

Abstract
Triple-negative breast cancer (TNBC) is a heterogeneous disease; gene expression (GE) analyses recently identified six distinct TNBC subtypes, each displaying a unique biology. Exploring novel approaches to treatment of these subtypes is critical, since less than 30% of women with metastatic breast cancer survive five years and virtually all women with metastatic TNBC will ultimately die of their disease despite systemic therapy. To date, not a single targeted therapy has been approved for the treatment of TNBC and cytotoxic chemotherapy remains the standard treatment. We will discuss the current and upcoming therapeutic strategies being explored in an attempt to “target” TNBC.
BACKGROUND
“Triple negative breast cancer” (TNBC) is a term used to identify the approximately 15% of invasive breast cancers which lack the expression of estrogen and progesterone receptor (ER/PR) and HER2 (ERBB2). TNBCs are generally of a higher grade, occur at a higher rate in young and African-American women, and most harbor a basal-like gene expression signature (1, 2). Patients with TNBC have an increased likelihood of distant recurrence and death compared with women with other types of breast cancer (3), as well as a tendency to develop visceral metastases early in the course of their disease. Improved approaches to treatment of these cancers is critical, since the median survival of patients with metastatic triple-negative breast cancer is only 13 months, and virtually all women with metastatic TNBC ultimately die of their disease despite systemic therapy (4).
TNBC SUBTYPING
Although the terms ‘triple negative’ (TN) and ‘basal-like’ are not synonymous, ~80% of clinical TNBCs (ER/PR/HER2-negative) classify as basal-like, based on PAM50 intrinsic subtype classification (5). Tumors arising in BRCA1 carriers have many similarities to basal-like sporadic breast tumors, including greater likelihood of being high-grade, ER/PR-negative, HER2-negative, and a high frequency of p53 mutations (6). Basal keratins are expressed by both sporadic basal-like tumors and tumors with BRCA1 mutations, and both groups cluster together by gene expression profiling (6). Other studies support these data, in which familial-BRCA1 breast cancers have shared features with a subset of sporadic tumors, indicating a common or similar etiology. Hallmarks of this “BRCAness” include basal-like phenotype (associated with the BRCA1 phenotype but not with the BRCA2 phenotype), ER-negativity, EGFR expression, c-MYC amplification, TP53 mutations, loss of RAD51-focus formation, extreme genomic instability and sensitivity to DNA-crosslinking agents (7). The clinical implications of the definition of this group of tumors with a “BRCAness” hallmark lies in its potential to influence the clinical management of these tumors, allowing for rational trials exploring the role of chemotherapy and biologic agents targeted towards DNA repair defects.
Using gene expression (GE) analyses, we recently identified distinct TNBC subtypes, each displaying a unique biology (8). The six TNBC subtypes include two basal-like (BL1 and BL2), an immunomodulatory (IM), a mesenchymal (M), a mesenchymal stem–like (MSL), and a luminal androgen receptor (LAR) subtype, the last being characterized by androgen receptor signaling (8). We further used GE analysis to identify TNBC cell lines representative of these subtypes. Predicted “driver” signaling pathways were pharmacologically targeted in these cell lines as proof-of-concept and to generate pre-clinical data to inform future clinical trial design.
We also performed a direct comparison of 374 TNBC samples extracted from 14 datasets to determine the relationship between the PAM50 intrinsic and TNBC molecular subtypes. As anticipated, the majority of the TNBC samples are indeed classified as basal-like (80.6%) followed by HER2 (0.2%), normal-like (14.6%), luminal B (3.5%) and luminal A (1.1%) by PAM50 (Figure 1A, modified from (9)). With exception to MSL and LAR, all other TNBC subtypes are primarily composed of the basal-like intrinsic subtype. MSL TNBCs are about 50% basal-like and the remainder is composed of normal-like (27.8%) and luminal B (13.9%). Unlike other subtypes, the LAR subtype is primarily classified as HER2 (74.3%) and Luminal B (14.3%) by PAM50 intrinsic subtyping (Figure 1B). Therefore, PAM50 intrinsic subtyping alone has the potential to classify ~75% of TNBCs that are AR+ as HER2+.
Figure 1. (9) TNBC subtype comparison to intrinsic PAM50 subtyping.
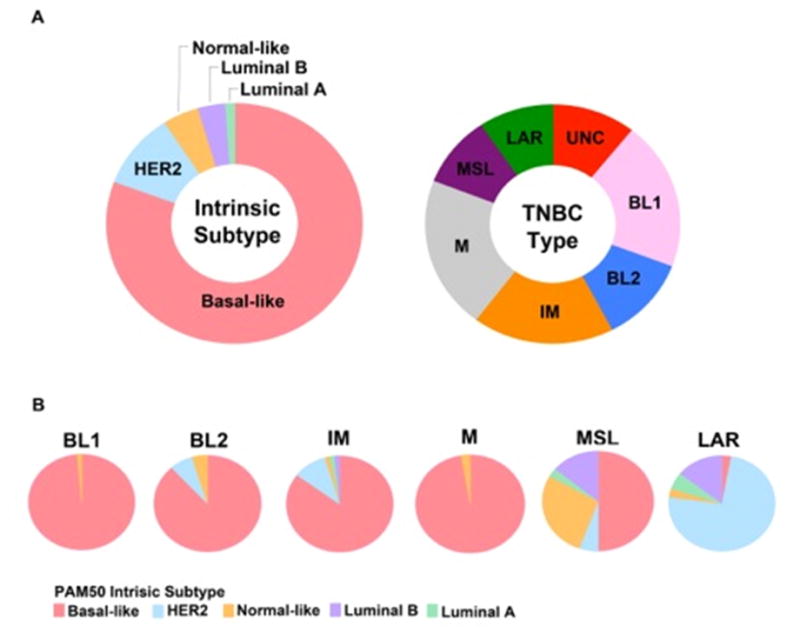
374 TNBC gene expression profiles from 14 datasets* were either subtyped using PAM50 (genefu, R package) or subtyped with TNBCtype. (A) Pie charts show the distribution of 374 TNBC samples using PAM50 intrinsic subtyping (left) or TNBCtype (right). (B) Pie charts show the intrinsic subtype composition of each of the TNBC subtypes. Basal-like 1 (BL1), basal-like 2 (BL2), immunomodulatory (IM), mesenchymal (M), mesenchymal stem-like, and luminal AR (LAR).
