

Abstract
Muscle plasticity is defined as the ability of a given muscle to alter its structural and functional properties in accordance with the environmental conditions imposed on it. As such, respiratory muscle is in a constant state of remodeling, and the basis of muscle’s plasticity is its ability to change protein expression and resultant protein balance in response to varying environmental conditions. Here, we will describe the changes of respiratory muscle imposed by extrinsic changes in mechanical load, activity, and innervation. Although there is a large body of literature on the structural and functional plasticity of respiratory muscles, we are only beginning to understand the molecular-scale protein changes that contribute to protein balance. We will give an overview of key mechanisms regulating protein synthesis and protein degradation, as well as the complex interactions between them. We suggest future application of a systems biology approach that would develop a mathematical model of protein balance and greatly improve treatments in a variety of clinical settings related to maintaining both muscle mass and optimal contractile function of respiratory muscles.
Introduction
The basic structure and function of respiratory muscles do not differ from those of other skeletal muscles. Skeletal muscle is the most abundant tissue in the human body, accounting for approximately 40% of total body mass. It gives us the ability to walk, run, breathe, talk, eat, and perform numerous other daily activities. In order to accomplish such a wide range of tasks, muscles, including respiratory muscles, are unique in their structure, fiber type composition and neural control.
From birth until death, skeletal muscle is in a constant state of remodeling in order to adjust to changes in load, activity, or innervation. This unique plasticity allows muscle to alter its structural and functional properties in accordance with its imposed environmental conditions. This is widely recognized in sports, where muscle changes imposed by training in athletes leads to obvious phenotypic modifications that optimize the specific performance of the muscle. The number of muscular contractions (activity) and the degree of loading appear to be the dominant stimuli for training-imposed muscle changes. For example, body builders perform low frequency, high load contractions that result in muscle growth (i.e., hypertrophy) and an increase in force-generating capacity. On the other hand, marathon runners perform high frequency, low load contractions that are not associated with hypertrophy, but cause muscle fibers to assume a more fatigue-resistant phenotype. Although genetic pre-disposition is also important, these adaptations, substantially contribute to the different physical attributes of body builders and marathon runners.
The importance of skeletal muscle, however, extends far beyond exercise physiology to many clinical applications and disease states. Musculoskeletal diseases such as age-related sarcopenia, cancer-induced cachexia, or congenital muscular disorders including dystrophies and lipid or glycogen storage diseases may result from a muscle’s inability to adapt to different stimuli. Just as there is not one exercise regimen for everyone, there is not just one treatment for musculoskeletal diseases. The variety of treatments used for these diseases highlights the complexity of skeletal muscle plasticity. Importantly, in general terms, the structure and function of respiratory muscles do not differ from those of other skeletal muscles; yet respiratory muscles serve a life-sustaining behavior: breathing. Thus, specific examination of respiratory muscle plasticity is the subject of great interest.
Changes in structural and functional properties of muscle are largely the consequence of altered protein expression in which either the amount or type of protein is altered to meet functional demands. Although the cellular-scale structural and functional changes related to skeletal muscle plasticity have been characterized for many disease states, molecular-scale changes in protein balance are not as well characterized. Since muscle is the largest reserve of protein in the body, any change in the balance between protein synthesis and protein degradation could have significant consequences not only for that specific muscle but for the system as a whole. Developments in molecular and cell biology have helped us begin to understand the mechanisms regulating changes in protein expression and balance, although many areas remain underexplored. The specific regulation of protein balance that will serve as the focus of this review has not been elucidated for many diseases or physiological states.
Respiratory Muscles
Respiratory muscles serve to implement the primary function of the lung: to provide gas exchange by supplying O2 and removing CO2 from the blood stream. The muscles involved in ventilation, the actual movement of air into the lungs, are called pump muscles. On the other hand, airway muscles are another group of respiratory muscles that control the caliber of upper and lower airways and are comprised of both skeletal (upper airways) and smooth (trachea and bronchi) muscles. Respiratory muscles must adapt to varying environmental and pathological conditions, and like other skeletal muscles, they are structurally plastic to allow functional adaptation. In this review, we will examine the basis of respiratory muscle plasticity with a focus on skeletal muscles.
Pump vs. airway muscles
There are two main types of skeletal muscles involved in respiration: pump muscles and upper airway muscles. The role of pump muscles is to move air into the lungs. The focus here will be on the major pump muscle, the diaphragm muscle, which is unique to mammals. The diaphragm muscle is a thin, dome-shaped sheet of muscle that is inserted into the lower ribs and serves as a septum separating the thoracic (pleural) and abdominal (peritoneal) compartments of the body.
Contraction of the diaphragm generates a negative intrathoracic pressure leading to inflation of the lungs (inspiration). With contraction, the diaphragm moves caudally, thereby increasing the vertical dimension of the thoracic cavity and exerting a force at the point of its attachment to the rib cage. This creates a decrease in pleural pressure and an increase in abdominal pressure, resulting in increased transdiaphragmatic pressure, i.e., the pressure difference between abdominal and thoracic cavities. The mechanics of diaphragm muscle contraction become more complicated in the zone of apposition, i.e., the region where the diaphragm is in direct contact with the inner aspect of the lower rib cage (157, 208). In this region, the increase in abdominal pressure by diaphragm muscle contraction may actually cause an increase in intrapleural pressure, contributing to ribcage expansion (143). This would cause a different loading to be applied to appositional components of the diaphragm compared to the main body of the diaphragm. This distinction is important; however, very little information regarding adaptive responses of these diaphragm components is available.
With relaxation of the diaphragm muscle, the recoil forces of the lung and chest wall cause a rapid decrease in transdiaphragmatic pressure and air moves out of the lungs, a process known as expiration. Thus, the diaphragm muscle serves as an active inspiratory pump for lung ventilation. Under certain conditions, post-inspiratory diaphragm activity can slow expiratory airflow through a “braking” action. These actions of the diaphragm are important for a number of controlled voluntary expiratory behaviors such as vocalization (126) as well as involuntary expulsive behaviors such as emesis, coughing or sneezing (147, 159, 160, 240).
In addition to the diaphragm muscle, which is responsible for lower rib cage expansion, there are other primary inspiratory muscles that are phasically recruited with resting ventilation: 1) parasternal intercostal muscles and 2) scalene muscles. The intercostal muscles form two layers that occupy each of the intercostal spaces. The lateral portion contains both internal and external intercostals, but the ventral and dorsal portions contain only internal intercostal muscles (47). The ventral intercostal muscles are called parasternal intercostals. Contraction of intercostal muscles displaces the ribs (49), but more importantly, intercostal activity stabilizes the ribcage, and thereby enhances diaphragmatic contraction and generation of transdiaphragmatic pressure. In addition, the scalene muscles are involved in inspiration. These muscles originate from the transverse processes of the lower five cervical vertebrae and insert on the upper surfaces of the first two ribs (47). Scalene muscle contraction in humans contributes to inspiration, even during minimal inspiratory efforts (45, 193). Although rare, in cases of bilateral diaphragm muscle paralysis, the remaining primary inspiratory muscles are capable of assuming sufficient inspiratory activity to maintain resting ventilation (118). In the more common clinical setting of chronic obstructive pulmonary disease (COPD), the ability of the diaphragm muscle to increase lung volume is often impaired, and the contribution of parasternal intercostal and scalene muscles becomes more obvious and significant (44). In addition to their respiratory function, parasternal intercostal and scalene muscles also contribute to posture, head position, and stabilization of the shoulder and trunk during movements of the upper extremity (46, 104, 131). Respiration may also be assisted by accessory inspiratory muscles, which are quiet during resting ventilation and only recruited under conditions of increased demand such as during exercise or due to pathological conditions (i.e., amyotrophic lateral sclerosis, emphysema) (88, 183). Accessory muscles include the sternocleidomastoid, external intercostal, and internal interosseous intercostal muscles (47, 67). Because of the multifunctional nature of these muscles, unambiguous interpretation of respiratory related plasticity is difficult.
Skeletal muscles are also involved in regulating upper airway patency muscle, i.e., controlling the caliber of the conduits for air movement into and out of the lungs. There are three major parts to the conductive airways: the upper airways constituting the nose, mouth and pharynx; the middle airways comprising the larynx, trachea and primary bronchi; and the lung airways comprising the more distal bronchi, bronchioles, alveolar ducts, alveolar sacs and alveoli. All of the airways must be patent in order to effectively move air into and out of the lungs. The airway muscles serve to actively regulate the diaphragm muscle of the airways and thereby decrease resistance to airflow during both inspiration and expiration, match ventilation of alveoli to their perfusion and prevent aspiration of foreign objects or noxious material into the lungs. In particular, skeletal muscles in the upper airways exhibit activation patterns that are coordinated with those inspiratory pump muscles in order to effectively couple low airway resistance to air inflow into the lungs during periods of reduced intrathoracic pressure. Furthermore, upper airway muscles involved in tongue protrusion show plasticity that resembles that of the diaphragm muscle. Specific aspects related to the plasticity of airway muscles will not be reviewed further.
Importance of plasticity of respiratory pump muscles
A unique property of all muscles is that even though they are highly specialized for specific functions, they are all able to modify their properties to meet changing functional demands. In 1958, John Eccles first used the term “plasticity” to refer to this malleability of muscle (60, 175), and today it is a commonly used term in muscle physiology. As such, muscle is in a constant state of remodeling to match changing functional demands. Physiological conditions such as exercise or respiratory disease may change the ventilatory demands required of respiratory muscles such as the diaphragm muscle. In addition, intrinsic changes in ventilatory demands associated with the continuum of life from early development to old age require muscle plasticity to meet these changing functional demands. During early postnatal development, compliance of the chest and lung decreases, altering the ability of the pump muscles to generate intrathoracic pressure (149, 171). Similarly, as a result of pathological conditions such as emphysema, asthma, and prolonged mechanical ventilation, the mechanics of respiratory muscle activation are impaired. Increased force generation by the diaphragm muscle is required to effect the negative intrathoracic pressures needed to sustain ventilation. Clearly, pump muscle plasticity is absolutely essential for life.
In order to understand how respiratory muscles respond to changing demands, it is necessary to understand their complex physiology and innervation, which will be described in the following sections.
Skeletal Muscle Physiology
The pump muscles involved in respiration are skeletal muscles. In mammals, the diaphragm muscle is the major inspiratory muscle and is vital as a ventilatory pump. Because of these functional demands, the diaphragm muscle is much more active than other skeletal muscles (e.g., limb muscles not involved in posture). For instance, the daily duty cycle (ratio of active to inactive times) is ~2% for the extensor digitorum longus muscle and ~14% for the soleus muscle (100). In contrast, the duty cycle of the diaphragm muscle is ~32–40% (123, 237). Beyond its high activity pattern, the diaphragm muscle is also unique because it is a non-weight bearing muscle, unlike other limb and postural muscles. However, the basic cellular and molecular structure and contractile function of respiratory muscles are similar to those of other skeletal muscles.
Skeletal muscle structure
Each skeletal muscle is composed of hundreds to thousands of individual, multinucleated cells called fibers. Each skeletal muscle cell is multinucleated, with the nuclei peripherally located in mature fibers. A single muscle fiber has a precise structure that is defined by myofibrils, which give skeletal muscle its striated appearance due to the arrangement of thick and thin filaments within myofibrils and the alignment of myofibrils within the muscle fiber (90, 140, 156). Myofibrils are composed of large polymerized protein molecules including myosin and actin, which are responsible for muscle contraction.
The sarcomere is the functional unit of the myofibril. Each sarcomere contains thick filaments and thin filaments, which are anchored to the Z-disc (Figure 1). The Z-disc passes crosswise across the myofibril and also from myofibril to myofibril, thus attaching the myofibrils to one another across the entire muscle fiber. Each myofibril in the muscle fiber is surrounded by sarcoplasmic reticulum, an intracellular membrane network that is involved in regulation of intracellular Ca2+ concentration. T-tubules are deep invaginations of the plasma membrane which allow depolarization of the membrane to quickly penetrate to the interior of the muscle cell. The sarcoplasmic reticulum is an intracellular network, whereas T-tubules are in contact with extracellular space.
Figure 1.
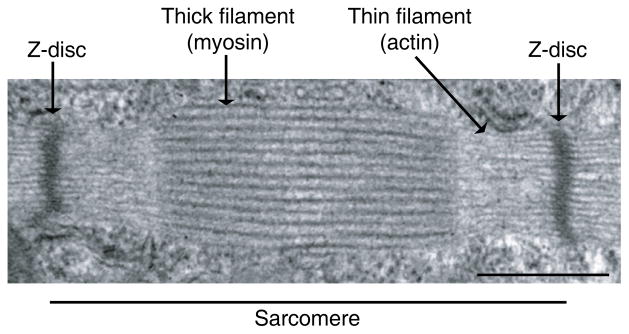
Transmission electron micrograph of a skeletal muscle sarcomere. The Z-disc defines the boundary of the sarcomere. The striations are formed by the highly organized arrangement of thick and thin filaments. Scale bar represents 500 nm.
Thick and thin filaments are highly organized. Thin filaments are composed of actin and the regulatory proteins tropomyosin and troponin. Thick filaments are made of myosin. Cross-bridges are small projections from the sides of the myosin filaments and protrude from the surfaces of myosin filaments. This actin-myosin interaction is the molecular basis for force generation (184). When muscle fibers contract, the thick and thin filaments do not change their size. Instead, the interaction between the myosin heads and actin pulls the thin filaments past the thick filaments, known as the sliding filament mechanism (107). In the relaxed state, the ends of actin filaments from two successive Z-discs barely overlap one another, but remain adjacent to the myosin filaments. In the contracted state, the actin filaments are pulled inward among the myosin filaments, such that their ends now overlap one another.
The ability of a muscle fiber to generate force and accomplish different functions is mainly determined by its myofibrillar protein composition, specifically myosin isoform composition. Myosin is a hexameric protein composed of two myosin heavy chains (MyHC) each with a molecular weight of about 200 kDa and four myosin light chains (MyLC) with molecular weights of about 20 kDa each. The heavy chains wrap around each other to form a double helix, known as the tail of the myosin molecule. One end of each heavy chain is a globular structure called the myosin head. There are also two light chains to each myosin head, and the light chains help control the function of the head during contraction.
Myosin molecules are arranged in the thick filament such that the tail points inward toward the center of the sarcomere, and the heads are on the outer ends of each thick filament. Part of each myosin molecule hangs off to the side in the head, providing an arm that extends the head outward away from the body. The arms and heads are known as cross-bridges. These cross-bridges are flexible at two points, where the arm leaves the body of the myosin filament and where the head attaches to the arm. The hinged heads contribute to the actual contraction process because they can bind and move along actin in the presence of Ca2+ and ATP. In addition to myosin and actin, myofibrils contain structural elements such as mitochondria and sarcoplasmic reticulum. Muscle contraction causes changes in the area of mitochondria and release of Ca2+ from the sarcoplasmic reticulum.
Several isoforms of myosin heavy and light chains exist. In the rat diaphragm muscle all four isoforms of MyHC are present: MyHCslow, MyHC2A, MyHC2X, MyHC2B (65, 240, 241, 249). In the human diaphragm muscle, MyHCslow, MyHC2A, and MyHC2X isoforms have been reported. There is debate as to whether there is persistent expression of embryonic and neonatal MyHC isoforms in the diaphragm muscle of adult humans (132, 168, 260). Regardless, differences in the expression and relative proportion of different MyHC isoforms determine the contractile and fatigue properties of the diaphragm muscle. Such differences have been extensively investigated across models of disease, injury or disuse.
Skeletal muscle function
In general, skeletal muscle function can be described by force generation and change in length.
Force generation
Sir Andrew Huxley introduced the first model of the sliding filament theory of muscle contraction in 1957 (107). According to this model of cross bridge cycling, the sliding during muscle contraction occurs when the myosin heads bind firmly to actin, bend at the junction of the head with the neck, and then detach. This “power stroke”, or transition of cross bridges from a force-generating to a non-force-generating state, requires hydrolysis of ATP. In this way, myosin is the motor of muscle contraction, and is able to convert the chemical energy of ATP into mechanical energy.
ATP binding causes the dissociation of myosin from actin. In the absence of ATP, myosin cannot dissociate from actin, and the muscles become stiff (known as rigor mortis in death). After ATP hydrolysis, a shape change is induced such that the myosin head is cocked. Cocking the myosin head puts it in line with a new binding site on the actin filament. Then myosin binds to actin and the power stroke occurs. This power stroke generates force and/or a change in length, pulling the thin filament toward the center of the sarcomere. Binding of another ATP molecule causes dissociation of myosin from actin, and the cycle repeats itself.
The amount of force produced by the muscle depends on the force generated by each cross-bridge and the proportion of cross-bridges that are active and ready to bind to actin. The force per cross-bridge depends on MyHC isoform composition, with MyHCslow-expressing fibers producing less force than the other MyHC isoforms (75–77). The proportion of cross-bridges in the force generating state depends upon the concentration of Ca2+ in the muscle cell cytoplasm. Thus, force generation is strongly affected by myosin and by Ca2+ regulation. Indirectly, force generation depends on the cross-sectional area (CSA) of a muscle fiber, since CSA reflects the number of cross-bridges that can be formed in parallel.
Change in length
The initial length of the contracting muscle (the length-force relationship) influences the proportion of active cross-bridges and thus alters the amount of force produced (83). Force generation is a function of the magnitude of overlap between actin and myosin filaments. A muscle that is highly stretched develops no force because there is no overlap between actin and myosin filaments. As the muscle length decreases, there is increasing overlap between actin and myosin. Thus, the amount of force generated by a muscle increases as sarcomere length decreases (82, 83).
Changes in length as a result of muscle contraction may occur with differing velocity. The velocity of shortening affects the function and ultimately the endurance of muscle since it takes a finite amount of time for cross-bridges to attach. As filaments slide past each other faster and faster (i.e. shortening with increasing velocity), force decreases due to the lower number of attached cross-bridges. On the other hand, as muscle velocity decreases, more cross-bridges have time to attach and generate force, and thus force increases. This is exemplified when someone lifts a light load compared to a heavy load; the light load can be moved much more quickly. These rapid movements, however, have very small muscle strengthening effects since the muscle forces are so low.
The expression of different MyHC isoforms is associated with varying contractile properties of muscle fibers and thus different maximum ATP consumption rates, with MyHC2B and MyHC2X having the highest ATP consumption rates followed by MyHC2A and MyHCslow (93, 209, 245). According to the Fenn effect, energy consumption increases with muscle power output or work performance, peaking during isovelocity shortening at a value corresponding to maximum power output (64). In order to meet the considerable range of ATP consumption demands of the muscle fiber, a reserve capacity for ATP production is required. The reserve capacity for ATP production of diaphragm muscle fibers expressing MyHC2X and MyHC2B is lower compared to fibers expressing MyHC2A and MyHCslow (209, 247). The greater fatigability of diaphragm muscle fibers expressing MyHC2X and MyHC2B may be in part due to this difference in reserve capacity for ATP consumption (65, 242).
Neuromotor control of the diaphragm muscle
Skeletal muscle is controlled by the central nervous system. Motor centers in the brain control the activity of motoneurons in the ventral horns of the spinal cord. These motoneurons synapse on skeletal muscle fibers such that each skeletal muscle fiber is innervated by a single motoneuron, but the same motoneuron usually also innervates other muscle fibers.
