

Abstract
Predicting absolute protein-ligand binding affinities remains a frontier challenge in ligand discovery and design. This becomes more difficult when ionic interactions are involved, because of the large opposing solvation and electrostatic attraction energies. In a blind test, we examined whether alchemical free energy calculations could predict binding affinities of 14 charged and 5 neutral compounds previously untested as ligands for a cavity binding site in Cytochrome C Peroxidase. In this simplified site, polar and cationic ligands compete with solvent to interact with a buried aspartate. Predictions were tested by calorimetry, spectroscopy, and crystallography. Of the 15 compounds predicted to bind, 13 were experimentally confirmed, while four compounds were false negative predictions. Predictions had an RMSE of 1.95 kcal/mol to the experimental affinities, and predicted poses had an average RMSD of 1.7 Å to the crystallographic poses. This test serves as a benchmark for these thermodynamically rigorous calculations at predicting binding affinities for charged compounds, and gives insights into the existing sources of error, which are primarily electrostatic interactions inside proteins. Our experiments also provide a useful set of ionic binding affinities in a simplified system for testing new affinity prediction methods.
Keywords: free energy calculations, electrostatics, ligand binding, molecular dynamics
1. Introduction
Predicting protein-ligand binding affinity from molecular structure is a fundamental challenge in biophysics, with particular importance in ligand discovery. Despite much effort over the last three decades1, the problem has resisted a general solution, and presents three major difficulties. First, many energetic terms contribute to affinity, including dispersion, electrostatics, hydrophobicity, and solvation, and each term can be much larger than the overall binding energy. Second, these terms depend in subtle ways on the environment of the binding site – buried or solvent exposed, rigid or flexible – making it difficult to design models which are transferrable from one protein to the next. Third, other critical energetic terms are “invisible”: while one can see each hydrogen bond in a protein-ligand complex structure, one cannot see the free energy cost of reorganizing the receptor from the unbound ensemble to the bound ensemble, or the change in conformational entropy of the ligand upon binding. This energetic complexity also makes it challenging to experimentally resolve individual energetic terms, leaving us with little insight for improving computational modeling.
Free energy calculations based on atomistic simulations have the potential to overcome the challenge of invisible energetic terms. These calculations compute affinities by sampling an ensemble of configurations from simulations of the bound state, the unbound state, and additional artificial states that form a complete and computationally efficient path between the end states2,3. Analyzing these simulations using statistical mechanics intrinsically accounts for the invisible energetic terms. Furthermore, by employing explicit solvent simulations, free energy calculations include atomistic details of the binding site’s solvation structure, which are lost in simpler methods. Despite their rigorous formulation, free energy calculations depend on empirical force fields, which still have many weaknesses4,5. Adequate sampling is extremely computationally intensive, which can necessitate using simpler force fields. Many conformational rearrangements – even simple rotamer flips – are difficult to sample when simulating many ligands6. This computational cost has limited the testing and improvement of free energy calculations, often to small numbers of ligands and highly simplified systems.
Here, we employ a model cavity site to present the first blind test of free energy calculations at predicting affinities for charged compounds. The site is a mutant of yeast Cytochome C Peroxidase (CCP), engineered to introduce a buried cavity binding site connected to the surface by a water channel (Figure 1)7. The “open cavity” site studied here is similar to an earlier, “closed cavity” mutant of the same protein (Figure S1), which has many known ligands and has been previously studied by retrospective free energy calculations8,9. Both sites contain an ionized aspartate, making them attractive for small cations. Simplified model sites like this open cavity present very few interactions for ligands. This makes it possible to experimentally isolate individual contributions to binding, overcoming the main difficulty of using experiments to refine computational models6,10. Similar model cavity sites in T4 lysozyme have proven valuable resources for testing affinity prediction methods, though these sites bind only neutral nonpolar and polar compounds.6,10,11,12,13,14 The CCP open cavity that we examine here complements these resources by enabling the study of charged compounds in a simplified, solvent exposed site. The open cavity is ideal for examining charged binding, because the ligands compete with solvent to interact with a single charged residue, many new ligands can be found for prospective testing, and because physical binding assays and x-ray crystallography enable a detailed comparison between theory and experiment for each ligand15,16.
Figure 1. The Cytochrome C Peroxidase (CCP) open cavity binding site bound to benzimidazole (1) (PDB Code 1KXM, ref. 7).

Figure generated using PyMOL 1.4.1, Schrödinger, LLC. (a) The binding site in the context of the full protein. The protein surface around the binding site is shown in yellow, illustrating the burial of the binding site and the channel between the ligand and the surface. (b) Close-up view of the binding site, with the Asp at left. (c) Stereo view of the miniature binding site model discussed in section 2.9.