*(GSE1456, GSE1561, GSE2034, GSE2109, GSE2990, GSE2603, GSE5327, GSE5460, GSE5847, GSE7390, GSE11121, GSE12276, GSE18864, GSE20194)
Adapted and reproduced from ref. 9 with permission from John Wiley & Sons, Ltd. and Lehmann, BD, Pietenpol, JA. Identification and use of biomarkers in treatment strategies for triple-negative breast cancer subtypes. J Pathol 2014;232:142-50. Copyright © 2013 Pathological Society of Great Britain and Ireland. Published by John Wiley & Sons, Ltd.
In order to determine potential clinical utility of assessing TNBC subtype, we generated a tool (TNBCtype) that determines the TNBC molecular subtype from GE profiles independent of platform (10). Recently, Masuda et al., performed a retrospective analysis on 130 TNBC cases treated with neoadjuvant adriamycin/ cytoxan/ taxol containing chemotherapy (11). While the overall pCR response was 28%, subtype specific responses differed substantially with the BL1 subtype achieving highest pCR rate (52%) and the BL2, LAR and MSL subtypes having the lowest response (0% and 10% 23%, respectively). Furthermore, TNBC subtype was shown to be an independent predictor of pCR status (p = 0.022) by a likelihood ratio test. We also used the TNBCtype tool (10) to subtype 163 primary cases in The Cancer Genome Atlas (TCGA) considered to be TNBC (Table 1 and Supplementary Table S1). In agreement with the work of Masuda et al. described above, we found a very similar distribution of subtypes and subtype-specific differences in survival. These findings and additional validation should guide differential use of chemotherapy-based regimens and alignment of patients with select TNBC subtypes to clinical trials investigating targeted therapies. There is a need for prospective validation of associated pCR rates amongst the TNBC subtypes and to determine if subtyping will be useful for predicting long-term patient outcome.
Table 1.
TNBC subtyping predictions for TCGA primary breast tumors
| TNBC Subtypea | # Samples (Percentage) | Median DFS (Months)b | Median OS (Months)b |
|---|---|---|---|
| BL1 | 27 (17%) | 20.1 | 21.1 |
| BL2 | 12 (7%) | 12.5 | 8.4 |
| IM | 30 (18%) | 22.7 | 24.8 |
| M | 39 (24%) | 9.1 | 9.5 |
| MSL | 10 (6%) | 13.9 | 20.9 |
| LAR | 14 (9%) | 4.4 | 5.7 |
| UNC | 31 (19%) | 22.0 | 24.9 |
|
| |||
| All TNBC | 163 (100%) | 11.8 | 15.2 |
Abbreviations: DFS, disease free survival; OS, overall survival.
TNBC subtype predictions for the TCGA primary tumors were made using RSEM genes normalized (upper quartile count = 1,000) RNA-seq abundance estimates (20130606 Firehose BRCA stddata run). TNBC cases were identified as previously described (8). Samples were filtered on ER, PR, and HER2 mRNA expression to identify TNBC tumors and outliers were removed after principal component analysis. The TNBCtype tool (10) was used to make TNBC subtype predictions for the 163 TNBC samples.
Survival data for TCGA TNBC cases was obtained from the cBioPortal (www.cbioportal.org) on 12-19-2013.
ON THE HORIZON
Distinguishing one subtype of TNBC from another from a histological point of view is challenging. Based on the fact that the different TNBC subtypes seem to have distinct responses to a given therapy, as exemplified by Masuda et al. retrospective analysis (11), we postulate that it is inappropriate to treat all TNBC in a similar fashion. The features of TNBC described in this review, in relation to possible therapeutic targets, will be discussed herein, and are summarized in Figure 2.
Figure 2. Triple-negative breast cancer: potential therapeutic targets.
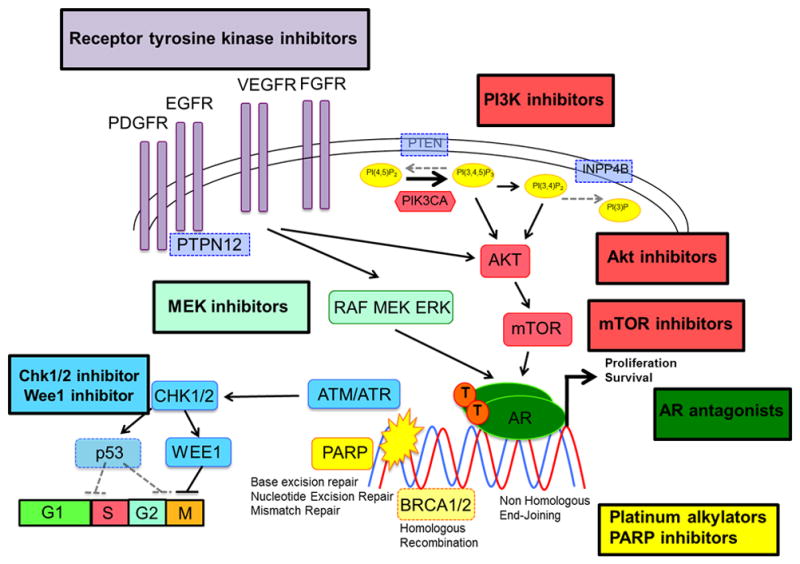
Current and future potential therapeutic targets in TNBC include: modulation of homologous recombination repair mechanism deficiency (platinum alkylators, PARP inhibitors [yellow], Chk1/2 inhibitors [blue]), modulation of p53 family signaling (PI3K/Akt/mTOR inhibitors [red], Chk1/2 inhibitors, Wee1 inhibitors [blue]), modulation of androgen receptor (PI3K/Akt/mTOR inhibitors [red], AR antagonists [dark green]), modulation of MAPK/MEK pathway (MEK inhibitors [light green]).
