

SUMMARY
B7-H1 (PD-L1) on immune cells plays an important role in T cell coinhibition by binding its receptor PD-1. Here we show that both human and mouse intestinal epithelium expressed B7-H1 and that B7-H1-deficient mice were highly susceptible to dextran sodium sulfate- or trinitrobenzenesulfonic acid-induced gut injury. B7-H1 deficiency during intestinal inflammation led to high mortality and morbidity, which were associated with severe pathological manifestations in the colon, including loss of epithelial integrity and overgrowth of commensal bacteria. Results from bone marrow chimeric and knock-out mice showed B7-H1 expressed on intestinal parenchyma, but not on hematopoietic cells, controlled intestinal inflammation in an adaptive immunity-independent fashion. Finally, we demonstrated that B7-H1 dampened intestinal inflammation by inhibiting TNF-α production and by stimulating IL-22 from CD11c+CD11b+ lamina propria cells. Thus, our data uncover a new mechanism by which intestinal tissue-expressed B7-H1 functions as an essential ligand for innate immune cells to prevent gut inflammation.
INTRODUCTION
The gut lumen hosts 90% of the microorganisms in the human body. This microbiota community benefits the host by extracting energy and nutrients from food, by preventing colonization of pathogenic species (Hooper and Gordon, 2001), and by regulating immune cell function (Maloy and Powrie, 2011). The epithelial intestinal barrier is an essential boundary that precludes bacterial entry and maintains mucosal homeostasis. Deregulation of intestinal homeostasis, with concomitant aberrant activation of mucosal innate and adaptive immunity, can result in gut injury, inflammation, and inflammatory bowel disease (IBD). However, the fundamental pathophysiologic mechanisms underlying IBD are still unclear.
The interaction between the B7 family members and their receptors provides critical costimulation and coinhibition, which regulate T cell function. In addition to the long-established pathway of B7-1/B7-2-CD28/CTLA-4, the interaction between B7-H1 (PD-L1) (Dong et al., 1999; Freeman et al., 2000), a member of the B7 family, and its receptor PD-1, a member of the CD28 family, has a major role in inhibiting T cell responses (Freeman et al., 2000) and in inducing CD8 T cell exhaustion during viral infections (Day et al., 2006). Since B7-H1 is mainly expressed on immune cells and PD-1 is expressed on activated T cells, research on this pathway has primarily focused on T cell coinhibition; nonetheless, the role of the B7-H1/PD-1 pathway in T cell-mediated intestinal inflammation and IBD remains largely unknown. A few earlier works report that the administration of anti-B7-H1 suppresses intestinal inflammation (Kanai et al., 2003), while the loss of PD-1/PD-L1 signaling leads to expansion of gut antigenspecific CD8 T cells (Reynoso et al., 2009) and the PD-1 blockade in SIV-infected monkeys enhances repair of gut-associated junctions (Shetty et al., 2012). B7-H1 can also be detected on some tissue cells (Liang et al., 2003), but its function is largely unexplored. Here we identified epithelium-expressed B7-H1 as a key regulator of intestinal inflammation and colitis by inhibiting innate immune cells.
RESULTS
B7-H1 Protects From Mortality and Morbidity in Two Models of Intestinal Injury
To investigate B7-H1 function in gut immunity we chose a chemical model of intestinal injury utilizing oral administration of dextran sodium sulfate (DSS) that injures the colonic epithelium (Okayasu et al., 1990) and triggers potent inflammatory responses (Rakoff-Nahoum et al., 2004). B7-H1-deficient (B7-H1−/−) mice showed much higher mortality and morbidity (weight loss, anal bleeding, diarrhea and anal erosion scores) upon DSS administration (2%; wt/vol) for 6 days (Figure 1A) than wild-type (WT) mice. While less than 20% of B7-H1−/− mice survived, more than 70% of WT mice remained alive. Higher DSS concentration (4%) for a shorter time (5 days) provided similar results with an earlier onset of disease (data not shown). We extended studies to a 2,4,6-trinitrobenzenesulfonic acid (TNBS)-induced colitis model and obtained similar results (Figure S1A). Thus, B7-H1 is critical for controlling intestinal epithelial injury and inflammation.
Figure 1. B7-H1−/− Mice are Hypersensitive to DSS-induced Intestinal Inflammation.