Motor unit organization and classification
The functional element of muscle neuromotor control is the motor unit, which consists of a single motoneuron and the group of muscle fibers it innervates (139, 234). Muscle fibers within the motor unit are synchronously activated by an action potential. Based on the velocity of shortening, motor units can be classified into slow-twitch and fast-twitch motor units. Slow-twitch motor units have the smallest CSA and lowest velocity of shortening. These are recruited during tasks that require precise movements but low force or power. Fast-twitch motor units have a larger CSA and higher shortening velocity. They are recruited when high power output is needed. In general, small muscles that react quickly and whose function must be very precise have few muscle fibers per motor unit. Large muscles such as the soleus, which do not require fine control, may have several hundred muscle fibers in a motor unit (120).
By considering the mechanical and fatigue characteristics of the innervated muscle fibers, four groups of motor units are generally described (Figure 2): 1) slow-twitch, fatigue resistant (type S); 2) fast-twitch, fatigue resistant (type FR); 3) fast-twitch, fatigue intermediate (type FInt); and 4) fast-twitch, fatigable (type FF) (27, 29, 30, 65, 241). Muscle fibers within a motor unit generally show similar properties allowing for classification of motor unit types.
Figure 2.

Four different types of motor units - slow-twitch, fatigue resistant (type S), fast-twitch, fatigue resistant (type FR), fast-twitch, fatigue intermediate (type FInt), and fast-twitch, fatigable (type FF) - are classified based on contractile and fatigue properties of innervated muscle fibers (MyHCSlow, MyHC2A, MyHC2X, and MyHC2B). The speed of contraction varies between the motor units. Modified from Mantilla and Sieck (148), with permission.
Muscle fiber type classification
The classification of muscle fiber type has changed throughout the years. The classification schemes represent the continuum of muscle fiber properties. As early as the 1800s, Ranvier observed that the gross appearance of muscle fibers ranged from white to deep red, reflecting the amount of myoglobin content (120). Slow muscles with a reddish appearance because of their greater myoglobin content were called “red”. Conversely, fast muscles store less myoglobin and thus were called “white”.
Another classification scheme is based on the energy metabolism of the muscle fiber (179). This scheme is a combination of myofibrillar ATPase-based and histochemical metabolic enzyme-based classification (8, 173). Histochemical myosin ATPase fiber typing classifies muscle fibers as type I or type II, corresponding to slow- and fast-twitch muscle fibers, respectively. By analyzing enzymes reflecting metabolic pathways that are either aerobic/oxidative or anaerobic/glycolytic (177), this classification scheme was extended into three fiber types: slow-twitch oxidative (SO), fast-twitch oxidative glycolytic (FOG), and fast-twitch glycolytic (FG).
With the development of antibodies against specific MyHC isoforms, muscle fiber type is now commonly classified based on myosin heavy chain (MyHC) isoform expression. Muscle fibers are classified into the following four types: 1) type I (expressing MyHCslow), 2) type IIa (expressing MyHC2A), 3) type IIb (expressing MyHC2B), and 4) type IIx (expressing MyHC2X; Figure 2) (21, 180, 223, 225–227, 241, 248, 249).
Motor unit recruitment
The classic model of neuromotor control by Liddell and Sherrington (139) predicts that motor units are recruited in an orderly fashion in order to generate a wide range of forces. Type S motor units are recruited first, followed in order by FR, FInt, and FF motor units. The mechanism for this orderly recruitment was expanded by the Henneman “size principle,” which provides a recruitment order based on the size of motoneurons and their intrinsic electrophysiological properties (97, 99). Smaller motoneurons with higher membrane input resistance and slower axonal conduction velocity are recruited first for a given synaptic input (27, 28, 98). As such, a relationship between motor unit type, motor unit maximum tetanic force, and recruitment order has been observed. Smaller motoneurons innervate slow-twitch muscle fibers (type I), which produce lower maximum tetanic force and display greater fatigue resistance during repetitive stimulation (Figure 2). Larger motoneurons innervate fast-twitch muscle fibers, which produce larger tetanic forces and display varying fatigue resistance. Among the fast-twitch motor units, type FR have the lowest recruitment threshold, followed by type FInt and FF motor units (27, 256). Size-related motor unit recruitment order is directly related to mechanical and fatigue properties of the muscle unit fibers.
Consistent with the Henneman size principle described earlier, several studies have shown an orderly recruitment of motor units in the diaphragm muscle. During spontaneous breathing in the cat, phrenic motoneurons that were recruited first had slower axonal conduction velocities than motoneurons that were subsequently recruited (54, 55). In several muscles, motoneuron properties relate their size and type. Type S motor units comprise motoneurons that are the smallest (exhibiting the highest input resistance), most excitable (i.e., lowest rheobase) and slowest in axonal conduction velocities within a specific motor pool (27, 278). In agreement, only ~23% of all phrenic motoneurons are recruited during inspiration (112), which is the approximate proportion of type S and FR motor units in the cat diaphragm muscle (65, 241). Thus, phrenic motoneurons that are normally recruited with respiration would comprise type S or FR motor units, and those not normally recruited with respiration belong to type FF or FInt motor units (240). Although diaphragm muscle motor unit recruitment order may be determined by specific synaptic input (101, 102, 163), motoneuron size (54, 55, 112) and motor unit type (235–237, 240) are important in determining recruitment order.
Further studies were performed to estimate the proportion of the motor unit pool in the cat and hamster diaphragm muscle (235, 237, 240) and in the rat diaphragm muscle (147) that would be recruited during ventilatory and non-ventilatory behaviors. Diaphragm muscle forces were estimated by measuring transdiaphragmatic pressure (Pdi) compared to the maximum Pdi generated by bilateral phrenic nerve stimulation (Pdimax). Motor unit recruitment was estimated by assuming that there is an orderly recruitment of motor units with type S and FR units recruited first, and that all motor units of a particular type would be recruited before the next type is recruited. In addition, maximal force production by each motor unit were based on previous studies where specific force (force per CSA) in single diaphragm muscle fibers, the CSA and proportion of fibers of each type and the number of muscle fibers per motoneuron were determined (61, 65, 73, 75–77, 136, 147, 161, 187, 236, 242, 243, 280). Figure 3 shows that the Pdi generated during quiet breathing (eupnea) was ~21% of Pdimax in the rat diaphragm muscle. Normal ventilatory behaviors could thus be accomplished by the activation of type S and FR units alone. When ventilatory drive was increased by creating a hypoxic and hypercapnic environment, Pdi increased to ~28% of Pdimax, and could still be accomplished by recruiting type S and FR units. Further recruitment of type FInt units was necessary during the extreme ventilatory effort caused by total airway occlusion, in which Pdi increased to 63% of Pdimax. Only during expulsive non-ventilatory behaviors (e.g., sneezing and coughing) where Pdi generated is near maximal would the recruitment of type FF units be required. Similar findings were previously reported in cats and hamsters (235, 237, 238, 240). Thus it was concluded that normal ventilatory demands of the diaphragm muscle are met by recruitment of only fatigue-resistant motor units (type S and FR), suggesting that a large portion of diaphragm muscle motor units remain fairly inactive during typical ventilation and are recruited only when short duration, high force efforts are necessary.
Figure 3.

Motor unit recruitment model in the rat diaphragm muscle during ventilatory and non-ventilatory behaviors, based on a model developed previously in cats and hamsters by Sieck and Fournier (239, 240). Motor units are recruited in a specific order (type S → type FR → type FInt → type FF) to accomplish the required forces. From Mantilla et al. (147), with permission.
The large reserve capacity of the diaphragm muscle might provide a first level of adaptation in response to disease or injury. Indeed, the Pdi generated during eupnea was estimated at ~10% of Pdimax in humans (238). Disease or injury associated with muscle atrophy or weakness (39, 132) might go unnoticed while this reserve capacity is restricted in normal ventilation (244). Only during expulsive behaviors may the reduced functional capacity of the diaphragm muscle become evident. Specific adaptations of skeletal muscle fibers thus are important in providing the functional limits of the muscle response to the environmental conditions that are imposed on it.
Skeletal muscle plasticity
Skeletal muscle is in a constant state of remodeling to match the changing functions required of it. Intrinsic or “natural” changes are common, such as development and aging. Skeletal muscle can also respond to altered function imposed by extrinsic changes, for example the environmental cues of mechanical loading, activity, and innervation. These cues are not independent of each other, and if one is altered, the others are likely also affected. This section will focus on these mediators of muscle plasticity.
Normal skeletal muscle is usually at “status quo” with an activity level, mechanical load, and innervation that is typical for it. We see this “status quo” exemplified by typical fiber size and mechanical properties of each muscle. Since each muscle of the body is unique and has a different fiber-type composition, the response to demands is different for different muscles. The basis of the response, however, is maintenance of the balance of protein synthesis and degradation. This will be described in detail later. Some examples of load-, activity-, and innervation-induced plasticity are given in this section.
Developmental changes
As we grow, bones lengthen and muscles change naturally. Muscle lengthening is achieved by addition of sarcomeres to the ends of the muscle fibers, a process that is reversible. A change in muscle length affects velocity and shortening capacity, but does not change the amount of force that can be generated by the muscle. In addition, the mechanical output of muscle during growth is altered by increasing muscle fiber diameter by hypertrophy. Hypertrophy adds more sarcomeres in parallel within the muscle fiber, which increases the amount of force the muscle generates but does not affect the velocity of shortening. Skeletal muscles do not typically form new muscle fibers, a process called hyperplasia.
In addition to changes in mechanical output of muscle, the fiber-type composition of muscle inherently changes. For example, inherent diaphragm muscle plasticity is reflected by transitions in MyHC isoform during postnatal development, when the replacement of neonatal myosin with adult slow and fast myosin occurs rapidly in the perinatal period. In humans, the proportion of MyHCslow increases from 9% at less than 37 weeks gestational age to 25% at birth, and reaches the adult level (55%) during the second postnatal year (119). In rats the proportion of MyHCslow increases from 7% at birth to 18% in adult (71). Furthermore, with aging there is a progressive loss of muscle mass, leading to a reduction in muscle performance and probable loss of function. Atrophy of type II fibers leads to a larger fraction of slow-twitch muscle mass and is evident by the slower contraction and relaxation times in older muscle (138, 165).
Changes in load
Many studies have examined muscle plasticity in limb muscles, where it is easier to control changes in load and activity. Two different types of loading can be imposed on muscle: 1) dynamic, elastic loading; and 2) static, resistive loading. Experiments involving hindlimb unloading typically reflect a change in a static, resistive load (i.e., hind-limb suspension or exposure to a microgravity environment). Muscle length is an important determinant of the changes induced with loading. Experiments in animals involving hind-limb suspension, which unloads hind-limb muscles, and in microgravity conditions such as spaceflight have indicated a change from slow fibers to fibers co-expressing slow and fast fiber phenotypes (33, 179, 180). Biochemically, these co-expressing fibers are a mix of slow and fast fibers; functionally, they possess properties of both slow and fast fiber types. In addition, studies on animals and humans with spinal cord injury have demonstrated a similar shift from slow fibers to co-expressing slow and fast fibers (31, 283). It is important to note that the fiber type transitions that occur after decreased load do not include a complete shift from slow to fast fibers, but rather result in a mixed fiber type. These unloading conditions also result in muscle atrophy (267); in fact significant atrophy in humans has been observed after only 5 days in spaceflight (19).
Because of the unique properties of the diaphragm muscle, its response to changes in load or activity will differ greatly compared to that of limb muscles. It is difficult to impose load changes on the diaphragm, as the only normal loads that the diaphragm experiences are extrinsic recoil forces from the lungs and chest wall during inspiration. In contrast to hindlimb muscles, length changes are unlikely in diaphragm muscle. With unilateral diaphragm muscle paralysis, it has been suggested that passive stretch of the affected side is imposed by continued activity of the contralateral hemidiaphragm and thus contributes to increased protein synthesis observed immediately after denervation (85, 279). However, passive length changes imposed on the paralyzed (denervated) hemidiaphragm were examined in rabbits using piezoelectric crystals (279). Although small changes in passive length were observed with contraction of the contralateral side, passive length never exceeded optimal length and did not result in measurable muscle strain.
Inspiratory resistive loading provides an experimental method to impose extrinsic diaphragm muscle loading by increasing airway resistance, but this is not synonymous to increased resistive load of the diaphragm muscle per se (as would occur in limb muscles). Instead it is more synonymous to elastic loading of diaphragm muscle. In contrast, conditions such as scoliosis that decrease chest wall compliance would impose resistive loading to diaphragm muscle (124). Nonetheless, muscle mass and fiber CSA increase modestly after inspiratory resistive loading imposed either by placing a band around the trachea for 24–28 weeks (190) or by inserting a resistance to inspiratory air flow for 8 weeks (14, 70, 202). Although the structural changes appear to be fiber-type dependent, the results also seem to be time-dependent. For example, 24–28 weeks of inspiratory resistive loading in rats resulted in hypertrophy of MyHCslow-expressing fibers (190), but 8 weeks of resistive loading resulted in hypertrophy of MyHC2A-expressing fibers (202). This response also appears to be load-dependent, where a small inspiratory resistive load resulted in hypertrophy of only MyHC2A-expressing fibers, but a high resistive load caused hypertrophy in all diaphragm muscle fiber types (14, 202). No studies have quantified changes in diaphragm muscle contractile protein content after inspiratory resistive loading. Gea et al. showed an increase in expression of MyHCslow mRNA after 4 days of resistive loading, as compared to no change in mRNA expression for the other MyHC isoforms (70). In general, these structural and functional changes associated with inspiratory resistive loading demonstrate the complexity of diaphragm muscle plasticity, since changes in load most likely go together with changes in activity. Unfortunately it is very difficult to control for changes in activity in the above model.
A unique clinical condition associated with altered diaphragm muscle load is emphysema, which is associated with dynamic hyperinflation and results in shortening of diaphragm muscle by 30–40% and resultant decrease in diaphragm muscle maximum specific force (50, 137, 167). These alterations cause loss of elastic lung recoil and lower maximal expiratory airflow (158). Since the diaphragm muscle must overcome the load imposed by recoil forces of the lung and chest wall, changes in the mechanical properties of the lung or chest wall will directly affect diaphragm muscle load. Importantly, after chronic hyperinflation, diaphragm muscle adapts to the hyperinflated state by reducing the number of sarcomeres in series and shifting the length-tension curve to a shorter, optimal length, whereby the force-generating capacity is partially restored (48, 63). Interventions such as lung volume reduction surgery (LVRS) have been used to resect the lung emphysematous tissue and indirectly partially recover the lung elastic recoil (40), however LVRS results in short-term diaphragm muscle fiber injury and impaired contractility due to acute diaphragm muscle fiber stretch (134). Nevertheless, the studies evaluating LVRS have been purely descriptive, and the mechanisms underlying improved respiratory function are not yet known.
Changes in activity
A common activity-dependent example is plasticity induced by different exercise training regimens. Endurance training, e.g. marathon running, consists of high frequency, low load contractions (e.g., 50–80 contractions/minute at ~30% maximum voluntary contraction of the calf muscle) (200). This typically does not result in muscle hypertrophy because the level of force production is relatively low. Within type II fibers there is a transformation from type IIb to IIa fibers, thereby providing efficient properties of energy use (6). Type I fibers may become faster, allowing the slow fibers to contract at a rate fast enough for the exercise (275). Thus, when exercise of a submaximal intensity and duration is performed in a trained state, the exercise usually becomes more economical to perform.
Resistance training, e.g. weight lifting, consists of low frequency, high load contractions (e.g., less than 15 contractions/minute at greater than 60% maximum voluntary contraction) (34). When a long-term load is imposed, all fibers hypertrophy (34). In addition to hypertrophying, type IIa fibers acquire phenotypic characteristics of type I fibers (210), and type IIb fibers shifted to type IIa/IIb hybrid fibers (34). In human studies, resistance training caused a decrease in number of type IIx fibers (103). However, by expanding the relative size and number of slow-twitch fibers, the muscle is better able to withstand the chronic overload while retaining functional properties of type IIx and IIb fibers.
These examples show that fiber type transitions in response to mechanical load and activity is dependent on 1) the magnitude of force and 2) the manner in which it is imposed (continuous vs intermittent). The physique of marathon runners is drastically different than body builders because of skeletal muscle’s ability to adapt to its required function.
Given that the diaphragm muscle is very active compared to other skeletal muscles, significant effects of inactivity are expected. It is important to note, however, that inactivity alone does not control muscle mass. As stated, diaphragm muscle has a high duty cycle that gives it a pattern of activity very different from other muscles, such as hind-limb muscles. If activity alone were important, we would expect that since type S and type FR motor units are most active, then type I and type IIa fibers would be the biggest. However, this is not the case, and this suggests that other environmental cues play a role in addition to activity.
A clinically relevant example of diaphragm muscle inactivity is mechanical ventilation. In this model respiratory muscles do not contract and the ventilator provides ventilatory support. Studies in animals have shown that short-term controlled mechanical ventilation (e.g. 6–18 hours) is enough to cause significant structural and functional plasticity in diaphragm muscle. Myofibrillar protein and CSA were reduced in all fiber types (233), apoptosis was induced (258), protein synthesis was decreased (232), and protein degradation was increased (51). The relative proportion of MyHC isoform expression, however, did not change after 1 or 3 days of controlled mechanical ventilation (192, 221, 222, 233). Controlled mechanical ventilation for 18 to 69 hours in brain-dead organ donors showed significant atrophy of diaphragm muscle fibers, an increase in oxidative stress, and activation of degradation pathways (132, 185). It is apparent that controlled mechanical ventilation causes diaphragm muscle plasticity, but it is unlikely that these changes are due to unloading or inactivity alone (209, 244). Importantly, controlled mechanical ventilation mode, which provides a mechanical breath on preset timing, is not a commonly used mode clinically (186, 244). Pressure-assist or volume-assist modes that are initiated by pressure or flow triggers are more common clinically and require some diaphragmatic work. In fact, flow-triggered assist-mode ventilation in rats attenuated the force loss induced by controlled mechanical ventilation, as well as maintained control levels of degradation pathways (222). The extent of plasticity largely depends on which mode is used.
Several models of inactivity have been developed to determine its effects on structural and functional diaphragm muscle properties, and the models described here will be: 1) cervical spinal cord hemisection; 2) unilateral phrenic denervation; and 3) tetrodotoxin nerve blockade. All three of these models result in diaphragm muscle paralysis (161, 209, 280). One model of inactivity is cervical spinal cord hemisection (SH) at C2, where ipsilateral descending excitatory drive to phrenic motoneurons is removed (Figure 4) (161, 281). In this model, communication between phrenic motoneurons and diaphragm muscle fibers remains intact, but motoneurons are inactive. Since both phrenic motoneurons and diaphragm muscle fibers are inactive, the activity patterns remain matched. Unilateral phrenic denervation (DNV) also induces inactivity and unloading of the diaphragm muscle; however, the nerve-derived trophic influence stemming from the motoneurons is completely eliminated with DNV but is preserved with SH. Phrenic motoneurons remain active but are unable to communicate with diaphragm muscle fibers. Diaphragm muscle inactivity can also be induced by placing a cuff around the phrenic nerve and chronically infusing tetrodotoxin (TTX) in order to block axonal propagation of action potentials. Like the SH model, communication between phrenic motoneurons and diaphragm muscle fibers remains intact. The difference between SH and TTX models, however, is that phrenic motoneuron activity is absent after SH but actually increased after TTX. The difference between DNV and TTX models is that axoplasmic flow of nerve-derived trophic factors is preserved after TTX but interrupted after DNV (209, 280).
