

Abstract
The PI 3-K and Akt signaling pathway regulates all phenotypes that contribute to progression of human cancers, including breast cancer. Akt mediates signal relay by phosphorylating numerous substrates, which are causally implicated in responses such as cell growth, survival, metabolic reprograming and migration and invasion. Here we identify a new Akt substrate, the adherens junction protein Afadin, that is phosphorylated by Akt at Ser1718. We show that under conditions of physiological IGF-1 signaling and oncogenic PI 3-K and Akt, Afadin is phosphorylated by all Akt isoforms, and that this phosphorylation elicits a relocalization of Afadin from adherens junctions to the nucleus. Phosphorylation of Afadin also results in a marked increase in breast cancer cell migration that is dependent on Ser1718 phosphorylation. We also observe nuclear localization in breast cancer tissues, indicating that regulation of Afadin by the PI 3-K and Akt pathway has pathophysiological significance.
Implications
Phosphorylation of the adhesion protein Afadin by Akt downstream of the PI 3-K pathway, leads to re-distribution of Afadin and controls cancer cell migration.
Keywords: Afadin, Akt, phosphorylation, cell migration, breast cancer
Introduction
The phosphoinositide 3-kinase (PI 3-K) and Akt signaling pathway orchestrates virtually all aspects of epithelial and tumor cell behavior, from initial transformation to dysplasia and ultimately the dissemination of cancer cells to distant metastatic sites (1). In addition, mutations in genes that encode proteins that are rate-limiting for transducing the PI 3-K and Akt signaling are frequent mutated in human cancers. This is most evident in breast cancer, whereby according to molecular subtype, the most frequent genetic lesions are oncogenic mutations in the p110α PI 3-K catalytic subunit, PIK3CA, inactivation or loss of heterozygosity of the tumor suppressors PTEN and INPP4B and amplification or somatic activating mutations in one of the three Aktgenes AKT1, AKT2 and AKT3 (2,3). All of these lesions ultimately result in hyperactivation of Akt and phosphorylation of downstream substrates that transduce the signal to secondary effector pathways and in turn the modulation of phenotypes associated with malignancy, including cell growth, proliferation, survival, metabolic reprogramming, and cell migration and invasion (4). Moreover, since most of the proteins that function to transduce PI 3-K and Akt signaling are enzymes with catalytic pockets, this pathway is highly druggable and numerous phase I and II clinical trials are underway with small molecule inhibitors targeting PI 3-K or Akt isoforms for single agent or combination therapy, including in breast cancer (5).
Increased Akt activity is detected in aggressive human breast cancers and is associated with poor prognosis and higher probability of relapse accompanied by distant metastases in patients (6–8). The ability of cancer cells to migrate requires signals which lead to the rearrangement of the actin cytoskeleton as well as proteolysis of the extracellular matrix (9,10). Importantly, molecular genetic as well as in vivo studies have demonstrated that Akt isoforms play unique roles in modulating breast cancer cell invasion leading to metastatic dissemination, such that Akt2 is a metastasis enhancer, whereas Akt1 either does not promote metastasis or can actually block this process and thus function as a suppressor (11,12). Yet in other cell types and tissues, Akt isoforms either have no specificity in modulating cell migration, or even have opposing roles to those identified in epithelial cells, such as that reported for fibroblast migration (9). Regardless, the signaling specificity attributed to Akt isoforms highlights the importance of a complete understanding of the mechanism that govern cancer cell phenotypes such as invasive migration and metastasis, if specific drugs are to be developed for effective cancer therapy. In terms of mechanisms that explain the function of Aktin the control of migration, invasion and metastasis, a number of specific substrates have been identified recently. These include the actin-bundling protein palladin, a unique Akt1 substrate that functions to mediate the inhibitor activity of this Akt isoform in cell migration (13). Other substrates include girdin, that following phosphorylation accumulates in the leading edges of migrating cells and is essential for the integrity of the actin cytoskeleton and cell migration (9). Also included in this list are ACAP1, whose phosphorylation controls the recycling of integrin-β1 and cell migration, and the G-protein coupled receptor EDG-1 that is required for endothelial cell chemotaxis (14,15). Recent global phospho-proteomic studies from cancer cell lines and tissues have identified thousands of novel phosphoproteins with phosphorylation sites that conform to the optimal Akt consensus motif, RxRxxS/T, greatly accelerating the discovery of Akt targets that transduce the signal (16).
Afadin, a tumor suppressor-like protein encoded by the MLLT4 gene, is a multi-domain F-actin-binding protein that is expressed in epithelial cells, neurons, fibroblasts and endothelial cells (17,18). There exist two splice variants: l-Afadin and s-Afadin (18). The longer splice variant, l-Afadin (herein referred to as Afadin unless otherwise specified) has two Ras associating domains, a Forkhead associating domain, a Dilute domain, a PDZ domain, three proline-rich domains and the F-actin binding domain at the carboxyl-terminus (see Fig. 1A). s-Afadin, the shorter splice variant, lacks the F-actin binding domain and the third proline-rich domain and its expression is restricted to neuronal tissues (19). Human s-Afadin is identical to the gene product of AF6, an ALL-1 fusion partner involved in acute myeloid leukemia (20,21).
Figure 1. Akt phosphorylates Afadin at Ser1718.

A. Schematic of s-Afadin and l-Afadin showing domains and putative Akt consensus phosphorylation site at Ser1718 in l-Afadin. The phosphorylation site is evolutionarily conserved. Numbers on the left indicate the amino acid residue of the phosphorylation site. RA1,2, Ras associating domains 1 and 2; FHA, Forkhead associating domain; DIL, dilute domain; PDZ, PDZ domain; Pro, proline rich domain. Figure adapted from ref. (24). B. Expression of Afadin in breast cancer cell lines and HeLa cells evaluated by immunoblotting with total Afadin antibody. β-actin served as loading control. C. MCF10A cells were serum starved, pre-treated with the indicated inhibitors and stimulated with IGF-1 for 20 min. Whole cell lysates were immunoblotted with the indicated antibodies. p85 served as loading control. D. HeLa cells were transfected with vector control (pEGFP-N1), wild-type Afadin, Myc-Afadin-Ser1718Ala (S1718A) or Myc-s-Afadin (Afadin short form), serum starved and stimulated with IGF-1 for 20 min. Myc immunoprecipitates were immunoblotted with pAfadin antibody, and whole cell lysates immunoblotted with the indicated antibodies. p85 served as loading control. Results are representative of at least three independent experiments.
Afadin is localized at epithelial adherens junctions (18), consisting of two adhesion complexes, the Nectin-Afadin and the E-cadherin-Catenin complexes (20). The role of Afadin in the adherens junction complex is not completely understood. Afadin interacts with cell adhesion molecules, cytoskeletal components, signaling molecules and is generally considered to function as an adaptor protein. Knockout of Afadin in mice results in embryonic lethality due to disorganization of the ectoderm, impaired migration of the mesoderm and impaired gastrulation. Moreover, loss of cell polarity due to improperly assembled adherens junction and tight junctions is observed (17,19,20,22). Afadin has also been shown to regulate integrin-mediated cell adhesion and cell migration, although it appears that the function of Afadin in positively or negatively regulating cell motility is context-dependent (23–27).