Modeling charged binding is much more challenging than modeling neutral binding17. Charged molecules are highly solvated; as an example, the gas to water transfer free energy of anilinium is −70 kcal/mol, compared with −5 kcal/mol for aniline18. Computational models must balance these powerful solvent-ligand interactions against equally powerful protein-ligand interactions. Furthermore, while neutral ligands primarily interact with the receptor surface19, charged interactions depend on the environment above and below the surface, because the environment determines how strongly electrostatic interactions are screened20,21,22. In simpler models of binding based on the Poisson-Boltzmann equation, the amount of screening by the receptor is a separate parameter for fitting: the so-called “dielectric constant”22,16,23. Free energy calculations are the main method where this screening is not directly parameterized, and instead depends on the ability of the fully flexible receptor and solvent to reorganize in response to charge. However, few studies have tested whether free energy calculations actually can reproduce charged binding affinities,24,25,26,27,28, and none have undertaken prospective tests.
Unlike retrospective studies, successful prospective tests must sample many protein conformations and ligand orientations because the true bound pose is unknown, if the ligand binds at all. Decoy poses, rarely sampled in retrospective calculations, can lead to incorrect affinities. Ligand parameterization methods must be highly reliable, because no experimental data is available beforehand to validate specific parameters. Convergence problems – which may be invisible in energetic data – must be carefully identified and corrected in advance, without knowledge of the correct affinity or structure. Here, we attempt to overcome these challenges in the CCP open cavity, to investigate the following five questions: (1) Can free energy calculations prospectively distinguish between ligands and nonbinders in a charged site? (2) Can free energy calculations quantitatively predict affinities for charged compounds, and (3) for neutral compounds in a charged site? (4) Do accurate affinity predictions result from correctly identifying the ligand’s crystallographic binding pose? (5) What do our experiments tell us about improving computer simulations? The site’s simplicity should make it possible to trace prediction failures to specific force field terms, so that the experiments can refine the theory.
2. Results
2.1 Blind Binding Affinity Prediction
We performed two successive rounds of blind affinity prediction to the open cavity. In the first round, we selected four known cationic ligands of the similar, previously studied “closed cavity” mutant15,16, and predicted their affinities for the open cavity. After comparing this first round of predictions to the experimental results, we made the second round of predictions. The second round included fifteen compounds: ten cations that had never been tested in either cavity site (open or closed), and five neutral compounds. All compounds were selected from a docking screen of 650,000 fragment-like compounds in the ZINC database29, and were chosen to explore a wide range of chemotypes, including single and double rings, amines, diamines, amidines, alcohols, and various heterocycles (Table 1). Predicting absolute affinities for these diverse compounds resembles an early-stage ligand discovery problem, and is presumably more difficult than predicting effects of perturbations on a common scaffold, as is common in the field30,31,32. All compounds fit well into the binding site by docking, but many were ranked poorly: several charged compounds were selected with ranks higher than 10,000, and several neutral compounds were selected from the bottom 60% of the database.
Table 1.
Experimental affinities and free energy calculation blind predictions for compounds tested in the CCP open cavity.
| Round 1 Compound | Exper. ΔGbind (kcal/mol) |
Predicted ΔGbind (kcal/mol) |
Predicted pose RMSD (Å) |
PDB Code | |
|---|---|---|---|---|---|
| 1 | 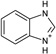 |
−5.8a | −5.8 ± 0.1b | 1.1c | 1KXM |
| 2 | 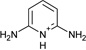 |
−5.8 ± 0.2 | −5.1 ± 0.2 | 0.6 | 4JM8 |
| 3 |  |
−5.1 ± 0.2 | −4.8 ± 0.2 | 1.9 | 4JM5 |
| 4 | −4.4 ± 0.2 | −2.2 ± 0.2 | 3.1 | 4JM6 | |
| 5 | 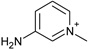 |
−3.4 ± 0.4 | −1.1 ± 0.2 | 2.9 (1st) 0.5 (2nd) |
4JM9 |
| Round 2 Compound | Exper. ΔGbind (kcal/mol) |
Predicted ΔGbind (kcal/mol) |
Predicted pose RMSD (Å) |
PDB Code | |
| 6 |  |
−7.1 ± 0.2 | −4.2 ± 0.3 | 0.6 | 4JQM |
| 7 | 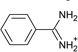 |
−6.6 ± 0.2 | −3.3 ± 0.2 | 0.5 | 4JPU |
| 8 | 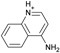 |
−5.8 ± 0.2 | −5.8 ± 0.3 | 0.4 | 4JQJ |
| 9 | 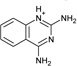 |
−5.7 ± 0.2 | −4.7 ± 0.4 | 0.9 | 4JPT |
| 10 | −4.8 ± 0.2 | −7.9 ± 0.4 | 1.0 | 4JPL | |
| (Corrected Tyr) | −4.8 ± 0.2 | −5.9 ± 0.4d | 3.2e | 4JPL | |
| 11 |  |
−4.7 ± 0.2 | −2.3 ± 0.3 | 1.1 | 4JQK |
| 12 |  |
> −3.9 | −3.7 ± 0.4 | naf | na |
| 13 | 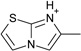 |
> −3.9 | −2.8 ± 0.2 | na | na |
| 14 | 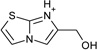 |
> −3.0 | −4.4 ± 0.5 | na | na |
| 15 | NB, > 3.0 | −0.9 ± 0.5 | na | na | |
| Neutral Compound | Exper. ΔGbind (kcal/mol) |
Predicted ΔGbind (kcal/mol) |
Predicted pose RMSD (Å) |
PDB Code | |
| 16 | > −3.3 | −4.6 ± 0.3 | 1.0 | 4JMW | |
| 17 | > −3.3 | −4.8 ± 0.2 | 2.8 | 4JMA | |
| 18 | > −3.3 | −2.6 ± 0.4 | 2.8 (1st) 0.8 (2nd) |
4JQN | |
| 19 | NB, > −3.9 | −6.8 ± 0.3 | na | na | |