Figure 4.

Comparison of experimental models (SH, DNV, and TTX) for the study of activity-induced diaphragm muscle plasticity. Adapted from Mantilla and Sieck (148), with permission.
Comparison of the following functional and structural changes induced by all three of these models is useful in determining the importance of muscle inactivity. If diaphragm muscle inactivity is the only cause of plasticity, then all three models of inactivity should cause similar structural and functional changes. The following sections, however, will show that DNV and TTX induce structural and functional changes, but SH induces minimal changes. Something is being preserved in the SH model but is being disrupted in DNV and TTX models. The difference is that with DNV and TTX, a positive trophic influence is removed, but with SH this trophic influence is preserved (150, 209). This suggests that inactivity alone does not cause structural and functional plasticity. Importantly these differences are also evident in the axon terminals at motor end-plates in the diaphragm muscle (146, 188, 189, 246), consistent with a nerve-derived influence on muscle adaptations. The specific structural and functional changes associated with each model will be described in detail next.
SH-induced inactivity
Electromyographic and electroneurographic recordings performed during surgery and at the terminal experiment confirmed that SH inactivates both phrenic motoneurons and diaphragm muscle (161, 188). At 2 weeks after SH, the only fibers that had a change in CSA were MyHCslow-expressing fibers, with an increase in CSA (~28% increase, Figure 5) (161, 280). Metabolic properties, as measured by changes in succinic dehydrogenase (SDH), were affected by SH. MyHCslow- and MyHC2A-expressing fibers had a decrease in SDH activity after SH, whereas MyHC2X- and MyHC2B-expressing fibers were unaffected (280). This indicates that activation history is an important determinant of mitochondrial function in type I and IIa fibers. Since type I and/or type IIa fibers are normally recruited during breathing, it follows that inactivity would have a greater impact on these motor units.
Figure 5.

Cross-sectional area (CSA) adaptations to 14 days of inactivity induced by spinal hemisection (SH), unilateral denervation (DNV), or tetrodotoxin nerve block (TTX), among fibers expressing different MyHC isoforms. Mean and SE of the fiber CSA are plotted according to fiber type. * Significantly different from the control group (p < 0.05 for all comparisons). Adapted from Miyata et al. (161), with permission.
DNV- and TTX-induced inactivity
In contrast to the minimal changes in CSA after SH, by 2 weeks after DNV or TTX, the CSA of MyHCslow- and MyHC2A-expressing fibers displayed a 60–70% increase in CSA, whereas CSA decreased by 40% in MyHC2X- and by 60% in MyHC2B-expressing fibers (Figure 5) (72, 161, 280). Despite the increase in CSA of MyHCslow- and MyHC2A-expressing fibers, there was a ~30% decrease in MyHCslow and MyHC2A protein by 3 days after DNV and persisting through 2 weeks after DNV (72). MyHC2X and MyHC2B protein decreased by 1 day after DNV (60–80% decrease) and remained decreased through 2 weeks after DNV (72). Since muscle mass is changing after SH, DNV, and TTX, there is likely a change in muscle protein balance. The pathways regulating protein balance have not been fully explored for such atrophy conditions, but will be described in the subsequent section.
Metabolic properties were more affected by DNV and TTX than SH. Following 2 weeks of inactivity, SDH activity was decreased in all fiber types, and to a larger degree than after SH (280). Contractile properties also changed. Maximum specific force decreased by 2 weeks after DNV or TTX (161). Force normalized to half-sarcomere MyHC content was unchanged in MyHCslow-expressing fibers, but decreased in MyHC2A- and MyHC2X-expressing fibers (74).
In addition, 3 days of DNV induces activation of satellite cell mitotic activity (84). These are small cells located beneath the basal lamina that play a role in recovery of muscle fibers from injury by differentiating into myoblasts and fusing with existing myofibers in order to repair muscle or increase its size (43, 229, 277). Although satellite cell activation is not required for muscle hypertrophy (15), it is involved in the diaphragm muscle response to DNV (84). A function of satellite cells is to maintain myonuclear domain (MND) size under conditions that stimulate muscle fiber hypertrophy (152, 155, 207, 211). MND size is also regulated by satellite cells in conditions of atrophy by deletion of myonuclei (1). At 2 weeks after DNV, the total number of myonuclei per fiber did not change, but MND size of MyHCslow- expressing fibers increased, and MND size of MyHC2X and MyHC2B-expressing fibers decreased (2) Similarly, corticosteroid-induced muscle atrophy caused a decrease in MND size of only MyHC2X and MyHC2B-expressing fibers (268). In addition, satellite cells may migrate to other fibers and play a role in co-expression of MyHC isoforms in single fibers as a means of plasticity (59). There is a significant increase in fibers co-expressing both MyHCslow and MyHC2A after diaphragm muscle denervation (68, 280)and hindlimb denervation (224).
These results taken together with the effects of SH suggest that diaphragm muscle inactivity alone is not responsible for structural and functional plasticity. Changes after DNV or TTX compared with SH demonstrate the impact of phrenic motoneuron innervation and trophic influences. In addition, these studies show that the morphological and contractile responses are highly dependent on diaphragm muscle fiber type and on duration of inactivity.
Changes in trophic environment
Seminal studies by Buller et al. in 1960 were the first to demonstrate the impact of innervation in determining skeletal muscle properties (24–26). Cross-innervation of fast-twitch and slow-twitch muscle with a nerve that normally innervates the opposite muscle type resulted in contractile and metabolic adaptations matching the newly established innervation.
It is known that motoneurons innervating slow-twitch muscles discharge at a low frequency, and those innervating fast-twitch muscles discharge at a high frequency (215). Based on this, the contractile speed of skeletal muscle was also found to be affected by motoneuron-specific impulse patterns generated by chronic electrical stimulation, where a tonic stimulus pattern mimicking the impulse pattern of a type S motor unit had a slowing effect on contraction and relaxation times of fast-twitch muscles (176, 178, 215). Since then, many animal studies have used chronic low-frequency (~10 Hz) stimulation to induce fast-to-slow muscle transformation (179, 181). In addition, Reid et al. found that chronic electrical stimulation counteracted some of the muscle fiber atrophy caused by tetrodotoxin nerve blockade (TTX) alone in the soleus and extensor digitorum longus (EDL) muscles (198). These studies show that innervation plays an important part in establishing and maintaining contraction properties of muscle.
These studies do not address the overall question of what is causing innervation-induced plasticity. It is unknown if the removal of nerve-derived trophic factors itself, or if the removal of a certain pattern of activity previously maintained by the nerve causes structural and functional changes in the muscle. Based on the findings of Reid et al., using electrical stimulation to mimic the muscle’s “normal” pattern of activity did not completely counteract the atrophy caused by TTX-induced nerve block (198). TTX altered the nerve-derived trophic influence, and this has an impact on muscle mass more than activity pattern.
Based on the changes induced by DNV compared to SH and TTX, it is apparent that neural influences are likely essential in maintaining diaphragm muscle contractile properties (89). One potential nerve-derived trophic factor is neuregulin (NRG), which contributes to the regulation of muscle fiber growth by increasing protein synthesis and possibly altering the balance between protein synthesis and degradation (96, 151). NRG contains an epidermal growth factor (EGF)-like domain that signals by stimulating the ErbB family of receptor tyrosine kinases expressed in motoneurons, Schwann cells, and skeletal muscle fibers (262, 284). Its signal transduction in muscle requires the phosphorylation of ErbB receptors, followed by activation of downstream pathways including PI3K/Akt and MAPK/Erk pathways (36). NRG isoforms are synthesized as transmembrane proteins, called pro-NRGs, with the EGF domain located outside of the cell. Pro-NRGs are cleaved extracellularly to release the diffusible, mature NRG (142). Neurotrophic factors such as brain-derived neurotrophic factor (BDNF) and glial-derived neurotrophic factor (GDNF) trigger the release of presynaptic NRG from nerve terminals, thus making more available for cleavage and release (62, 141). BDNF and GDNF are stored in vesicles and released with activity (58, 81). Other factors mediating pro-NRG cleavage are proteases (116, 144), some of which can also be released in an activity-dependent manner (91). However, it is possible that other factors influence protease activity, such as mechanical strain (57) or muscle fiber type, thus activity alone is likely not responsible for NRG cleavage and release. NRG controls acetylcholine receptor synthesis at the neuromuscular junction (111, 141), but its effect on contractile proteins such as MyHC has not been reported. It is possible that DNV results in loss of nerve-derived NRG and thus shifts the balance of protein synthesis and degradation towards net protein breakdown, leading to loss of contractile proteins such as MyHC.
Molecular pathways
The previous sections discuss skeletal muscle’s ability to adapt to altered demand via changes in structural and functional properties. These changes are largely the consequence of altered protein expression in which either the amount or type of protein is altered to meet functional demands. Although the cellular-scale structural and functional changes related to skeletal muscle plasticity have been characterized for many physiological states, the molecular-scale protein changes are not as well characterized.
Proteins are in a continuous state of being produced (synthesis) and then degraded (degradation). The balance between protein synthesis and degradation determines if there is either a net loss (atrophy) or net gain (hypertrophy) in muscle mass. Changes in muscle mass and protein balance are common adaptations to spinal cord injury, denervation, exercise, etc. For example, the unilateral diaphragm muscle DNV model caused both protein synthesis and protein degradation to increase, with the increase in degradation outweighing the increase in synthesis and leading to a decrease in net protein balance by 5 days after DNV (3) (Figure 6). Based on a previous study by Geiger et al. (72), a correlation between MyHC protein and mRNA levels after DNV was not straightforward, with decreased MyHC2B protein expression 14 days after DNV associated with unchanged MyHC2B mRNA levels, for example. Although MyHC gene transcription and translation may change differentially after DNV, these results suggested that posttranslational changes are likely responsible for change in diaphragm muscle MyHC protein expression and overall protein balance.
Figure 6.

DNV-induced change in rat diaphragm muscle net protein balance as determined by performing parallel but separate incubations of strips from the same diaphragm muscle for protein synthesis and protein degradation measurements. Mean and SE of the percent change relative to sham controls are plotted over time after DNV. * Significantly different from the sham control group at the same DNV time-point, † Significantly different from 1 and 3 days after DNV, # Significantly different from 5 days after DNV (p < 0.05 for all comparisons). Figure from Argadine et al. (3), with permission.
Despite developments in molecular and cell biology that have aided in the understanding of mechanisms regulating atrophy and hypertrophy, the specific regulation of protein balance has not been elucidated for many diseases or physiological states. In fact until recently, most of the research has focused on specific molecular pathways that regulate protein synthesis and a distinct set of pathways that regulate protein degradation. The pathways are complex in themselves, and recent studies have suggested that there are multiple levels at which these pathways are interrelated, creating an extremely complex system (4, 216, 217, 255).
Pathways regulating protein synthesis
The increase in protein synthesis that is associated with muscle hypertrophy is thought to involve Akt signaling (79, 216), because: 1) Akt phosphorylation increases (17); and 2) expression of an activated form of Akt is sufficient to cause hypertrophy (204, 205, 257) and prevent atrophy of denervated skeletal muscles (17). The downstream branches of the Akt pathway are mammalian target of rapamycin (mTOR) and glycogen synthase kinase-3β (GSK3β) (Figure 7). p70S6 kinase (p70S6K) is an effector of mTOR and is thought of as a marker of increased protein synthesis (5, 35, 109, 219). The rate of protein synthesis is primarily affected by downstream changes in translation initiation factors such as eukaryotic initiation factor 2B (eIF2B), eukaryotic initiation factor 4E (eIF4E), and eIF4E-binding protein 1 (4EBP1) (122, 270). In addition to Akt, the p44/42 MAPK (ERK) pathway influences translational control through eIF4E (231). Aside from responding to changes in growth factor signaling, respiratory muscle responds to metabolic demands as well. As such, mTOR is inhibited by the energy sensor AMP-activated protein kinase (AMPK) (94, 125). Importantly, when evaluating the regulation of protein synthesis, it is necessary to study all of these signaling pathways at the same time, due to the complexity and the interrelation of all the individual pathways and relevant proteins.
Figure 7.

Simplified model of signaling pathways regulating protein synthesis and degradation. Arrows denote activating events, whereas perpendicular lines denote inhibitory events. The solid lines indicate direct activation. The dashed lines indicate indirect activation, whereby intermediate steps are involved but are not specified in this schematic. Protein synthesis is regulated by protein kinase B (Akt), p44/42 MAPK (ERK) and AMP-activated protein kinase (AMPK), resulting in activation of the downstream targets mammalian target of rapamycin (mTOR), glycogen synthase kinase-3β (GSK3β), MAPK-interacting kinases 1/2 (MNK1/2), p70S6 kinase (p70S6K), eIF4E-binding protein 1 (4EBP1), and eukaryotic initiation factors 2B and 4E (eIF2B and eIF4E). Conversely, Akt is responsible for the phosphorylation status of forkhead box protein (FoxO). Upon phosphorylation by Akt, FoxO leaves the nucleus and becomes inactive, thus preventing protein degradation. If Akt activity is suppressed, FoxO becomes dephosphorylated, translocates to the nucleus, and exerts its transcriptional effects on atrogenes to induce protein degradation through the ubiquitin-proteasome pathway. Figure from Argadine et al. (4), with permission.
Signaling pathways related to protein synthesis have been evaluated after diaphragm muscle DNV and after acute short-term nutritional deprivation (4, 133, 135). The following section will describe three specific pathways that affect protein synthesis in relation to these models: p70S6K, eIF2B, and eIF4E.
p70S6K pathway
The serine-threonine kinase Akt family (also known as protein kinase B, PKB) has three members that are expressed in a tissue specific manner (38, 113). In skeletal muscle, Akt1 and Akt2 are expressed at higher levels compared to Akt3, which is mainly expressed in the brain. Phosphorylation and activation of Akt is induced by IGF-1, insulin, and other ligands (96, 204). After receptor binding, such ligands recruit the activity of phosphoinositide-3 kinase (PI3K). Akt is then targeted to the plasma membrane and becomes phosphorylated. Activation of Akt has been found to phosphorylate a wide range of substrates, including proteins that induce protein synthesis, gene transcription, glucose metabolism, synaptic signaling, cell proliferation, and block apoptosis (154, 269).
Akt-induced activation of mTOR is a key regulator of protein synthesis (95, 270), since: 1) muscle hypertrophy results in increased mTOR phosphorylation (199), and 2) mTOR inhibition with rapamycin blocks overload hypertrophy and growth in regenerating muscles (17, 170). In contrast to Akt, AMPK inhibits mTOR. AMPK is a cellular sensor of energy balance that responds to changes in the intracellular ratio of AMP:ATP (94). AMPK can be activated pharmacologically by 5-aminoimidazole-4-carboxamide-1-β-d-ribofuranoside (AICAR) (42). Activation of AMPK with the drug AICAR has previously been shown to inhibit protein synthesis by deactivating both Akt and mTOR (18, 121, 276).
mTOR is a member of two multiprotein complexes: mTOR Complex 1 (mTORC1) and mTOR Complex 2 (mTORC2). mTOR is phosphorylated at Ser2448 (mTORC1) via the PI3 kinase/Akt signaling pathway and autophosphorylated at Ser2481 (mTORC2) (41, 166, 174). The influence of mTOR on translation and ultimately protein synthesis is commonly mediated by mTORC1-induced phosphorylation of p70S6K (95, 109, 270) and 4EBP1 (78, 231). Most of the research on protein synthesis regulation has focused on mTORC1; however, mTORC2 also appears to play a role in regulating the actin cytoskeleton and in activating Akt (174, 218, 220). Furthermore, there is cross-talk between mTORC1 and mTORC2, so activation of mTORC2 may subsequently activate mTORC1 and increase protein synthesis (153). The influence of mTOR on protein synthesis is mediated in part by its phosphorylation of p70S6K (95, 109, 270). A compensatory hypertrophy model of the plantaris muscle resulted in increased phosphorylation of Akt and downstream targets p70S6K and 4EBP1 (17).
In the diaphragm muscle DNV model, Akt and downstream effectors mTOR and p70S6K were not responsible for the initial increase in protein synthesis, although they likely contributed to the maintenance of elevated synthesis (4). After short-term nutritional deprivation, phosphorylation of Akt, mTOR, and p70S6K decreased (133).
eIF2B pathway
Initiation factor eIF2B functions as a regulator of translation initiation by mediating the recycling of eukaryotic initiation factor-2 (122, 191). GSK3β phosphorylates eIF2B and results in its inactivation (273, 274). Akt acts through GSK3β to promote protein synthesis whereby Akt phosphorylates GSK3β, leading to its inactivation and diminishing its inhibitory effect on the initiation factor eIF2B (110, 231). In addition, ERK1/2 also phosphorylates and inactivates GSK3β through MAP kinase activated protein (MAPKAP) kinase-l (214). Expressing a dominant-negative form of GSK3β resulted in hypertrophy in cultured C2C12 differentiated myotubes (204), and a compensatory hypertrophy model of the plantaris muscle caused GSK3β phosphorylation and inactivation (17).
In the diaphragm muscle DNV model, GSK3β phosphorylation increased by 3 days after DNV, due to ERK1/2 activation rather than Akt activation (4). Increased GSK3β phosphorylation may contribute to elevated protein synthesis through its release of eIF2B or reduced inhibition of mTOR (108), in conjunction with eIF4E. GSK3β phosphorylation increased as well after short-term nutritional deprivation (133).
eIF4E pathway
eIF4E is thought to be a rate-limiting initiation factor and plays a major role in determining global translation rates (122, 194). One way in which eIF4E is regulated is through binding to 4EBP1, a translation repressor protein (206, 231). Interaction of eIF4E with 4EBP1 prevents cap-dependent translation. This inhibition is relieved by mTOR-induced phosphorylation of 4EBP1 (23, 78), which disrupts this interaction and results in initiation of cap-dependent translation through eIF4E (172, 199).
In addition, eIF4E is regulated through the MAPK-ERK1/2 pathway (231). ERK1/2 activates MAPK-interacting kinases 1/2 (MNK1/2), leading to phosphorylation of eIF4E independently of 4EBP1 phosphorylation (195, 271, 272).
In the diaphragm muscle DNV model, phosphorylation of 4EBP1 and thus mTOR did not contribute to activation of eIF4E (4). Instead, the increased phosphorylation of ERK1/2 likely activated eIF4E and was responsible for both the initial elevation and the sustained level of protein synthesis after DNV.
Pathways regulating protein degradation
Pioneering studies on gene expression profiling after muscle atrophy were performed separately by the groups of Goldberg and Glass and provided major contributions to the study of atrophy mechanisms (16, 80). The studies compared gene expression in different models of muscle atrophy (i.e., diabetes, cancer cachexia, chronic renal failure, fasting, and denervation) and determined the commonly up- or down-regulated genes as atrophy-related genes, or atrogenes (212). The two most induced genes were two muscle-specific ubiquitin ligases, atrogin-1/MAFbx and MuRF1, which are upregulated in atrophy models and are responsible for increased protein degradation through the ubiquitin-proteasome system (16, 80). These two genes are currently considered the best markers for muscle atrophy, although the upstream pathways are not completely elucidated. This section will discuss pathways upstream and downstream of these atrogenes that are involved in regulation of protein degradation, namely transcription factors, ubiquitin-proteasome, autophagy-lysosome, and metabolic processes.