Here, we show that Afadin is a substrate of Akt whose phosphorylation leads to its relocalization form the plasma membrane to the nucleus, concomitant with an enhancement of breast epithelial and cancer cell migration.
Materials and Methods
Cell culture
HEK293T, HeLa, MCF7, MDA-MB-231, MDA-MB-453, MDA-MB-468, SKBR3, BT-549, ZR-75-1, MCF10A, T47D and ZR-75-30 were obtained from the American Type Culture Collection (ATCC), and authenticated using Short Tandem Repeat (STR) profiling. Cells were maintained in culture not more than 6 months. Cells were routinely screened for mycoplasma contamination. Cell lines were maintained as follows: HEK293T, HeLa, MCF7, MDA-MB-231, MDA-MB-453 and MDA-MB-468, Dulbecco’s modified Eagle’s medium (DMEM) (Cellgro; Manassas, VA) supplemented with 10% fetal bovine serum (FBS) (HyClone; Waltham, MA); SKBR3, McCoy’s 5A medium (Cambrex; East Rutherford, NJ) supplemented with 10% FBS; BT-549 and ZR-75-1, RPMI 1640 (Cellgro) supplemented with 10% FBS; MCF10A DMEM/Ham’s F12 supplemented with 5% equine serum (GIBCO; Carlsbad, CA), 10 μg/ml Insulin (Sigma-Aldrich; St. Louis, MO) 500 ng/ml hydrocortisone (Sigma-Aldrich), 20 ng/ml EGF (R&D Systems; Minneapolis, MN), and 100 ng/ml cholera toxin (List Biological Labs; Campbell, CA); T47D and ZR-75-30 in RPMI 1640 supplemented with 10% FBS and 10 μg/ml Insulin.
Growth Factors and Inhibitors
Cells were stimulated with recombinant human IGF-1 (R&D Systems) at a final concentration of 100 ng/ml for 20 min unless otherwise specified. CGK733 (Sigma-Aldrich) was added to cells for 4 hrin a final concentration of 10 μM prior to IGF1 stimulation. All other inhibitors were added to cells for 20 min prior to stimulation at the following final concentrations: wortmannin (Sigma-Aldrich), 100 nM; BEZ235 (Cayman Chemical company; Ann Harbor, MI) and MK2206 (Active Biochem; Maplewood, NJ), 1 μM; A66 (Symansis; Temecula, CA), 0.7 μM. Rapamycin (Sigma-Aldrich), 100 nM; PF4708671 (Sigma-Aldrich),10 μM. GSK650394 (R&D Systems),5 μM; Cycloheximide (Sigma-Aldrich) was used for 3–6 hr at 20 μg/ml.
Antibodies
Anti-phospho-Afadin Ser1718 antibody, anti-pan-Akt antibody, anti-Akt1, anti-Akt2, anti-Akt3, anti-phospho-Akt Ser473, anti-GSK3β, anti-phospho-GSK3β Ser9, anti-p110α, anti-S6K, anti-phospho-S6K Thr389, anti E-cadherin, anti-NDRG1, anti-phospho-NDRG1 Thr346, anti-CENP-A, anti-NuP98, anti-Fibrillarin and anti-Histone H3 were from Cell Signaling Technology (Danvers, MA). Anti-Myc antibody, anti-Tubulin and anti-lamin A/C antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-Afadin antibodies used for immunoblotting were from Bethyl laboratories (Montgomery, TX) and used for immunofluorescence from BD Biosciences (Franklin Lakes, NJ). Horseradish peroxidase-conjugated anti-mouse and anti-rabbit immunoglobulin G (IgG) antibodies were from Chemicon (Billerica, MA). Cy3-conjugated anti-mouse IgG antibody and Alexa-Flour 488 anti-rabbit antibody were from Jackson ImmonoResearch Laboratories (West-Grove, PA). Anti-β-actin monoclonal antibody was purchased from Sigma-Aldrich. Anti-HA monoclonal antibody was purified from the 12CA5 hybridoma. The anti-p85 antibody has been described (28).
Plasmids
The Afadin cDNA constructs pEGFP-N2-AF6i3-Myc and pEGFP-N2-AF6i1-Myc were a gift from Mihaela Lorger and have been described (24). ShRNA rescue mutants were generated by introduction of 6 silent mutations using the following primer: 5′ GGAACG CCA GCG TCT TTT TTC ACA AGG ACA GGA CGT CTC TAA TAA AGT GAA AGC TTC TCG 3′. The shRNA resistant mutants with phosphorylation site mutations were generated by site directed mutagenesis using the following primers: S1718A: 5′ GAA CGC CAG CGT CTT TTT GCA CAA GGA CAG GAC G 3′; S1718D: 5′ ACA TTC AAG GAA CGC CAG CGT CTT TTT GAT CAA GGA CAG GAC GTC 3′; S1718E: 5′ ACA TTC AAG GAA CGC CAG CGT CTT TTT GAG CAA GGA CAG GAC GTC 3′. All sequences were verified by sequencing. pcDNA3-Myr-HA-Akt1, pcDNA3-Myr-HA-Akt2, pcDNA3-Myr-HA-Akt3 from William Sellers (Addgene plasmids 9008, 9016, 9017) (29). HA-GSK3β has been described (30). JP1520-GFP, JP1520-PIK3CA-WT-HA, JP1520-PIK3CA-H1047R-HA, JP1520-PIK3CA-E545K-HA were from Joan Brugge (Addgene plasmids 14570, 14571, 15572) (31).
RNA Interference
For shRNA-silencing, a set of single-stranded oligonucleotides encoding the Akt1 or Akt2 target shRNA and its complement were previously described (11). Akt3, sense, 5′ CCG GCT GCC TTG GAC TAT CTA CAT TCT CGA GAA TGT AGA TAG TCC AAG GCA GTT TTT G 3′; Akt3 antisense, 5′ AAT TCA AAA ACTGCCTTGGACTATCTACATTCTCGA GAA TGT AGA TAG TCC AAG GCA G 3′ (Sigma-Aldrich). For silencing of Afadin, specific sequences for l-Afadin were used: shAfadin #2, sense, 5′ CCG GAA GGT CAA GAT GTA TCC AAT ACT CGA GTA TTG GAT ACA TCT TGA CCT TTT TTT G 3′; shAfadin #2, antisense: 5′ AAT TCA AAA AAA GGT CAA GAT GTA TCC AAT ACT CGA GTA TTG GAT ACA TCT TGA CCT T 3′; shAfadin #3, sense: 5′ CCG GAA ACT TGA CAT TCA AGG AAC GCT CGA GCG TTC CTT GAA TGT CAA GTT TTT TTT G 3′; shAfadin #3, antisense: 5′ AAT TCA AAA AAA ACT TGA CAT TCA AGG AAC GCT CGA GCG TTC CTT GAA TGT CAA GTT T 3′. The oligonucleotide pair for each target was annealed and inserted into pLKO. To produce lentiviral supernatants, 293T cells were co-transfected with control or shRNA-containing pLKO vectors, VSVG, and psPAX2 for 48 hr.
In Vitro Kinase Assays
HeLa cells were transfected with Myc-Afadin-wild type or Myc-Afadin-S1718A. 24 hr after transfection, cells were serum starved for 16 hr. Afadin was immunoprecipitated from cell extracts and incubated with 500 ng recombinant Akt1 or Akt2 (Cell Signaling Technology) in the presence of 250 μM cold ATP in a kinase buffer for 1 hr at 30°C. The kinase reaction was terminated by addition of SDS-PAGE sample buffer.