| 20 | NB, > −3.0 | −5.2 ± 0.3 | na | na | |
NB = no evidence of binding, to a given maximum concentration. Round One predictions were made using a scaling factor of +/− 0.981, calibrated using benzimidazole (compound 1). Round Two predictions were made using a scaling factor of 0.986, calibrated based on the five Round One compounds. Pose RMSDs were calculated using a representative snapshot of the MD ensemble for the dominant pose, measured against the nearest ligand structure observed by crystallography after aligning the MD protein to the crystal protein. Experimental uncertainties were calculated as the standard deviation of three or more independent measurements, or as a 20% error in Kd if only two experiments were conducted or if the experimental agreement was within 20%.
From ref. 7.
Not a blind prediction, used to calibrate the original scaling factor.
Not a blind prediction, but the RMSD to the most favored benzimidazole pose of several decoys considered.
Not a blind prediction; based on re-simulating the badly equilibrated 4-azaindole pose retrospectively.
Not a blind prediction, but based on using the most favorable 4-azaindole pose after retrospectively resimulating the badly equilibrated 4-azaindole pose.
Not applicable (no structure collected).
2.2 Experimental
The 14 cationic and 5 neutral compounds were tested for binding to the open cavity using both isothermical titration calorimetry (ITC) and UV-Vis titration of the heme Soret band, as described previously15,16 (Figure 2). Data collected are an average of two or more independent experiments, each run in duplicate. Binding isotherms from ITC were readily modeled and affinities were reproducible within 0.2 kcal/mol. For ligands too weak for ITC, UV-Vis measurements clearly demonstrated binding at the measured concentration. All compounds except 4-aminobenzamidine (15) and two neutral compounds (19 and 20) were found to bind (Table 1). To characterize these interactions at atomic resolution, we determined crystal structures with 13 different compounds (Figure 3, SI Table S4). These structures were determined to resolutions between 1.2 and 1.6 Å; both initial Fo-Fc and refined 2Fo-Fc electron density enabled unambiguous placement of the ligand in one or two orientations.
Figure 2. Representative binding affinity data.

(left) Binding affinity determination for compound 2 by low C-value isothermal titration calorimetry (right) Characteristic UV-Vis red-shift upon binding of compound 14. black: protein only, red: 5 mM ligand.
Figure 3. Comparison of predicted to experimental binding modes.

Predicted (magenta) and experimentally observed (cyan) structures of CCP open cavity ligands, with the observed protein conformation in white. In some structures, the flexible loop moves upon ligand binding; the apo structure of this loop is shown in black for reference in these cases. Hydrogen bonds are drawn to clarify which atoms are in contact and do not represent a specific distance cutoff. a–f: Compounds with correct pose predictions. g–h: Compounds where two poses were predicted to be within kBT in energy, and the most favorable pose (magenta) was not observed experimentally while the second most favorable pose matched the experimental (cyan) pose. i–k: Compounds with close, but not correct predictions. l–m: Compounds with incorrect predictions. n: Stereo view of 2Fo-Fc density around compound 6 at 1σ. Refinement statistics in SI Results Table S4. Figure generated using PyMOL 1.4.1, Schrödinger, LLC.
2.3 Free energy calculations protocol
Alchemical free energy calculations were used to predict all binding affinities using an existing protocol6,10, modified to account for the difficulty of modeling charged compounds. Free energies of charge insertion require long timescales to converge. We therefore divided each charged binding affinity calculation into two steps. First, a charged “reference” compound was inserted into the binding site using long simulations. Second, the reference compound was transformed into the (likewise charged) “target” compound using shorter simulations. Together, these sum to the free energy of inserting the target compound into the binding site. Three reference compound calculations (averaged together) were used to efficiently calculate the binding affinities for the fifteen charged “target” compounds (see Methods and SI Methods). Because the binding site is partially open to solvent, we observed that water molecules readily re-filled the site when compounds were fully decoupled, unlike in sites where conformational changes are required for access to solvent.33
Charged affinities can also be sensitive to the overall receptor protonation state, which depends on pH. We therefore computed the free energy of inserting the charged reference compounds at three different overall receptor charge states: net −5, net neutral, and net +9 (the binding site Asp was ionized in each case). This “effective pH” had an expected small34 (1 kcal/mol) effect on the charge insertion free energy (Figure S2). We used the results from the net +9 receptor to be consistent with our experiments, which are performed at pH 4.5, below the protein pI (measured at 4.9–5.2535). Finally, charge insertion calculations in periodic boundary simulations can introduce significant finite size artifacts36,37; we examined these in detail and corrected our results to be independent of the simulated box size, as detailed in SI Results and SI Methods. Here, this correction amounts to a +1 kcal/mol offset to all affinities for the cations and does not affect the ordering or relative affinities of the cations.
