resistance to antiandrogen therapy. Nat Med. 2004;10:33–39. doi: 10.1038/nm972. [DOI] [PubMed] [Google Scholar]
- 3.Taplin ME, Balk SP. Androgen receptor: a key molecule in the progression of prostate cancer to hormone independence. J Cell Biochem. 2004;91:483–490. doi: 10.1002/jcb.10653. [DOI] [PubMed] [Google Scholar]
- 4.Brooke GN, Parker MG, Bevan CL. Mechanisms of androgen receptor activation in advanced prostate cancer: differential co-activator recruitment and gene expression. Oncogene. 2008;27:2941–2950. doi: 10.1038/sj.onc.1210955. [DOI] [PubMed] [Google Scholar]
- 5.Araki S, Omori Y, Lyn D, Singh RK, Meinbach DM, Sandman Y, Lokeshwar VB, Lokeshwar BL. Interleukin-8 is a molecular determinant of androgen independence and progression in prostate cancer. Cancer Res. 2007;67:6854–6862. doi: 10.1158/0008-5472.CAN-07-1162. [DOI] [PubMed] [Google Scholar]
- 6.Debes JD, Schmidt LJ, Huang H, Tindall DJ. p300 mediates androgen-independent transactivation of the androgen receptor by interleukin 6. Cancer Res. 2002;62:5632–5636. [PubMed] [Google Scholar]
- 7.Yeh S, Lin HK, Kang HY, Thin TH, Lin MF, Chang C. From HER2/Neu signal cascade to androgen receptor and its coactivators: a novel pathway by induction of androgen target genes through MAP kinase in prostate cancer cells. Proc Natl Acad Sci U S A. 1999;96:5458–5463. doi: 10.1073/pnas.96.10.5458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dehm SM, Schmidt LJ, Heemers HV, Vessella RL, Tindall DJ. Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res. 2008;68:5469–5477. doi: 10.1158/0008-5472.CAN-08-0594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guo Z, Yang X, Sun F, Jiang R, Linn DE, Chen H, Kong X, Melamed J, Tepper CG, Kung HJ, Brodie AM, Edwards J, Qiu Y. A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth. Cancer Res. 2009;69:2305–2313. doi: 10.1158/0008-5472.CAN-08-3795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hu R, Dunn TA, Wei S, Isharwal S, Veltri RW, Humphreys E, Han M, Partin AW, Vessella RL, Isaacs WB, Bova GS, Luo J. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res. 2009;69:16–22. doi: 10.1158/0008-5472.CAN-08-2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sun S, Sprenger CC, Vessella RL, Haugk K, Soriano K, Mostaghel EA, Page ST, Coleman IM, Nguyen HM, Sun H, Nelson PS, Plymate SR. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J Clin Invest. 2010;120:2715–2730. doi: 10.1172/JCI41824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mostaghel EA, Montgomery B, Nelson PS. Castration-resistant prostate cancer: Targeting androgen metabolic pathways in recurrent disease. Urol Oncol. 2009;27:251–257. doi: 10.1016/j.urolonc.2009.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Knudsen KE, Penning TM. Partners in crime: deregulation of AR activity and androgen synthesis in prostate cancer. Trends Endocrinol Metab. 2010;21:315–324. doi: 10.1016/j.tem.2010.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ryan CJ, Smith MR, Fong L, Rosenberg JE, Kantoff P, Raynaud F, Martins V, Lee G, Kheoh T, Kim J, Molina A, Small EJ. Phase I clinical trial of the CYP17 inhibitor abiraterone acetate demonstrating clinical activity in patients with castration-resistant prostate cancer who received prior ketoconazole therapy. J Clin Oncol. 2010;28:1481–1488. doi: 10.1200/JCO.2009.24.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, Miller MD, de Wit R, Mulders P, Chi KN, Shore ND, Armstrong AJ, Flaig TW, Flechon A, Mainwaring P, Fleming M, Hainsworth JD, Hirmand M, Selby B, Seely L, de Bono JS. Increased Survival with Enzalutamide in Prostate Cancer after Chemotherapy. N Engl J Med. 2012;367:1187–1197. doi: 10.1056/NEJMoa1207506. [DOI] [PubMed] [Google Scholar]