Transcription factors FoxO and NF-κB
Signaling pathways upregulated during muscle atrophy induce two families of transcription factors: FoxO and NF-κB. FoxO belongs to the “forkhead box” family of transcription factors. Three mammalian isoforms have been identified: FoxO1, FoxO3a, and FoxO4 (12, 13). When Akt is activated, it phosphorylates FoxO and promotes the export of FoxO from the nucleus to the cytoplasm, where FoxO is unable to act on promoters (22, 250). Thus, FoxO is essentially inactivated by Akt. Conversely, upon inactivation of Akt (which commonly occurs during muscle atrophy), FoxO becomes dephosphorylated and is imported back to the nucleus where it can exert its transcriptional activity. The mechanisms by which FoxO is imported and exported have been described in detail and are outside the scope of this section (263).
The role of FoxO in protein degradation has been shown by several studies in atrophy models. Myotube atrophy caused by fasting or treatment with the glucocorticoid dexamethasone leads to dephosphorylation of Akt, FoxO1, and FoxO3a and upregulation of atrogin-1/MAFbx mRNA (217, 251). When the PI3K/Akt pathway is inhibited by LY294002 treatment of myotubes, atrogin-1/MAFbx is upregulated (130, 213) and FoxO1 is localized to the nucleus and thus “active” (127, 255). When transfected in skeletal muscles in vivo, FoxO3a is sufficient to promote atrogin-1/MAFbx expression and muscle atrophy (217). FoxO1 transgenic mice display decreased muscle mass, suggesting that FoxO is sufficient to blunt postnatal growth (117). Conversely, overexpression of a constitutively active form of Akt, prevents activation of atrophy pathways induced by glucocorticoid treatment of C2C12 myotubes by inhibiting FoxO transcription factors MuRF1 and MAFbx (255).
After diaphragm muscle DNV, an immediate decrease in Akt phosphorylation was followed by nuclear translocation of FoxO1 protein, subsequently leading to increased total protein ubiquitination and protein degradation (3, 4).
Nuclear factor κB (NF-κB) transcription factors play a role in immunity and inflammation. Nuclear translocation of NF-κB is regulated by the kinases IKKα and IKKβ. Many models of hindlimb muscle atrophy induce NF-κB signaling and transcriptional activity, namely unloading (105, 106, 264), immobilization (230), and denervation (32). NF-κB activation is sufficient to cause muscle atrophy in vivo (32, 264) and necessary for muscle atrophy caused by cachexia or disuse (32, 115, 264). NF-κB-mediated muscle atrophy is likely due to its transcriptional regulation of MuRF1, since mice overexpressing IKKβ have significantly increased MuRF1 mRNA expression but no change in atrogin-1/MAFbx (32). In addition, transcription factors NF-κB and FoxO have a cumulative effect on muscle atrophy, since inhibiting IKKα, IKKβ, and FoxO completely blocked muscle atrophy after immobilization (197).
Ubiquitin-proteasome pathway
The proteasome is a large (2000 kDa) complex made up of more than 50 subunits and comprising as much as 1% of the total cell protein (9). Ubiquitin is a short peptide that can be conjugated to specific protein substrates. A chain of ubiquitin molecules is built onto the substrate, and this chain marks the protein substrate for degradation through the proteasome where the substrate is proteolyzed into small peptides (182). The addition of ubiquitin to a substrate is a highly regulated process that requires three distinct enzymes: E1 ubiquitin-activating enzyme, E2 ubiquitin-conjugating enzyme, and E3 ubiquitin-ligating enzyme. The E3 ubiquitin ligases provide substrate specificity.
The role of the ubiquitin-proteasome pathway in muscle atrophy has been well established. Inhibition of the proteasome blocks atrophy-induced increases in protein breakdown (259). The amount of polyubiquitin conjugation per total protein measured increases after atrophy (52). Furthermore, the atrogenes previously described, MuRF1 and MAFbx, have been shown to encode E3 ubiquitin ligases (16). Importantly, both ubiquitin ligases were shown to be necessary for the atrophy response to hind-limb DNV (16). Overexpression of FoxO3 has been shown to induce ubiquitin-proteasome pathway in myotubes (282) and regulate atrogin-1 and MuRF1 activity and mRNA expression (203, 217). In contrast, overexpression of constitutively active FoxO1 does not increase atrogin-1 or MuFR1 expression, suggesting that FoxO1 and FoxO3 signal through different mechanisms (255).
It was previously thought that the ubiquitin-proteasome could not break down myofibrillar protein (129, 253). Important recent findings, however, show that MyHC is indeed degraded by the ubiquitin-proteasome pathway (37, 253). In fact, MuRF1 was identified as a key modulator of MyHC protein levels (37). In addition, caspase-3 activation was found to be a mechanism involved in the initial steps of myofibrillar degradation, where it was responsible for the initial cleavage of actin (56, 130). Although the cellular signaling mechanisms that activate the ubiquitin-proteasome pathway are not completely known, there is no question that this pathway plays a large role in regulating protein degradation.
Diaphragm muscle DNV resulted in an increase in total protein ubiquitination starting at 5 days after DNV and remaining elevated through 14 days after DNV (3). This is consistent with an increase in protein degradation at 5 days after DNV. DNV had no effect on caspase-3 activity, suggesting that the ubiquitin-proteasome pathway is responsible for the diaphragm muscle response to DNV (3).
Autophagy-lysosome system
The autophagy-lysosome system is responsible for removing dysfunctional organelles and unfolded proteins via lysosomes (216). The lysosomal protease cathepsin-L is upregulated during muscle wasting (53, 128), and the autophagy system is activated during fasting in skeletal muscle (162). In addition, autophagy is activated after sciatic denervation-induced atrophy (66). It has recently been shown that the ubiquitin-proteasome and autophagy-lysosome pathways are interrelated via FoxO3. Expression of the autophagy-related genes LC3, GABARAP, and BNIP3 is regulated by FoxO3, and FoxO3 is sufficient and required to activate autophagic protein breakdown (145, 282).
Metabolic processes
While AMPK has been previously shown to inhibit protein synthesis by downregulating mTOR (18, 69, 254), it has recently has been shown that AMPK is also involved in protein degradation. A connection between AMPK and FoxO3a has been suggested, where through an unknown mechanism AMPK phosphorylation enhances FoxO3a-dependent transcription without affecting FoxO3a subcellular localization (86, 87). Furthermore, in vitro studies using myotubes showed that activation of AMPK with AICAR (42) stimulated myofibrillar protein degradation and increased atrogin-1/MAFbx expression through FoxO1 and FoxO3a activation (164, 169, 261). When protein degradation was elevated after diaphragm muscle DNV, there was no evidence of AMPK activation (4). Thus, more work is needed to determine if metabolic sensors such as AMPK may play a role in regulating protein degradation in concert with FoxO.
Activation of AMPK is also responsible for muscle atrophy caused by mitochondrial remodeling (203). Mitochondria are crucial for energy storage and constantly adapt to changes in cellular needs. Mitochondria change shape during fasting, denervation, and overexpression of constitutively active FoxO3 (203). Their shape and number depend on the balance between fusion and fission processes. Mitochondrial fission segregates dysfunctional components of the mitochondrial network, and removes them via autophagy (11). In this way, mitochondrial fission processes induce muscle atrophy through energy stress and subsequent AMPK activation. AMPK activation triggers FoxO3-induced signaling through the ubiquitin-proteasome and autophagy-lysosome pathways (203).
Interaction of protein synthesis and degradation pathways
The molecular-scale changes that affect net protein balance are time-dependent, inter-related, and highly complex. In addition, no single physiologic trigger is responsible for net protein balance changes. For example, the effect of diaphragm muscle DNV on net protein balance could be triggered by changes in mechanical load, muscle activity, and/or innervation.
Since the diaphragm muscle is paralyzed, mechanical load decreases instantly after DNV. Although the contralateral side of diaphragm muscle still contracts, passive mechanical stress is not responsible for the muscle fiber adaptations associated with DNV (279). Unloading of a highly active muscle may, however, remove muscle-derived trophic signals such as IGF-1, which signals through PI3K/Akt (7).
Similarly, changes in muscle activity also arise immediately after DNV. There is a spontaneous transient (5 min) increase in muscle activity (161) after the nerve injury induced by DNV, likely due to muscle fasciculations (252). This is followed by the expected reduction in muscle activity after DNV (161). These changes in muscle activity may result in decreased energy consumption, reflected by altered AMPK signaling (114).
In contrast, the removal of nerve-derived trophic influence is delayed after DNV. In fact, trophic factors from the distal phrenic nerve stump may not be eliminated until 24 hours after DNV (4, 20). NRG is one possible growth factor that could be removed after DNV. NRG alters protein synthesis via an Akt-dependent mechanism (96), so changes in PI3K/Akt signaling may reflect a reduction in NRG. The MAPK-Erk1/2 pathway is activated by growth factor signaling as well (92).
It is obvious that diaphragm muscle DNV is a dynamic model. The concerted evaluation of molecular signaling changes that occur after DNV and the subsequent changes in net protein balance will provide important insight into other conditions that affect muscle mass including normal developmental changes such as exercise and aging as well as pathological conditions (e.g., cachexia, chronic lung disease, prolonged mechanical ventilation).
The understanding of signaling mechanisms associated with protein synthesis and degradation regulation has led to the use of several therapeutic strategies to counter muscle atrophy. Based on evidence that the Akt pathway induces hypertrophy (79, 216) and inhibits activation of FoxO transcription factors MuRF1 and MAFbx (255), an intramuscular injection of IGF-1 has been used to activate Akt and prevent glucocorticoid and denervation-induced muscle atrophy (255). IGF-1 inhibited denervation atrophy by suppressing atrogenes MAFbx and MuRF1, thus rescuing denervation-induced muscle loss.
The ubiquitin-proteasome pathway has also been targeted to therapeutically reduce muscle atrophy. The proteasome inhibitor Velcade (also known as bortezomib) is approved for use in humans, specifically to treat multiple myeloma and exhibits relatively mild toxicity (201). Treatment of denervated rats (sciatic nerve) with Velcade significantly reduced atrophy in the soleus muscle and reduced ubiquitin mRNA expression to control levels in denervated soleus muscle (10). The use of Velcade in a congestive heart failure model of diaphragm muscle weakness (266) and in a COPD animal model (265) resulted in decreased proteasome activities and improved diaphragm muscle contractility, showing that reduction of muscle atrophy with Velcade translates into preservation of muscle strength.
Future of systems biology
Traditional intracellular signaling diagrams, such as that in Figure 7, are less than satisfactory in representing the complexity of protein balance. Importantly, these diagrams do not indicate highly contextual changes in signaling pathways that are the result of several confounding and time-dependent triggers involved in the regulation of protein balance. Furthermore, these diagrams do not show the indirect relationships between protein expression and downstream effectors. For example, Akt does not directly phosphorylate mTOR, thus the activation arrow is essentially a “black box” containing multiple intermediate steps such as TSC2 inhibition, Rheb inhibition, etc. Although we are far from elucidating all of the mechanisms in this network, approaches that take a systems biology approach of looking at each part and viewing how the parts interact will help advance the field.
The dynamic and complex nature of protein balance makes it a prime candidate for a systems biology approach. Systems biology focuses on the systematic study of complex interactions in biological systems (196). One goal of systems biology is to generate models that can explain or predict cellular behavior, leading to generation of new biological hypotheses. The system we are addressing here is a stochastic process, in which the system’s subsequent state is determined both by the process’s predictable actions and by random elements. This is in contrast to a deterministic process, in which randomness is not involved in the development of future states of the system, thus producing the same output for a given starting condition.
There are four steps in a systems biology approach, with the purpose being to create a model to predict cellular behavior (196). These steps are outlined in Figure 8. Previous studies of muscle atrophy and hypertrophy, as described earlier, help identify the components of each module, i.e., relevant proteins. In addition, component interactions such as links between proteins and cause-and-effect links can be identified from literature. Information about links between proteins primarily comes from literature, with some links being well established (i.e., PI3K-Akt) and other links only previously hypothesized (i.e., AMPK-FoxO). Using this information about network components and their interactions, a model of protein balance can be developed, as in Figure 7. Note that this model contains solid lines and dashed lines. The solid lines represent direct activation, i.e., linear relationships, for example growth factor signaling directly activates PI3K. The dashed lines represent indirect activation or nonlinear relationships, whereby intermediate steps are involved but are not specified in this model, for example Akt indirectly activates mTOR through inhibition of TSC2 and Rheb. As such, this model greatly simplifies the actual signaling scheme, and future models can be developed to incorporate the multiple steps involved in each “indirect activation” step.
Figure 8.

Steps in a systems biology approach. The first two steps use data to identify components and interactions in order to generate a reconstruction. The last steps generate a mathematical model to predict network behavior.
Next, a beginning step to developing a numerical model is to use flux balance analysis of the system (228). This approach is similar to Kirchoff’s law of circuit design, whereby the sum of all the currents going into a node must equal the sum of the currents going out. A stoichiometric matrix can be constructed, based on the number of elements in the network and the stoichiometric coefficients of the “reactions”. A flux balance can be found for each element within the network in order to yield the dynamic mass balance equations that interconnect the individual elements. For example, Akt and AMPK interconnect at mTOR. Akt positively regulates mTOR (positive flux), and AMPK negatively regulates it (negative flux). Since the sum of the “currents” going in has to equal that going out, we assume Akt − AMPK = mTOR. Stoichiometric coefficients can be given to each protein as such. This approach could connect all of the key regulatory proteins with each other and with protein synthesis/degradation in order to determine protein balance regulation in this dynamic model.
Knowing how protein balance is regulated and how to change this balance if necessary is essential for determining any therapeutic course of action for treatment. Much work is left in elucidating the complex regulation of protein balance. Expansion of the systems biology approach for making a mathematical model of protein balance would greatly improve treatments in a multitude of clinical settings by determining how to optimally redesign the cellular network (i.e., which proteins/pathways to upregulate or downregulate, timing of treatment, etc.) to produce the desired outcomes.
Conclusion
Respiratory muscle is in a constant state of remodeling in order to adapt to altered demand. Muscle plasticity is exemplified by structural and functional changes, and the basis of these cellular-scale changes are molecular-scale changes such as the balance between protein synthesis and degradation. Since muscle is the largest protein reserve in the body, any change in protein balance can affect the whole system. As such, protein balance has not been characterized for many disease states, although it is undoubtedly altered.
Our understanding of the molecular mechanisms underlying muscle plasticity has developed tremendously in recent years. Importantly, highly contextual changes in signaling pathways are the result of several inter-related and time-dependent triggers involved in the regulation of net protein balance. A single molecular trigger is not responsible for changes in net protein balance. Thus, protein balance in skeletal muscle depends on multiple synthetic/degradation pathways that should be jointly studied. The challenge ahead is development of a mathematical model of protein balance, which would greatly improve treatments in a variety of clinical settings by determining how to optimally redesign the cellular network to produce the desired outcome of maintaining both muscle mass and optimal contractile function.