Transwell migration assay
Cells (1 × 105) in serum-free medium containing 0.1% BSA were added to upper chambers of transwell filters (8 μm pore size; BD Biosciences) in triplicate. NIH 3T3 -cell-conditioned medium, or growth medium from MCF10A was added to lower chambers. After 2–16 hr incubation at 37°C, non-migrated cells were removed and cells that had migrated to the bottom of the filters were fixed and stained using the Hema-3 stain set Protocol (Fisher Scientific; Pittsburgh, PA).
Immunofluorescence
Cells plated on coverslips were fixed with 2% paraformaldehyde for 10 min, permeabilized with 0.5% Triton X-100 and blocked with 1% BSA in 20 mM Tris-HCl (pH 7.5) for 20 min, then incubated with antibodies for 1 hr (anti-Afadin, anti-pAfadin, anti-pAkt S473, anti-E-cadherin, anti-CENP-A, anti-NuP98, anti-Fibrillarin, anti-Histone H3; 1:200). After washing twice with phosphate-buffered saline (PBS), cells were incubated with Cy3-conjugated anti-mouse IgG antibody or Alexa-flour 488 anti-Rabbit IgG for 1 hr. F-actin was visualized with Alexa Fluor 647-conjugated phalloidin (Life Technologies; Grand Island, NY). Cells were then rinsed twice with PBS and mounted with Prolong Gold antifade reagent-4,6-diamidino-2-phenylindole (DAPI) (Life Technologies). Images of cells were acquired using a fluorescence microscope (Nikon Eclipse Ti) and digital image analysis software (NIS-Elements, Nikon). Magnification plan Apo VC 60x/1.40 oil. Experiments to determine staining with phospho-Afadin S1718 antibody always included co-staining with total Afadin antibody in order to ensure specificity of staining.
Immunoblotting and Immunoprecipitation
Cells were lysed in RIPA as previously described (13). Lysates were resolved on 6–10% acrylamide gels by SDS-PAGE and transferred to PVDF membrane (EMD Millipore; Billerica MA). The blots were blocked in TBST buffer (10 mM Tris-HCl [pH 8], 150 mM NaCl, 0.2% Tween 20) containing 5% (w/v) non-fat dry milk for 30 min and then incubated with the specific primary antibody diluted in blocking buffer at 4°C for 16 hr. Membranes were washed three times in TBST and incubated with horseradish peroxidase-conjugated secondary antibody for 1 hr at room temperature. Membranes were washed three times and developed using enhanced chemiluminescence substrate (EMD Millipore). For immunoprecipitation, lysates were incubated with 1–2 μg antibody for 2–4 hr at 4°C followed by incubation with 15 μl protein A/G Sepharose beads (Amersham Biosciences; Pittsburgh). Immune complexes were washed with NETN buffer (0.5% NP-40, 1 mM EDTA, 20 mM Tris-HCl [pH 8], 100mMNaCl). Precipitates were resolved by SDS-PAGE.
Subcellular Fractionation
Cells were fractionated using the Subcellular protein fractionation kit for cultured cells (Thermo Fisher Scientific; Rockford, IL) according the manufacturer’s instructions.
Tissue Microarrays
Tissue microarrays (TMA) containing normal breast tissue (2 cores per case) and invasive breast cancer (2 cores per case) were constructed from archival FFPE breast tissue specimens obtained from Beth Israel Deaconess Medical Center under an institutionally-approved IRB protocol for discarded de-identified tissues. Double immunofluorescence for Afadin and E-cadherin was performed. Antigen retrieval was performed by boiling the slides for 10 min in 10mM sodium citrate pH6 with a pressure cooker. The sections were then incubated with 1mg/ml sodium borohydride (MP Biomedicals; Solon, OH) for 5 min at room temperature. Sections were incubated with 5% normal donkey serum (Jackson ImmunoResearch) for one hr at room temperature. Slides were then incubated with mouse anti-Afadin (1:100, BD Biosciences) and Rabbit anti-E-Cadherin (1:100, Cell Signaling Technology) overnight at 4°C. The slides were washed and incubated with Alexa 488 conjugated Donkey anti-rabbit or anti-mouse secondary antibodies (Jackson ImmunoResearch Lab, 1:200). Samples were then washed and mounted with Prolong Gold anti-fade mounting media containing DAPI (Invitrogen). We digitally acquired 98 microscopic images of normal breast tissue and 98 microscopic images of invasive breast cancer at 63 X magnification. Features were extracted from the digital images in ImageJ and statistical analyses were performed using Jython and R. To determine the statistical significance of the difference in Afadin nuclear localization score in normal breast as compared with invasive breast cancer we performed a two-sided Student’s T-test.
Results
Akt phosphorylates Afadin at Ser1718
Global phosphoproteomic analyses have revealed that the adherens junction protein Afadin is phosphorylated at serine 1718 (Ser1718) (16), in a sequence within the actin-binding domain that conforms to the optimal Akt consensus motif RXRXXS/T (32). The motif surrounding Ser1718 is evolutionarily conserved from Drosophila to mammals (Fig. 1A). Since the PI 3-K/Aktpathway modulates all phenotypes associated with breast cancer and does so by phosphorylating substrate proteins to transduce the signal, we evaluated Afadin protein expression in breast cancer cell lines. Afadin is highly expressed in various breast cancer cell lines, including basal and luminal molecular subtypes as well as the non-tumorigenic line MCF10A (Fig. 1B).
To determine whether Afadin is a substrate of Akt, MCF10A (Fig. 1C) and HeLa cells (Supplementary Fig. S1A) were serum-starved and stimulated with IGF-1. Stimulation leads to phosphorylation of Afadin at Ser1718 as detected by a phospho-specific anti-pSer1718 antibody. Ser1718 phosphorylation induced by IGF-1 is significantly inhibited by wortmannin (a pan-PI 3-K inhibitor), BEZ-235 (a dual PI 3-K and TORC1 inhibitor), A66 (a p110α specific inhibitor) and MK2206 (an allosteric pan-Akt inhibitor) (33–36) (Fig. 1C). By contrast, Ser1718 phosphorylation is not blocked by Rapamycin (an mTOR inhibitor), GSK650394 (an SGK (serum and glucocorticoid-induced kinase) inhibitor), PF4708671 (an S6K1 (p70 S6-kinase-1) inhibitor) or CGK733 (an ATM/ATR (Ataxia Telangiectasia Mutated/ATM-related (ATR)) inhibitor) (37–41). Akt phosphorylates Afadin specifically at Ser1718 since a Ser1718Ala (S1718A) mutant is not phosphorylated in response to IGF-1, and no additional Akt consensus motifs are found in the Afadin amino acid sequence. Moreover, the short isoform of Afadin (s-Afadin)is not phosphorylated in response to IGF-1, (Fig. 1D), consistent with the fact that it lacks the Ser1718 motif (Fig. 1A)
To determine whether one or more Akt isoform can phosphorylate Afadin in cells, Akt1, Akt2 and Akt3 were silenced using specific shRNA’s introduced into MCF10A cells (Fig. 2A) and HeLa cells (Supplementary Fig. S1B). Silencing individual Akt isoforms partially attenuates Ser1718 phosphorylation, whereas combination silencing of Akt1, Akt2 and Akt3 leads to a complete abrogation of the pSer1718 signal (16% in pLKO versus 100% in pLKO cell stimulated with IGF-1; Akt1 silencing (104%), silencing Akt2 (45%), silencing Akt3 (25%) and combined Akt1/Akt2/Akt3 silencing (34%), normalized relative to total Afadin). Moreover, co-expression of constitutively active, myristoylated Akt1, Akt2 and Akt3 alleles leads to enhanced Afadin Ser1718 phosphorylation (Fig. 2B), and purified recombinant Akt1 or Akt2 can directly phosphorylate Afadin at Ser1718 in an in vitro protein kinase assay (Fig. 2C). Similarly, co-expression of the oncogenic PIK3CA alleles H1047R and E545K that stimulate hyperactivation of Akt also induces Afadin Ser1718 phosphorylation in MCF10A cells (Fig. 2D) and HeLa cells (Supplementary Fig. S1C). In aggregate, these data demonstrate that Afadin is phosphorylated by all Akt isoforms downstream of PI 3-K, but is not a substrate for other AGC kinases including SGKs and S6K1.