PLATINUM AGENTS
Platinum salts, including carboplatin and cisplatin, lead to DNA cross-link strand breaks, which may be especially important in cells which are deficient in homologous recombination repair mechanisms such as BRCA mutated cells and TNBC. Leong et al.(12) reported a p63-dependent tumor survival pathway that mediates cisplatin sensitivity, specifically in TNBC cells grown in vitro. Extending this observation to the clinical setting, Rocca et al.(13) conducted a retrospective analysis of core biopsies of breast cancer patients treated with neoadjuvant cisplatin-based chemotherapy in breast cancer and showed that administration of cisplatin without anthracyclines yielded a higher rate of pCR in patients with p63-positive tumors. Analysis of tumor tissue from ongoing cisplatin-based neoadjuvant trials will allow further analysis of the relationship of the molecular features of various TNBC subtypes and cisplatin sensitivity.
In a small phase II study (29 patients), Silver et al. showed activity of neoadjuvant cisplatin as a single agent in the treatment of patients with locally advanced triple-negative breast cancers. The observed pCR was 22%, and 50% of patients had a partial response, and 14% had a complete response (14). In another small study, 9 of 10 patients with stage I-III breast cancer harboring BRCA1 mutations achieved a pathological complete remission after neoadjuvant therapy with cisplatin (15).
More recently, solid evidence of the activity of platinum agents in TNBC was provided by large phase II randomized trials in the neoadjuvant setting: the GeparSixto trial, in its TNBC subset, compared neoadjuvant paclitaxel, doxorubicin, and bevacizumab with (159 patients) or without (161 patients) carboplatin. The pCR rate improved from 37.9% to 58.7% with the addition of carboplatin. Biomarker studies in this trial are underway to determine whether certain subsets of TNBC benefit more from the addition of carboplatin (16) and should provide significant insights to the genomic alterations that mediate sensitivity to platinum agents. CALGB40603 (NCT00861705) is a randomized phase II trial with a 2 × 2 factorial design that explored the addition of carboplatin +/− bevacizumab to neoadjuvant weekly paclitaxel followed by dose-dense AC in in 443 patients with stage II/III TNBC(17). The pCR rate improved from 41% to 54% with the addition of carboplatin; bevacizumab had no added benefit. It is important to note that neither one of these studies was powered to detect disease-free or overall survival benefit.
PARP INHIBITION
ATM, BRCA1 and TP53 are critical genes in the DNA damage response signaling (DDR) pathway which could play a role during breast tumorigenesis. BRCA1 mutations are rare in sporadic tumors, but nevertheless, high-grade breast cancers display a high frequency of loss of heterozigosity (LOH) and/or abnormal expression of ATM, BRCA1 and TP53. Multi-genetic analyses show that BRCA1 abnormalities are independently associated with high-grade tumors (18). Both BRCA1 and BRCA2 contribute to DNA repair and transcriptional regulation in response to DNA damage, and regulate other genes involved in DNA repair, cell cycle and apoptosis (19). Increasing evidence supports a role for BRCA1 in double-strand DNA break repair in part through its interaction with RAD51 and the Fanconi anemia proteins (20, 21). Cell lines deficient in BRCA1 or other components of the Fanconi anemia-BRCA1 pathway are more sensitive to x-ray induced damage and DNA crosslinking agents such as cisplatin (20, 22).
Poly(ADP-ribose) polymerase 1 (PARP1) is an enzyme critical to the base excision repair pathway and is key for repair of single-strand DNA breaks. Inhibition of this enzyme by RNA interference or with chemical inhibitors leads to severe, highly selective toxicity in BRCA1 and BRCA2-defective cells, with selectivity being several-fold higher that for conventional chemotherapy drugs (23). This leads to chromosomal instability, cell cycle arrest and subsequent apoptosis, most likely due to persistence of DNA lesions normally repaired by homologous recombination (24). Sensitivity to PARP inhibition depends on homologous recombination deficiency and not on inherited BRCA1 or BRCA2 deficiency per se (24). Therefore, use of PARP1 inhibitors as a therapeutic strategy in the treatment of sporadic breast cancers with “BRCAness,” including basal-like breast cancers, may be a promising approach. There are at least three potential roles of PARP inhibitors in cancer treatment: sensitization to chemotherapy and radiotherapy, synthetic lethality in patients with hereditary mutations in BRCA1/2 genes (inherited defect in homologous recombination) and, finally, leveraging of putative ‘BRCA-like’ defective tumors and defects in DNA repair such as those seen in TNBC.
Significant single agent activity was recently reported with the PARP inhibitor olaparib in patients with BRCA-deficient metastatic breast cancer. Overall responses ranged from 22% (100 mg bid) to 41% (400 mg bid) with minimal toxicity (25). Olaparib as a single agent was also evaluated in a phase II study in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or TNBC(26). In this trial, however, no confirmed objective responses were seen in the 26 patients with TNBC.
A phase II, open-label, two-arm randomized, safety and efficacy trial (Study 20070102) investigated gemcitabine/carboplatin with or without iniparib in patients with metastatic TNBC. The final analysis of 123 randomized patients showed that addition of iniparib to gemcitabine/carboplatin improved the clinical benefit rate from 33.9% to 55.7% (p=0.015) and ORR from 32.3% to 52.5% (p=0.023). The addition of iniparib prolonged the median progression free survival (PFS) from 3.6 to 5.9 months (hazard ratio [HR], 0.59; p=0.012) and the median overall survival (OS) from 7.7 to 12.3 months (HR, 0.57; p=0.014)(27). A subsequent phase III study evaluating gemcitabine/carboplatin ± iniparib with similar inclusion criteria as the phase II study did not meet the pre-specified criteria for significance for the co-primary endpoints of OS and PFS. Interestingly, the results of a pre-specified analysis in patients treated in the second- and third-line setting showed an improvement in OS and PFS, consistent with what was seen in the smaller phase II study (28).
More recently, a single arm phase II study of neoadjuvant gemcitabine, carboplatin, and iniparib in patients with TNBC or BRCA 1/2 mutation associated breast cancer showed impressive responses, especially in BRCA 1/2 carriers (29). A homologous recombination deficiency (HRD) assay was developed as part of this study to identify non-BRCA1/2 mutation carriers with “BRCA-like” cancers who may benefit from DNA repair-targeted treatment strategies. Higher HRD scores were significantly associated with higher rates response. Of note, more recent pre-clinical data have shown that iniparib does not possess characteristics typical of the PARP inhibitor class. Investigations into potential targets of iniparib and its metabolites are ongoing, and additional targets are under investigation.