References
- 1.Allen DL, Monke SR, Talmadge RJ, Roy RR, Edgerton VR. Plasticity of myonuclear number in hypertrophied and atrophied mammalian skeletal muscle fibers. J Appl Physiol. 1995;78:1969–1976. doi: 10.1152/jappl.1995.78.5.1969. [DOI] [PubMed] [Google Scholar]
- 2.Aravamudan B, Mantilla CB, Zhan WZ, Sieck GC. Denervation effects on myonuclear domain size of rat diaphragm fibers. J Appl Physiol. 2006;100:1617–1622. doi: 10.1152/japplphysiol.01277.2005. [DOI] [PubMed] [Google Scholar]
- 3.Argadine HM, Hellyer NJ, Mantilla CB, Zhan WZ, Sieck GC. The effect of denervation on protein synthesis and degradation in adult rat diaphragm muscle. J Appl Physiol. 2009;107:438–444. doi: 10.1152/japplphysiol.91247.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Argadine HM, Mantilla CB, Zhan WZ, Sieck GC. Intracellular Signaling Pathways Regulating Net Protein Balance Following Diaphragm Muscle Denervation. Am J Physiol Cell Physiol. 2011;300:C318–C327. doi: 10.1152/ajpcell.00172.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baar K, Esser K. Phosphorylation of p70(S6k) correlates with increased skeletal muscle mass following resistance exercise. Am J Physiol. 1999;276:C120–127. doi: 10.1152/ajpcell.1999.276.1.C120. [DOI] [PubMed] [Google Scholar]
- 6.Baldwin KM, FH Effects of different activity and inactivity paradigms on myosin heavy chain gene expression in striated muscle. J Appl Physiol. 2001;90:345–357. doi: 10.1152/jappl.2001.90.1.345. [DOI] [PubMed] [Google Scholar]
- 7.Bamman MM, Shipp JR, Jiang J, Gower BA, Hunter GR, Goodman A, McLafferty CL, Jr, Urban RJ. Mechanical load increases muscle IGF-I and androgen receptor mRNA concentrations in humans. Am J Physiol Endocrinol Metab. 2001;280:E383–390. doi: 10.1152/ajpendo.2001.280.3.E383. [DOI] [PubMed] [Google Scholar]
- 8.Barnard RJ, Edgerton VR, Furukawa T, Peter JB. Histochemical, biochemical, and contractile properties of red, white, and intermediate fibers. Am J Physiol. 1971;220:410–414. doi: 10.1152/ajplegacy.1971.220.2.410. [DOI] [PubMed] [Google Scholar]
- 9.Baumeister W, Walz J, Zuhl F, Seemuller E. The proteasome: paradigm of a self-compartmentalizing protease. Cell. 1998;92:367–380. doi: 10.1016/s0092-8674(00)80929-0. [DOI] [PubMed] [Google Scholar]
- 10.Beehler BC, Sleph PG, Benmassaoud L, Grover GJ. Reduction of skeletal muscle atrophy by a proteasome inhibitor in a rat model of denervation. Exp Biol Med (Maywood) 2006;231:335–341. doi: 10.1177/153537020623100315. [DOI] [PubMed] [Google Scholar]
- 11.Benard G, Karbowski M. Mitochondrial fusion and division: Regulation and role in cell viability. Semin Cell Dev Biol. 2009;20:365–374. doi: 10.1016/j.semcdb.2008.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Biggs WH, 3rd, Cavenee WK, Arden KC. Identification and characterization of members of the FKHR (FOX O) subclass of winged-helix transcription factors in the mouse. Mamm Genome. 2001;12:416–425. doi: 10.1007/s003350020002. [DOI] [PubMed] [Google Scholar]
- 13.Biggs WH, 3rd, Meisenhelder J, Hunter T, Cavenee WK, Arden KC. Protein kinase B/Akt-mediated phosphorylation promotes nuclear exclusion of the winged helix transcription factor FKHR1. Proc Natl Acad Sci U S A. 1999;96:7421–7426. doi: 10.1073/pnas.96.13.7421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bisschop A, Gayan-Ramirez G, Rollier H, Gosselink R, Dom R, de Bock V, Decramer M. Intermittent inspiratory muscle training induces fiber hypertrophy in rat diaphragm. Am J Respir Crit Care Med. 1997;155:1583–1589. doi: 10.1164/ajrccm.155.5.9154861. [DOI] [PubMed] [Google Scholar]
- 15.Blaauw B, Canato M, Agatea L, Toniolo L, Mammucari C, Masiero E, Abraham R, Sandri M, Schiaffino S, Reggiani C. Inducible activation of Akt increases skeletal muscle mass and force without satellite cell activation. FASEB J. 2009;23:3896–3905. doi: 10.1096/fj.09-131870. [DOI] [PubMed] [Google Scholar]
- 16.Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, Clarke BA, Poueymirou WT, Panaro FJ, Na E, Dharmarajan K, Pan ZQ, Valenzuela DM, DeChiara TM, Stitt TN, Yancopoulos GD, Glass DJ. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science. 2001;294:1704–1708. doi: 10.1126/science.1065874. [DOI] [PubMed] [Google Scholar]
- 17.Bodine SC, Stitt TN, Gonzalez M, Kline WO, Stover GL, Bauerlein R, Zlotchenko E, Scrimgeour A, Lawrence JC, Glass DJ, Yancopoulos GD. Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nat Cell Biol. 2001;3:1014–1019. doi: 10.1038/ncb1101-1014. [DOI] [PubMed] [Google Scholar]
- 18.Bolster DR, Crozier SJ, Kimball SR, Jefferson LS. AMP-activated protein kinase suppresses protein synthesis in rat skeletal muscle through down-regulated mammalian target of rapamycin (mTOR) signaling. J Biol Chem. 2002;277:23977–23980. doi: 10.1074/jbc.C200171200. [DOI] [PubMed] [Google Scholar]
- 19.Booth FW, Criswell DS. Molecular events underlying skeletal muscle atrophy and the development of effective countermeasures. Int J Sports Med. 1997;18 (Suppl 4):S265–269. doi: 10.1055/s-2007-972723. [DOI] [PubMed] [Google Scholar]
- 20.Bray JJ, Harris AJ. Dissociation between nerve-muscle transmission and nerve trophic effects on rat diaphragm using type D botulinum toxin. J Physiol. 1975;253:53–77. doi: 10.1113/jphysiol.1975.sp011179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brooke MH, Kaiser KK. Muscle fiber types: how many and what kind? Arch Neurol. 1970;23:369–379. doi: 10.1001/archneur.1970.00480280083010. [DOI] [PubMed] [Google Scholar]
- 22.Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–868. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- 23.Brunn GJ, Hudson CC, Sekulic A, Williams JM, Hosoi H, Houghton PJ, Lawrence JC, Jr, Abraham RT. Phosphorylation of the translational repressor PHAS-I by the mammalian target of rapamycin. Science. 1997;277:99–101. doi: 10.1126/science.277.5322.99. [DOI] [PubMed] [Google Scholar]
- 24.Buller AJ, Eccles JC, Eccles RM. Interactions between motoneurones and muscles in respect of the characteristic speeds of responses. J Physiol (London) 1960;150:417–439. doi: 10.1113/jphysiol.1960.sp006395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Buller AJ, Kean AJC, Ranatunga KW. The force-velocity characteristics of cat fast and slow-twitch skeletal muscle following cross-innervation. J Physiol (London) 1971;213:66P–67P. [PubMed] [Google Scholar]
- 26.Buller AJ, Mommaerts WF, Seraydarian K. Enzymic properties of myosin in fast and slow twitch muscles of the cat following cross-innervation. J Physiol (London) 1969;205:581–597. doi: 10.1113/jphysiol.1969.sp008984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Burke RE. Motor units: anatomy, physiology and functional organization. In: Peachey LD, editor. Handbook of Physiology. The Nervous System. Motor Control. Bethesda, MD: Am Physiol Soc; 1981. pp. 345–422. [Google Scholar]
- 28.Burke RE, Fleshman JW, Glenn LL, Lev-Tov A, O’Donovan MJ, Pinter MJ. An HRP study of the relation between cell size and motor unit type in cat ankle extensor motoneurons. J Comp Neurol. 1982;209:17–28. doi: 10.1002/cne.902090103. [DOI] [PubMed] [Google Scholar]
- 29.Burke RE, Levine DN, Tsairis P, Zajac FE., 3rd Physiological types and histochemical profiles in motor units of the cat gastrocnemius. J Physiol (Lond) 1973;234:723–748. doi: 10.1113/jphysiol.1973.sp010369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Burke RE, Levine DN, Zajac FE., 3rd Mammalian motor units: physiological-histochemical correlation in three types in cat gastrocnemius. Science. 1971;174:709–712. doi: 10.1126/science.174.4010.709. [DOI] [PubMed] [Google Scholar]
- 31.Burnham R, Martin T, Stein R, Bell G, MacLean I, Steadward R. Skeletal muscle fibre type transformation following spinal cord injury. Spinal Cord. 1997;35:86–91. doi: 10.1038/sj.sc.3100364. [DOI] [PubMed] [Google Scholar]
- 32.Cai D, Frantz JD, Tawa NE, Jr, Melendez PA, Oh BC, Lidov HG, Hasselgren PO, Frontera WR, Lee J, Glass DJ, Shoelson SE. IKKbeta/NF-kappaB activation causes severe muscle wasting in mice. Cell. 2004;119:285–298. doi: 10.1016/j.cell.2004.09.027. [DOI] [PubMed] [Google Scholar]
- 33.Caiozzo VJ, Haddad F, Baker MJ, Herrick RE, Prietto N, Baldwin KMg. Microgravity-induced transformations of myosin isoforms and contractile properties of skeletal muscle. J Appl Physiol. 1996;81:123–132. doi: 10.1152/jappl.1996.81.1.123. [DOI] [PubMed] [Google Scholar]
- 34.Campos GE, Luecke TJ, Wendeln HK, Toma K, Hagerman FC, Murray TF, Ragg KE, Ratamess NA, Kraemer WJ, Staron RS. Muscular adaptations in response to three different resistance-training regimens: specificity of repetition maximum training zones. Eur J Appl Physiol. 2002;88:50–60. doi: 10.1007/s00421-002-0681-6. [DOI] [PubMed] [Google Scholar]
- 35.Chen J. Novel regulatory mechanisms of mTOR signaling. Curr Top Microbiol Immunol. 2004;279:245–257. doi: 10.1007/978-3-642-18930-2_14. [DOI] [PubMed] [Google Scholar]
- 36.Citri A, Skaria KB, Yarden Y. The deaf and the dumb: the biology of ErbB-2 and ErbB-3. Exp Cell Res. 2003;284:54–65. doi: 10.1016/s0014-4827(02)00101-5. [DOI] [PubMed] [Google Scholar]
- 37.Clarke BA, Drujan D, Willis MS, Murphy LO, Corpina RA, Burova E, Rakhilin SV, Stitt TN, Patterson C, Latres E, Glass DJ. The E3 Ligase MuRF1 degrades myosin heavy chain protein in dexamethasone-treated skeletal muscle. Cell Metab. 2007;6:376–385. doi: 10.1016/j.cmet.2007.09.009. [DOI] [PubMed] [Google Scholar]
- 38.Coffer PJ, Woodgett JR. Molecular cloning and characterisation of a novel putative protein-serine kinase related to the cAMP-dependent and protein kinase C families. Eur J Biochem. 1991;201:475–481. doi: 10.1111/j.1432-1033.1991.tb16305.x. [DOI] [PubMed] [Google Scholar]
- 39.Cohen CA, Zagelbaum G, Gross D, Roussos C, Macklem PT. Clinical manifestations of inspiratory muscle fatigue. Am J Med. 1982;73:308–316. [PubMed] [Google Scholar]
- 40.Cooper JD, Trulock EP, Triantafillou AN, Patterson GA, Pohl MS, Deloney PA, Sundaresan RS, Roper CL. Bilateral pneumectomy (volume reduction) for chronic obstructive pulmonary disease. J Thorac Cardiovasc Surg. 1995;109:106–116. doi: 10.1016/S0022-5223(95)70426-4. discussion 116-109. [DOI] [PubMed] [Google Scholar]
- 41.Copp J, Manning G, Hunter T. TORC-specific phosphorylation of mammalian target of rapamycin (mTOR): phospho-Ser2481 is a marker for intact mTOR signaling complex 2. Cancer Res. 2009;69:1821–1827. doi: 10.1158/0008-5472.CAN-08-3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Corton JM, Gillespie JG, Hawley SA, Hardie DG. 5-aminoimidazole-4-carboxamide ribonucleoside. A specific method for activating AMP-activated protein kinase in intact cells? Eur J Biochem. 1995;229:558–565. doi: 10.1111/j.1432-1033.1995.tb20498.x. [DOI] [PubMed] [Google Scholar]
- 43.Darr KC, Schultz D. Exercise-induced satellite cell activation in growing and mature skeletal muscle. J Appl Physiol. 1987;63:1816–1821. doi: 10.1152/jappl.1987.63.5.1816. [DOI] [PubMed] [Google Scholar]
- 44.De Troyer A. Effect of hyperinflation on the diaphragm. Eur Respir J. 1997;10:708–713. [PubMed] [Google Scholar]
- 45.De Troyer A, Estenne M. Coordination between rib cage muscles and diaphragm during quiet breathing in humans. J Appl Physiol. 1984;57:899–906. doi: 10.1152/jappl.1984.57.3.899. [DOI] [PubMed] [Google Scholar]
- 46.De Troyer A, Farkas GA. Contribution of the rib cage inspiratory muscles to breathing in baboons. Respir Physiol. 1994;97:135–146. doi: 10.1016/0034-5687(94)90021-3. [DOI] [PubMed] [Google Scholar]
- 47.De Troyer A, Kirkwood PA, Wilson TA. Respiratory action of the intercostal muscles. Physiol Rev. 2005;85:717–756. doi: 10.1152/physrev.00007.2004. [DOI] [PubMed] [Google Scholar]
- 48.Decramer M. Hyperinflation and respiratory muscle interaction. Eur Respir J. 1997;10:934–941. [PubMed] [Google Scholar]
- 49.Decramer M, De Troyer A. Respiratory changes in parasternal intercostal length. J Appl Physiol. 1984;57:1254–1260. doi: 10.1152/jappl.1984.57.4.1254. [DOI] [PubMed] [Google Scholar]
- 50.Decramer M, Xi JT, Reid MB, Kelly S, Macklem PT, Demedts M. Relationship between diaphragm length and abdominal dimensions. J Appl Physiol. 1986;61:1815–1820. doi: 10.1152/jappl.1986.61.5.1815. [DOI] [PubMed] [Google Scholar]
- 51.Deruisseau KC, Kavazis AN, Deering MA, Falk DJ, Van Gammeren D, Yimlamai T, Ordway GA, Powers SK. Mechanical ventilation induces alterations of the ubiquitin-proteasome pathway in the diaphragm. J Appl Physiol. 2005;98:1314–1321. doi: 10.1152/japplphysiol.00993.2004. [DOI] [PubMed] [Google Scholar]
- 52.DeRuisseau KC, Kavazis AN, Deering MA, Falk DJ, Van Gammeren D, Yimlamai T, Ordway GA, Powers SK. Mechanical ventilation induces alterations of the ubiquitin-proteasome pathway in the diaphragm. J Appl Physiol. 2005;98:1314–1321. doi: 10.1152/japplphysiol.00993.2004. [DOI] [PubMed] [Google Scholar]
- 53.Deval C, Mordier S, Obled C, Bechet D, Combaret L, Attaix D, Ferrara M. Identification of cathepsin L as a differentially expressed message associated with skeletal muscle wasting. Biochem J. 2001;360:143–150. doi: 10.1042/0264-6021:3600143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dick TE, Kong FJ, Berger AJ. Correlation of recruitment order with axonal conduction velocity for supraspinally driven diaphragmatic motor units. J Neurophysiol. 1987;57:245–259. doi: 10.1152/jn.1987.57.1.245. [DOI] [PubMed] [Google Scholar]
- 55.Dick TE, Kong FJ, Berger AJ. Recruitment order of diaphragmatic motor units obeys Hennemans’s size principle. In: Sieck GC, Gandevia SC, Cameron WE, editors. Respiratory Muscles and Their Neuromotor Control. New York: Alan R. Liss; 1987. pp. 249–261. [Google Scholar]
- 56.Du J, Wang X, Miereles C, Bailey JL, Debigare R, Zheng B, Price SR, Mitch WE. Activation of caspase-3 is an initial step triggering accelerated muscle proteolysis in catabolic conditions. J Clin Invest. 2004;113:115–123. doi: 10.1172/JCI200418330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dudhia J, Scott CM, Draper ER, Heinegard D, Pitsillides AA, Smith RK. Aging enhances a mechanically-induced reduction in tendon strength by an active process involving matrix metalloproteinase activity. Aging Cell. 2007;6:547–556. doi: 10.1111/j.1474-9726.2007.00307.x. [DOI] [PubMed] [Google Scholar]
- 58.Dupont-Versteegden EE, Houle JD, Dennis RA, Zhang J, Knox M, Wagoner G, Peterson CA. Exercise-induced gene expression in soleus muscle is dependent on time after spinal cord injury in rats. Muscle Nerve. 2004;29:73–81. doi: 10.1002/mus.10511. [DOI] [PubMed] [Google Scholar]
- 59.Dusterhoft S, Pette D. Satellite cells from slow rat muscle express slow myosin under appropriate culture conditions. Differentiation. 1993;53:25–33. doi: 10.1111/j.1432-0436.1993.tb00642.x. [DOI] [PubMed] [Google Scholar]
- 60.Eccles JC. Problems of plasticity and organization at simplest levels of mammalian central nervous system. Perspect Biol Med. 1958;1:379–396. doi: 10.1353/pbm.1958.0034. [DOI] [PubMed] [Google Scholar]
- 61.Enad JG, Fournier M, Sieck GC. Oxidative capacity and capillary density of diaphragm motor units. J Appl Physiol. 1989;67:620–627. doi: 10.1152/jappl.1989.67.2.620. [DOI] [PubMed] [Google Scholar]
- 62.Esper RM, Loeb JA. Neurotrophins induce neuregulin release through protein kinase Cdelta activation. J Biol Chem. 2009;284:26251–26260. doi: 10.1074/jbc.M109.002915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Farkas GA, Roussos C. Diaphragm in emphysematous hamsters: sarcomere adaptability. J Appl Physiol. 1983;54:1635–1640. doi: 10.1152/jappl.1983.54.6.1635. [DOI] [PubMed] [Google Scholar]
- 64.Fenn WO. A quantitative comparison between the energy liberated and the work performed by the isolated sartorius muscle of the frog. J Physiol (London) 1923;58:175–203. doi: 10.1113/jphysiol.1923.sp002115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fournier M, Sieck GC. Mechanical properties of muscle units in the cat diaphragm. J Neurophysiol. 1988;59:1055–1066. doi: 10.1152/jn.1988.59.3.1055. [DOI] [PubMed] [Google Scholar]
- 66.Furuno K, Goodman MN, Goldberg AL. Role of different proteolytic systems in the degradation of muscle proteins during denervation atrophy. J Biol Chem. 1990;265:8550–8557. [PubMed] [Google Scholar]
- 67.Gandevia SC, McKenzie DK, Plassman BL. Activation of human respiratory muscles during different voluntary manoeuvres. J Physiol. 1990;428:387–403. doi: 10.1113/jphysiol.1990.sp018218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gauthier GF, Hobbs AW. Effects of denervation on the distribution of myosin isozymes in skeletal muscle fibers. Exp Neurol. 1982;76:331–346. doi: 10.1016/0014-4886(82)90213-8. [DOI] [PubMed] [Google Scholar]
- 69.Gautsch TA, Anthony JC, Kimball SR, Paul GL, Layman DK, Jefferson LS. Availability of eIF4E regulates skeletal muscle protein synthesis during recovery from exercise. Am J Physiol. 1998;274:C406–414. doi: 10.1152/ajpcell.1998.274.2.C406. [DOI] [PubMed] [Google Scholar]
- 70.Gea J, Hamid Q, Czaika G, Zhu E, Mohan-Ram V, Goldspink G, Grassino A. Expression of myosin heavy-chain isoforms in the respiratory muscles following inspiratory resistive breathing. Am J Respir Crit Care Med. 2000;161:1274–1278. doi: 10.1164/ajrccm.161.4.99040103. [DOI] [PubMed] [Google Scholar]