Figure 2. PI 3-K and Akt Signaling Promotes Afadin Phosphorylation in breast cancer cells.

A. Akt1, Akt2 and Akt3 shRNA transduced alone or in combination in MCF10A cells. Cells were stimulated with IGF-1 and whole cell lysates immunoblotted with the indicated antibodies. B. MCF10A cells transfected with control vector or myristoylated Akt alleles (MyrAkt1,2,3), cells were serum starved for 16 hr and whole cell lysates immunoblotted with the indicated antibodies. C. In vitro kinase assay using wild type Afadin, Afadin Ser1718Ala (S1718A) or control immunoprecipitated with Myc antibody. Akt substrate GSK-3β expressed in HeLa cells and immunoprecipitated with HA antibody and served as a positive control. Precipitates were incubated with purified recombinant Akt1 or Akt2. Whole cell lysates were immunoblotted with the indicated antibodies. D. MCF10A cells infected with vector control, PIK3CA wild type, PIK3CA H1047R, PIK3CA E545K, cells serum starved and whole cell lysates immunoblotted with the indicated antibodies. In all cases p85 served as loading control. Results are representative of at least three independent experiments.
Phosphorylation of Afadin at Ser1718 promotes nuclear localization
Since Afadin is an adherens junction protein, we next evaluated the consequence of Ser1718 phosphorylation by Akton cellular localization. Using immunofluorescence of IGF-1-stimulated MCF10A cells, we detect phosphorylated, activated Akt (pS473) within 20 min of stimulation (Supplementary Fig. S2). Under these conditions, total Afadin shows a predominantly membrane-restricted localization. However, within 1 hr of stimulation, Afadin membrane localization is significantly diminished, concomitant with the appearance of nuclear localization as evidenced by co-staining with DAPI (Fig. 3A and Supplementary Fig. S2). Nuclear localization of Afadin is most evident by 6 hr post-stimulation, with a punctate nuclear staining pattern (Fig. 3A, IGF-1, 6hr). Nuclear translocation of total Afadin is dependent on PI 3-K and Akt activity, since wortmannin, MK2206, A66 and BEZ235 block nuclear localization in favor of membrane localization (Fig. 3A and Supplementary Fig. S3A). Quantification of the nuclear translocation in response to IGF-1 and Akt inhibitor is depicted in the bar graph (Fig. 3A). The pSer1718 antibody also reveals Afadin nuclear localization in response to IGF-1 stimulation (Fig. 3B, quantification shown in the corresponding bar graph).
Figure 3. IGF-1 Stimulation Promotes Afadin Nuclear Localization.

A. MCF10A cells were serum starved and stimulated with IGF-1 for 1 or 6 hr, either alone or following treatment with MK2206 (1μM). Immunofluorescence was performed with the indicated antibodies. Quantification of the nuclear staining is presented in the bar graph. (Student T-test, * p<0.05, **p<0.01).
B. MCF10A cells serum starved or stimulated for 6 hr with IGF-1, and immunofluorescence performed with the indicated antibodies. In all cases nuclei were stained with DAPI. Images are representative of multiple fields, and of at least three independent experiments. Quantification of the nuclear staining is presented in the bar graph. (Student T-test, **p<0.01). Magnified single channel staining for Afadin is presented in Supplementary Fig S4.
To explore the contribution of Ser1718 in plasma membrane to nucleus translocation, an shRNA silencing and rescue experiment was performed. MCF10A cells were transduced with Afadin shRNA and non-silenceable wild-type, Ser1718Ala (S1718A)and Ser1718Glu (S1718E) mutants transiently expressed. As predicted, in full serum conditions, wild-type Afadin is localized to the plasma membrane and nucleus, Ser1718Ala Afadin is localized predominantly to the plasma membrane, whereas the phosphomimetic Ser1718Glu mutant shows an exclusively nuclear localization (Fig. 4A, quantification shown in the corresponding bar graph). Moreover, co-expression of constitutively activated, myristoylated Akt1, Akt2 or Akt3 alleles also promotes Afadin nuclear localization compared to control cells (Fig. 4B and corresponding bar graph, and Supplementary Fig. S3B). Therefore, Akt signaling promotes the relocalization of Afadin from the plasma membrane to the nucleus in a manner that depends on Ser1718 phosphorylation.
Figure 4. Phosphorylation of Afadin Promotes Nuclear Relocalization.

A. MCF10A cells infected with control, pLKO or Afadin shRNA, and transfected with wild-type (WT) Afadin, Ser1718Ala (S1718A) or Ser1718Glu (S1718E) and immunofluorescence performed with the indicated antibodies. Quantification of the nuclear staining of Afadin is presented in the graph (** p<0.01, Student T-test). B. MCF10A cells transfected with pcDNA3 or myristoylated Akt1 alleles and immunofluorescence performed with the indicated antibodies. In all cases nuclei were stained with DAPI. Quantification of the nuclear staining of Afadin is presented in the bar graph (* P<0.05, Student T-test). Images are representative of multiple fields, and of at least three independent experiments. Magnified single channel staining for Afadin is presented in Supplementary Fig S4.
To further explore the mechanism of Afadin nuclear translocation, cell fractionation was performed. In agreement with the immunofluorescence data, IGF-1 stimulation of MCF10A cells results in a time-dependent decrease of total Afadin from the cytoplasm and membrane compartments, concomitant with an increase of Afadin in the nuclear compartment, most dramatically evident 6 hr post-stimulation (Fig. 5A, left panels). Treatment with the Akt inhibitor MK2206 attenuates this translocation (Fig. 5A, right panels). We also evaluated the consequence of Afadin phosphorylation on protein stability. Serum-starved cells were stimulated over time with IGF-1 in the presence or absence of the protein synthesis inhibitor cycloheximide as well as Akt inhibitor. As observed in Fig. 5B, prolonged treatment of cells with MK 2006 results in a reduction of total Afadin expression, which is further enhanced in the presence of cyclohexamide. Similar results are observed when the same cells are examined by immunofluorescence (Fig. 5C and corresponding bar graph). These data indicate that Akt signaling, in addition to promoting nuclear relocalization, also promotes stabilization of Afadin.