MODULATION OF p53 FAMILY SIGNALING
The p53 family of transcription factors (p53, p63 and p73) are key regulators of signaling pathways that regulate developmental and tumor suppressive processes (30). The p53 tumor suppressor is mutated in ~30% of breast cancers (31) and shows a strong association with the basal-like subgroup (32).
The transcription factor p73 plays critical roles during development and tumorigenesis. It exhibits sequence identity and structural homology with p53 and can engage p53-like tumor-suppressive programs. Rosenbluth et al. defined the p73 genomic binding profile and demonstrated its modulation by rapamycin, an inhibitor of mammalian target of rapamycin (mTOR) and inducer of p73. Rapamycin selectively increases p73 occupancy at a subset of its binding sites. In addition, multiple determinants of p73 binding, activity, and function are evident and modulated by mTOR (33). Interestingly, the combination of mTOR inhibitor, paclitaxel and cisplatin can synergistically regulate the p73/p63 signaling axis and promote apoptosis in breast cancer cells.
Considering the above preclinical data, our group hypothesized that drugs that either negatively modulate p63 and/or activate p73 (such as an mTOR inhibitor, paclitaxel and cisplatin combination)would promote increased apoptosis in TNBC. We recently completed a large randomized phase II study of neoadjuvant cisplatin and paclitaxel with or without everolimus for 3 months in 145 patients with stage II and III TNBC (NCT00930930). Despite no difference in pCR rate in both arms (38%), the pCR rate seen with this short course of taxane/platinum chemotherapy backbone, which did not contain anthracyclines, was similar to a more extensive, anthracycline/ taxane containing chemotherapy regimen (34). TNBC subtyping, DNA mutations and alterations, as well as markers of proliferation, apoptosis, PI3K/mTOR and DNA damage response signaling are part of the correlative studies being done to investigate a molecular signature or biomarker predictive of benefit from the paclitaxel/cisplatin ± everolimus combination in TNBC.
PI3K INHIBITION
Exome sequencing of TNBC identifies TP53 (62%) as the most frequently mutated gene in TNBC, followed by alterations in phosphatidylinositol-3 kinase (PI3K) pathway that include PIK3CA (10.2%) and PTEN (9.6%) mutations (35).
Our preclinical data show that a significant fraction of TNBC cells lines are sensitive to PI3K inhibitors. The LAR subtype cell lines in particular have a high rate of PIK3CA activating mutations and exhibit strong sensitivity to PI3K inhibitors, as well as to androgen blockers such as bicalutamide (8). The co-evolution of PIK3CA mutations with AR-dependency is similar to ER-positive breast cancers frequently contain PIK3CA mutations (36, 37). Our studies of combinations of PI3K-inhibitors and cisplatin show either additive or synergistic decreases in tumor viability, with significant decreases in pAKT and pS6 levels and a concomitant elevation in cleaved PARP. Of note, PI3K inhibition of TNBC cells has been shown to sensitize cells to DNA damaging agents by, in effect, creating a BRCA deficient state (38).
Other studies provide additional preclinical rationale for the combined use of a DNA damaging agent with PI3K inhibitors (39, 40). These studies confirm that in addition to regulating cellular processes including growth, metabolism, and survival (41), PI3K also stabilizes double strand breaks by interacting with the HR complex (42). PI3K blockade promotes HR deficiency by down-regulating BRCA1/2 and thus sensitizing BRCA-proficient tumors to PARP inhibition. The elevated levels of pAKT and pS6 are consistent with activated PI3K in the TNBC tumors. To capitalize on these findings, a phase I study of the pan-PI3K inhibitor BKM120 (Novartis®) in combination with the PARP inhibitor olaparib in patients with metastatic TNBC is ongoing. BKM120 would be expected to create a BRCA mutant-like tumor state, thus making it susceptible to PARP inhibition.
Altogether, these data suggest that targeting the PI3K pathway could be clinically relevant in TNBC. Our group is now initiating a clinical trial in which therapy for patients with metastatic TNBC will be determined by androgen receptor status. Patients with tumors who express androgen receptor by immunohistochemistry will receive the anti-androgen bicalutamide with a PI3K inhibitor, and patients who have androgen receptor negative TNBC will be randomized to chemotherapy with cisplatin or cisplatin with a PI3K inhibitor.
MEK INHIBITION
Hoeflich et al. have shown that a large number of basal-like breast cancer cell lines are sensitive to MEK inhibition (43). Several of these have either a BRAF, HRAS, KRAS mutation; however, the majority of these cell lines show up-regulation of the RAS/MEK pathway without an identified oncogenic mutation. Interestingly, loss of the tumor suppressor phosphatase PTEN(which occurs in 29% of TNBC(44)), results in up-regulation of the PI3K/Akt pathway, and is associated with lack of response to MEK inhibitors (43).
Balko et al. found that DUSP4 loss, a negative regulator of ERK1 and ERK2, is associated with basal-like breast cancers and Ras-ERK activation (45). In existing microarray datasets, DUSP4 mRNA levels were lowest in basal-like breast cancers. Moreover, in a dataset of 286 breast cancers from patients who did not receive adjuvant therapy, low DUSP4 expression predicted for shorter recurrence-free survival time. DUSP4 deficiency was associated with Ras-ERK pathway activation in chemotherapy-refractory basal-like breast cancers. In addition, DUSP4 mRNA expression was inversely correlated with Ras-ERK pathway score in a series of 230 primary breast cancers. Thus, loss of DUSP4 may be a crucial biomarker in identifying activation of the Ras-ERK pathway in basal-like breast cancers (45).
In summary, the pre-clinical data above suggest that TNBC with PTEN expression and low expression of DUSP4 have a dependency on Ras-ERK activation, and therefore could be sensitive to clinical trials combining MEK inhibitors with chemotherapy.
CONCLUSION
TNBC is a very heterogeneous disease, for which a number of therapeutic strategies are being explored. Studies described above clearly indicate that TNBC cannot be treated in a uniform fashion; for instance: TNBCs that have a basal-like genotype are more likely sensitive to DNA crosslinking agents, such as platinum-based chemotherapy and PARP inhibitors. On the other hand, the androgen-receptor expressing tumors may derive greater benefit from a combination of an androgen-blocker and a PI3K inhibitor. Numerous experimental approaches are underway with the goal of identifying “targets” in TNBC, with PI3K inhibitors, MEK inhibitors, HSP-90 inhibitors, histone deacetylase inhibitors, PD-1 (programmed death 1) inhibitors, etc. under consideration or currently being investigated in the clinical setting (Table 2).