- 71.Geiger PC, Bailey JP, Mantilla CB, Zhan WZ, Sieck GC. Mechanisms underlying myosin heavy chain expression during development of the rat diaphragm muscle. J Appl Physiol. 2006;101:1546–1555. doi: 10.1152/japplphysiol.00221.2006. [DOI] [PubMed] [Google Scholar]
- 72.Geiger PC, Bailey JP, Zhan WZ, Mantilla CB, Sieck GC. Denervation-induced changes in myosin heavy chain expression in the rat diaphragm muscle. J Appl Physiol. 2003;95:611–619. doi: 10.1152/japplphysiol.00862.2002. [DOI] [PubMed] [Google Scholar]
- 73.Geiger PC, Cody MJ, Han YS, Hunter LW, Zhan WZ, Sieck GC. Effects of hypothyroidism on maximum specific force in rat diaphragm muscle fibers. J Appl Physiol. 2002;92:1506–1514. doi: 10.1152/japplphysiol.00095.2001. [DOI] [PubMed] [Google Scholar]
- 74.Geiger PC, Cody MJ, Macken RL, Bayrd ME, Sieck GC. Effect of unilateral denervation on maximum specific force in rat diaphragm muscle fibers. J Appl Physiol. 2001;90:1196–1204. doi: 10.1152/jappl.2001.90.4.1196. [DOI] [PubMed] [Google Scholar]
- 75.Geiger PC, Cody MJ, Macken RL, Bayrd ME, Sieck GC. Mechanisms underlying increased force generation by rat diaphragm muscle fibers during development. J Appl Physiol. 2001;90:380–388. doi: 10.1152/jappl.2001.90.1.380. [DOI] [PubMed] [Google Scholar]
- 76.Geiger PC, Cody MJ, Macken RL, Sieck GC. Maximum specific force depends on myosin heavy chain content in rat diaphragm muscle fibers. J Appl Physiol. 2000;89:695–703. doi: 10.1152/jappl.2000.89.2.695. [DOI] [PubMed] [Google Scholar]
- 77.Geiger PC, Cody MJ, Sieck GC. Force-calcium relationship depends on myosin heavy chain and troponin isoforms in rat diaphragm muscle fibers. J Appl Physiol. 1999;87:1894–1900. doi: 10.1152/jappl.1999.87.5.1894. [DOI] [PubMed] [Google Scholar]
- 78.Gingras AC, Kennedy SG, O’Leary MA, Sonenberg N, Hay N. 4E-BP1, a repressor of mRNA translation, is phosphorylated and inactivated by the Akt(PKB) signaling pathway. Genes Dev. 1998;12:502–513. doi: 10.1101/gad.12.4.502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Glass DJ. Signalling pathways that mediate skeletal muscle hypertrophy and atrophy. Nat Cell Biol. 2003;5:87–90. doi: 10.1038/ncb0203-87. [DOI] [PubMed] [Google Scholar]
- 80.Gomes MD, Lecker SH, Jagoe RT, Navon A, Goldberg AL. Atrogin-1, a muscle-specific F-box protein highly expressed during muscle atrophy. Proc Natl Acad Sci U S A. 2001;98:14440–14445. doi: 10.1073/pnas.251541198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gomez-Pinilla F, Ying Z, Roy RR, Molteni R, Edgerton VR. Voluntary exercise induces a BDNF-mediated mechanism that promotes neuroplasticity. J Neurophysiol. 2002;88:2187–2195. doi: 10.1152/jn.00152.2002. [DOI] [PubMed] [Google Scholar]
- 82.Gordon AM, Huxley AF, Julian FJ. Tension development in highly stretched vertebrate muscle fibres. J Physiol. 1966;184:143–169. doi: 10.1113/jphysiol.1966.sp007908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gordon AM, Huxley AF, Julian FJ. The variation in isometric tension with sarcomere length in vertebrate muscle fibres. J Physiol. 1966;184:170–192. doi: 10.1113/jphysiol.1966.sp007909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gosselin LE, Brice G, Carlson B, Prakash YS, Sieck GC. Changes in satellite cell mitotic activity during acute period of unilateral diaphragm denervation. J Appl Physiol. 1994;77:1128–1134. doi: 10.1152/jappl.1994.77.3.1128. [DOI] [PubMed] [Google Scholar]
- 85.Gosselin LE, Sieck GC, Aleff RA, Martinez DA, Vailas AC. Changes in diaphragm muscle collagen gene expression after acute unilateral denervation. J Appl Physiol. 1995;79:1249–1254. doi: 10.1152/jappl.1995.79.4.1249. [DOI] [PubMed] [Google Scholar]
- 86.Greer EL, Dowlatshahi D, Banko MR, Villen J, Hoang K, Blanchard D, Gygi SP, Brunet A. An AMPK-FOXO pathway mediates longevity induced by a novel method of dietary restriction in C. elegans. Curr Biol. 2007;17:1646–1656. doi: 10.1016/j.cub.2007.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Greer EL, Oskoui PR, Banko MR, Maniar JM, Gygi MP, Gygi SP, Brunet A. The energy sensor AMP-activated protein kinase directly regulates the mammalian FOXO3 transcription factor. J Biol Chem. 2007;282:30107–30119. doi: 10.1074/jbc.M705325200. [DOI] [PubMed] [Google Scholar]
- 88.Gregory SA. Evaluation and management of respiratory muscle dysfunction in ALS. NeuroRehabilitation. 2007;22:435–443. [PubMed] [Google Scholar]
- 89.Gundersen K. Determination of muscle contractile properties: the importance of the nerve. Acta Physiol Scand. 1998;162:333–341. doi: 10.1046/j.1365-201X.1998.0336e.x. [DOI] [PubMed] [Google Scholar]
- 90.Guyton AC, Hall JE. Textbook of Medical Physiology. 10. Philadelphia, PA: W.B. Saunders Company; 2000. [Google Scholar]
- 91.Haas TL, Milkiewicz M, Davis SJ, Zhou AL, Egginton S, Brown MD, Madri JA, Hudlicka O. Matrix metalloproteinase activity is required for activity-induced angiogenesis in rat skeletal muscle. Am J Physiol Heart Circ Physiol. 2000;279:H1540–1547. doi: 10.1152/ajpheart.2000.279.4.H1540. [DOI] [PubMed] [Google Scholar]
- 92.Halevy O, Cantley LC. Differential regulation of the phosphoinositide 3-kinase and MAP kinase pathways by hepatocyte growth factor vs. insulin-like growth factor-I in myogenic cells. Exp Cell Res. 2004;297:224–234. doi: 10.1016/j.yexcr.2004.03.024. [DOI] [PubMed] [Google Scholar]
- 93.Han YS, Geiger PC, Cody MJ, Macken RL, Sieck GC. ATP consumption rate per cross bridge depends on myosin heavy chain isoform. J Appl Physiol. 2003;94:2188–2196. doi: 10.1152/japplphysiol.00618.2002. [DOI] [PubMed] [Google Scholar]
- 94.Hardie DG, Carling D. The AMP-activated protein kinase--fuel gauge of the mammalian cell? Eur J Biochem. 1997;246:259–273. doi: 10.1111/j.1432-1033.1997.00259.x. [DOI] [PubMed] [Google Scholar]
- 95.Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18:1926–1945. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- 96.Hellyer NJ, Mantilla CB, Park EW, Zhan WZ, Sieck GC. Neuregulin-dependent protein synthesis in C2C12 myotubes and rat diaphragm muscle. Am J Physiol Cell Physiol. 2006;291:C1056–1061. doi: 10.1152/ajpcell.00625.2005. [DOI] [PubMed] [Google Scholar]
- 97.Henneman E. Relation between size of neurons and their susceptibility to discharge. Science. 1957;126:1345–1346. doi: 10.1126/science.126.3287.1345. [DOI] [PubMed] [Google Scholar]
- 98.Henneman E, Mendell LM. Functional organization of motoneuron pool and its inputs. In: Brookhart JM, Mountcastle VB, editors. Handbook of Physiology. Bethesda: American Physiological Society; 1981. pp. 423–507. [Google Scholar]
- 99.Henneman E, Somjen G, Carpenter DO. Functional significance of cell size in spinal motoneurons. J Neurophysiol. 1965;28:560–580. doi: 10.1152/jn.1965.28.3.560. [DOI] [PubMed] [Google Scholar]
- 100.Hensbergen E, Kernell D. Daily durations of spontaneous activity in cat’s ankle muscles. Exp Brain Res. 1997;115:325–332. doi: 10.1007/pl00005701. [DOI] [PubMed] [Google Scholar]
- 101.Hilaire G, Gauthier P, Monteau R. Central respiratory drive and recruitment order of phrenic and inspiratory laryngeal motoneurones. Respir Physiol. 1983;51:341–359. doi: 10.1016/0034-5687(83)90028-2. [DOI] [PubMed] [Google Scholar]
- 102.Hilaire G, Monteau R, Khatib M. Determination of recruitment order of phrenic motoneurons. In: Sieck GC, Gandevia SC, Cameron WE, editors. Respiratory Muscles and Their Neuromotor Control. New York: Alan R. Liss; 1987. pp. 249–261. [Google Scholar]
- 103.Holm L, Reitelseder S, Pedersen TG, Doessing S, Petersen SG, Flyvbjerg A, Andersen JL, Aagaard P, Kjaer M. Changes in muscle size and MHC composition in response to resistance exercise with heavy and light loading intensity. J Appl Physiol. 2008;105:1454–1461. doi: 10.1152/japplphysiol.90538.2008. [DOI] [PubMed] [Google Scholar]
- 104.Hudson AL, Butler JE, Gandevia SC, De Troyer A. Interplay between the inspiratory and postural functions of the human parasternal intercostal muscles. J Neurophysiol. 2010;103:1622–1629. doi: 10.1152/jn.00887.2009. [DOI] [PubMed] [Google Scholar]
- 105.Hunter RB, Kandarian SC. Disruption of either the Nfkb1 or the Bcl3 gene inhibits skeletal muscle atrophy. J Clin Invest. 2004;114:1504–1511. doi: 10.1172/JCI21696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hunter RB, Stevenson E, Koncarevic A, Mitchell-Felton H, Essig DA, Kandarian SC. Activation of an alternative NF-kappaB pathway in skeletal muscle during disuse atrophy. FASEB J. 2002;16:529–538. doi: 10.1096/fj.01-0866com. [DOI] [PubMed] [Google Scholar]
- 107.Huxley AF. Muscle structure and theories of contraction. Prog Biophysics Biophys Chem. 1957;7:255–318. [PubMed] [Google Scholar]
- 108.Inoki K, Ouyang H, Zhu T, Lindvall C, Wang Y, Zhang X, Yang Q, Bennett C, Harada Y, Stankunas K, Wang CY, He X, MacDougald OA, You M, Williams BO, Guan KL. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell. 2006;126:955–968. doi: 10.1016/j.cell.2006.06.055. [DOI] [PubMed] [Google Scholar]
- 109.Jacinto E, Hall MN. Tor signalling in bugs, brain and brawn. Nat Rev Mol Cell Biol. 2003;4:117–126. doi: 10.1038/nrm1018. [DOI] [PubMed] [Google Scholar]
- 110.Jefferson LS, Fabian JR, Kimball SR. Glycogen synthase kinase-3 is the predominant insulin-regulated eukaryotic initiation factor 2B kinase in skeletal muscle. Int J Biochem Cell Biol. 1999;31:191–200. doi: 10.1016/s1357-2725(98)00141-1. [DOI] [PubMed] [Google Scholar]
- 111.Jo SA, Zhu X, Marchionni MA, Burden SJ. Neuregulins are concentrated at nerve-muscle synapses and activate ACh-receptor gene expression. Nature. 1995;373:158–161. doi: 10.1038/373158a0. [DOI] [PubMed] [Google Scholar]
- 112.Jodkowski JS, Viana F, Dick TE, Berger AJ. Electrical properties of phrenic motoneurons in the cat: correlation with inspiratory drive. J Neurophysiol. 1987;58:105–124. doi: 10.1152/jn.1987.58.1.105. [DOI] [PubMed] [Google Scholar]
- 113.Jones PF, Jakubowicz T, Pitossi FJ, Maurer F, Hemmings BA. Molecular cloning and identification of a serine/threonine protein kinase of the second-messenger subfamily. Proc Natl Acad Sci U S A. 1991;88:4171–4175. doi: 10.1073/pnas.88.10.4171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Jorgensen SB, Richter EA, Wojtaszewski JF. Role of AMPK in skeletal muscle metabolic regulation and adaptation in relation to exercise. J Physiol. 2006;574:17–31. doi: 10.1113/jphysiol.2006.109942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Judge AR, Koncarevic A, Hunter RB, Liou HC, Jackman RW, Kandarian SC. Role for IkappaBalpha, but not c-Rel, in skeletal muscle atrophy. Am J Physiol Cell Physiol. 2007;292:C372–382. doi: 10.1152/ajpcell.00293.2006. [DOI] [PubMed] [Google Scholar]
- 116.Kalinowski A, Plowes NJ, Huang Q, Berdejo-Izquierdo C, Russell RR, Russell KS. Metalloproteinase-dependent cleavage of neuregulin and autocrine stimulation of vascular endothelial cells. FASEB J. 2010;24:2567–2575. doi: 10.1096/fj.08-129072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kamei Y, Miura S, Suzuki M, Kai Y, Mizukami J, Taniguchi T, Mochida K, Hata T, Matsuda J, Aburatani H, Nishino I, Ezaki O. Skeletal muscle FOXO1 (FKHR) transgenic mice have less skeletal muscle mass, down-regulated Type I (slow twitch/red muscle) fiber genes, and impaired glycemic control. J Biol Chem. 2004;279:41114–41123. doi: 10.1074/jbc.M400674200. [DOI] [PubMed] [Google Scholar]
- 118.Katagiri M, Young RN, Platt RS, Kieser TM, Easton PA. Respiratory muscle compensation for unilateral or bilateral hemidiaphragm paralysis in awake canines. J Appl Physiol. 1994;77:1972–1982. doi: 10.1152/jappl.1994.77.4.1972. [DOI] [PubMed] [Google Scholar]
- 119.Keens TG, Bryan AC, Levison H, Ianuzzo CD. Developmental pattern of muscle fiber types in human ventilatory muscles. J Appl Physiol. 1978;44:909–913. doi: 10.1152/jappl.1978.44.6.909. [DOI] [PubMed] [Google Scholar]
- 120.Kernell D. The motoneurone and its muscle fibres. Oxford, New York: Oxford University Press; 2006. [Google Scholar]
- 121.King TD, Song L, Jope RS. AMP-activated protein kinase (AMPK) activating agents cause dephosphorylation of Akt and glycogen synthase kinase-3. Biochem Pharmacol. 2006;71:1637–1647. doi: 10.1016/j.bcp.2006.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kleijn M, Scheper GC, Voorma HO, Thomas AA. Regulation of translation initiation factors by signal transduction. Eur J Biochem. 1998;253:531–544. doi: 10.1046/j.1432-1327.1998.2530531.x. [DOI] [PubMed] [Google Scholar]
- 123.Kong FJ, Berger AJ. Firing properties and hypercapnic responses of single phrenic motor axons in the rat. J Appl Physiol. 1986;61:1999–2004. doi: 10.1152/jappl.1986.61.6.1999. [DOI] [PubMed] [Google Scholar]
- 124.Koumbourlis AC. Scoliosis and the respiratory system. Paediatr Respir Rev. 2006;7:152–160. doi: 10.1016/j.prrv.2006.04.009. [DOI] [PubMed] [Google Scholar]
- 125.Lantier L, Mounier R, Leclerc J, Pende M, Foretz M, Viollet B. Coordinated maintenance of muscle cell size control by AMP-activated protein kinase. FASEB J. 2010;24:3555–3561. doi: 10.1096/fj.10-155994. [DOI] [PubMed] [Google Scholar]
- 126.Larson CR, Yajima Y, Ko P. Modification in activity of medullary respiratory-related neurons for vocalization and swallowing. J Neurophysiol. 1994;71:2294–2304. doi: 10.1152/jn.1994.71.6.2294. [DOI] [PubMed] [Google Scholar]
- 127.Latres E, Amini AR, Amini AA, Griffiths J, Martin FJ, Wei Y, Lin HC, Yancopoulos GD, Glass DJ. Insulin-like growth factor-1 (IGF-1) inversely regulates atrophy-induced genes via the phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin (PI3K/Akt/mTOR) pathway. J Biol Chem. 2005;280:2737–2744. doi: 10.1074/jbc.M407517200. [DOI] [PubMed] [Google Scholar]
- 128.Lecker SH, Jagoe RT, Gilbert A, Gomes M, Baracos V, Bailey J, Price SR, Mitch WE, Goldberg AL. Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. Faseb J. 2004;18:39–51. doi: 10.1096/fj.03-0610com. [DOI] [PubMed] [Google Scholar]
- 129.Lecker SH, Solomon V, Mitch WE, Goldberg AL. Muscle protein breakdown and the critical role of the ubiquitin-proteasome pathway in normal and disease states. J Nutr. 1999;129:227S–237S. doi: 10.1093/jn/129.1.227S. [DOI] [PubMed] [Google Scholar]
- 130.Lee SW, Dai G, Hu Z, Wang X, Du J, Mitch WE. Regulation of muscle protein degradation: coordinated control of apoptotic and ubiquitin-proteasome systems by phosphatidylinositol 3 kinase. J Am Soc Nephrol. 2004;15:1537–1545. doi: 10.1097/01.asn.0000127211.86206.e1. [DOI] [PubMed] [Google Scholar]
- 131.Legrand A, Cappello M, De Troyer A. Response of the inspiratory intercostal [correction of intercoastal] muscles to increased inertial loads. Respir Physiol. 1995;102:17–27. doi: 10.1016/0034-5687(95)00051-e. [DOI] [PubMed] [Google Scholar]
- 132.Levine S, Nguyen T, Taylor N, Friscia ME, Budak MT, Rothenberg P, Zhu J, Sachdeva R, Sonnad S, Kaiser LR, Rubinstein NA, Powers SK, Shrager JB. Rapid disuse atrophy of diaphragm fibers in mechanically ventilated humans. N Engl J Med. 2008;358:1327–1335. doi: 10.1056/NEJMoa070447. [DOI] [PubMed] [Google Scholar]
- 133.Lewis MI, Bodine SC, Kamangar N, Xu X, Da X, Fournier M. Effect of severe short-term malnutrition on diaphragm muscle signal transduction pathways influencing protein turnover. J Appl Physiol. 2006;100:1799–1806. doi: 10.1152/japplphysiol.01233.2005. [DOI] [PubMed] [Google Scholar]
- 134.Lewis MI, Fournier M, Da X, Li H, Mosenifar Z, McKenna RJ, Jr, Cohen AH. Short-term influences of lung volume reduction surgery on the diaphragm in emphysematous hamsters. Am J Respir Crit Care Med. 2004;170:753–759. doi: 10.1164/rccm.200402-181OC. [DOI] [PubMed] [Google Scholar]
- 135.Lewis MI, Li H, Huang ZS, Biring MS, Cercek B, Fournier M. Influence of varying degrees of malnutrition on IGF-I expression in the rat diaphragm. J Appl Physiol. 2003;95:555–562. doi: 10.1152/japplphysiol.00916.2002. [DOI] [PubMed] [Google Scholar]
- 136.Lewis MI, Sieck GC. Effect of acute nutritional deprivation on diaphragm structure and function. J Appl Physiol. 1990;68:1938–1944. doi: 10.1152/jappl.1990.68.5.1938. [DOI] [PubMed] [Google Scholar]
- 137.Lewis MI, Zhan WZ, Sieck GC. Adaptations of the diaphragm in emphysema. J Appl Physiol. 1992;72:934–943. doi: 10.1152/jappl.1992.72.3.934. [DOI] [PubMed] [Google Scholar]
- 138.Lexell J, Taylor CC, Sjostrom M. What is the cause of the ageing atrophy? Total number, size and proportion of different fiber types studied in whole vastus lateralis muscle from 15- to 83-year-old men. J Neurol Sci. 1988;84:275–294. doi: 10.1016/0022-510x(88)90132-3. [DOI] [PubMed] [Google Scholar]
- 139.Liddell EGT, Sherrington CS. Recruitment and some other factors of reflex inhibition. Proc Roy Soc Lond (Biol) 1925;97:488–518. [Google Scholar]
- 140.Lieber RL. Skeletal muscle structure, function, & plasticity: the physiological basis of rehabilitation. Baltimore, MD: Lippincott Williams & Wilkins; 2002. [Google Scholar]