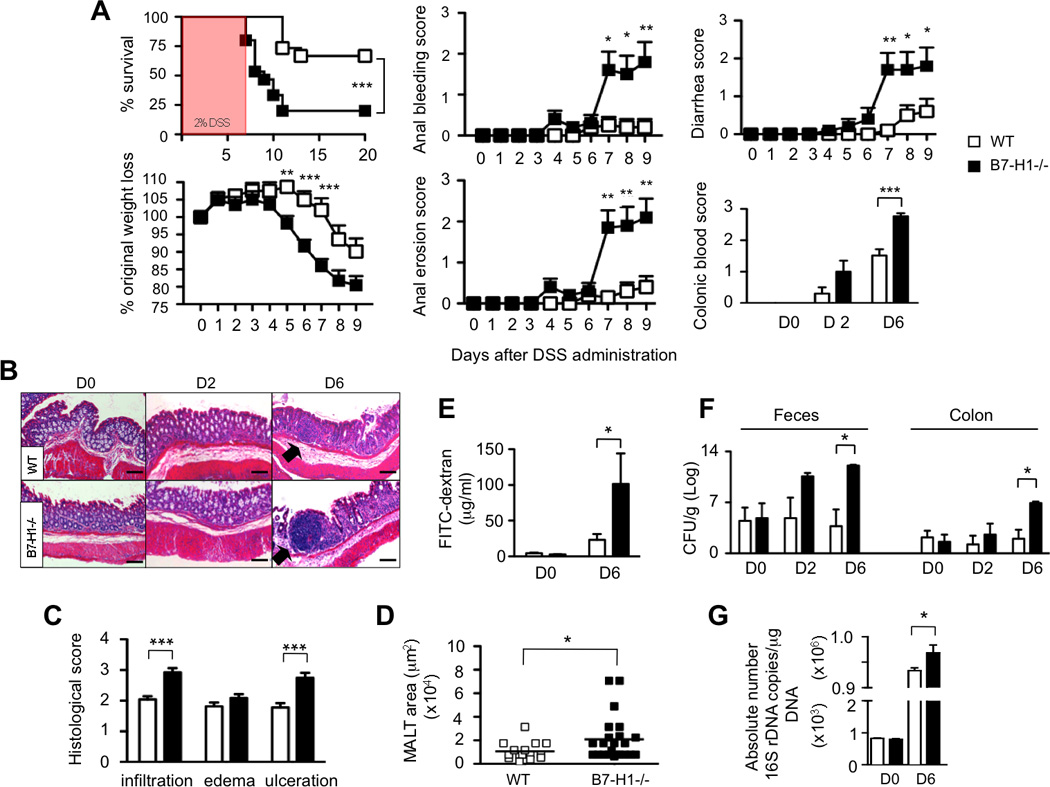
(A) Wild-type (WT) and B7-H1−/− mice were fed with 2% DSS in drinking water for 7 days. Survival, percentage of original weight loss, anal bleeding, diarrhea, anal erosion and colonic blood scores were monitored. Survival data are represented as Kaplan-Meier curves, ***P<0.001; Log-rank test. Data were pooled from two independent experiments (n=15/group). *P<0.05; **P<0.01; ***P<0.001; Student’s t test.
(B) HE-stained colon tissue of DSS-treated WT and B7-H1−/− mice (D0, D2, D6).
(C, D) Histopathological scoring of infiltrating leukocytes, edema, ulceration and MALT area (n=20 MALTs/mouse) in WT and B7-H1−/− mice (D6) (scale bars, 100µm; arrows indicate MALTs). ***P<0.001; Student’s t test.
(E) DSS-fed mice were gavaged (D0, D6) with FITC-dextran. 3h later FITC-fluorescence was determined in sera. Data are pooled from two independent experiments (n=6–9). *P<0.05; Student’s t test.
(F) CFU in feces and colon of DSS-fed mice (D0, D2, D6); *P<0.05; Mann-Whitney U test.
(G) qPCR analysis of 16S rDNA copies for the whole bacterial kingdom in the feces of DSS-treated (D0, D6) WT and B7-H1−/− mice. Data were pooled from two independent experiments, n=3–4. *P<0.05; Student’s t test.
We dissected the causes of death and morbidity in B7-H1−/− mice by histopathological analyses of colon tissues. While colonic bleeding in B7-H1−/− mice occurred earlier than in WT mice (Figure 1A), both group of mice became anemic at day 6 of DSS treatment (data not shown). Hematoxylin/eosin (HE) analyses confirmed that B7-H1−/− mice displayed more severe ulceration, extensive epithelium erosion and cellular infiltration (Figure 1B, C). In addition, we found an enlargement of distinct lymphoid aggregations known as “mucosal associated lymphoid tissues” (MALTs) beneath the epithelium in B7-H1−/− mice (Figure 1B, D), which correlates with severe pathological changes (Laukoetter et al., 2007). Neither naïve B7-H1−/− nor naïve WT mice showed signs of colonic inflammation or tissue damage.
Augmented mucosal permeability and intestinal barrier dysfunction are associated with colitis development in mice (Garrett et al., 2007) and IBD patients (Cobrin and Abreu, 2005). Therefore, we investigated the role of B7-H1 in maintaining gut epithelial integrity by orally gavaging mice with FITC-dextran (Garrett et al., 2007). We found a 4-fold increase of FITC-dextran levels in sera of B7-H1−/− mice at day 6 of DSS treatment (Figure 1E), suggesting that B7-H1 is important for regulation of intestinal permeability during gut injury.
Intestinal microbial populations vary enormously between IBD patients and healthy individuals (Hooper and Gordon, 2001), therefore we analyzed their concentration and composition. After DSS administration, B7-H1−/− mice had higher bacteria colony-forming units (CFU) not only in feces and colon (Figure 1F), but also in mesenteric lymph nodes (MLN), spleen and liver (data not shown) than WT mice, suggesting the presence of a bacterial systemic dissemination in B7-H1−/− mice. Quantitative analysis of 16S rDNA confirmed that B7-H1−/− mice had an increased number of total bacteria than WT mice (Figure 1G), but none of the specific intestinal microbiota groups analyzed accounted for this increase (Figure S1B). Altogether, our findings demonstrate that B7-H1 is required to maintain gastrointestinal integrity during gut inflammation and to avoid commensal bacteria overgrowth.
B7-H1 Expression on Intestinal Parenchyma Confers Protection
B7-H1 is constitutively expressed on a wide range of immune and tissue cells and is upregulated after activation (Freeman et al., 2000; Liang et al., 2003). As the colon is the site of DSS-induced disease, we examined B7-H1 expression in the colon. Single cell suspensions from colon of naïve and DSS-treated mice were analyzed by flow cytometry to detect hematopoietic cells (Cytokeratin− CD45+) and parenchymal cells such as epithelial cells (Cytokeratin+CD45−) or myofibroblasts and pericytes (Cytokeratin−CD45−) (Mifflin et al., 2011). We found B7-H1 was expressed on both colonic parenchymal and hematopoietic cells from naïve and DSS-treated WT mice and it was moderately upregulated on Cytokeratin−CD45+ cells at D6 of DSS treatment (Figure 2A). B7-H1 expression was also increased on colon of IBD patients (Figure 2B).