- 16.Kaku T, Hitaka T, Ojida A, Matsunaga N, Adachi M, Tanaka T, Hara T, Yamaoka M, Kusaka M, Okuda T, Asahi S, Furuya S, Tasaka A. Discovery of orteronel (TAK-700), a naphthylmethylimidazole derivative, as a highly selective 17,20-lyase inhibitor with potential utility in the treatment of prostate cancer. Bioorg Med Chem. 2011;19:6383–6399. doi: 10.1016/j.bmc.2011.08.066. [DOI] [PubMed] [Google Scholar]
- 17.Bruno RD, Vasaitis TS, Gediya LK, Purushottamachar P, Godbole AM, Ates-Alagoz Z, Brodie AM, Njar VC. Synthesis and biological evaluations of putative metabolically stable analogs of VN/124-1 (TOK-001): head to head anti-tumor efficacy evaluation of VN/124-1 (TOK-001) and abiraterone in LAPC-4 human prostate cancer xenograft model. Steroids. 2011;76:1268–1279. doi: 10.1016/j.steroids.2011.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li Y, Chan SC, Brand LJ, Hwang TH, Silverstein KA, Dehm SM. Androgen Receptor Splice Variants Mediate Enzalutamide Resistance in Castration-Resistant Prostate Cancer Cell Lines. Cancer Res. 2013;73:483–489. doi: 10.1158/0008-5472.CAN-12-3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mostaghel EA, Marck BT, Plymate SR, Vessella RL, Balk S, Matsumoto AM, Nelson PS, Montgomery RB. Resistance to CYP17A1 inhibition with abiraterone in castration-resistant prostate cancer: induction of steroidogenesis and androgen receptor splice variants. Clin Cancer Res. 2011;17:5913–5925. doi: 10.1158/1078-0432.CCR-11-0728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jones JO, Bolton EC, Huang Y, Feau C, Guy RK, Yamamoto KR, Hann B, Diamond MI. Non-competitive androgen receptor inhibition in vitro and in vivo. Proc Natl Acad Sci U S A. 2009;106:7233–7238. doi: 10.1073/pnas.0807282106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jones JO, Diamond MI. A cellular conformation-based screen for androgen receptor inhibitors. ACS Chem Biol. 2008;3:412–418. doi: 10.1021/cb800054w. [DOI] [PubMed] [Google Scholar]
- 22.Lomenick B, Hao R, Jonai N, Chin RM, Aghajan M, Warburton S, Wang J, Wu RP, Gomez F, Loo JA, Wohlschlegel JA, Vondriska TM, Pelletier J, Herschman HR, Clardy J, Clarke CF, Huang J. Target identification using drug affinity responsive target stability (DARTS) Proc Natl Acad Sci U S A. 2009;106:21984–21989. doi: 10.1073/pnas.0910040106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thorne CA, Hanson AJ, Schneider J, Tahinci E, Orton D, Cselenyi CS, Jernigan KK, Meyers KC, Hang BI, Waterson AG, Kim K, Melancon B, Ghidu VP, Sulikowski GA, LaFleur B, Salic A, Lee LA, Miller DM, 3rd, Lee E. Small-molecule inhibition of Wnt signaling through activation of casein kinase 1alpha. Nat Chem Biol. 2010;6:829–836. doi: 10.1038/nchembio.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gregory CW, Johnson RT, Jr, Mohler JL, French FS, Wilson EM. Androgen receptor stabilization in recurrent prostate cancer is associated with hypersensitivity to low androgen. Cancer Res. 2001;61:2892–2898. [PubMed] [Google Scholar]