- 141.Loeb JA, Hmadcha A, Fischbach GD, Land SJ, Zakarian VL. Neuregulin expression at neuromuscular synapses is modulated by synaptic activity and neurotrophic factors. J Neurosci. 2002;22:2206–2214. doi: 10.1523/JNEUROSCI.22-06-02206.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Loeb JA, Susanto ET, Fischbach GD. The neuregulin precursor proARIA is processed to ARIA after expression on the cell surface by a protein kinase C-enhanced mechanism. Mol Cell Neurosci. 1998;11:77–91. doi: 10.1006/mcne.1998.0676. [DOI] [PubMed] [Google Scholar]
- 143.Loring SH, Mead J. Action of the diaphragm on the rib cage inferred from a force-balance analysis. J Appl Physiol. 1982;53:756–760. doi: 10.1152/jappl.1982.53.3.756. [DOI] [PubMed] [Google Scholar]
- 144.Luo X, Prior M, He W, Hu X, Tang X, Shen W, Yadav S, Kiryu-Seo S, Miller R, Trapp BD, Yan R. Cleavage of neuregulin-1 by BACE1 or ADAM10 protein produces differential effects on myelination. J Biol Chem. 2011;286:23967–23974. doi: 10.1074/jbc.M111.251538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P, Burden SJ, Di Lisi R, Sandri C, Zhao J, Goldberg AL, Schiaffino S, Sandri M. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 2007;6:458–471. doi: 10.1016/j.cmet.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 146.Mantilla CB, Rowley KL, Zhan WZ, Fahim MA, Sieck GC. Synaptic vesicle pools at diaphragm neuromuscular junctions vary with motoneuron soma, not axon terminal, inactivity. Neurosci. 2007;146:178–189. doi: 10.1016/j.neuroscience.2007.01.048. [DOI] [PubMed] [Google Scholar]
- 147.Mantilla CB, Seven YB, Zhan WZ, Sieck GC. Diaphragm motor unit recruitment in rats. Respir Physiol Neurobiol. 2010;173:101–106. doi: 10.1016/j.resp.2010.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Mantilla CB, Sieck GC. Invited review: Mechanisms underlying motor unit plasticity in the respiratory system. J Appl Physiol. 2003;94:1230–1241. doi: 10.1152/japplphysiol.01120.2002. [DOI] [PubMed] [Google Scholar]
- 149.Mantilla CB, Sieck GC. Key Aspects of Phrenic Motoneuron and Diaphragm Muscle Development during the Perinatal Period. J Appl Physiol. 2008 doi: 10.1152/japplphysiol.01192.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Mantilla CB, Sieck GC. Neuromuscular adaptations to respiratory muscle inactivity. Respir Physiol Neurobiol. 2009;169:133–140. doi: 10.1016/j.resp.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Mantilla CB, Sieck GC. Trophic factor expression in phrenic motor neurons. Respir Physiol Neurobiol. 2008;164:252–262. doi: 10.1016/j.resp.2008.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Mantilla CB, Sill RV, Aravamudan B, Zhan WZ, Sieck GC. Developmental effects on myonuclear domain size of rat diaphragm fibers. J Appl Physiol. 2008;104:787–794. doi: 10.1152/japplphysiol.00347.2007. [DOI] [PubMed] [Google Scholar]
- 153.Martelli AM, Tazzari PL, Evangelisti C, Chiarini F, Blalock WL, Billi AM, Manzoli L, McCubrey JA, Cocco L. Targeting the phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin module for acute myelogenous leukemia therapy: from bench to bedside. Curr Med Chem. 2007;14:2009–2023. doi: 10.2174/092986707781368423. [DOI] [PubMed] [Google Scholar]
- 154.Matsui T, Nagoshi T, Rosenzweig A. Akt and PI 3-kinase signaling in cardiomyocyte hypertrophy and survival. Cell Cycle. 2003;2:220–223. [PubMed] [Google Scholar]
- 155.McCall GE, Allen DL, Linderman JK, Grindeland RE, Roy RR, Mukku VR, Edgerton VR. Maintenance of myonuclear domain size in rat soleus after overload and growth hormone/IGF-I treatment. J Appl Physiol. 1998;84:1407–1412. doi: 10.1152/jappl.1998.84.4.1407. [DOI] [PubMed] [Google Scholar]
- 156.McMahon T. Muscles, reflexes and locomotion. Princeton, NJ: Princeton Univ. Press; 1984. [Google Scholar]
- 157.Mead J, Loring SH. Analysis of volume displacement and length changes of the diaphragm during breathing. J Appl Physiol. 1982;53:750–755. doi: 10.1152/jappl.1982.53.3.750. [DOI] [PubMed] [Google Scholar]
- 158.Mead J, Turner JM, Macklem PT, Little JB. Significance of the relationship between lung recoil and maximum expiratory flow. J Appl Physiol. 1967;22:95–108. doi: 10.1152/jappl.1967.22.1.95. [DOI] [PubMed] [Google Scholar]
- 159.Milano S, Grelot L, Bianchi AL, Iscoe S. Discharge patterns of phrenic motoneurons during fictive coughing and vomiting in decerebrate cats. J Appl Physiol. 1992;73:1626–1636. doi: 10.1152/jappl.1992.73.4.1626. [DOI] [PubMed] [Google Scholar]
- 160.Miller AD, Nonaka S, Lakos SF, Tan LK. Diaphragmatic and external intercostal muscle control during vomiting: Behavior of inspiratory bulbospinal neurons. J Neurophysiol. 1990;63(1):31–36. doi: 10.1152/jn.1990.63.1.31. [DOI] [PubMed] [Google Scholar]
- 161.Miyata H, Zhan WZ, Prakash YS, Sieck GC. Myoneural interactions affect diaphragm muscle adaptations to inactivity. J Appl Physiol. 1995;79:1640–1649. doi: 10.1152/jappl.1995.79.5.1640. [DOI] [PubMed] [Google Scholar]
- 162.Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell. 2004;15:1101–1111. doi: 10.1091/mbc.E03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Monteau R, Khatib M, Hilaire G. Central determination of recruitment order: intracellular study of phrenic motoneurons. Neurosci Lett. 1985;56:341–346. doi: 10.1016/0304-3940(85)90266-6. [DOI] [PubMed] [Google Scholar]
- 164.Nakashima K, Yakabe Y. AMPK activation stimulates myofibrillar protein degradation and expression of atrophy-related ubiquitin ligases by increasing FOXO transcription factors in C2C12 myotubes. Biosci Biotechnol Biochem. 2007;71:1650–1656. doi: 10.1271/bbb.70057. [DOI] [PubMed] [Google Scholar]
- 165.Narici MV, Bordini M, Cerretelli P. Effect of aging on human adductor pollicis muscle function. J Appl Physiol. 1991;71:1277–1281. doi: 10.1152/jappl.1991.71.4.1277. [DOI] [PubMed] [Google Scholar]
- 166.Nave BT, Ouwens M, Withers DJ, Alessi DR, Shepherd PR. Mammalian target of rapamycin is a direct target for protein kinase B: identification of a convergence point for opposing effects of insulin and amino-acid deficiency on protein translation. Biochem J. 1999;344(Pt 2):427–431. [PMC free article] [PubMed] [Google Scholar]
- 167.Newman S, Road J, Bellemare F, Clozel JP, Lavigne CM, Grassino A. Respiratory muscle length measured by sonomicrometry. J Appl Physiol. 1984;56:753–764. doi: 10.1152/jappl.1984.56.3.753. [DOI] [PubMed] [Google Scholar]
- 168.Nguyen T, Shrager J, Kaiser L, Mei L, Daood M, Watchko J, Rubinstein N, Levine S. Developmental myosin heavy chains in the adult human diaphragm: coexpression patterns and effect of COPD. J Appl Physiol. 2000;88:1446–1456. doi: 10.1152/jappl.2000.88.4.1446. [DOI] [PubMed] [Google Scholar]
- 169.Nystrom GJ, Lang CH. Sepsis and AMPK Activation by AICAR Differentially Regulate FoxO-1, -3 and -4 mRNA in Striated Muscle. Int J Clin Exp Med. 2008;1:50–63. [PMC free article] [PubMed] [Google Scholar]
- 170.Pallafacchina G, Calabria E, Serrano AL, Kalhovde JM, Schiaffino S. A protein kinase B-dependent and rapamycin-sensitive pathway controls skeletal muscle growth but not fiber type specification. Proc Natl Acad Sci U S A. 2002;99:9213–9218. doi: 10.1073/pnas.142166599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Papastamelos C, Panitch HB, England SE, Allen JL. Developmental changes in chest wall compliance in infancy and early childhood. J Appl Physiol. 1995;78:179–184. doi: 10.1152/jappl.1995.78.1.179. [DOI] [PubMed] [Google Scholar]
- 172.Pause A, Belsham GJ, Gingras AC, Donze O, Lin TA, Lawrence JC, Jr, Sonenberg N. Insulin-dependent stimulation of protein synthesis by phosphorylation of a regulator of 5′-cap function. Nature. 1994;371:762–767. doi: 10.1038/371762a0. [DOI] [PubMed] [Google Scholar]
- 173.Peter JB, Barnard RJ, Edgerton VR, Gillespie CA, Stempel KE. Metabolic profiles of three fiber types of skeletal muscle in guinea pigs and rabbits. Biochemistry. 1972;11:2627–2633. doi: 10.1021/bi00764a013. [DOI] [PubMed] [Google Scholar]
- 174.Peterson RT, Beal PA, Comb MJ, Schreiber SL. FKBP12-rapamycin-associated protein (FRAP) autophosphorylates at serine 2481 under translationally repressive conditions. J Biol Chem. 2000;275:7416–7423. doi: 10.1074/jbc.275.10.7416. [DOI] [PubMed] [Google Scholar]
- 175.Pette D. Historical Perspectives: plasticity of mammalian skeletal muscle. J Appl Physiol. 2001;90:1119–1124. doi: 10.1152/jappl.2001.90.3.1119. [DOI] [PubMed] [Google Scholar]
- 176.Pette D, Muller W, Leisner E, Vrbova G. Time dependent effects on contractile properties, fibre population, myosin light chains and enzymes of energy metabolism in intermittently and continuously stimulated fast twitch muscles of the rabbit. Pflugers Arch. 1976;364:103–112. doi: 10.1007/BF00585177. [DOI] [PubMed] [Google Scholar]
- 177.Pette D, Peuker H, Staron RS. The impact of biochemical methods for single muscle fibre analysis. Acta Physiol Scand. 1999;166:261–277. doi: 10.1046/j.1365-201x.1999.00568.x. [DOI] [PubMed] [Google Scholar]
- 178.Pette D, Smith ME, Staudte HW, Vrbova G. Effects of long-term electrical stimulation on some contractile and metabolic characteristics of fast rabbit muscles. Pflugers Arch. 1973;338:257–272. doi: 10.1007/BF00587391. [DOI] [PubMed] [Google Scholar]
- 179.Pette D, Staron RS. Mammalian skeletal muscle fiber type transitions. Int Rev Cytol. 1997;170:143–223. doi: 10.1016/s0074-7696(08)61622-8. [DOI] [PubMed] [Google Scholar]
- 180.Pette D, Staron RS. Myosin isoforms, muscle fiber types, and transitions. Microsc Res Tech. 2000;50:500–509. doi: 10.1002/1097-0029(20000915)50:6<500::AID-JEMT7>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 181.Pette D, Vrbova G. Adaptation of mammalian skeletal muscle fibers to chronic electrical stimulation. Rev Physiol Biochem Pharmacol. 1992;120:115–202. doi: 10.1007/BFb0036123. [DOI] [PubMed] [Google Scholar]
- 182.Pickart CM. Ubiquitin in chains. Trends Biochem Sci. 2000;25:544–548. doi: 10.1016/s0968-0004(00)01681-9. [DOI] [PubMed] [Google Scholar]
- 183.Pinto S, de Carvalho M. Motor responses of the sternocleidomastoid muscle in patients with amyotrophic lateral sclerosis. Muscle Nerve. 2008;38:1312–1317. doi: 10.1002/mus.21109. [DOI] [PubMed] [Google Scholar]
- 184.Plowman S, Smith D. Exercise physiology for health, fitness, and performance. Boston, Mass: Allyn & Bacon; 1997. [Google Scholar]
- 185.Powers SK, Kavazis AN, Levine S. Prolonged mechanical ventilation alters diaphragmatic structure and function. Crit Care Med. 2009;37:S347–353. doi: 10.1097/CCM.0b013e3181b6e760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Powers SK, Shanely RA, Coombes JS, Koesterer TJ, McKenzie M, Van Gammeren D, Cicale M, Dodd SL. Mechanical ventilation results in progressive contractile dysfunction in the diaphragm. J Appl Physiol. 2002;92:1851–1858. doi: 10.1152/japplphysiol.00881.2001. [DOI] [PubMed] [Google Scholar]
- 187.Prakash YS, Mantilla CB, Zhan WZ, Smithson KG, Sieck GC. Phrenic motoneuron morphology during rapid diaphragm muscle growth. J Appl Physiol. 2000;89:563–572. doi: 10.1152/jappl.2000.89.2.563. [DOI] [PubMed] [Google Scholar]
- 188.Prakash YS, Miyata H, Zhan WZ, Sieck GC. Inactivity-induced remodeling of neuromuscular junctions in rat diaphragmatic muscle. Muscle Nerve. 1999;22:307–319. doi: 10.1002/(sici)1097-4598(199903)22:3<307::aid-mus3>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 189.Prakash YS, Zhan WZ, Miyata H, Sieck GC. Adaptations of diaphragm neuromuscular junction following inactivity. Acta Anat (Basel) 1995;154:147–161. doi: 10.1159/000147762. [DOI] [PubMed] [Google Scholar]
- 190.Prezant DJ, Aldrich TK, Richner B, Gentry EI, Valentine DE, Nagashima H, Cahill J. Effects of long-term continuous respiratory resistive loading on rat diaphragm function and structure. J Appl Physiol. 1993;74:1212–1219. doi: 10.1152/jappl.1993.74.3.1212. [DOI] [PubMed] [Google Scholar]
- 191.Proud CG. Protein phosphorylation in translational control. Curr Top Cell Regul. 1992;32:243–369. doi: 10.1016/b978-0-12-152832-4.50008-2. [DOI] [PubMed] [Google Scholar]
- 192.Racz GZ, Gayan-Ramirez G, Testelmans D, Cadot P, De Paepe K, Zador E, Wuytack F, Decramer M. Early changes in rat diaphragm biology with mechanical ventilation. Am J Respir Crit Care Med. 2003;168:297–304. doi: 10.1164/rccm.200206-541OC. [DOI] [PubMed] [Google Scholar]
- 193.Raper AJ, Thompson WT, Jr, Shapiro W, Patterson JL., Jr Scalene and sternomastoid muscle function. J Appl Physiol. 1966;21:497–502. doi: 10.1152/jappl.1966.21.2.497. [DOI] [PubMed] [Google Scholar]
- 194.Rau M, Ohlmann T, Morley SJ, Pain VM. A reevaluation of the cap-binding protein, eIF4E, as a rate-limiting factor for initiation of translation in reticulocyte lysate. J Biol Chem. 1996;271:8983–8990. doi: 10.1074/jbc.271.15.8983. [DOI] [PubMed] [Google Scholar]
- 195.Raught B, Gingras AC. eIF4E activity is regulated at multiple levels. Int J Biochem Cell Biol. 1999;31:43–57. doi: 10.1016/s1357-2725(98)00131-9. [DOI] [PubMed] [Google Scholar]
- 196.Reed JL, Palsson BO. Systems biology: a four-step process. In: Chien S, Chen PC, Fung YC, editors. An introductory text to bioengineering. Singapore: World Scientific Publishing Co; 2008. pp. 387–399. [Google Scholar]
- 197.Reed SA, Senf SM, Cornwell EW, Kandarian SC, Judge AR. Inhibition of IkappaB kinase alpha (IKKalpha) or IKKbeta (IKKbeta) plus forkhead box O (Foxo) abolishes skeletal muscle atrophy. Biochem Biophys Res Commun. 2011;405:491–496. doi: 10.1016/j.bbrc.2011.01.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Reid B, Martinov VN, Nja A, Lomo T, Bewick GS. Activity-dependent plasticity of transmitter release from nerve terminals in rat fast and slow muscles. J Neurosci. 2003;23:9340–9348. doi: 10.1523/JNEUROSCI.23-28-09340.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Reynolds THt, Bodine SC, Lawrence JC., Jr Control of Ser2448 phosphorylation in the mammalian target of rapamycin by insulin and skeletal muscle load. J Biol Chem. 2002;277:17657–17662. doi: 10.1074/jbc.M201142200. [DOI] [PubMed] [Google Scholar]
- 200.Richardson D, Shewchuk R. Effects of contraction force and frequency on postexercise hyperemia in human calf muscles. J Appl Physiol. 1980;49:649–654. doi: 10.1152/jappl.1980.49.4.649. [DOI] [PubMed] [Google Scholar]
- 201.Richardson PG, Barlogie B, Berenson J, Singhal S, Jagannath S, Irwin D, Rajkumar SV, Srkalovic G, Alsina M, Alexanian R, Siegel D, Orlowski RZ, Kuter D, Limentani SA, Lee S, Hideshima T, Esseltine DL, Kauffman M, Adams J, Schenkein DP, Anderson KC. A phase 2 study of bortezomib in relapsed, refractory myeloma. N Engl J Med. 2003;348:2609–2617. doi: 10.1056/NEJMoa030288. [DOI] [PubMed] [Google Scholar]
- 202.Rollier H, Bisschop A, Gayan-Ramirez G, Gosselink R, Decramer M. Low load inspiratory muscle training increases diaphragmatic fiber dimensions in rats. Am J Respir Crit Care Med. 1998;157:833–839. doi: 10.1164/ajrccm.157.3.9512103. [DOI] [PubMed] [Google Scholar]
- 203.Romanello V, Guadagnin E, Gomes L, Roder I, Sandri C, Petersen Y, Milan G, Masiero E, Del Piccolo P, Foretz M, Scorrano L, Rudolf R, Sandri M. Mitochondrial fission and remodelling contributes to muscle atrophy. EMBO J. 2010;29:1774–1785. doi: 10.1038/emboj.2010.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Rommel C, Bodine SC, Clarke BA, Rossman R, Nunez L, Stitt TN, Yancopoulos GD, Glass DJ. Mediation of IGF-1-induced skeletal myotube hypertrophy by PI(3)K/Akt/mTOR and PI(3)K/Akt/GSK3 pathways. Nat Cell Biol. 2001;3:1009–1013. doi: 10.1038/ncb1101-1009. [DOI] [PubMed] [Google Scholar]
- 205.Rommel C, Clarke BA, Zimmermann S, Nunez L, Rossman R, Reid K, Moelling K, Yancopoulos GD, Glass DJ. Differentiation stage-specific inhibition of the Raf-MEK-ERK pathway by Akt. Science. 1999;286:1738–1741. doi: 10.1126/science.286.5445.1738. [DOI] [PubMed] [Google Scholar]
- 206.Rose AJ, Bisiani B, Vistisen B, Kiens B, Richter EA. Skeletal muscle eEF2 and 4EBP1 phosphorylation during endurance exercise is dependent on intensity and muscle fiber type. Am J Physiol Regul Integr Comp Physiol. 2009;296:R326–333. doi: 10.1152/ajpregu.90806.2008. [DOI] [PubMed] [Google Scholar]
- 207.Rosenblatt JD, Yong D, Parry DJ. Satellite cell activity is required for hypertrophy of overloaded adult rat muscle. Muscle Nerve. 1994;17:608–613. doi: 10.1002/mus.880170607. [DOI] [PubMed] [Google Scholar]
- 208.Roussos C. Function and fatigue of respiratory muscles. Chest. 1985;88:124S–132S. doi: 10.1378/chest.88.2_supplement.124s. [DOI] [PubMed] [Google Scholar]
- 209.Rowley KL, Mantilla CB, Sieck GC. Respiratory muscle plasticity. Respir Physiol Neurobiol. 2005;147:235–251. doi: 10.1016/j.resp.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 210.Roy RR, Baldwin KM, Martin TP, Chimarusti SP, Edgerton VR. Biochemical and physiological changes in overloaded rat fast- and slow-twitch ankle extensors. J Appl Physiol. 1985;59:639–646. doi: 10.1152/jappl.1985.59.2.639. [DOI] [PubMed] [Google Scholar]
- 211.Roy RR, Monke SR, Allen DL, Edgerton VR. Modulation of myonuclear number in functionally overloaded and exercised rat plantaris fibers. J Appl Physiol. 1999;87:634–642. doi: 10.1152/jappl.1999.87.2.634. [DOI] [PubMed] [Google Scholar]
- 212.Sacheck JM, Hyatt JP, Raffaello A, Jagoe RT, Roy RR, Edgerton VR, Lecker SH, Goldberg AL. Rapid disuse and denervation atrophy involve transcriptional changes similar to those of muscle wasting during systemic diseases. Faseb J. 2007;21:140–155. doi: 10.1096/fj.06-6604com. [DOI] [PubMed] [Google Scholar]