Figure 2. B7-H1 is Expressed in Inflamed Colon and is Required on the Parenchyma for Protection Against DSS-induced Colitis.
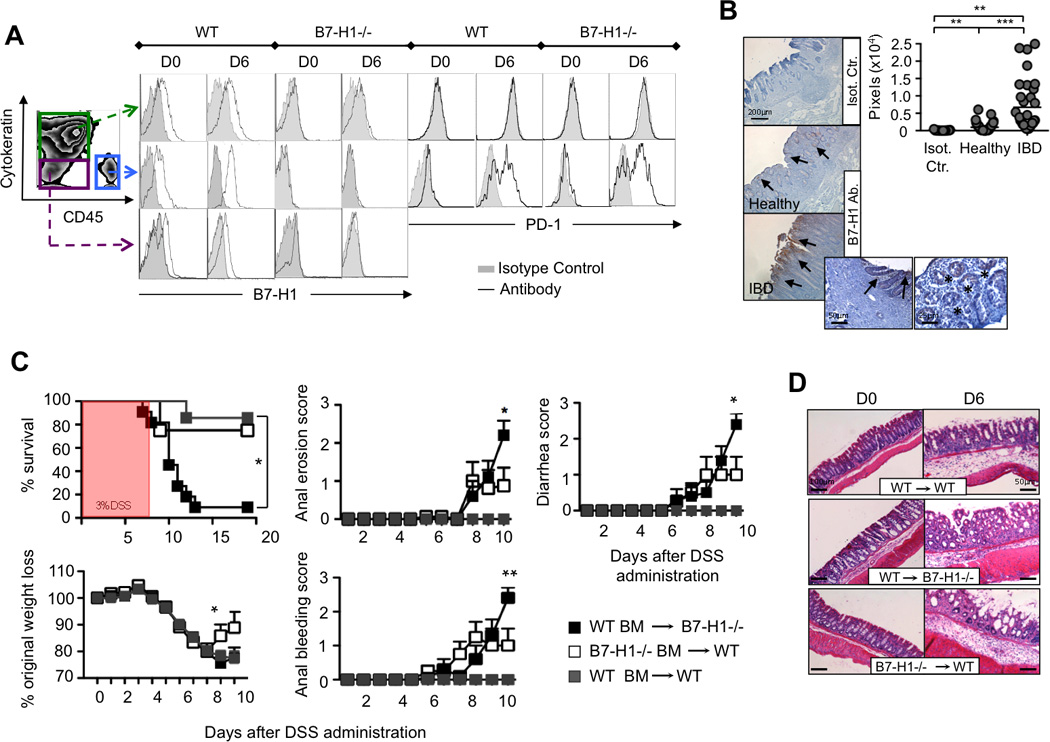
(A) FACS plots of single cell suspensions from colons of DSS-fed mice (D0, D6) representing hematopoietic cells (Cytokeratin-CD45+), epithelial cells (Cytokeratin+CD45-) and myofibroblast or pericytes (Cytokeratin-CD45-) to detect B7-H1 and PD-1 expression (open histograms), whereas shaded histograms indicate isotype control staining, n=4.
(B) Immunohistochemistry staining for B7-H1 in human colon tissue from healthy donors and IBD patients, and relative quantification. Isotype control was used. Arrows and stars indicate focal expression of B7-H1; scale bars: 200, 50 and 25 µm. Quantification is represented as amount of pixels positively stained for B7-H1 by Velocity® software. Data are representative of two independent experiments, n=3. **P<0.01; ***P<0.001; Student’s t test.
(C) Three groups of bone-marrow chimera mice [B7-H1−/− mice reconstituted with WT BM (WT BM→B7-H1−/−), WT mice reconstituted with B7-H1−/− BM (B7-H1−/− BM→WT) or WT BM (WT BM→WT)] were treated with 3% DSS in drinking water for 6 days. Survival, percentage of original weight loss, anal bleeding, diarrhea and anal erosion score were monitored. Data represent one of three independent experiments, n=7–10. Survival data are represented as Kaplan-Meier curve (*P <.05; log-rank test). *P<0.05; **P<0.01; Student’s t test.
(D) HE staining of colon tissue sections of chimera colons (D0, D6). Scale bars: 100 and 50 µm.
PD-1, the receptor for B7-H1, is not expressed on parenchymal cells, but is mainly expressed on activated T and B cells (Agata et al., 1996), on exhausted CD8 T cells (Barber et al., 2006; Day et al., 2006) and on dendritic cells (DCs) (Yao et al., 2009) or monocytes/macrophages (Huang et al., 2009) during infection. We detected PD-1 on colonic hematopoietic cells of both WT and B7-H1−/− mice before and after DSS administration, respectively (Figure 2A).
We next asked whether B7-H1 on hematopoietic cells and/or parenchymal cells was required to inhibit the pathogenic process during DSS-induced disease. We generated B7-H1 bone marrow (BM) chimera mice with control groups (Figure S2A). Upon DSS administration, B7-H1−/− mice reconstituted with WT BM developed significantly worse clinical symptoms as compared to WT mice reconstituted with B7-H1−/− BM or with WT BM (Figure 2C). Only 10% of mice survived in the first group vs 70%–80% in the other two groups. The first group also showed severe histopathological changes in colon tissue sections stained with HE (Figure 2D). These findings, together with the B7-H1 expression pattern on colonic cell populations, suggest that B7-H1 on parenchyma, but not on hematopoietic cells, confers protection from intestinal injury and inflammation.