Figure 5. Afadin Phosphorylation Modulates Protein Stability.

A. MCF10A cells serum starved for 18 hr and stimulated with IGF-1 for 3 or 6 hr. Alternatively, cells in full serum were treated with MK2206 for 3 or 6 hr. Cytoplasmic and nuclear extracts were prepared and immunoblotted with the indicated antibodies. B and C, MCF10A cells serum starved for 18 hr and stimulated for 3 or 6 hr with IGF-1 either alone or with 20μg/ml cycloheximide for 3 or 6 hr. Alternatively, cells in full serum were treated with MK2206 as indicated. Whole cell lysates were blotted with the indicated antibodies (B) or immunofluorescence was performed (C) Quantification of the nuclear staining of Afadin is presented in the bar graph (* p<0.05, Student T-test). Results are representative of multiple fields and at least three independent experiments. Magnified single channel staining for Afadin is presented in Supplementary Fig S4.
Phosphorylation of Afadin at Ser1718 enhances migration and perturbs cell-to cell adhesion
We next reasoned that since Akt promotes relocalization of Afadin from adherens junctions to the nucleus, this would likely have a profound impact on cell adhesion and cell migration, phenotypes that are dependent on intact adherens junctions. In this context, previous studies have shown that Afadin shRNA enhances migration of MCF7, SK-BR3 and MDA-MB-231 cells (27). Yet in other studies Afadin silencing reportedly enhances cell adhesion in T cells (42). The contribution of Afadin to cell migration is therefore likely to be context dependent. In BT549 and MDA-MB-468 breast cancer cells, that express high levels of Afadin and exhibit PI 3-K pathway activation due to PTEN inactivation and consequently predominantly nuclear localized Afadin (Supplementary Fig S5A and S5B), silencing Afadin with specific shRNA leads to a profound inhibition of cell migration (Supplementary Fig. S5C). Conversely, expression of wild-type or phospho-mimetic Ser1718Asp (S1718D) or Ser1718Glu (S1718E)Afadin in T47D cells, that do not express Afadin, leads to enhanced cell migration in a manner that is not phenocopied by the Ser1718Ala mutant (Fig. 6A). The cellular localization of these mutants expressed in T47D cells is in agreement with the localization observed in MCF10A cells (Supplementary Fig. S6 compared to Fig. 4A). Consistent with the finding that Afadin promotes cell migration of breast cancer cells, silencing Afadin in MCF10A cells profoundly blocks migration in a manner that is partially rescued by re-expression of wild-type and Ser1718Asp (S1718D), but not Ser1718Ala (S1718A) mutants (Fig. 6B). Taken together, these results demonstrate that Afadin promotes breast cancer cell migration in a manner that depends, at least in part, on Ser1718 phosphorylation mediated by Akt.
Figure 6. Afadin Phosphorylation Promotes Cell Migration.

A. T47D transfected with wild-type (WT) Afadin, Ser1718Ala (S1718A), Ser1718Asp (S1718D) or Ser1718Glu (S1718E)Afadin. Transwell cell migration was assessed. In all cases data are represented as mean ± SEM. *p<0.05, **p<0.01 (Student T-test). B. MCF10A cells infected with Afadin shRNA and transfected with wild-type (WT), Ser1718Ala (S1718A) or Ser1718Asp (S1718D) mutants. Transwell cell migration was evaluated. In all cases data are represented as mean ± SEM. **p<0.01 (Student T-test). C. MCF10A cells transduced with shRNA with or without transfection of Afadin Ser1718Asp (SD) were evaluated by immunofluorescence with the indicated antibodies. Results are representative of multiple fields and at least three independent experiments. D. Immunofluorescence of MCF10A cells was performed on growing cells using the indicated nuclear markers antibodies (green channel), Afadin antibodies (red channel) and DAPI staining (blue channel). Results are representative of multiple fields and three independent experiments.
Finally, since Afadin is localized to adherens junctions (20), we evaluated the consequence of Afadin relocalization on cell to cell adhesion using E-cadherin staining measured by immunofluorescence. In control serum starved MCF10A cells, both Afadin and E-cadherin show restricted membrane localization with defined cell to cell adhesion (Fig. 6C). By contrast, in cells transduced with Afadin shRNA, E-cadherin staining is significantly disrupted. A similar phenotype is observed in cells in which Afadin is silenced and the nuclear localized Ser1718Asp (S1718D) mutant is re-expressed (Fig. 6C, controls of wild type Afadin, S1718A and S1718E Afadin are shown in Supplementary Fig. S7). We conclude that membrane-localized Afadin is required for maintaining intact adherens junctions and productive cell to cell adhesion, such that loss of membrane localization and nuclear relocalization disrupts adhesion, concomitant with an increase in cell migration.
In order to address the specific nuclear compartment that Afadin localizes to, we performed co-localization experiments using a number of established nuclear markers: CENP-A, a centromere marker; NuP98, a nuclear envelope marker; Fibrillarin, a nucleolar marker; and Histone H3, a nucleosome or chromatin marker (Fig. 6D). The punctate nuclear pattern of Afadin does not colocalize with any of these markers. Future studies will address the specific nuclear compartment that Afadin localizes to, and the importance of this localization for the nuclear function of Afadin.
Afadin localization in breast cancer
We next assessed Afadin localization in human breast cancer. Tissue microarrays containing normal breast epithelium and invasive breast cancer tissue obtained from archival pathology specimens from 49 patients were used for localization evaluated by immunofluorescence using Afadin and E-cadherin staining. The quantification protocol and analysis is summarized in Supplementary Fig. S8. We identified significantly increased nuclear localization of Afadin in invasive breast cancer as compared to normal breast, with 53% higher nuclear localization in invasive breast cancer (mean Afadin nuclear localization score in normal = 0.019 vs. mean Afadin nuclear localization score in cancer = 0.029; p, 0.02). Fig. 7 shows representative images of normal breast tissue and invasive breast cancer specimens. From these data we conclude that the nuclear localization of Afadin, regulated by phosphorylation at Ser1718 by the Akt pathway, is clinically relevant for breast cancer progression.
Figure 7. Afadin Localization in Breast Cancer.
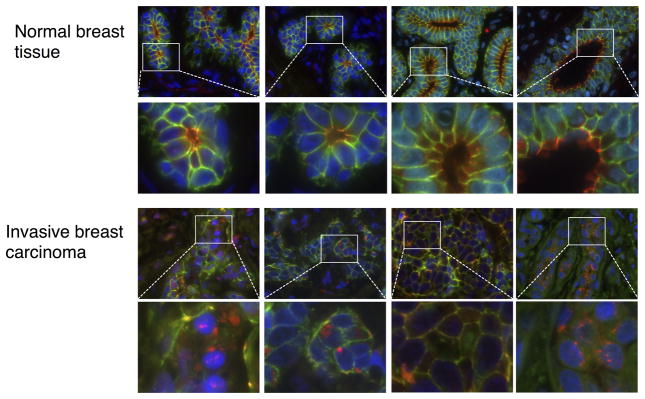
Tissue microarrays containing normal and invasive breast cancer from surgical specimens were subjected to immunofluorescence with the following antibodies: Afadin (red channel), E-cadherin (green channel) and DAPI (blue channel). Digital images were acquired with 63 X magnification. Representative images from normal tissue and invasive breast cancer are shown.