2.4 Round One: Prediction of closed cavity ligands to the open cavity
For the Round One blind predictions, we simulated a control ligand benzimidazole (compound 1)7, and the four closed cavity ligands (compounds 2–5) with unknown affinities for the open cavity. The initial free energy calculations on these ligands produced affinities which were unexpectedly strong. The control simulation of benzimidazole produced an affinity of −7.5 kcal/mol, more favorable than its known affinity by 1.7 kcal/mol. Three of the four other compounds were, at first, predicted to bind more strongly to the open cavity than their known affinities for the closed cavity, which would go against a previously observed trend7. Because these affinities seemed implausible, we changed our protocol before comparing predictions with experiments.
Standard molecular dynamics simulations like ours lack explicit electronic polarizability. The absence of explicit polarizability reduces the protein dielectric response, in which electronic polarizability can play a substantial role, but has little effect on the solvent dielectric response, where molecular reorientation dominates the dielectric response. This reduced protein dielectric response increases the overall strength of electrostatic interactions in proteins. This is especially true in buried protein environments like our binding site, where little solvent is nearby to provide screening38,39. This strongly affects ionic interactions, because the ionic net charges (+1/−1) have not been “parameterized” (in the manner of partial charges) to reflect interaction strengths38,39. Increasing the protein dielectric response is expected to weaken binding in this site, because this response would have a larger (favorable) effect on the unbound state (negatively charged binding site) compared with the bound state (negatively charged binding site neutralized by a cationic ligand). We hypothesized that correcting for these effects would improve our predicted affinities, which we expected were overly strong (though they had not yet been compared with experiment). We therefore changed the force field by scaling the net charge on the ligand and binding site aspartate from +1/−1 to smaller, non-integral (equal and oppositely signed) values. This weakens the electrostatic interactions between the ligands and the binding site hydrogen bond acceptors, and especially with the binding site aspartate. Charge scaling was only applied to the ligand when in complex with the protein, because the simulated solvent environment already includes the full amount of dielectric relaxation expected for an aqueous environment. Non-integral net charges have previously proven helpful in modeling charged interactions38,39,40,41,42, and other similar scaling methods have been previously applied43.
We used the known affinity of benzimidazole to determine a scaled charge of +/− 0.981 (for the Asp and ligand) for the first set of predictions. Though these scaled predictions, described below, are still prospective – the correct affinities and poses are not known in advance – the scaled predictions are based on some prior knowledge; e.g. the affinity of benzimidazole. While charge scaling has been suggested as an useful tool to account for missing electronic screening38,39, it is admittedly empirical and cannot quantitatively replicate the effect that true electronic polarizability would have. However, if the scaled predictions are successful, it will demonstrate free energy calculations are predictive in this system when a uniform reduction in the strength of electrostatic interactions is applied. It will also suggest that the simulated reorganization of the protein and solvent do not sufficiently screen ionic interactions in this system, and additional screening is needed. Note that scaling the charges to +/− 0.981 does not correspond to an applied dielectric of 1.04 (as would be calculated by 1/0.981^2). This is because scaling exclusively weakens interactions, while an increased dielectric weakens some interactions via screening and strengthens others via enhanced dipoles, though it is argued in refs. 38 and 39 that the screening effects dominate for charge-charge interactions.
We computed and tested blind predictions based on this charge scaling for our Round One compounds. The scaled free energy calculations correctly ranked the four compounds and benzimidazole by affinity (Table 1). The root-mean-squared error (RMSE) for the four blind predictions is 1.64 kcal/mol, and the mean signed deviation (MSD) is +1.38 kcal/mol. Had we not applied scaling, our predictions would have ranked the compounds incorrectly, and the four predicted affinities would have been less accurate (RMSE 2.33 kcal/mol) and too favorable (MSD −1.70 kcal/mol) (Table S1). If, retrospectively, we treat the charge scaling as a fit parameter for all five compounds (instead of using only the known ligand benzimidazole), we obtain a scaling factor of +/− 0.986. This retrospective factor preserves the correct ranking and minimizes the RMSE for all five compounds (0.94 kcal/mol), with an MSD of +0.36 kcal/mol (Table S1).