Effect of B7-H1 on Intestinal Epithelium Homeostasis
To understand how intestinal epithelium-expressed B7-H1 regulates intestinal injury and inflammation, we considered two possible mechanisms: cell-intrinsic alterations and cell-extrinsic interactions. A possible cell-intrinsic mechanism could be a homeostatic imbalance of intestinal epithelium in the absence of B7-H1. We hypothesized that intestinal epithelium-expressed B7-H1 controls intestinal injury by affecting proliferation or by impairing apoptosis of intestinal epithelial cells. To assess the proliferation we used an immunofluorescence staining for Ki67 and the 5-bromo-2'-deoxyuridine (BrdU) assay. Our data showed a slightly higher baseline level (D0) of proliferation in B7-H1−/− mice (Figure S2B), which quickly disappeared at D2 and did not correlate with an alteration of the intestinal epithelial permeability (Figure 1E) or with colonic hyperplasia (data not shown). However, upon inflammation (D6), both B7-H1−/− and WT colonic cells exhibited a similar epithelial proliferative capacity (Figure S2B).
Another possible cell-intrinsic mechanism accounting for the dysregulation of intestinal epithelial cells in B7-H1−/− mice is increased cell death. Analyses of apoptosis and necrosis in colon epithelial cells showed an increased number of both apoptotic and necrotic epithelial cells in B7-H1−/− mice at D6 of DSS-treatment (Figures 3A, B). Altogether these results demonstrated that B7-H1 did not affect tissue repair processes after an injurious insult but it prevents apoptosis and necrosis of intestinal epithelial cells during gut inflammation.
Figure 3. B7-H1 Prevents Intestinal Cell Death and Adaptive Immunity is not Needed for B7-H1 Function.
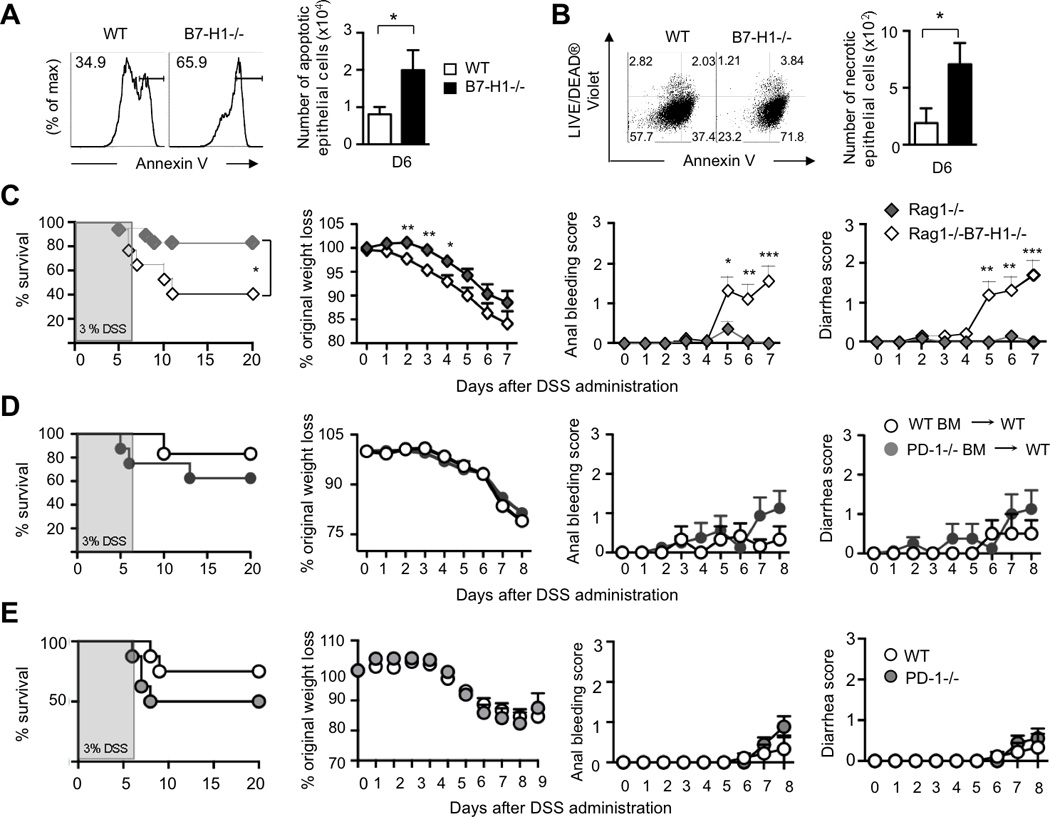
(A) FACS plots for Annexin V+ epithelial cells and quantification of apoptotic epithelial cells in DSS-fed (D6) WT and B7-H1−/− mice. *P<0.05; Student’s t test.
(B) Dot plots and quantification of necrotic epithelial cells double positive for Annexin V and LIVE/DEAD® Violet marker from DSS-fed (D6). Data represent one of three independent experiments; n=3/group. *P<0.05; Student’s t test.
(C) B7-H1−/−Rag1−/− and Rag1−/− were treated with 3% DSS for 6 days and monitored for survival, body weight loss, anal bleeding and diarrhea scores. Data are pooled from two independent experiments, n=17. (D) WT mice were reconstituted with PD-1−/− (PD-1−/−BM→WT) or WT bone marrow cells (WT BM→WT) and then treated with 3% DSS for 6 days. Survival, percentage of weight loss, anal bleeding and diarrhea scores were monitored. Data are representative of two independent experiments, n=6–8.
(E) WT and PD-1−/− mice were fed with 3% DSS in drinking water for 6 days and monitored for survival, body weight loss, anal bleeding and diarrhea score. Data are representative of four independent experiments, n=9. Survival data are showed as Kaplan-Meier curves (*P<0.05; log-rank test). Data represent means ± S.E.M. *P<0.05; **P<0.01; ***P<0.001; Student’s t test.