Discussion
We have identified and characterized a new substrate of Akt, the adherens junction protein Afadin. This finding adds to the list of the over 200 currently identified substrates of Akt kinases that transduce the PI 3-K/Akt signal to a plethora of biological and pathophysiological responses, particularly in the context of cancer (43). We have shown that Akt phosphorylates Afadin at Ser1718 in a motif that is evolutionarily conserved, indicating that this phosphorylation has evolved to modulate a key biological event. We have shown that physiological signaling in non-tumorigenic MCF10A cells stimulated with IGF-1 leads to Afadin phosphorylation, and that in breast cancer cell lines harboring pathway mutations such as oncogenic PIK3CA and PTEN inactivation, Afadin is phosphorylated in an Akt-dependent manner. Moreover, although several other AGC kinases such as a S6K and SGKs have an overlapping optimal consensus phosphorylation motif to Akt, only Akt is capable of phosphorylating Afadin in cells. Although a number of Akt isoform-specific have recently been identified, Afadin does not appear to be an isoform-specific substrate, at least in the breast cancer cell lines tested here.
Since mutations in genes that encode proteins in the PI 3-K and Akt signaling pathway are among the most common and frequent in human cancers, especially breast cancer, there are currently numerous clinical trials targeting both PI 3-K and Akt for therapeutic benefit. Hyperactivation of Akt due to oncogenic PIK3CA mutations as well as amplification and somatic mutations in Akt genes are common events in breast cancer etiology, and have been shown to result in cell transformation and cancer progression using mouse models. While the mechanisms by which PI 3-K and Akt promote cell transformation are well understood, the mechanisms by which this pathway promotes cancer progression at the level of tumor dissemination, invasion and metastasis are not as well characterized. In this context, it is now well-established that Akt isoforms play differential roles in promoting breast cancer cell invasion and metastasis, whereby Akt1 does not enhance metastasis or can actually function as an invasion and metastasis suppressor, yet Akt2 promotes invasive migration leading to metastatic dissemination (12,44). A role for Akt3 in breast cancer progression has yet to be defined. The mechanistic basis for the differential role of Akt isoforms in mediating breast cancer progression is likely complex, and involves both differential intracellular localization as well as accessibility to specific substrates that control cell migration and invasion. A number of such substrates have been identified, including the actin-bundling protein palladin, Girdin and ACAP1 (9,13,14). In this context, it is interesting to note that Ser1718 lies within a previously-identified actin-binding domain (18), and as such it is possible that Ser1718 phosphorylation may modulate actin binding to promote cell migration, though this remains to be determined. Regardless, identifying the specific mechanisms by which the Akt pathway controls the phosphorylation of substrates that mediate cell migration is critical for a complete understanding of the contribution of this pathway in cancer progression, and in turn the development of drugs to target Akt kinases therapeutically.
We have shown that phosphorylation of Afadin promote relocalization from adherens junctions to the nucleus. This is most evident when evaluating Afadin localization by immunofluorescence, whereby IGF-1 stimulation leads to a relocalization of total and pSer1718 Afadin from the plasma membrane to the nucleus (Fig. 3). Similarly, a Ser1718Ala non-phosphorylatable mutant is membrane restricted and moreover a phosphomimetic Ser1718Asp mutant is constitutively localized to punctate nuclear structures in breast epithelial cells (Fig. 4). Most importantly, this localization phenocopies cell migration, whereby the Ser1718Ala cannot rescue the deficit in Transwell migration induced by Afadin shRNA, whereas the Ser1718Asp mutant can (Fig. 6). We conclude that Afadin phosphorylation at Ser1718 by Akt promotes cell migration, concomitant with a relocalization from the membrane to the nucleus. Although localization of Afadin to the nucleus is dependent on productive Akt signaling and was observed in all experiments and in many cells visualized by immunofluorescence, it was not a quantitative event observed in 100% of the cell population. This is not surprising, however, since previous studies have reported cell-to-cell variability with respect to the activation status of Akt in a population of cells as result of PIK3CA heterogeneity (45). The proposed model is a bimodal distribution of Akt activation that is an invariable characteristic of exponentially growing cells. Limiting Akt activity to only 20%–30% of cells in a population serves, according to this study, two related purposes: it prevents senescence and maintains suboncogenic levels of PI 3-K activity in large populations.
What is more surprising is the relocalization of an adherens junction protein from the membrane to the nucleus in response to a single posttranslational modification, most obviously identified by the localization pattern of the Ser1718Asp and Ser1718Glu mutant Afadin. However there is some precedent to Afadin nuclear localization, since the short form of Afadin, s-Afadin, has been shown to be a dual-residency protein that localizes to the nucleus and to the plasma membrane in a manner dependent on growth factor signaling (46). In this study l-Afadin was not detected in the nucleus, although this is likely due to distinct experimental conditions and antibodies used to detect Afadin localization. What specifically mediates the translocation of Afadin from adherens junctions to the nucleus remains to be defined, and likely involves a multi-step process of nuclear import and retention.
However nuclear translocation is associated with an increase in breast epithelial and cancer cell migration. There are likely to be several mechanisms by which changes in Afadin localization mediate cell migration, since knocking out Afadin using specific shRNA decreases cell migration. At the same time, expression of wild-type or Ser1718Asp mutant Afadin that is nuclear-restricted robustly enhances cell migration (Fig. 6B), indicating the simple removal of Afadin from adherens junction is not the only mechanism that affords cell migration. How a nuclear localized Afadin promotes cell migration remains to be determined, but could involve the induction of a transcriptional program. Although there is no evidence that Afadin can function as a transcriptional co-activator, this is reminiscent of β-catenin, also a component of adherens junctions that upon Wnt signaling translocates to the nucleus and functions as a transcriptional co-activator for the TCF/LEF transcription factor complex, and that in turn initiates a range responses including the epithelial to mesenchymal transition (EMT) (47). Whether Afadin functions in a similar manner remains to be determined. However, it is intriguing to note that either knocking out Afadin with shRNA or expression of a nuclear restricted Afadin mutant (Ser1718Asp) results in significant disruption of E-cadherin staining at the membrane (Fig. 6C).
Interestingly, loss of Afadin has been suggested to be a marker of poor prognosis in breast cancer, such that loss of Afadin actually promotes cell migration of MCF7, MDA-MB-231 and SKBR3 cells as measured in non-directional wound healing assays (27). In our studies performed in Transwell assays, in MCF10A, BT549 and MDA-MB-468 specific Afadin shRNA suppresses cell migration towards chemoattractants, and this can be effectively rescued by introduction of wild-type or phosphomimetic Afadin alleles (Fig. 6B). Therefore, the specific contribution of Akt signaling to Afadin and in turn cell migration is likely to be highly context-dependent, including the level of Afadin expression as well as the genetic background, in particular PI 3-K/Aktpathway mutations.
In summary, our study identifies Afadin as a new substrate of Akt that mediates cell migration in a manner that is dependent on cellular localization. Moreover, we show that nuclear-localized Afadin is a feature of human tumors as evident from localization studies from tissue microarrays. We propose that the phosphorylation of Afadin, an adherens junction protein that is traditionally thought to reside exclusively at cell to cell adhesions and whose phosphorylation modulates cell migration, is a previously uncharacterized mechanism by which the Akt pathway promotes cancer progression. In this context, although Afadin was originally defined as a “tumor-suppressor-like” protein, it may also serve to function as a “tumor-promoting” protein at least in situations of pathophysiological PI 3-K and Akt signaling.