2.5 Round Two: Prediction of novel compounds to the open cavity
For Round Two, we changed the charge scaling based on the available Round One results, and made blind predictions of the affinities of the ten new cationic compounds using a scaling factor of +/− 0.986. Predicted affinities for the nine ligands (one of the ten did not bind) were less accurate than in Round One (RMSE 2.10 kcal/mol), and predictions for four compounds –4-aminoquinazoline (6), benzamidine (7), 4-azaindole (10), and 2-amino-1-(2-hydroxyethyl)pyridinium (11) – had errors larger than 2 kcal/mol (Table 1, Figure 4 a). The free energy calculations correctly predicted the non-binder 4-aminobenzamidine (15) to be a non-binder and to have the weakest affinity of the set.
Figure 4. (a,b,c) Agreement between free energy calculations and experimental binding affinities.

Nonbinders have been assigned a Kd of 1M (ΔGbind = 0 kcal/mol), with error bars showing the highest tested concentration. Both panels show results corrected for the 4-azaindole convergence error; other outliers discussed in the text are labeled by compound number. (a) Scaled results a scaled charge of +/− 0.986. Red: retrospective results for Round 1 compounds (for prospective results see Table 1). Blue: prospective results for Round 2 compounds. (b) Unscaled results for all compounds. Red: Round 1 compounds. Blue: Round 2 compounds. Black: Neutral compounds. (c) Unscaled results in the Miniature Binding Site. Black circles: cations, open squares: neutral compounds. Values from Table 1 and Table S1. (d) Errors in unscaled binding predictions on cations correlate with the gas-to-solution transfer (hydration) free energy. Solid circles = ligands, open circle = nonbinder (4-aminobenzamidine, 15), assigned a Kd of 1M (ΔGbind = 0 kcal/mol). Compounds 6 (4-aminoquinazoline) and 7 (benzamidine) are labeled to indicate that they are minor outliers from the trend (though they are included in the fit), which is consistent with their scaled predictions being unsuccessful outliers.
After comparing with the experiments, we discovered an unusual convergence failure in the 4-azaindole (10) prediction. Each predicted affinity results from the sum of contributions from multiple ligand poses44. For 4-azaindole, the two most favorable poses had predicted affinities of −7.9 and −5.7 kcal/mol. During equilibration of the most favorable pose, Tyr 225 flipped toward the binding site, creating new protein-ligand contacts. Critically, Tyr 225 never returned to its apo conformation once the ligand was decoupled, so the energy cost of moving Tyr 225 was never accounted for, causing an overly favorable prediction for this pose. If we eliminate the contribution of this pose from the overall predicted 4-azaindole affinity, the prediction changes from −7.9 kcal/mol to −5.7 kcal/mol, in reasonable agreement with the experimental affinity of −4.8 kcal/mol. Repeating the equilibration of the original best pose starting from the apo Tyr 225 conformation did not reproduce the flip, which we expect is a high-energy state and is not observed in any crystal structure of CCP. Retrospectively recalculating the affinity of the original best pose with a corrected Tyr position leads to a similar prediction of −5.9 kcal/mol. The other three overly favorable predictions – 4-aminoquinazoline (6), benzamidine (7), and 2-amino-1-(2-hydroxyethyl)pyridinium (11) – did not result from clear convergence errors, and we discuss these later.
To test whether free energy calculations could predict affinities for neutral compounds in a charged site, we made predictions for five neutral compounds using the standard force field. We did not apply charge scaling to the neutral compounds because we expected that the absence of polarizability would mainly overstabilize charge-charge interactions and have less effect on charge-neutral interactions38,39. The calculations predicted four of the five neutral compounds to bind too strongly, and both nonbinders were false positive predictions (Table 1). The simulations without charge scaling thus overstabilized both charged binding and neutral binding.
2.6 Pose Prediction
Of the sixteen new ligands, we determined high-resolution structures for thirteen of the complexes (Figure 3). For six of these thirteen, the free energy calculations favored one single ligand pose by more than kBT, and this pose was observed in the experimental structure (RMSD < 1.1 Å, Table 1, Figure 3 a – f). These correct pose predictions included the three compounds with the largest errors in affinity prediction: 4-aminoquinazoline (6), benzamidine (7), and 2-amino-1-(2-hydroxyethyl)pyridinium (11). While these affinity predictions were too weak by more than 2 kcal/mol, the correct pose predictions suggest that the energy function, rather than pose sampling, was responsible for these errors.
For compounds 5 and 18, the most favorable ligand pose was not observed in the experimental structures (Figure 3 g, h). However, the second most favorable pose did match the observed structure (RMSD < 0.8 Å, Table 1), and was predicted to be within kBT of the most favorable predicted pose. The more favorable predicted poses may be considered computational decoys, though the error is small energetically (0.4 kcal/mol for 5 and 0.6 kcal/mol for 18).