Protective Function of B7-H1 is not Dependent on Adaptive Immunity
We next explored a cell-extrinsic mechanism by testing the hypothesis that intestinal parenchymal cells expressed B7-H1 could interact with immune cells and, as a consequence, reduce intestinal inflammation. Both innate and adaptive immunity contribute to intestinal inflammation (Maloy and Powrie, 2011). Therefore we analyzed immune cell populations in the colon and MLN, and found no difference in the number of T and B cells, NK/NKT cells, macrophages, DCs, neutrophils, myeloid and innate lymphoid cells (Figure S3A). To assess if components of the adaptive immune system including T and B cells could participate in the B7-H1-mediated protection, we generated Rag-1−/−B7-H1−/− mice and found that these mice exhibited significant higher mortality and morbidity than Rag-1−/− mice after DSS administration (Figure 3C). Compared to WT mice reconstituted with WT BM, WT mice reconstituted with PD-1−/− BM seemed to be slightly more sensitive to DSS-induced colitis without reaching significant difference (Figure 3D), which correlates with no significant difference between PD-1−/− mice and WT mice during DSS treatment (Figure 3E). However, we cannot exclude the involvement of PD-1 on radioresistant PD-1 positive tissue resident innate cells.
These unexpected findings demonstrated that B7-H1 delivered protection from intestinal injury and inflammation in the absence of adaptive immunity.
B7-H1 Inhibits TNF-α Production and Enhances IL-22 Production from CD11c+CD11b+ LP Inflamed Cells
TNF-α has been shown to have a major role in the pathogenesis of IBD (Dharmani et al., 2011). Thus, we measured cytokines in the colon and found B7-H1−/− colons produced about 3-fold more TNF-α than WT colons at D6 of DSS-treatment (Figure 4A); whereas the levels of cytokines IL-6, IL-2p70, IL-4, IFN-γ and MCP-1 were comparable (Figure S3B).
Figure 4. TNF-α Blockade Rescues B7-H1−/− Mice from Intestinal Inflammation and B7-H1-Expressing IECs Promote IL-22 Production.
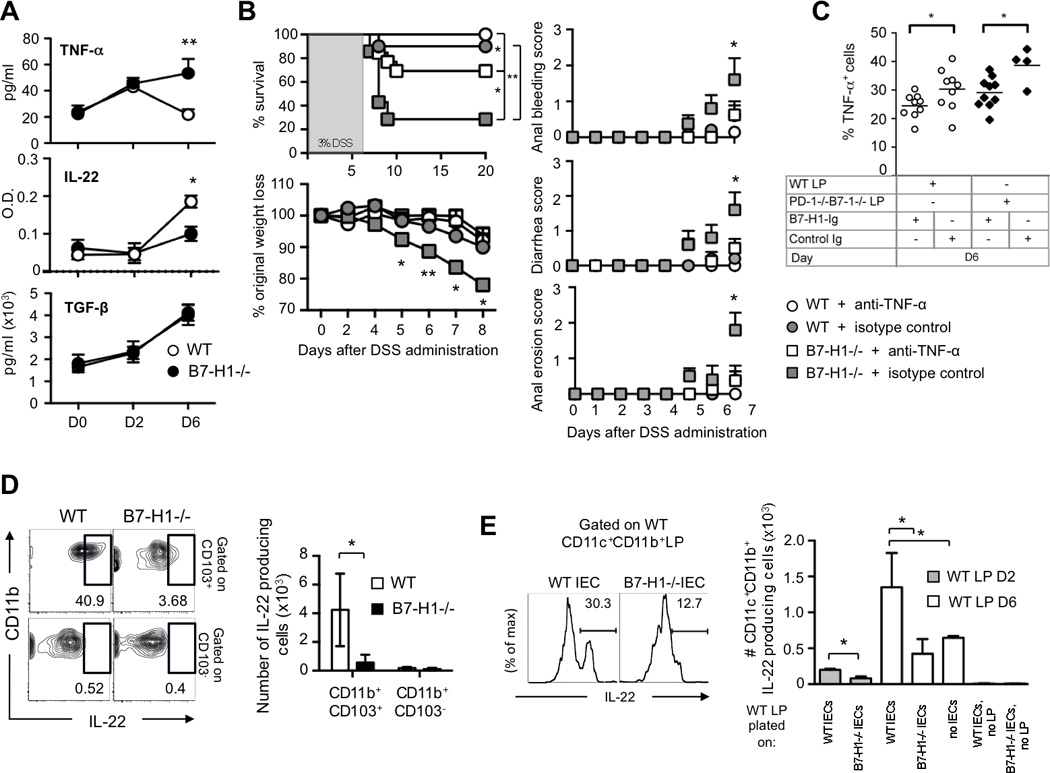
(A) Production of TNF, IL-22 and TGF-β in supernatant of colons from DSS-fed (D0, D2, D6) mice. Data are pooled from three independent experiments (n=4).
(B) WT and B7-H1−/− mice were intraperitoneally injected with anti-TNF-α blocking antibody or isotype control or PBS starting at D0 of 3% DSS treatment and every other day until D20. Survival, percentage of original weight loss, anal bleeding, diarrhea and anal erosion were monitored. Data were pooled from two independent experiments (n=8/group). Survival data are represented as Kaplan-Meier curves. **P<0.01; ***P<0.001; Log-rank test; *P<0.05; **P<0.01; Student’s t test.
(C) LP cells were isolated from WT or PD-1−/−B7-1−/− mice on day 6 post DSS treatment and incubated with plate-bound B7-H1-Ig or control-Ig proteins for 24h. LP cells were stained for CD11c+CD11b+ cells to detect percentage of intracellular TNF-α production by flow cytometry. Data are pooled from at least two independent experiments. Each symbol represents an individual mouse. Data represent means ± S.E.M. *P<0.05; Student’s t test.