Supplementary Material
Acknowledgments
Financial support: This work was supported by grant BC097703 from the Breast Cancer Research Program fellowship, Department of Defense (to S.E.); by NIH grant R01CA092644 (to A.T.); and by an award from the Klarman Family Foundation (to A.H.B.).
The authors thank Mihaela Lorger for providing the Afadin expression plasmids, Lay-Hong Ang for TMA staining and members of the Toker laboratory for discussions.
Footnotes
Conflict of interest disclosure: No potential conflicts of interest are disclosed.
Authors Contributions’
Conception and design: A. Toker, S. Elloul
Acquisition of data: S. Elloul
Analysis and Interpretation of data (biochemical assays, cell biology, immunofluorescence): S. Elloul
Analysis and Interpretation of data (immunofluorescence): D. Kedrin
Analysis and Interpretation of data (tissue microarrays): S. Elloul, N. W. Knoblauch, A.H. Beck
Writing, review and/or revision of manuscript: A. Toker and S. Elloul
Study supervision: A. Toker
References
- 1.Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–7. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 2.Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov. 2009;8:627–44. doi: 10.1038/nrd2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhao JJ, Liu Z, Wang L, Shin E, Loda MF, Roberts TM. The oncogenic properties of mutant p110alpha and p110beta phosphatidylinositol 3-kinases in human mammary epithelial cells. Proc Natl Acad Sci US A. 2005;102:18443–8. doi: 10.1073/pnas.0508988102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- 5.Rodon J, Dienstmann R, Serra V, Tabernero J. Development of PI3K inhibitors: lessons learned from early clinical trials. Nat Rev Clin Oncol. 2013;10:143–53. doi: 10.1038/nrclinonc.2013.10. [DOI] [PubMed] [Google Scholar]
- 6.Sun M, Wang G, Paciga JE, Feldman RI, Yuan ZQ, Ma XL, et al. AKT1/PKBalpha kinase is frequently elevated in human cancers and its constitutive activation is required for oncogenic transformation in NIH3T3 cells. Am J Pathol. 2001;159:431–7. doi: 10.1016/s0002-9440(10)61714-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Staal SP. Molecular cloning of the akt oncogene and its human homologues AKT1 and AKT2: amplification of AKT1 in a primary human gastric adenocarcinoma. Proc Natl Acad Sci US A. 1987;84:5034–7. doi: 10.1073/pnas.84.14.5034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pérez-Tenorio G, Stål O Southeast Sweden Breast Cancer Group. Activation of AKT/PKB in breast cancer predicts a worse outcome among endocrine treated patients. Br J Cancer. 2002;86:540–5. doi: 10.1038/sj.bjc.6600126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Enomoto A, Murakami H, Asai N, Morone N, Watanabe T, Kawai K, et al. Akt/PKB regulates actin organization and cell motility via Girdin/APE. Dev Cell. 2005;9:389–402. doi: 10.1016/j.devcel.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 10.Park BK, Zeng X, Glazer RI. Akt1 induces extracellular matrix invasion and matrix metalloproteinase-2 activity in mouse mammary epithelial cells. Cancer Research. 2001;61:7647–53. [PubMed] [Google Scholar]
- 11.Irie HY, Pearline RV, Grueneberg D, Hsia M, Ravichandran P, Kothari N, et al. Distinct roles of Akt1 and Akt2 in regulating cell migration and epithelial-mesenchymal transition. J Cell Biol. 2005;171:1023–34. doi: 10.1083/jcb.200505087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chin YR, Toker A. Function of Akt/PKB signaling to cell motility, invasion and the tumor stroma in cancer. Cellular Signalling Elsevier Inc. 2009;21:470–6. doi: 10.1016/j.cellsig.2008.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chin YR, Toker A. The Actin-Bundling Protein Palladin Is an Akt1-Specific Substrate that Regulates Breast Cancer Cell Migration. Molecular Cell Elsevier Ltd. 2010;38:333–44. doi: 10.1016/j.molcel.2010.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li J, Ballif BA, Powelka AM, Dai J, Gygi SP, Hsu VW. Phosphorylation of ACAP1 by Akt regulates the stimulation-dependent recycling of integrin beta1 to control cell migration. Dev Cell. 2005;9:663–73. doi: 10.1016/j.devcel.2005.09.012. [DOI] [PubMed] [Google Scholar]
- 15.Lee MJ, Thangada S, Paik JH, Sapkota GP, Ancellin N, Chae SS, et al. Akt-mediated phosphorylation of the G protein-coupled receptor EDG-1 is required for endothelial cell chemotaxis. Molecular Cell. 2001;8:693–704. doi: 10.1016/s1097-2765(01)00324-0. [DOI] [PubMed] [Google Scholar]
- 16.Moritz A, Li Y, Guo A, Villen J, Wang Y, MacNeill J, et al. Akt-RSK-S6 Kinase Signaling Networks Activated by Oncogenic Receptor Tyrosine Kinases. Sci Signal. 2010;3:ra64–4. doi: 10.1126/scisignal.2000998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Takai Y, Ikeda W, Ogita H, Rikitake Y. The immunoglobulin-like cell adhesion molecule nectin and its associated protein afadin. Annu Rev Cell Dev Biol. 2008;24:309–42. doi: 10.1146/annurev.cellbio.24.110707.175339. [DOI] [PubMed] [Google Scholar]
- 18.Mandai K, Nakanishi H, Satoh A, Obaishi H, Wada M, Nishioka H, et al. Afadin: A novel actin filament-binding protein with one PDZ domain localized at cadherin-based cell-to-cell adherens junction. J Cell Biol. 1997;139:517–28. doi: 10.1083/jcb.139.2.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ikeda W, Nakanishi H, Miyoshi J, Mandai K, Ishizaki H, Tanaka M, et al. Afadin: A key molecule essential for structural organization of cell-cell junctions of polarized epithelia during embryogenesis. J Cell Biol. 1999;146:1117–32. doi: 10.1083/jcb.146.5.1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Takai Y. Nectin and afadin: novel organizers of intercellular junctions. Journal of Cell Science. 2003;116:17–27. doi: 10.1242/jcs.00167. [DOI] [PubMed] [Google Scholar]
- 21.Prasad R, Gu Y, Alder H, Nakamura T, Canaani O, Saito H, et al. Cloning of the ALL-1 fusion partner, the AF-6 gene, involved in acute myeloid leukemias with the t (6;11) chromosome translocation. Cancer Research. 1993;53:5624–8. [PubMed] [Google Scholar]
- 22.Komura H, Ogita H, Ikeda W, Mizoguchi A, Miyoshi J, Takai Y. Establishment of cell polarity by afadin during the formation of embryoid bodies. Genes Cells. 2008;13:79–90. doi: 10.1111/j.1365-2443.2007.01150.x. [DOI] [PubMed] [Google Scholar]