Table 2.
Targeted therapies clinical trials in triple-negative breast cancer
| Study Design | Clinical Trial | Type of inhibitor | Patient Population | Clinicaltrials.gov |
|---|---|---|---|---|
| Phase I | Phase I of BKM120/ Olaparib for Triple Negative Breast Cancer or High Grade Serous Ovarian Cancer | pan-PI3K PARP |
Patients with Triple Negative Breast Cancer or High Grade Serous Ovarian Cancer | NCT01623349 |
| Phase II | A Trial of BKM120 (a PI3K Inhibitor) in Patients With Triple Negative Metastatic Breast Cancer | pan-PI3K | Triple Negative Metastatic Breast Cancer | NCT01629615 |
| A Trial of cisplatin +/− GDC0941 (a PI3K Inhibitor) in Patients with Androgen- Receptor Negative TNBC | NCT01918306 | |||
| Phase Ib/II | Everolimus (RAD001) and Carboplatin in Pretreated Metastatic Breast Cancer | TORC1 inhibitor | HER2 negative MBC | NCT00930475 |
| Phase II | Cisplatin and Paclitaxel With or Without Everolimus in Treating Patients With Stage II or Stage III Breast Cancer | Stage II/III TNBC | NCT00930930 | |
| Phase I | Dinaciclib and Epirubicin Hydrochloride in Treating Patients With Metastatic Triple- Negative Breast Cancer | CDK inhibitor | Metastatic TNBC | NCT01624441 |
| Phase II | Safety and Efficacy Study of Enzalutamide in Patients with Advancer, AR+ TNBC | Androgen blocker | AR+ Metastatic TNBC | NCT01889238 |
| Phase II | Re-expression of ER in Triple Negative Breast Cancers | HDAC inhibitor | Metastatic TNBC | NCT01194908 |
| Azacitidine and Entinostat in Treating Patients With Advanced Breast Cancer | NCT01349959 | |||
| Entinostat and Anastrozole in Treating Postmenopausal Women With Triple- Negative Breast Cancer That Can Be Removed by Surgery | Stage I – III TNBC | NCT01234532 | ||
| Carboplatin and Paclitaxel Albumin- Stabilized Nanoparticle Formulation With or Without Vorinostat in Treating Women With Breast Cancer That Can Be Removed by Surgery | Stage II/III TNBC | NCT00616967 | ||
| Phase I | Olaparib in Combination With Carboplatin for Refractory or Recurrent Women’s Cancers | PARP inhibitor | Women’s cancers; males with BRCA mutations | NCT01237067 |
| AZD2281 Plus Carboplatin to Treat Breast and Ovarian Cancer | Breast and ovarian metastatic cancers | NCT01445418 | ||
| Study to Assess the Safety and Tolerability of a PARP Inhibitor in Combination With Carboplatin and/or Paclitaxel | TNBC and ovarian metastatic cancers | NCT00516724 | ||
| Phase I of BKM120/Olaparib for Triple Negative Breast Cancer or High Grade Serous Ovarian Cancer | TNBC and ovarian metastatic cancers | NCT01623349 | ||
| Veliparib With Radiation Therapy in Patients With Inflammatory or Loco- regionally Recurrent Breast Cancer | Inflammatory or Loco-regionally Recurrent Breast Cancer | NCT01477489 | ||
| Phase I/II | A Study of Oral Rucaparib in Patients With gBRCA Mutation Breast or Ovarian Cancer, or Other Solid Tumor | gBRCA Mutation Breast or Ovarian Cancer | NCT01482715 | |
| Phase II | A Phase 2 Study of Standard Chemotherapy Plus BSI-201 (a PARP Inhibitor) in the Neoadjuvant Treatment of Triple Negative Breast Cancer | Stage II/III TNBC |
NCT00813956 | |
| A Study Evaluating INIPARIB in Combination With Chemotherapy to Treat Triple Negative Breast Cancer Brain Metastasis | TNBC with brain mets | NCT01173497 | ||
| Phase II Study of AZD2281 in Patients With Known BRCA Mutation Status or Recurrent High Grade Ovarian Cancer or Patients With Known BRCA Mutation Status/Triple Neg Breast Cancer | TNBC and ovarian metastatic cancers; BRCA mt cancers | NCT00679783 | ||
| PARP Inhibition for Triple Negative Breast Cancer (ER-/PR-/HER2-)With BRCA1/2 Mutations | TNBC metastatic BRCA mt cancers | NCT01074970 | ||
| ABT-888 With Cyclophosphamide in Refractory BRCA-Positive Ovarian, Primary Peritoneal or Ovarian High- Grade Serous Carcinoma, Fallopian Tube Cancer, Triple-Negative Breast Cancer, and Low-Grade Non-Hodgkin’s Lymphoma | Metastatic TNBC | NCT01306032 | ||
| Study of SAR240550 (BSI-201) in Combination With Gemcitabine/Carboplatin, in Patients With Metastatic Triple Negative Breast Cancer | NCT01045304 | |||
| Two Regimens of SAR240550/Weekly Paclitaxel and Paclitaxel Alone as Neoadjuvant Therapy in Triple Negative Breast Cancer Patients | Stage II/III TNBC | NCT01204125 | ||
| ABT-888 and Temozolomide for Metastatic Breast Cancer and BRCA1/2 Breast Cancer | Metastatic Breast Cancer and BRCA1/2 Breast Cancer | NCT01009788 | ||
| Rucaparib(CO-338; Formally Called AG- 014699 or PF-0136738) in Treating Patients With Locally Advanced or Metastatic Breast Cancer or Advanced Ovarian Cancer | Metastatic breast, ovarian and BRCA mt cancers | NCT00664781 |
However, TNBC is clearly a complex disease. As such, it is likely that its biology involves multiple redundancies and pathway crosstalk. If only one pathway is selectively inhibited, the efficacy of the therapeutic strategy would likely be undermined by activation of a compensatory pathway. Therefore, it is not surprising that to date, not a single targeted therapy has been approved for treatment of TNBC, where cytotoxic chemotherapy remains the standard treatment. Combining two or more targeted agents may be required for a more rational and optimal approach to TNBC treatment. Efforts in this direction are evidenced by novel clinical trials involving different/ complementary pathway inhibitors, such as phase I and II studies combining PI3K inhibitors with PARP inhibitors or with androgen blockers, platinum agents with PI3K or mTOR inhibitors or HDAC inhibitors, etc. (Table 2).