(D) FACS plots and numbers of CD11b+CD103+ and CD11b+CD103− LP cells producing IL-22 from WT and B7-H1−/− mice at day 6 of DSS-treatment. Data represent one of two independent experiments, n=3. Data represent means ± S.E.M. *P<0.05; Student’s t test.
(E) FACS plots and quantification of intracellular IL-22 production from CD11c+CD11b+ inflamed LP cells. LP cells were isolated from WT mice (D6 of DSS treatment) and co-cultured with naïve IEC of WT or B7-H1−/− mice for 24h. Numbers above bracketed lines indicate percentage of IL-22-producing cells. Data are pooled from two independent experiments (n=3). *P<0.05; Student’s t test.
Next, we evaluated innate immune cell activity in the colon. Intracellular cytokine staining of inflamed LP cells (D6) revealed that CD11c+CD11b+ cells in B7-H1−/− mice significantly enhanced TNF-α production (Figure S4A) despite a similar expression of surface activation markers in both groups of mice (data not shown). Treatment with anti-TNF-α neutralizing antibody reduced by 50% the mortality in B7-H1−/− mice as compared to isotype-treated mice (Figure 4B). These data indicate that TNF-α is one of the major causes of mortality in B7-H1−/− mice, suggesting a cell-extrinsic mechanism where B7-H1 might inhibit TNF-α production from CD11c+CD11b+ LP cells. To test this possibility, we generated a B7-H1-Ig fusion protein and found that plate-bound B7-H1-Ig protein, but not control Ig protein, suppressed TNF-α production from both WT and PD-1−/−B7-1−/− inflamed LP cells (Figure 4C) as B7-1 has been found as an additional binding molecule (Butte et al., 2007). These data indicate the PD-1/B7-H1 pathway alone or the B7-1/B7-H1 pathway alone may not significantly contribute to the disease onset in B7-H1−/− mice.
IL-22 induces innate immune response during mucosal infections (Aujla et al., 2008) and promotes colonic epithelial cell restitution including induction of anti-microbial peptides such as Reg3-γ (Zheng et al., 2008) and β-defensins (Wolk et al., 2004). We investigated whether the cell-extrinsic mechanism so far observed could promote a release of protective factors like IL-22 and TGF-β. We found WT colon supernatants had higher levels of IL-22 than B7-H1−/− samples, but no difference for TGF-β was detected (Figure 4A). Interestingly, we discovered IL-22 production was strongly produced by WT CD11b+CD103+ dendritic cells, which have a tolerogenic phenotype (Maloy and Powrie, 2011), but not from CD11b+CD103− DCs (Figure 4D). Finally, we found, in a co-culture assay, WT primary intestinal epithelial cells (IEC) were able to stimulate IL-22 production from WT CD11c+CD11b+ LP cells significantly better than B7-H1−/− IEC (Figure 4E). These data proved B7-H1 is required on the intestinal epithelium to dampen the inflammation by CD11c+CD11b+ LP cells in this disease model. Our results provide a new mechanism by which B7-H1 expressed on parenchyma colon reduces inflammation through inhibition of TNF-α and by promoting IL-22 production from CD11c+CD11b+ LP cells. Altogether, our results demonstrate that tissue-expressed B7-H1 is an essential regulator in the control of intestinal inflammation.
DISCUSSION
Beside the classical co-inhibitory function of the B7-H1/PD-1 pathway in T cell activation and immune pathology (Keir et al., 2008), very few data are available on the role of tissue-expressed B7-H1. Here we reported a novel role for B7-H1 in reducing tissue pathology during gut injury as demonstrated by severe morbidity and mortality in B7-H1−/− mice upon DSS or TNBS treatment.
We revealed two unexpected functions for B7-H1. First, B7-H1-mediated protection during intestinal inflammation minimally required the expression of PD-1 on hematopoietic cells as the morbidity and mortality observed in PD1−/− chimera mice was slightly increased but was not statistically different to that of control chimera mice. Second, B7-H1 expressed on tissue cells, but not on hematopoietic cells, was essential for reducing gut pathogenesis because chimera mice lacking B7-H1 expression on parenchyma exhibited an increased mortality and morbidity during DSS-induced colitis, whereas chimera mice lacking B7-H1 expression on hematopoietic cells showed minimal signs of disease. These results, together with other reports in type 1 diabetes (Keir et al., 2006) and LCMV infection (Mueller et al., 2010), highlight an emerging role for tissue-expressed B7-H1 in the control of peripheral immune responses. In addition, we found that adaptive immune response was not required for B7-H1 protection, as Rag-1−/−B7-H1−/− mice developed more severe gut pathogenesis than control Rag-1−/− mice. These data suggest that B7-H1 controls intestinal inflammation through innate immunity, which is in line with previous reports on the role of innate immunity in DSS-induced colitis (Garrett et al., 2007; Rakoff-Nahoum et al., 2004; Zaki et al., 2010).
TNF-α has been recognized as a master cytokine in the onset of DSS-induced colitis (Dharmani et al., 2011) and anti-TNF-α therapy inhibits IEC apoptosis in patients with IBD (Marini et al., 2003). The increased TNF-α production observed in inflamed B7-H1−/− colons corresponded with the peak of mortality and intestinal epithelial apoptosis and the disease was successfully ameliorated by TNF-α blockade, indicating that B7-H1 is one of the major inhibitors of the TNF-α production in this disease model. An in vitro evidence of the B7-H1 inhibitory effect was provided using B7-H1-Ig fusion protein, which impaired the TNF-α production of inflamed CD11c+CD11b+ LP cells isolated from WT or PD-1−/−B7-1−/− mice. These data showed not only that B7-H1 inhibited innate cells but also that the B7-H1/PD-1 pathway alone or the B7-H1/B7-1 pathway alone might not primarily involved. In addition to PD-1 and B7-1, new studies also demonstrate unidentified receptor(s) for B7-H1 (Xu et al., 2013). As the multi-interactions of those molecules could raise some difficulties for the data interpretation of these different pathways, further investigations on the specific functions of those molecules are warranted to better understand their specific roles in the maintenance of intestinal peripheral tolerance.