- 213.Sacheck JM, Ohtsuka A, McLary SC, Goldberg AL. IGF-I stimulates muscle growth by suppressing protein breakdown and expression of atrophy-related ubiquitin ligases, atrogin-1 and MuRF1. Am J Physiol Endocrinol Metab. 2004;287:E591–601. doi: 10.1152/ajpendo.00073.2004. [DOI] [PubMed] [Google Scholar]
- 214.Saito Y, Vandenheede JR, Cohen P. The mechanism by which epidermal growth factor inhibits glycogen synthase kinase 3 in A431 cells. Biochem J. 1994;303 (Pt 1):27–31. doi: 10.1042/bj3030027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Salmons S, Vrbova G. The influence of activity on some contractile characteristics of mammalian fast and slow muscles. J Physiol. 1969;201:535–549. doi: 10.1113/jphysiol.1969.sp008771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Sandri M. Signaling in muscle atrophy and hypertrophy. Physiology (Bethesda) 2008;23:160–170. doi: 10.1152/physiol.00041.2007. [DOI] [PubMed] [Google Scholar]
- 217.Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, Picard A, Walsh K, Schiaffino S, Lecker SH, Goldberg AL. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell. 2004;117:399–412. doi: 10.1016/s0092-8674(04)00400-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Sarbassov DD, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol. 2004;14:1296–1302. doi: 10.1016/j.cub.2004.06.054. [DOI] [PubMed] [Google Scholar]
- 219.Sarbassov DD, Ali SM, Sabatini DM. Growing roles for the mTOR pathway. Curr Opin Cell Biol. 2005;17:596–603. doi: 10.1016/j.ceb.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 220.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 221.Sassoon CS, Caiozzo VJ, Manka A, Sieck GC. Altered diaphragm contractile properties with controlled mechanical ventilation. J Appl Physiol. 2002;92:2585–2595. doi: 10.1152/japplphysiol.01213.2001. [DOI] [PubMed] [Google Scholar]
- 222.Sassoon CS, Zhu E, Caiozzo VJ. Assist-control mechanical ventilation attenuates ventilator-induced diaphragmatic dysfunction. Am J Respir Crit Care Med. 2004;170:626–632. doi: 10.1164/rccm.200401-042OC. [DOI] [PubMed] [Google Scholar]
- 223.Schiaffino S, Ausoni S, Gorza L, Saggin I, Gundersen K, Lomo T. Myosin heavy chain isoforms and velocity of shortening of type 2 skeletal muscle fibres. Acta Physiol Scand. 1988;134:575–576. doi: 10.1111/j.1748-1716.1998.tb08539.x. [DOI] [PubMed] [Google Scholar]
- 224.Schiaffino S, Gorza L, Pitton G, Saggin L, Ausoni S, Sartore S, Lomo T. Embryonic and neonatal myosin heavy chain in denervated and paralyzed rat skeletal muscle. Dev Biol. 1988;127:1–11. doi: 10.1016/0012-1606(88)90183-2. [DOI] [PubMed] [Google Scholar]
- 225.Schiaffino S, Gorza L, Sartore S, Saggin L, Ausoni S, Vianello M, Gundersen K, Lomo T. Three myosin heavy chain isoforms in type 2 skeletal muscle fibres. J Muscle Res Cell Motil. 1989;10:197–205. doi: 10.1007/BF01739810. [DOI] [PubMed] [Google Scholar]
- 226.Schiaffino S, Reggiani C. Molecular diversity of myofibrillar proteins: Gene regulation and functional significance. Physiol Rev. 1996;76:371–423. doi: 10.1152/physrev.1996.76.2.371. [DOI] [PubMed] [Google Scholar]
- 227.Schiaffino S, Reggiani C. Myosin isoforms in mammalian skeletal muscle. J Appl Physiol. 1994;77:493–501. doi: 10.1152/jappl.1994.77.2.493. [DOI] [PubMed] [Google Scholar]
- 228.Schilling CH, Edwards JS, Palsson BO. Toward metabolic phenomics: analysis of genomic data using flux balances. Biotechnol Prog. 1999;15:288–295. doi: 10.1021/bp9900357. [DOI] [PubMed] [Google Scholar]
- 229.Schultz E, Jaryszak DL, Valliere CR. Response of satellite cells to focal skeletal muscle injury. Muscle And Nerve. 1985;8:217–222. doi: 10.1002/mus.880080307. [DOI] [PubMed] [Google Scholar]
- 230.Senf SM, Dodd SL, McClung JM, Judge AR. Hsp70 overexpression inhibits NF-kappaB and Foxo3a transcriptional activities and prevents skeletal muscle atrophy. FASEB J. 2008;22:3836–3845. doi: 10.1096/fj.08-110163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Shah OJ, Anthony JC, Kimball SR, Jefferson LS. 4E-BP1 and S6K1: translational integration sites for nutritional and hormonal information in muscle. Am J Physiol Endocrinol Metab. 2000;279:E715–729. doi: 10.1152/ajpendo.2000.279.4.E715. [DOI] [PubMed] [Google Scholar]
- 232.Shanely RA, Van Gammeren D, Deruisseau KC, Zergeroglu AM, McKenzie MJ, Yarasheski KE, Powers SK. Mechanical ventilation depresses protein synthesis in the rat diaphragm. Am J Respir Crit Care Med. 2004;170:994–999. doi: 10.1164/rccm.200304-575OC. [DOI] [PubMed] [Google Scholar]
- 233.Shanely RA, Zergeroglu MA, Lennon SL, Sugiura T, Yimlamai T, Enns D, Belcastro A, Powers SK. Mechanical ventilation-induced diaphragmatic atrophy is associated with oxidative injury and increased proteolytic activity. Am J Respir Crit Care Med. 2002;166:1369–1374. doi: 10.1164/rccm.200202-088OC. [DOI] [PubMed] [Google Scholar]
- 234.Sherrington CS. Some functional problems attaching to convergence. Proc R Soc Lond (Biol) 1929;105:332–362. [Google Scholar]
- 235.Sieck GC. Diaphragm motor units and their response to altered use. Sem Respir Med. 1991;12:258–269. [Google Scholar]
- 236.Sieck GC. Diaphragm muscle: structural and functional organization. Clin Chest Med. 1988;9:195–210. [PubMed] [Google Scholar]
- 237.Sieck GC. Neural control of the inspiratory pump. NIPS. 1991;6:260–264. [Google Scholar]
- 238.Sieck GC. Physiological effects of diaphragm muscle denervation and disuse. In: Fanburg BL, Sicilian L, editors. Clinics in Chest Medicine: Respiratory Dysfunction in Neuromuscular Disease. Philadelphia, PA: W. B. Saunders Company; 1994. pp. 641–659. [PubMed] [Google Scholar]
- 239.Sieck GC, Fournier M. Developmental aspects of diaphragm muscle cells: Structural and functional organization. In: Haddad GG, Farber JP, editors. Developmental Neurobiology of Breathing. New York: Marcel Dekker; 1991. pp. 375–428. [Google Scholar]
- 240.Sieck GC, Fournier M. Diaphragm motor unit recruitment during ventilatory and nonventilatory behaviors. J Appl Physiol. 1989;66:2539–2545. doi: 10.1152/jappl.1989.66.6.2539. [DOI] [PubMed] [Google Scholar]
- 241.Sieck GC, Fournier M, Enad JG. Fiber type composition of muscle units in the cat diaphragm. Neurosci Lett. 1989;97:29–34. doi: 10.1016/0304-3940(89)90134-1. [DOI] [PubMed] [Google Scholar]
- 242.Sieck GC, Fournier M, Prakash YS, Blanco CE. Myosin phenotype and SDH enzyme variability among motor unit fibers. J Appl Physiol. 1996;80:2179–2189. doi: 10.1152/jappl.1996.80.6.2179. [DOI] [PubMed] [Google Scholar]
- 243.Sieck GC, Lewis MI, Blanco CE. Effects of undernutrition on diaphragm fiber size, SDH activity, and fatigue resistance. J Appl Physiol. 1989;66:2196–2205. doi: 10.1152/jappl.1989.66.5.2196. [DOI] [PubMed] [Google Scholar]
- 244.Sieck GC, Mantilla CB. Effect of mechanical ventilation on the diaphragm. N Engl J Med. 2008;358:1392–1394. doi: 10.1056/NEJMe0801226. [DOI] [PubMed] [Google Scholar]
- 245.Sieck GC, Prakash YS. Cross bridge kinetics in respiratory muscles. Eur Respir J. 1997;10:2147–2158. doi: 10.1183/09031936.97.10092147. [DOI] [PubMed] [Google Scholar]
- 246.Sieck GC, Prakash YS. Morphological adaptations of neuromuscular junctions depend on fiber type. Can J Appl Physiol. 1997;22:197–230. doi: 10.1139/h97-014. [DOI] [PubMed] [Google Scholar]
- 247.Sieck GC, Prakash YS, Han YS, Fang YH, Geiger PC, Zhan WZ. Changes in actomyosin ATP consumption rate in rat diaphragm muscle fibers during postnatal development. J Appl Physiol. 2003;94:1896–1902. doi: 10.1152/japplphysiol.00617.2002. [DOI] [PubMed] [Google Scholar]
- 248.Sieck GC, Roy RR, Powell P, Blanco C, Edgerton VR, Harper RM. Muscle fiber type distribution and architecture of the cat diaphragm. J Appl Physiol. 1983;55:1386–1392. doi: 10.1152/jappl.1983.55.5.1386. [DOI] [PubMed] [Google Scholar]
- 249.Sieck GC, Zhan WZ, Prakash YS, Daood MJ, Watchko JF. SDH and actomyosin ATPase activities of different fiber types in rat diaphragm muscle. J Appl Physiol. 1995;79:1629–1639. doi: 10.1152/jappl.1995.79.5.1629. [DOI] [PubMed] [Google Scholar]
- 250.Skurk C, Maatz H, Kim HS, Yang J, Abid MR, Aird WC, Walsh K. The Akt-regulated forkhead transcription factor FOXO3a controls endothelial cell viability through modulation of the caspase-8 inhibitor FLIP. J Biol Chem. 2004;279:1513–1525. doi: 10.1074/jbc.M304736200. [DOI] [PubMed] [Google Scholar]
- 251.Smith IJ, Alamdari N, O’Neal P, Gonnella P, Aversa Z, Hasselgren PO. Sepsis increases the expression and activity of the transcription factor Forkhead Box O 1 (FOXO1) in skeletal muscle by a glucocorticoid-dependent mechanism. Int J Biochem Cell Biol. 2010;42:701–711. doi: 10.1016/j.biocel.2010.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Smith JW, Thesleff S. Spontaneous activity in denervated mouse diaphragm muscle. J Physiol. 1976;257:171–186. doi: 10.1113/jphysiol.1976.sp011362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Solomon V, Goldberg AL. Importance of the ATP-ubiquitin-proteasome pathway in the degradation of soluble and myofibrillar proteins in rabbit muscle extracts. J Biol Chem. 1996;271:26690–26697. doi: 10.1074/jbc.271.43.26690. [DOI] [PubMed] [Google Scholar]
- 254.Stephens TJ, Chen ZP, Canny BJ, Michell BJ, Kemp BE, McConell GK. Progressive increase in human skeletal muscle AMPKalpha2 activity and ACC phosphorylation during exercise. Am J Physiol Endocrinol Metab. 2002;282:E688–694. doi: 10.1152/ajpendo.00101.2001. [DOI] [PubMed] [Google Scholar]
- 255.Stitt TN, Drujan D, Clarke BA, Panaro F, Timofeyva Y, Kline WO, Gonzalez M, Yancopoulos GD, Glass DJ. The IGF-1/PI3K/Akt pathway prevents expression of muscle atrophy-induced ubiquitin ligases by inhibiting FOXO transcription factors. Mol Cell. 2004;14:395–403. doi: 10.1016/s1097-2765(04)00211-4. [DOI] [PubMed] [Google Scholar]
- 256.Stuart DG, Enoka RM. Motoneurons, motor units, and the size principle. In: Rosenberg RN, editor. The Clinical Neurosciences. New York: Churchill Livingstone; 1983. pp. 471–517. [Google Scholar]
- 257.Takahashi A, Kureishi Y, Yang J, Luo Z, Guo K, Mukhopadhyay D, Ivashchenko Y, Branellec D, Walsh K. Myogenic Akt signaling regulates blood vessel recruitment during myofiber growth. Mol Cell Biol. 2002;22:4803–4814. doi: 10.1128/MCB.22.13.4803-4814.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Tang H, Lee M, Budak MT, Pietras N, Hittinger S, Vu M, Khuong A, Hoang CD, Hussain SN, Levine S, Shrager JB. Intrinsic apoptosis in mechanically ventilated human diaphragm: linkage to a novel Fos/FoxO1/Stat3-Bim axis. FASEB J. 2011;25:2921–2936. doi: 10.1096/fj.11-183798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Tawa NE, Jr, Odessey R, Goldberg AL. Inhibitors of the proteasome reduce the accelerated proteolysis in atrophying rat skeletal muscles. J Clin Invest. 1997;100:197–203. doi: 10.1172/JCI119513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Tikunov BA, Mancini D, Levine S. Changes in myofibrillar protein composition of human diaphragm elicited by congestive heart failure. J Mol Cell Cardiol. 1996;28:2537–2541. doi: 10.1006/jmcc.1996.0245. [DOI] [PubMed] [Google Scholar]
- 261.Tong JF, Yan X, Zhu MJ, Du M. AMP-activated protein kinase enhances the expression of muscle-specific ubiquitin ligases despite its activation of IGF-1/Akt signaling in C2C12 myotubes. J Cell Biochem. 2009;108:458–468. doi: 10.1002/jcb.22272. [DOI] [PubMed] [Google Scholar]
- 262.Trinidad JC, Fischbach GD, Cohen JB. The Agrin/MuSK signaling pathway is spatially segregated from the neuregulin/ErbB receptor signaling pathway at the neuromuscular junction. J Neurosci. 2000;20:8762–8770. doi: 10.1523/JNEUROSCI.20-23-08762.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Van Der Heide LP, Hoekman MF, Smidt MP. The ins and outs of FoxO shuttling: mechanisms of FoxO translocation and transcriptional regulation. Biochem J. 2004;380:297–309. doi: 10.1042/BJ20040167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Van Gammeren D, Damrauer JS, Jackman RW, Kandarian SC. The IkappaB kinases IKKalpha and IKKbeta are necessary and sufficient for skeletal muscle atrophy. FASEB J. 2009;23:362–370. doi: 10.1096/fj.08-114249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.van Hees H, Ottenheijm C, Ennen L, Linkels M, Dekhuijzen R, Heunks L. Proteasome inhibition improves diaphragm function in an animal model for COPD. Am J Physiol Lung Cell Mol Physiol. 2011;301:L110–116. doi: 10.1152/ajplung.00396.2010. [DOI] [PubMed] [Google Scholar]
- 266.van Hees HW, Li YP, Ottenheijm CA, Jin B, Pigmans CJ, Linkels M, Dekhuijzen PN, Heunks LM. Proteasome inhibition improves diaphragm function in congestive heart failure rats. Am J Physiol Lung Cell Mol Physiol. 2008;294:L1260–1268. doi: 10.1152/ajplung.00035.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Vandenburgh H, Chromiak J, Shansky J, Del Tatto M, Lemaire J. Space travel directly induces skeletal muscle atrophy. Faseb J. 1999;13:1031–1038. doi: 10.1096/fasebj.13.9.1031. [DOI] [PubMed] [Google Scholar]
- 268.Verheul AJ, Mantilla CB, Zhan WZ, Bernal M, Dekhuijzen PN, Sieck GC. Influence of corticosteroids on myonuclear domain size in the rat diaphragm muscle. J Appl Physiol. 2004;97:1715–1722. doi: 10.1152/japplphysiol.00625.2003. [DOI] [PubMed] [Google Scholar]
- 269.Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- 270.Wang X, Proud CG. The mTOR pathway in the control of protein synthesis. Physiology (Bethesda) 2006;21:362–369. doi: 10.1152/physiol.00024.2006. [DOI] [PubMed] [Google Scholar]
- 271.Waskiewicz AJ, Flynn A, Proud CG, Cooper JA. Mitogen-activated protein kinases activate the serine/threonine kinases Mnk1 and Mnk2. Embo J. 1997;16:1909–1920. doi: 10.1093/emboj/16.8.1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Waskiewicz AJ, Johnson JC, Penn B, Mahalingam M, Kimball SR, Cooper JA. Phosphorylation of the cap-binding protein eukaryotic translation initiation factor 4E by protein kinase Mnk1 in vivo. Mol Cell Biol. 1999;19:1871–1880. doi: 10.1128/mcb.19.3.1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Welsh GI, Miller CM, Loughlin AJ, Price NT, Proud CG. Regulation of eukaryotic initiation factor eIF2B: glycogen synthase kinase-3 phosphorylates a conserved serine which undergoes dephosphorylation in response to insulin. FEBS Lett. 1998;421:125–130. doi: 10.1016/s0014-5793(97)01548-2. [DOI] [PubMed] [Google Scholar]
- 274.Welsh GI, Proud CG. Glycogen synthase kinase-3 is rapidly inactivated in response to insulin and phosphorylates eukaryotic initiation factor eIF-2B. Biochem J. 1993;294 (Pt 3):625–629. doi: 10.1042/bj2940625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Widrick JJ, Trappe SW, Blaser CA, Costill DL, Fitts RH. Isometric force and maximal shortening velocity of single muscle fibers from elite master runners. Am J Physiol. 1996;271:c666–c675. doi: 10.1152/ajpcell.1996.271.2.C666. [DOI] [PubMed] [Google Scholar]
- 276.Williamson DL, Bolster DR, Kimball SR, Jefferson LS. Time course changes in signaling pathways and protein synthesis in C2C12 myotubes following AMPK activation by AICAR. Am J Physiol Endocrinol Metab. 2006;291:E80–89. doi: 10.1152/ajpendo.00566.2005. [DOI] [PubMed] [Google Scholar]
- 277.Zammit PS, Partridge TA, Yablonka-Reuveni Z. The skeletal muscle satellite cell: the stem cell that came in from the cold. J Histochem Cytochem. 2006;54:1177–1191. doi: 10.1369/jhc.6R6995.2006. [DOI] [PubMed] [Google Scholar]
- 278.Zengel JE, Reid SA, Sypert GW, Munson JB. Membrane electrical properties and prediction of motor-unit type of medial gastrocnemius motoneurons in the cat. J Neurophysiol. 1985;53(5):1323–1344. doi: 10.1152/jn.1985.53.5.1323. [DOI] [PubMed] [Google Scholar]
- 279.Zhan WZ, Farkas GA, Schroeder MA, Gosselin LE, Sieck GC. Regional adaptations of rabbit diaphragm muscle fibers to unilateral denervation. J Appl Physiol. 1995;79:941–950. doi: 10.1152/jappl.1995.79.3.941. [DOI] [PubMed] [Google Scholar]
- 280.Zhan WZ, Miyata H, Prakash YS, Sieck GC. Metabolic and phenotypic adaptations of diaphragm muscle fibers with inactivation. J Appl Physiol. 1997;82:1145–1153. doi: 10.1152/jappl.1997.82.4.1145. [DOI] [PubMed] [Google Scholar]
- 281.Zhan WZ, Sieck GC. Adaptations of diaphragm and medial gastrocnemius muscles to inactivity. J Appl Physiol. 1992;72:1445–1453. doi: 10.1152/jappl.1992.72.4.1445. [DOI] [PubMed] [Google Scholar]
- 282.Zhao J, Brault JJ, Schild A, Cao P, Sandri M, Schiaffino S, Lecker SH, Goldberg AL. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab. 2007;6:472–483. doi: 10.1016/j.cmet.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 283.Zhong H, Roy RR, Siengthai B, Edgerton VR. Effects of inactivity on fiber size and myonuclear number in rat soleus muscle. J Appl Physiol. 2005;99:1494–1499. doi: 10.1152/japplphysiol.00394.2005. [DOI] [PubMed] [Google Scholar]
- 284.Zhu X, Lai C, Thomas S, Burden SJ. Neuregulin receptors, erbB3 and erbB4, are localized at neuromuscular synapses. EMBO J. 1995;14:5842–5848. doi: 10.1002/j.1460-2075.1995.tb00272.x. [DOI] [PMC free article] [PubMed] [Google Scholar]