It is generally established that patients with breast cancer who achieve a pathological complete remission (lack of residual disease in both breast and axilla) after neoadjuvant therapy exhibit a good long-term outcome (46). A high residual disease burden in the post-treatment, surgically-excised cancer has been shown to correlate with a high rate of recurrence and death (47, 48). More specifically, at least 40% of patients with TNBC who do not achieve a pCR after anthracycline and taxane–based neoadjuvant chemotherapy will have a recurrence within 36 months (49). However, approximately 30% of TNBC treated with anthracycline and taxane–based chemotherapy will have a pCR after treatment (49), and consistent with above data, achieving a pCR to neoadjuvant chemotherapy in this group of patients has been shown to be a strong positive prognostic factor. Patients with TNBC that complete neoadjuvant therapy and have no clinical evidence of metastatic disease after surgical excision of the cancer, regardless of their residual disease burden (RDB), are usually observed without further treatment. This conduct might not be appropriate for patients at a very high risk of early recurrence such as those with a high residual disease burden in the residual drug-resistant tumor. However, the appropriate therapy for those patients is unknown, and personalized treatment strategies using adjuvant therapies that molecularly target tumor-specific dependencies are sorely needed. The inter-tumor heterogeneity of TNBCs before and after neoadjuvant chemotherapy underscores the need for powerful and broad molecular approaches to identify actionable molecular alterations and, in turn, better inform personalized therapy of this aggressive disease. Incorporation of these approaches into clinical studies and eventually standards of care will aid in the prioritization of patients with residual disease after neoadjuvant chemotherapy into rational adjuvant studies. The post-neoadjuvant treatment setting could be a valuable source for clinical trials initiated to align patients with treatments best suited to target their cancer subtypes.
In summary, TNBC is a complex disease. Its relative uncommonness, aggressiveness and impressive heterogeneity have been only a few of the challenges researchers and clinicians face in making strides against this disease. Therefore, future clinical trial design for TNBC should focus on a) continued efforts to select appropriate targeted therapies based on TNBC subtyping; b) continued efforts for tissue collection in the post-neoadjuvant setting and metastatic setting for a better understanding of the relevant pathways that are associated with TNBC pathogenesis and therapeutic resistance; and c) combination of “complementary” pathway inhibitors to maximize efficacy, and minimize therapeutic resistance.
Supplementary Material
Acknowledgments
Support: Breast Cancer and the Breast Cancer Specialized Program of Research Excellence (SPORE) grant 2P50 CA098131-06 (IAM, VGA, JAP) and Susan G. Komen for the Cure grant SAC110030 (BDL)
Footnotes
Conflict of Interest: I. Mayer: commercial research support from Novartis V.G. Abramson, B.D. Lehmann, J.A. Pietenpol: none
References
- 1.Dent R, Trudeau M, Pritchard KI, Hanna WM, Kahn HK, Sawka CA, et al. Triple-negative breast cancer: clinical features and patterns of recurrence. Clin Cancer Res. 2007;13(15 Pt 1):4429–34. doi: 10.1158/1078-0432.CCR-06-3045. Epub 2007/08/03. [DOI] [PubMed] [Google Scholar]
- 2.Carey LA, Perou CM, Livasy CA, Dressler LG, Cowan D, Conway K, et al. Race, breast cancer subtypes, and survival in the Carolina Breast Cancer Study. JAMA. 2006;295(21):2492–502. doi: 10.1001/jama.295.21.2492. Epub 2006/06/08. [DOI] [PubMed] [Google Scholar]
- 3.Millikan RC, Newman B, Tse CK, Moorman PG, Conway K, Dressler LG, et al. Epidemiology of basal-like breast cancer. Breast Cancer Res Treat. 2008;109(1):123–39. doi: 10.1007/s10549-007-9632-6. Epub 2007/06/21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kassam F, Enright K, Dent R, Dranitsaris G, Myers J, Flynn C, et al. Survival Outcomes for Patients with Metastatic Triple-Negative Breast Cancer: Implications for Clinical Practice and Trial Design. Clinical Breast Cancer. 2009;9(1):29–33. doi: 10.3816/CBC.2009.n.005. [DOI] [PubMed] [Google Scholar]
- 5.Bertucci F, Finetti P, Cervera N, Esterni B, Hermitte F, Viens P, et al. How basal are triple-negative breast cancers? Int J Cancer. 2008;123:236–40. doi: 10.1002/ijc.23518. [DOI] [PubMed] [Google Scholar]
- 6.Matros E, Wang ZC, Lodeiro G, Miron A, Iglehart JD, Richardson AL. BRCA1 promoter methylation in sporadic breast tumors: relationship to gene expression profiles. Breast Cancer Res Treat. 2005;91(2):179–86. doi: 10.1007/s10549-004-7603-8. Epub 2005/05/04. [DOI] [PubMed] [Google Scholar]
- 7.Turner N, Tutt A, Ashworth A. Hallmarks of ‘BRCAness’ in sporadic cancers. Nat Rev Cancer. 2004;4(10):814–9. doi: 10.1038/nrc1457. [DOI] [PubMed] [Google Scholar]
- 8.Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011;121(7):2750–67. doi: 10.1172/JCI45014. Epub 2011/06/03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lehmann BD, Pietenpol JA. Identification and use of biomarkers in treatment strategies for triple negative breast cancer subtypes. J Pathol. 2013 doi: 10.1002/path.4280. Epub 2013/10/12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen X, Li J, Gray WH, Lehmann BD, Bauer JA, Shyr Y, et al. TNBCtype: A Subtyping Tool for Triple-Negative Breast Cancer. Cancer informatics. 2012;11:147–56. doi: 10.4137/CIN.S9983. Epub 2012/08/09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Masuda H, Bagggerly K, Wang Y. Differential pathologic complete response rates after neoadjuvant chemotherapy among molecular subtypes of triple-negative breast cancer. J Clin Oncol. 2013;31(suppl):abstr 1005. [Google Scholar]