For three additional ligands, the dominant pose in the free energy calculations was similar to a pose observed in the experimental structure: the prediction for 2,4-diaminopyrimidine (4) (RMSD 3.1 Å) is flipped 180° from an experimentally observed pose, making an extra hydrogen bond to Asp 233 instead of to Leu 177; the prediction for 2,4-diaminoquinazoline (9) (0.9 Å) contains an extra water molecule, and the prediction for 3-fluorocatechol (17) (2.8 Å) correctly places the hydroxyls but misplaces the fluorine (Figure 3 i,j,k). In the remaining two cases (4-azaindole (10) and 2-amino-5-methylthiazole (3)), the simulations predicted a decoy pose as most favorable. For 4-azaindole, the original predicted pose actually matched one of two poses observed in the crystal structure, but after fixing the simulation to remove the Tyr 225 flip, this crystallographic pose became 0.8 kcal/mol less favorable than the decoy pose. For 2-amino-5-methylthiazole, the pose observed in the crystal structure was unlike any predicted pose, and was never sampled (Figure 3 l,m).
Most of the experimental structures showed conformational changes in the Gly 189-Ala 192 loop. For compounds 2, 3, 4, 6, 7, and 10, the backbone carbonyl of Gly 190 flips toward the ligand, creating a new electrostatic interaction (Figure 3). As part of the predictions protocol, we explicitly sampled this ‘flipped-in’ conformation for the latter three ligands, but did not find it to be favorable for any of them. Other loop movements are observed in the experimental structures for compounds 5, 6, 8, 9, 10, 11, and the existing structure of benzimidazole (1), which were also not captured in the free energy calculations. The failure of the free energy calculations to prefer these alternate loop conformations could contribute to the incorrectly weak predicted affinities for compounds 5, 6, 7, and 11. However, the other six compounds have accurately predicted affinities (using charges scaled to +/− 0.986) despite failing to reproduce the loop conformation observed in the crystal structures.
2.7 Charge parameters explain specific failures
To investigate the compounds with inaccurate affinity predictions, we retrospectively calculated affinities for all compounds using new ligand charges, derived using electrostatic potential (ESP) fitting to gas-phase ab initio quantum calculations (Table S1, Figure S3). Like the original results, the ESP results overstabilized binding before charge scaling (Table S1). By applying a scaling factor of +/− 0.992 to the cations, the binding affinities using ESP charges became very similar to the original scaled AM1-BCC binding affinities (RMSD 0.8 kcal/mol). Despite this small difference, the ESP charges substantially improve the agreement with experiment. The ESP binding affinity for benzamidine (7) is 1.8 kcal/mol more favorable than the original prediction, reducing that compound’s error to +1.5 kcal/mol. The next largest outliers - 4-aminoquinazoline (6) and 2-amino-1-(2-hydroxyethyl)pyridinium (11) - are improved as well. The ESP charges also predict a 2.5 kcal/mol weaker affinity for the neutral compound 19, which was originally a false positive. The original partial charges may have thus contributed substantially to error in the predictions.
2.8 Charge scaling corrected a systematic error in the force field model
How accurate would the predictions have been using the force field as-is, without charge scaling? The RMSE for all charged affinities, including benzimidazole and after correcting for 4-azaindole, would have been 2.33 kcal/mol (Table S1 and Figure 4 b), compared to 1.57 kcal/mol for all charged affinities using a +/− 0.986 scaling factor (Figure 4 a). These unscaled affinities are too favorable, with an MSD of −1.48 kcal/mol, compared with +0.72 kcal/mol after scaling. All three nonbinders – 4-aminobenzamidine (15) and the neutral compounds 19 and 20 – are false positive predictions for binding according to the force field without scaling.
To test if these overly favorable affinities from the unscaled force field resulted from electrostatics, we compared the error in each compound’s simulated affinity with its simulated gas-to-solution transfer (hydration) free energy, which measures polarity. For charged compounds, there is a clear trend that as a compound becomes more polar, its unscaled, simulated binding affinity becomes more incorrect (Figure 4 d), though this trend disappears once charges are scaled (Figure S4 a). For neutral compounds, the same trend appears, though only as a result of the nonbinders (Figure S4 b). Because the errors become systematically larger as compounds become more polar, the force-field electrostatics are likely at fault.
Are the predicted hydration free energies themselves incorrect? To examine this, we computed hydration free energies for 10 additional organic cations which have experimentally measured values18. The simulated results agreed with experiment without systematic errors (Figure S5 and Table S2), and similar results have been shown for neutral compounds45. This indicates that within the force field, the protein-ligand electrostatic interactions, not the solvent-ligand interactions, cause binding to be systematically too strong.
2.9 The buried environment can explain the systematic error
What physical factors caused the force-field to overstabilize protein-ligand electrostatic interactions? Understanding these factors could lead to more transferrable force fields, avoiding the need for the empirical scaling employed here. One particular factor could be the buried nature of the binding site. In a buried site like the open CCP cavity, electronic polarizability contributes significantly to the screening which weakens electrostatic interactions, but conventional, non-polarizable simulations lack this contribution17,38,39. However, the missing effect could still be small, because protein flexibility, waters in the nearby channel, and water on the protein surface all still screen electrostatics. To directly examine the amount of screening in the buried environment, we created a new ‘miniature’ binding site in silico, made of only the cavity residues surrounding the ligand (Figure 1 c), and held in place by harmonic restraints (Figure S6 b,c). We then surrounded these residues with explicit solvent, to compare binding affinities in the less polarizable protein with the more polarizable solvated miniature site.