Different subsets of intestinal dendritic cells are known to play crucial roles in the maintenance of intestinal tolerance by inducing tolerogenic T cell responses including increasing TGF-β and retinoic acid levels (Maloy and Powrie, 2011). Our data showed that while TGF-β was not differentially regulated, IL-22 levels were significantly increased in mice expressing B7-H1. We further proved that IL-22 production was strongly impaired when B7-H1−/− IEC were culture with DSS-inflamed LP. These data revealed a new role for B7- H1 expressed on IEC by regulating IL-22 production for the epithelial restoration process. Thus a lack of B7-H1 could skew the mucosal intestinal responses toward a pro-inflammatory phenotype leading to increased inflammation and injury. Strategies that specifically enhance the activation of B7-H1 in the parenchyma would be beneficial for therapeutic control of IBD.
EXPERIMENTAL PROCEDURES
Mice
B7-H1−/− and PD-1−/− mice were previously described (Dong et al., 2004). C57BL/6, Rag1−/− on C57BL/6 background, and B7-1−/− on C57BL/6 background mice were purchased from Jackson and bred at the Albert Einstein Animal Facility. Rag1−/−B7-H1−/− and PD-1−/−B7-1−/− mice were obtained by intercrossing Rag1−/− with B7-H1−/− and PD-1−/− with B7-1−/− mice, respectively. Mice were housed in the same room in specific pathogen-free facility and used for studies at 7–9 week-old under protocols approved by the Institutional Animal Care and Use Committee.
Experimental colitis models
DSS-mediated colitis was induced by oral administration of 2, 3 and 4% (w/v) of DSS (M.W. 36,000–50,000, MP Biomedicals, LLC) in drinking water for 7, 6 and 5 days, respectively, ad libitum; then water was replaced until day 20. TNBS-mediated colitis was induced by intrarectal instillation of 2,4,6- trinitrobenzenesulfonic acid (TNBS) at 15mg/Kg body weight diluted in 50% ethanol at day 0. The intensity of colitis for both models was monitored daily and clinical parameters were determined as follows: anal erosion (score 0–3; 0=normal; 1=mild; 2=moderate; 3=severe), anal bleeding(score 0–3: 0=normal; 1=mild; 2=moderate; 3=severe), diarrhea (score 0–3; 0=normal; 1=mild; 2=moderate; 3=severe) and percentage of body weight loss.
Statistical Analysis
Data were expressed as mean ± S.E.M. Student's t test or Mann-Whitney test were performed and P values < 0.05 using a 95% confidence interval were considered significant. Survival graphs were represented as Kaplan-Meier curves and analyzed with log-rank test. The GraphPad Prism statistical software program (GraphPad Software) was used for all analyses.
Other detailed experimental procedures and additional references can be found in Extended Experimental Procedures.
Supplementary Material
Highlights.
-
○
B7-H1−/− mice are more sensitive to DSS- and TNBS-induced colitis
-
○
Tissue-expressed B7-H1 induces protection from gut injury
-
○
Adaptive immunity does not contribute the B7-H1-mediated protection
-
○
B7-H1 suppresses TNF-mediated gut inflammation and promotes IL-22
ACKNOWLEDGEMENTS
We thank the Flow Cytometry Core and the Histotechnology and Comparative Pathology Facility of Albert Einstein College of Medicine, Vera Des Marais for the assistance on Volocity® software and Jordan Chinai for reading the manuscript. This work was supported by NIH DP2DK083076, GM094665, AI007289, DOD PC094137, NIH P30CA013330, P60DK020541, AI51519, P30AG038072, T32DK007218, T32DK007513 and T32GM007491.
L.S. designed the study, performed experiments, analyzed data and wrote the manuscript. K.G., K.A.H., Y.M.A and H.J. contributed to discussion. E.L.M., S.C.A., S.G.N. and L.C. provided knock-out mice. E.Y.L and Q.L. provided human colon tissue. X.Z. supervised the study and wrote the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
All authors declare no competing financial interests.