- 25.Diamond MI, Robinson MR, Yamamoto KR. Regulation of expanded polyglutamine protein aggregation and nuclear localization by the glucocorticoid receptor. Proc Natl Acad Sci U S A. 2000;97:657–661. doi: 10.1073/pnas.97.2.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou ZX, Wong CI, Sar M, Wilson EM. The androgen receptor: an overview. Recent Prog Horm Res. 1994;49:249–274. doi: 10.1016/b978-0-12-571149-4.50017-9. [DOI] [PubMed] [Google Scholar]
- 27.Godowski PJ, Picard D, Yamamoto KR. Signal transduction and transcriptional regulation by glucocorticoid receptor-LexA fusion proteins. Science. 1988;241:812–816. doi: 10.1126/science.3043662. [DOI] [PubMed] [Google Scholar]
- 28.Liu W, Wang J, Sauter NK, Pearce D. Steroid receptor heterodimerization demonstrated in vitro and in vivo. Proc Natl Acad Sci U S A. 1995;92:12480–12484. doi: 10.1073/pnas.92.26.12480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rogatsky I, Hittelman AB, Pearce D, Garabedian MJ. Distinct glucocorticoid receptor transcriptional regulatory surfaces mediate the cytotoxic and cytostatic effects of glucocorticoids. Mol Cell Biol. 1999;19:5036–5049. doi: 10.1128/mcb.19.7.5036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shaffer PL, Jivan A, Dollins DE, Claessens F, Gewirth DT. Structural basis of androgen receptor binding to selective androgen response elements. Proc Natl Acad Sci U S A. 2004;101:4758–4763. doi: 10.1073/pnas.0401123101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Berruti A, Dogliotti L, Terrone C, Cerutti S, Isaia G, Tarabuzzi R, Reimondo G, Mari M, Ardissone P, De Luca S, Fasolis G, Fontana D, Rossetti SR, Angeli A. Changes in bone mineral density, lean body mass and fat content as measured by dual energy x-ray absorptiometry in patients with prostate cancer without apparent bone metastases given androgen deprivation therapy. J Urol. 2002;167:2361–2367. discussion 2367. [PubMed] [Google Scholar]
- 32.Kearbey JD, Gao W, Fisher SJ, Wu D, Miller DD, Dalton JT. Effects of selective androgen receptor modulator (SARM) treatment in osteopenic female rats. Pharm Res. 2009;26:2471–2477. doi: 10.1007/s11095-009-9962-7. [DOI] [PubMed] [Google Scholar]
- 33.Culig Z. Androgen receptor cross-talk with cell signalling pathways. Growth Factors. 2004;22:179–184. doi: 10.1080/08977190412331279908. [DOI] [PubMed] [Google Scholar]
- 34.Mellinghoff IK, Vivanco I, Kwon A, Tran C, Wongvipat J, Sawyers CL. HER2/neu kinase-dependent modulation of androgen receptor function through effects on DNA binding and stability. Cancer Cell. 2004;6:517–527. doi: 10.1016/j.ccr.2004.09.031. [DOI] [PubMed] [Google Scholar]
- 35.Li Y, Alsagabi M, Fan D, Bova GS, Tewfik AH, Dehm SM. Intragenic rearrangement and altered RNA splicing of the androgen receptor in a cell-based model of prostate cancer progression. Cancer Res. 2011;71:2108–2117. doi: 10.1158/0008-5472.CAN-10-1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sleno L, Emili A. Proteomic methods for drug target discovery. Curr Opin Chem Biol. 2008;12:46–54. doi: 10.1016/j.cbpa.2008.01.022. [DOI] [PubMed] [Google Scholar]
- 37.Terstappen GC, Schlupen C, Raggiaschi R, Gaviraghi G. Target deconvolution strategies in drug discovery. Nat Rev Drug Discov. 2007;6:891–903. doi: 10.1038/nrd2410. [DOI] [PubMed] [Google Scholar]
- 38.Song LN, Herrell R, Byers S, Shah S, Wilson EM, Gelmann EP. Beta-catenin binds to the activation function 2 region of the androgen receptor and modulates the effects of the N-terminal domain and TIF2 on ligand-dependent transcription. Mol Cell Biol. 2003;23:1674–1687. doi: 10.1128/MCB.23.5.1674-1687.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen S, Wang J, Yu G, Liu W, Pearce D. Androgen and glucocorticoid receptor heterodimer formation. A possible mechanism for mutual inhibition of transcriptional activity. J Biol Chem. 1997;272:14087–14092. doi: 10.1074/jbc.272.22.14087. [DOI] [PubMed] [Google Scholar]