Simulated affinities to the miniature site were 1–5 kcal/mol weaker than simulated affinities to the full protein (Table S1). Surprisingly, the new affinities were in better agreement with the experimental affinities to the full protein (RMSE=1.63 kcal/mol) (Figure 4 c). Unlike the original results, binding is no longer too favorable, and no charge scaling is required. The weakened affinities result from the electrostatic component of the free energy cycle (Table S4). The weaker affinities in the miniature site demonstrate that electrostatic interactions in the simulated full protein binding site are only partially screened, and that additional polarizability – provided in this test by nearby water – could have led to more accurate predicted affinities without charge scaling. This result does not prove that missing polarizability is the primary cause of the systematic error – errors in force field calculations can rarely be linked to a single cause. However, it establishes that the environment has a sufficient impact on the binding affinities to potentially account for the error. If, instead, the miniature site affinities had been similar to those in the full protein, this would have demonstrated that electrostatics were already heavily screened in the original calculations and suggested that additional environmental polarizability would have little effect. While the miniature site is a useful reference point for examining the amount of screening in the full protein model, the miniature site environment is still different from the environment of a fully polarizable protein, being both less polarizable in the “first shell” and more polarizable outside the first shell.
3. Discussion
Predicting absolute binding affinities is a challenging goal even in a simplified site, requiring a careful balance of many energetic contributions and sampling of many configurations. Here, we examined a protein with only one known bound structure, and predicted affinities for nineteen unrelated charged and neutral compounds, all of which fit well into the site by docking but spanned three logs of affinity. Prospective tests of free energy calculations have not previously been attempted in charged systems, or even in systems with solvent in the binding site. Performing these calculations prospectively introduced numerous potential sources of error, but uncovering these errors is essential for developing free energy calculations as a robust tool for ligand design. Three key observations emerge from our study: (1) affinity prediction using scaled charges was only partially successful due to inadequate ligand partial charges, (2) free energy calculations with full (unscaled) charges systematically overstabilize binding in this buried, charged binding site, and (3) altering the polarizability of the environment can correct this error. Along with these observations, our experimental results in this simplified, yet revealing system offer a rich resource for further methods development and testing, especially for more sophisticated models of polarizability, models of flexible regions in proteins and their energy landscapes, and models of water energetics and its role in binding.
Eleven of the fourteen charged compounds were correctly classified as ligands or non-binders in the blind predictions, and the non-binder (15) was correctly ranked as the weakest potential ligand. However, even with charge scaling, affinity prediction was only marginally successful. The prospective RMSE for the fourteen charged compounds was 1.95 kcal/mol, which is large compared to the measured range of affinities (4 kcal/mol), though this small range understates the difficulty of the task because we did not know in advance whether any compound would bind and fall inside this range. Aside from one clear convergence failure (4-azaindole, 10), the largest errors in the predictions resulted from the ligand partial charges. Retrospective calculations using more advanced ESP charges corrected the largest outliers (benzamidine, 7, and 4-aminoquinazoline, 6) and produced an overall error of 1.45 kcal/mol for charged compounds. The AM1-BCC charges we originally employed have proven comparable to ESP charges in other studies46, but this charged and highly polar binding site demonstrates the importance of further accuracy in charge models. The blind predictions on neutral compounds were less successful, as the two strongest predictions were each false positives. We attribute this to a systematic error that affected all compounds when using the unmodified force field.
Binding affinities using the unmodified force field (without scaling) were systematically incorrect. However, hydration energies for both charged and neutral compounds are well reproduced by simulations, suggesting that protein-ligand interaction energies cause the error. The problem seems to be caused by electrostatics, because the size of the error correlates with the polarity of the compound. This is exactly the type of correlation which is most apparent in a model site like this open cavity, with few other factors complicating the results. Admittedly, there may be other sources of error, including in the Heme parameters or hidden convergence failures, and we cannot rule these out at this time. Still, in our view it is unlikely that these errors would systematically bias binding toward the most polar compounds. Previous relative binding free energy calculations using the CHARMM22 force field have also suggested electrostatic interactions are overly strong in buried sites8. CHARMM electrostatic parameters are derived differently from the AMBER99 parameters employed here, suggesting that these errors are not unique to one force field.
By calculating affinities to a miniature binding site surrounded by solvent, we showed that changing the polarizability of the environment around the binding site without changing protein-ligand contacts has a substantial impact on the predicted affinities. Because the protein model is not fully polarizable, the buried environment of the binding site may overestimate electrostatic interactions, which could account for the error we observed. Current force fields have been parameterized to match pure liquid properties and gas-phase electrostatic potentials. Our experimental results indicate that electrostatic interactions in a buried environment are weaker than these potentials predict. Here, we tried to capture this effect in the full protein by empirically reducing the charges – essentially faking polarization.