- 23.Su L, Hattori M, Moriyama M, Murata N, Harazaki M, Kaibuchi K, et al. AF-6 controls integrin-mediated cell adhesion by regulating Rap1 activation through the specific recruitment of Rap1GTP and SPA-1. J Biol Chem. 2003;278:15232–8. doi: 10.1074/jbc.M211888200. [DOI] [PubMed] [Google Scholar]
- 24.Lorger M. Regulation of epithelial wound closure and intercellular adhesion by interaction of AF6 with actin cytoskeleton. Journal of Cell Science. 2006;119:3385–98. doi: 10.1242/jcs.03027. [DOI] [PubMed] [Google Scholar]
- 25.Miyata M, Rikitake Y, Takahashi M, Nagamatsu Y, Yamauchi Y, Ogita H, et al. Regulation by Afadin of Cyclical Activation and Inactivation of Rap1, Rac1, and RhoA Small G Proteins at Leading Edges of Moving NIH3T3 Cells. Journal of Biological Chemistry. 2009;284:24595–609. doi: 10.1074/jbc.M109.016436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Severson EA, Lee WY, Capaldo CT, Nusrat A, Parkos CA. Junctional adhesion molecule A interacts with Afadin and PDZ-GEF2 to activate Rap1A, regulate beta1 integrin levels, and enhance cell migration. Mol Biol Cell. 2009;20:1916–25. doi: 10.1091/mbc.E08-10-1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fournier G, Cabaud O, Josselin E, Chaix A, la iuml de JAE, Isnardon D, et al. Loss of afadin, a marker of poor outcome in breast cancer, induces cell migration, invasiveness and tumor growth. Oncogene. 2011;30:3862–74. doi: 10.1038/onc.2011.106. [DOI] [PubMed] [Google Scholar]
- 28.Kapeller R, Toker A, Cantley LC, Carpenter CL. Phosphoinositide 3-kinase binds constitutively to alpha/beta-tubulin and binds to gamma-tubulin in response to insulin. J. Biol. Chem. 1995;270:25985–91. doi: 10.1074/jbc.270.43.25985. [DOI] [PubMed] [Google Scholar]
- 29.Ramaswamy S, Nakamura N, Vazquez F, Batt DB, Perera S, Roberts TM, et al. Regulation of G1 progression by the PTEN tumor suppressor protein is linked to inhibition of the phosphatidylinositol 3-kinase/Akt pathway. Proc Natl Acad Sci US A. 1999;96:2110–5. doi: 10.1073/pnas.96.5.2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ding VW, Chen RH, McCormick F. Differential regulation of glycogen synthase kinase 3beta by insulin and Wnt signaling. J Biol Chem. 2000;275:32475–81. doi: 10.1074/jbc.M005342200. [DOI] [PubMed] [Google Scholar]
- 31.Isakoff SJ. Breast Cancer-Associated PIK3CA Mutations Are Oncogenic in Mammary Epithelial Cells. Cancer Research. 2005;65:10992–1000. doi: 10.1158/0008-5472.CAN-05-2612. [DOI] [PubMed] [Google Scholar]
- 32.Obata T, Yaffe MB, Leparc GG, Piro ET, Maegawa H, Kashiwagi A, et al. Peptide and protein library screening defines optimal substrate motifs for AKT/PKB. J Biol Chem. 2000;275:36108–15. doi: 10.1074/jbc.M005497200. [DOI] [PubMed] [Google Scholar]
- 33.Powis G, Bonjouklian R, Berggren MM, Gallegos A, Abraham R, Ashendel C, et al. Wortmannin, a potent and selective inhibitor of phosphatidylinositol-3-kinase. Cancer Research. 1994;54:2419–23. [PubMed] [Google Scholar]
- 34.Serra V, Markman B, Scaltriti M, Eichhorn PJA, Valero V, Guzman M, et al. NVP-BEZ235, a dual PI3K/mTOR inhibitor, prevents PI3K signaling and inhibits the growth of cancer cells with activating PI3K mutations. Cancer Research. 2008;68:8022–30. doi: 10.1158/0008-5472.CAN-08-1385. [DOI] [PubMed] [Google Scholar]
- 35.Jamieson S, Flanagan JU, Kolekar S, Buchanan C, Kendall JD, Lee WJ, et al. A drug targeting only p110α can block phosphoinositide 3-kinase signalling and tumour growth in certain cell types. Biochem J. 2011;438:53–62. doi: 10.1042/BJ20110502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hirai H, Sootome H, Nakatsuru Y, Miyama K, Taguchi S, Tsujioka K, et al. MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol Cancer Ther. 2010;9:1956–67. doi: 10.1158/1535-7163.MCT-09-1012. [DOI] [PubMed] [Google Scholar]
- 37.Brown EJ, Albers MW, Shin TB, Ichikawa K, Keith CT, Lane WS, et al. A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature. 1994;369:756–8. doi: 10.1038/369756a0. [DOI] [PubMed] [Google Scholar]
- 38.Sherk AB, Frigo DE, Schnackenberg CG, Bray JD, Laping NJ, Trizna W, et al. Development of a small-molecule serum- and glucocorticoid-regulated kinase-1 antagonist and its evaluation as a prostate cancer therapeutic. Cancer Research. 2008;68:7475–83. doi: 10.1158/0008-5472.CAN-08-1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pearce LR, Alton GR, Richter DT, Kath JC, Lingardo L, Chapman J, et al. Characterization of PF-4708671, a novel and highly specific inhibitor of p70 ribosomal S6 kinase (S6K1) Biochem J. 2010;431:245–55. doi: 10.1042/BJ20101024. [DOI] [PubMed] [Google Scholar]
- 40.Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER, Hurov KE, Luo J, et al. ATM and ATR Substrate Analysis Reveals Extensive Protein Networks Responsive to DNA Damage. Science. 2007;316:1160–6. doi: 10.1126/science.1140321. [DOI] [PubMed] [Google Scholar]
- 41.Alao JP, Sunnerhagen P. The ATM and ATR inhibitors CGK733 and caffeine suppress cyclin D1 levels and inhibit cell proliferation. Radiat Oncol. 2009;4:51. doi: 10.1186/1748-717X-4-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang Z, Rehmann H, Price LS, Riedl J, Bos JL. AF6 negatively regulates Rap1-induced cell adhesion. J Biol Chem. 2005;280:33200–5. doi: 10.1074/jbc.M505057200. [DOI] [PubMed] [Google Scholar]
- 43.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–74. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dillon RL, Muller WJ. Distinct biological roles for the akt family in mammary tumor progression. Cancer Research. 2010;70:4260–4. doi: 10.1158/0008-5472.CAN-10-0266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yuan TL, Wulf G, Burga L, Cantley LC. Cell-to-cell variability in PI3K protein level regulates PI3K-AKT pathway activity in cell populations. Curr Biol. 2011;21:173–83. doi: 10.1016/j.cub.2010.12.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Buchert M, Poon C, King JAJ, Baechi T, D’Abaco G, Hollande F, et al. AF6/s-afadin is a dual residency protein and localizes to a novel subnuclear compartment. J Cell Physiol. 2007;210:212–23. doi: 10.1002/jcp.20853. [DOI] [PubMed] [Google Scholar]
- 47.Kim W, Kim M, Jho E-H. Wnt/β-catenin signalling: from plasma membrane to nucleus. Biochem J. 2013;450:9–21. doi: 10.1042/BJ20121284. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.