- 12.Leong CO, Vidnovic N, DeYoung MP, Sgroi D, Ellisen LW. The p63/p73 network mediates chemosensitivity to cisplatin in a biologically defined subset of primary breast cancers. J Clin Invest. 2007;117(5):1370–80. doi: 10.1172/JCI30866. Epub 2007/04/21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rocca A, Viale G, Gelber RD, Bottiglieri L, Gelber S, Pruneri G, et al. Pathologic complete remission rate after cisplatin-based primary chemotherapy in breast cancer: correlation with p63 expression. Cancer Chemother Pharmacol. 2008;61(6):965–71. doi: 10.1007/s00280-007-0551-3. Epub 2007/07/20. [DOI] [PubMed] [Google Scholar]
- 14.Silver DP, Richardson AL, Eklund AC, Wang ZC, Szallasi Z, Li Q, et al. Efficacy of neoadjuvant Cisplatin in triple-negative breast cancer. J Clin Oncol. 2010;28(7):1145–53. doi: 10.1200/JCO.2009.22.4725. Epub 2010/01/27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Byrski T, Huzarski T, Dent R, Gronwald J, Zuziak D, Cybulski C, et al. Response to neoadjuvant therapy with cisplatin in BRCA1-positive breast cancer patients. Breast Cancer Res Treat. 2009;115(2):359–63. doi: 10.1007/s10549-008-0128-9. Epub 2008/07/24. [DOI] [PubMed] [Google Scholar]
- 16.Von Minckwitz G, Schneeweiss A, Salat C. A randomized phase II trial investigating the addition of carboplatin to neoadjuvant therapy for triple-negative and HER2-positive early breast cancer (GeparSixto) J Clin Oncol. 2013;31(suppl):abst 1004. [Google Scholar]
- 17.Sikov W, Berry D, Perou C, Singh B, Cirrincione C, Tolaney S, et al. Impact of the addition of carboplatin (Cb) and/or bevacizumab (B) to neoadjuvant weekly paclitaxel (P) followed by dose-dense AC on pathologic complete response (pCR) rates in triple-negative breast cancer (TNBC): CALGB 40603 (Alliance). San Antonio Breast Cancer Symposium 2013. 2013; Oral Session 5:01; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ding SL, Sheu LF, Yu JC, Yang TL, Chen BF, Leu FJ, et al. Abnormality of the DNA double-strand-break checkpoint/repair genes, ATM, BRCA1 and TP53, in breast cancer is related to tumour grade. Br J Cancer. 2004;90(10):1995–2001. doi: 10.1038/sj.bjc.6601804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yoshida K, Miki Y. Role of BRCA1 and BRCA2 as regulators of DNA repair, transcription, and cell cycle in response to DNA damage. Cancer Sci. 2004;95(11):866–71. doi: 10.1111/j.1349-7006.2004.tb02195.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scully R, Chen J, Plug A, Xiao Y, Weaver D, Feunteun J, et al. Association of BRCA1 with Rad51 in mitotic and meiotic cells. Cell. 1997;88(2):265–75. doi: 10.1016/s0092-8674(00)81847-4. [DOI] [PubMed] [Google Scholar]
- 21.Taniguchi T, Garcia-Higuera I, Andreassen PR, Gregory RC, Grompe M, D’Andrea AD. S-phase-specific interaction of the Fanconi anemia protein, FANCD2, with BRCA1 and RAD51. Blood. 2002;100(7):2414–20. doi: 10.1182/blood-2002-01-0278. [DOI] [PubMed] [Google Scholar]
- 22.Bhattacharyya A, Ear US, Koller BH, Weichselbaum RR, Bishop DK. The breast cancer susceptibility gene BRCA1 is required for subnuclear assembly of Rad51 and survival following treatment with the DNA cross-linking agent cisplatin. J Biol Chem. 2000;275(31):23899–903. doi: 10.1074/jbc.C000276200. [DOI] [PubMed] [Google Scholar]
- 23.Turner N, Tutt A, Ashworth A. Targeting the DNA repair defect of BRCA tumours. Curr Opin Pharmacol. 2005;5(4):388–93. doi: 10.1016/j.coph.2005.03.006. [DOI] [PubMed] [Google Scholar]
- 24.Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917–21. doi: 10.1038/nature03445. Epub 2005/04/15. [DOI] [PubMed] [Google Scholar]
- 25.Tutt A, Robson M, Garber JE, Domchek SM, Audeh MW, Weitzel JN, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet. 2010;376(9737):235–44. doi: 10.1016/S0140-6736(10)60892-6. Epub 2010/07/09. [DOI] [PubMed] [Google Scholar]
- 26.Gelmon KA, Tischkowitz M, Mackay H, Swenerton K, Robidoux A, Tonkin K, et al. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: a phase 2, multicentre, open-label, non-randomised study. Lancet Oncol. 2011;12(9):852–61. doi: 10.1016/S1470-2045(11)70214-5. Epub 2011/08/25. [DOI] [PubMed] [Google Scholar]
- 27.O’Shaughnessy J, Osborne C, Pippen JE, Yoffe M, Patt D, Rocha C, et al. Iniparib plus chemotherapy in metastatic triple-negative breast cancer. N Engl J Med. 2011;364(3):205–14. doi: 10.1056/NEJMoa1011418. Epub 2011/01/07. [DOI] [PubMed] [Google Scholar]
- 28.O’Shaughnessy JSL, Danso MA, et al. A randomized phase III study of iniparib (BSI-201) in combination with gemcitabine/carboplatin (G/C) in metastatic triple-negative breast cancer (TNBC) J Clin Oncol. 2011:29. abstract #1007. [Google Scholar]
- 29.Telli M, Jensen K, Kurian A. PrECOG 0105: Final efficacy results from a phase II study of gemcitabine (G) and carboplatin (C) plus iniparib (BSI-201) as neoadjuvant therapy for triple-negative (TN) and BRCA1/2 mutation-associated breast cancer. J Clin Oncol. 2013;31(suppl):abstr 1003. doi: 10.1200/JCO.2014.57.0085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barbieri CE, Pietenpol JA. p63 and epithelial biology. Exp Cell Res. 2006;312(6):695–706. doi: 10.1016/j.yexcr.2005.11.028. [DOI] [PubMed] [Google Scholar]