REFERENCES
- Agata Y, Kawasaki A, Nishimura H, Ishida Y, Tsubata T, Yagita H, Honjo T. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int Immunol. 1996;8:765–772. doi: 10.1093/intimm/8.5.765. [DOI] [PubMed] [Google Scholar]
- Aujla SJ, Chan YR, Zheng M, Fei M, Askew DJ, Pociask DA, Reinhart TA, McAllister F, Edeal J, Gaus K, et al. IL-22 mediates mucosal host defense against Gram-negative bacterial pneumonia. Nat Med. 2008;14:275–281. doi: 10.1038/nm1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, Freeman GJ, Ahmed R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682–687. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- Butte MJ, Keir ME, Phamduy TB, Sharpe AH, Freeman GJ. Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity. 2007;27:111–122. doi: 10.1016/j.immuni.2007.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobrin GM, Abreu MT. Defects in mucosal immunity leading to Crohn's disease. Immunol Rev. 2005;206:277–295. doi: 10.1111/j.0105-2896.2005.00293.x. [DOI] [PubMed] [Google Scholar]
- Day CL, Kaufmann DE, Kiepiela P, Brown JA, Moodley ES, Reddy S, Mackey EW, Miller JD, Leslie AJ, DePierres C, et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature. 2006;443:350–354. doi: 10.1038/nature05115. [DOI] [PubMed] [Google Scholar]
- Dharmani P, Leung P, Chadee K. Tumor necrosis factor-alpha and Muc2 mucin play major roles in disease onset and progression in dextran sodium sulphate-induced colitis. PLoS One. 2011;6:e25058. doi: 10.1371/journal.pone.0025058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong H, Zhu G, Tamada K, Chen L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat Med. 1999;5:1365–1369. doi: 10.1038/70932. [DOI] [PubMed] [Google Scholar]
- Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, Fitz LJ, Malenkovich N, Okazaki T, Byrne MC, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192:1027–1034. doi: 10.1084/jem.192.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett WS, Lord GM, Punit S, Lugo-Villarino G, Mazmanian SK, Ito S, Glickman JN, Glimcher LH. Communicable ulcerative colitis induced by T-bet deficiency in the innate immune system. Cell. 2007;131:33–45. doi: 10.1016/j.cell.2007.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper LV, Gordon JI. Commensal host-bacterial relationships in the gut. Science. 2001;292:1115–1118. doi: 10.1126/science.1058709. [DOI] [PubMed] [Google Scholar]
- Huang X, Venet F, Wang YL, Lepape A, Yuan Z, Chen Y, Swan R, Kherouf H, Monneret G, Chung CS, Ayala A. PD-1 expression by macrophages plays a pathologic role in altering microbial clearance and the innate inflammatory response to sepsis. Proc Natl Acad Sci U S A. 2009;106:6303–6308. doi: 10.1073/pnas.0809422106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanai T, Totsuka T, Uraushihara K, Makita S, Nakamura T, Koganei K, Fukushima T, Akiba H, Yagita H, Okumura K, et al. Blockade of B7- H1 suppresses the development of chronic intestinal inflammation. J Immunol. 2003;171:4156–4163. doi: 10.4049/jimmunol.171.8.4156. [DOI] [PubMed] [Google Scholar]
- Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008;26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keir ME, Liang SC, Guleria I, Latchman YE, Qipo A, Albacker LA, Koulmanda M, Freeman GJ, Sayegh MH, Sharpe AH. Tissue expression of PD-L1 mediates peripheral T cell tolerance. J Exp Med. 2006;203:883–895. doi: 10.1084/jem.20051776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laukoetter MG, Nava P, Lee WY, Severson EA, Capaldo CT, Babbin BA, Williams IR, Koval M, Peatman E, Campbell JA, et al. JAM-A regulates permeability and inflammation in the intestine in vivo. J Exp Med. 2007;204:3067–3076. doi: 10.1084/jem.20071416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang SC, Latchman YE, Buhlmann JE, Tomczak MF, Horwitz BH, Freeman GJ, Sharpe AH. Regulation of PD-1, PD-L1, and PD-L2 expression during normal and autoimmune responses. Eur J Immunol. 2003;33:2706–2716. doi: 10.1002/eji.200324228. [DOI] [PubMed] [Google Scholar]
- Maloy KJ, Powrie F. Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature. 2011;474:298–306. doi: 10.1038/nature10208. [DOI] [PubMed] [Google Scholar]
- Marini M, Bamias G, Rivera-Nieves J, Moskaluk CA, Hoang SB, Ross WG, Pizarro TT, Cominelli F. TNF-alpha neutralization ameliorates the severity of murine Crohn's-like ileitis by abrogation of intestinal epithelial cell apoptosis. Proc Natl Acad Sci U S A. 2003;100:8366–8371. doi: 10.1073/pnas.1432897100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mifflin RC, Pinchuk IV, Saada JI, Powell DW. Intestinal myofibroblasts: targets for stem cell therapy. Am J Physiol Gastrointest Liver Physiol. 2011;300:G684–G696. doi: 10.1152/ajpgi.00474.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller SN, Vanguri VK, Ha SJ, West EE, Keir ME, Glickman JN, Sharpe AH, Ahmed R. PD-L1 has distinct functions in hematopoietic and nonhematopoietic cells in regulating T cell responses during chronic infection in mice. J Clin Invest. 2010;120:2508–2515. doi: 10.1172/JCI40040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okayasu I, Hatakeyama S, Yamada M, Ohkusa T, Inagaki Y, Nakaya R. A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology. 1990;98:694–702. doi: 10.1016/0016-5085(90)90290-h. [DOI] [PubMed] [Google Scholar]
- Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell. 2004;118:229–241. doi: 10.1016/j.cell.2004.07.002. [DOI] [PubMed] [Google Scholar]
- Reynoso ED, Elpek KG, Francisco L, Bronson R, Bellemare-Pelletier A, Sharpe AH, Freeman GJ, Turley SJ. Intestinal tolerance is converted to autoimmune enteritis upon PD11 ligand blockade. J Immunol. 2009;182:2102–2112. doi: 10.4049/jimmunol.0802769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shetty RD, Velu V, Titanji K, Bosinger SE, Freeman GJ, Silvestri G, Amara RR. PD-1 blockade during chronic SIV infection reduces hyperimmune activation and microbial translocation in rhesus macaques. J Clin Invest. 2012;122:1712–1716. doi: 10.1172/JCI60612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolk K, Kunz S, Witte E, Friedrich M, Asadullah K, Sabat R. IL-22 increases the innate immunity of tissues. Immunity. 2004;21:241–254. doi: 10.1016/j.immuni.2004.07.007. [DOI] [PubMed] [Google Scholar]
- Xu D, Fu HH, Obar JJ, Park JJ, Tamada K, Yagita H, Lefrancois L. A potential new pathway for PD-L1 costimulation of the CD8-T cell response to Listeria monocytogenes infection. PLoS One. 2013;8:e56539. doi: 10.1371/journal.pone.0056539. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.