- 40.Savory JG, Prefontaine GG, Lamprecht C, Liao M, Walther RF, Lefebvre YA, Hache RJ. Glucocorticoid receptor homodimers and glucocorticoid-mineralocorticoid receptor heterodimers form in the cytoplasm through alternative dimerization interfaces. Mol Cell Biol. 2001;21:781–793. doi: 10.1128/MCB.21.3.781-793.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Arkin MR, Wells JA. Small-molecule inhibitors of protein-protein interactions: progressing towards the dream. Nat Rev Drug Discov. 2004;3:301–317. doi: 10.1038/nrd1343. [DOI] [PubMed] [Google Scholar]
- 42.Neher TM, Bodenmiller D, Fitch RW, Jalal SI, Turchi JJ. Novel irreversible small molecule inhibitors of replication protein A display single-agent activity and synergize with cisplatin. Mol Cancer Ther. 2011;10:1796–1806. doi: 10.1158/1535-7163.MCT-11-0303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Estebanez-Perpina E, Arnold LA, Nguyen P, Rodrigues ED, Mar E, Bateman R, Pallai P, Shokat KM, Baxter JD, Guy RK, Webb P, Fletterick RJ. A surface on the androgen receptor that allosterically regulates coactivator binding. Proc Natl Acad Sci U S A. 2007;104:16074–16079. doi: 10.1073/pnas.0708036104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Andersen RJ, Mawji NR, Wang J, Wang G, Haile S, Myung JK, Watt K, Tam T, Yang YC, Banuelos CA, Williams DE, McEwan IJ, Wang Y, Sadar MD. Regression of castrate-recurrent prostate cancer by a small-molecule inhibitor of the amino-terminus domain of the androgen receptor. Cancer Cell. 2010;17:535–546. doi: 10.1016/j.ccr.2010.04.027. [DOI] [PubMed] [Google Scholar]
- 45.Gottlieb B, Lehvaslaiho H, Beitel LK, Lumbroso R, Pinsky L, Trifiro M. The Androgen Receptor Gene Mutations Database. Nucleic Acids Res. 1998;26:234–238. doi: 10.1093/nar/26.1.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chan SC, Li Y, Dehm SM. Androgen receptor splice variants activate AR target genes and support aberrant prostate cancer cell growth independent of the canonical AR nuclear localization signal. J Biol Chem. 2012;287:19736–19749. doi: 10.1074/jbc.M112.352930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Otto K, Vander Griend D, Conzen S, Szmulewitz R. Proceedings of the 103rd Annual Meeting of the American Association for Cancer Research; American Association for Cancer Research; Chicago, IL. 2012. pp Abstract nr 148. [Google Scholar]
- 48.Sprenkle PC, Fisch H. Pathologic effects of testosterone deprivation. Curr Opin Urol. 2007;17:424–430. doi: 10.1097/MOU.0b013e3282f0ebef. [DOI] [PubMed] [Google Scholar]
- 49.Lim M, Zhong C, Yang S, Bell AM, Cohen MB, Roy-Burman P. Runx2 regulates survivin expression in prostate cancer cells. Lab Invest. 2010;90:222–233. doi: 10.1038/labinvest.2009.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bolton EC, So AY, Chaivorapol C, Haqq CM, Li H, Yamamoto KR. Cell- and gene-specific regulation of primary target genes by the androgen receptor. Genes Dev. 2007;21:2005–2017. doi: 10.1101/gad.1564207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Feldman J, Goldwasser R, Mark S, Schwartz J, Orion I. A mathematical model for tumor volume evaluation using two-dimensions. J. Appl. Quant. Methods. 2009;4:455. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.