- 31.Borresen-Dale AL. TP53 and breast cancer. Hum Mutat. 2003;21(3):292–300. doi: 10.1002/humu.10174. [DOI] [PubMed] [Google Scholar]
- 32.Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. 2001;98(19):10869–74. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rosenbluth JM, Mays DJ, Jiang A, Shyr Y, Pietenpol JA. Differential regulation of the p73 cistrome by mammalian target of rapamycin reveals transcriptional programs of mesenchymal differentiation and tumorigenesis. Proc Natl Acad Sci U S A. 2011;108(5):2076–81. doi: 10.1073/pnas.1011936108. Epub 2011/01/20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mayer IA, Jovanovic B, Abramson VG, Mayer EL, Sanders ME, Bardia A, et al. A randomized phase II neoadjuvant study of cisplatin, paclitaxel with or without everolimus (an mTOR inhibitor) in patients with stage II/III triple-negative breast cancer (TNBC). San Antonio Breast Cancer Symposium 2013. 2013; Poster Discussion PD1-6.. [Google Scholar]
- 35.Shah SP, Roth A, Goya R, Oloumi A, Ha G, Zhao Y, et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature. 2012 doi: 10.1038/nature10933. Epub 2012/04/13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gonzalez-Angulo AM, Stemke-Hale K, Palla SL, Carey M, Agarwal R, Meric-Berstam F, et al. Androgen receptor levels and association with PIK3CA mutations and prognosis in breast cancer. Clin Cancer Res. 2009;15(7):2472–8. doi: 10.1158/1078-0432.CCR-08-1763. Epub 2009/03/12. [DOI] [PubMed] [Google Scholar]
- 37.Stemke-Hale K, Gonzalez-Angulo AM, Lluch A, Neve RM, Kuo WL, Davies M, et al. An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer Res. 2008;68(15):6084–91. doi: 10.1158/0008-5472.CAN-07-6854. Epub 2008/08/05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ibrahim YH, Garcia-Garcia C, Serra V, He L, Torres-Lockhart K, Prat A, et al. PI3K inhibition impairs BRCA1/2 expression and sensitizes BRCA-proficient triple-negative breast cancer to PARP inhibition. Cancer discovery. 2012;2(11):1036–47. doi: 10.1158/2159-8290.CD-11-0348. Epub 2012/08/24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ibrahim YH, Garcia-Garcia C, Serra V, He L, Torres-Lockhart K, Prat A, et al. PI3K inhibition impairs BRCA1/2 expression and sensitizes BRCA proficient triple negative breast cancer to PARP inhibition. Cancer discovery. 2012 doi: 10.1158/2159-8290.CD-11-0348. Epub 2012/08/24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Juvekar A, Burga LN, Hu H, Lunsford EP, Ibrahim YH, Balmana J, et al. Combining a PI3K inhibitor with a PARP inhibitor provides an effective therapy for a mouse model of BRCA1-related breast cancer. Cancer discovery. 2012 doi: 10.1158/2159-8290.CD-11-0336. Epub 2012/08/24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wong KK, Engelman JA, Cantley LC. Targeting the PI3K signaling pathway in cancer. Curr Opin Genet Dev. 2010;20(1):87–90. doi: 10.1016/j.gde.2009.11.002. Epub 2009/12/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kumar A, Fernandez-Capetillo O, Carrera AC. Nuclear phosphoinositide 3-kinase beta controls double-strand break DNA repair. Proc Natl Acad Sci U S A. 2010;107(16):7491–6. doi: 10.1073/pnas.0914242107. Epub 2010/04/07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hoeflich KP, O’Brien C, Boyd Z, Cavet G, Guerrero S, Jung K, et al. In vivo antitumor activity of MEK and phosphatidylinositol 3-kinase inhibitors in basal-like breast cancer models. Clin Cancer Res. 2009;15(14):4649–64. doi: 10.1158/1078-0432.CCR-09-0317. Epub 2009/07/02. [DOI] [PubMed] [Google Scholar]
- 44.Korse CM, Taal BG, Vincent A, van Velthuysen ML, Baas P, Buning-Kager JC, et al. Choice of tumour markers in patients with neuroendocrine tumours is dependent on the histological grade. A marker study of Chromogranin A, Neuron specific enolase, Progastrin-releasing peptide and cytokeratin fragments. Eur J Cancer. 2012;48(5):662–71. doi: 10.1016/j.ejca.2011.08.012. Epub 2011/09/29. [DOI] [PubMed] [Google Scholar]
- 45.Balko JM, Cook RS, Vaught DB, Kuba MG, Miller TW, Bhola NE, et al. Profiling of residual breast cancers after neoadjuvant chemotherapy identifies DUSP4 deficiency as a mechanism of drug resistance. Nat Med. 2012;18(7):1052–9. doi: 10.1038/nm.2795. Epub 2012/06/12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liedtke C, Mazouni C, Hess KR, Andre F, Tordai A, Mejia JA, et al. Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J Clin Oncol. 2008;26(8):1275–81. doi: 10.1200/JCO.2007.14.4147. Epub 2008/02/06. [DOI] [PubMed] [Google Scholar]
- 47.Jones RL, Salter J, A’Hern R, Nerurkar A, Parton M, Reis-Filho JS, et al. The prognostic significance of Ki67 before and after neoadjuvant chemotherapy in breast cancer. Breast Cancer Res Treat. 2009;116(1):53–68. doi: 10.1007/s10549-008-0081-7. Epub 2008/07/02. [DOI] [PubMed] [Google Scholar]
- 48.Guarneri V, Piacentini F, Ficarra G, Frassoldati A, D’Amico R, Giovannelli S, et al. A prognostic model based on nodal status and Ki-67 predicts the risk of recurrence and death in breast cancer patients with residual disease after preoperative chemotherapy. Ann Oncol. 2009 doi: 10.1093/annonc/mdn761. Epub 2009/02/18. [DOI] [PubMed] [Google Scholar]
- 49.von Minckwitz G, Untch M, Blohmer JU, Costa SD, Eidtmann H, Fasching PA, et al. Definition and impact of pathologic complete response on prognosis after neoadjuvant chemotherapy in various intrinsic breast cancer subtypes. J Clin Oncol. 2012;30(15):1796–804. doi: 10.1200/JCO.2011.38.8595. Epub 2012/04/18. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.