Our study has several notable limitations. First, the scaling approach is physically unappealing, and could not be used to correct affinities for neutral compounds. We applied it here for charged compounds to allow us to examine the advantages of free energy calculations–conformational sampling, explicit water molecules, and inclusion of the invisible energetic terms – without being held back by one major source of error. Still, the necessary scaling factors were purely empirical, and do not reflect a theoretical estimate of missing polarizability. Second, even with scaling, our purely prospective results could not rank ligands by affinity, and many errors were only discovered retrospectively. In part, this reflects the new complexity in this system compared with past prospective tests: for example, the strong electrostatics in this binding site revealed weaknesses in charge parameterization schemes which were adequate in other systems6. Third, our retrospective analysis itself – including the systematic error in electrostatics and the superiority of QM ESP charges – can be criticized as opportunistic, reflecting our available data on a limited number of compounds, even if the test set is relatively large for a free energy study. Further prospective testing can confirm or refute these hypotheses. Finally, we cannot prove that missing polarizability is the main cause of overly strong electrostatics. Instead, we can only show that the error in unscaled calculations is directly correlated with polarity, that a uniform reduction in ionic interactions partially corrects this, and that altering the polarizability of the binding site has a large enough effect on affinities to account for the error.
Previous blind predictive studies using this force field have suggested that free energy calculations can predict binding affinities for hydrophobic and polar compounds if the proper ligand pose is sampled and calculations converge6,10. By examining charged compounds in this study, we demonstrate that the same force field can produce systematically incorrect affinities even when these criteria are met. For this strongest class of molecular interactions, the environment surrounding the binding site plays a critical role in determining affinity, and the absence of polarizability in the force field may cause the error we observe. By employing a receptor-specific charge scaling procedure, we achieved quantitative affinity prediction for most compounds, and suggested likely reasons for failure in the remaining cases. While the calculations were only partially successful, quantitative affinity prediction is an aim beyond the ambition of simpler models of binding that do not include “invisible” energetic contributions. A key challenge for free energy calculations in the future will be designing models, polarizable or not, that can transferrably represent the strength of electrostatic interactions in both buried and solvent exposed environments.
4. Methods
4.1 Free Energy Simulation Approach
Absolute binding free energies for all cationic compounds to the protein were computing using a two-step process, in which a cationic reference compound is transferred into the binding site, and then transformed into the cationic ligand. This process is described by Eq. (1):
| (1) |
where P is the unbound protein, L is the unbound ligand, PL is the protein-ligand complex, and PR is the complex between the protein and the cationic reference topology which was identical for all ligands. The net charge of the system changes when calculating ΔGP → PR, but not when calculating ΔGPR + L → PL, because both R and L are charged. Because ΔGP → PR was identical for all ligands, only ΔGPR + L → PL needed to be computed for each individual ligand, using the procedure described in SI Methods sections 5b and 5c. ΔGP → PR was computed using Eq. (2):
| (2) |
where L is one of three cationic ligands (compounds 1, 3, and 5) employed for full absolute binding calculations, ΔGP + L → PL is the absolute binding free energy of L calculated using the procedure described in SI Methods section 5a, and ΔGPL → PR + L is the free energy of moving L from the protein to solution simultaneously with creating R in the binding site, using the procedure described in SI Methods section 5b and in ref. 47. The final ΔGP → PR we employed is the average of Eq. (2) using compounds 1, 3, and 5. For neutral compounds, all absolute affinities were computed using only an absolute binding AFE cycle, described in SI Methods section 5d. All binding calculations explicitly considered multiple ligand poses, and the final predicted affinity is the sum of the contributions from all poses44. Full details on how ligand poses were generated and sampled are given in SI Methods section 2.
Alchemical free energy calculations in the receptor were performed using Hamiltonian-exchange Langevin dynamics in the NVT ensemble. Absolute binding calculations were performed using a modified version of the thermodynamic cycle in Mobley, Chodera et al.44, described in SI Methods section 5a. The confine-and-release method48 was applied to sample binding site side chains, as described in SI Methods. Free energy differences were computed using the Bennett Acceptance Ratio49, Multistate Bennett Acceptance Ratio50, and the Zwanzig equation as described in SI Methods section 3.
4.2 Software
Simulations were performed using GROMACS51 3.3.4, 4.0.7, and the FEP branch of the GROMACS 4.5 git repository52, kindly provided by Michael Shirts. Ligand preparation was performed as in ref. 10.
4.3 Force fields
The protein was modeled using the AMBER99SB force field53, solvated in TIP3P water. The Generalized Amber Force Field (GAFF) was used for ligand parameters54, with AM1-BCC partial charges55,56 generated using the OEChem toolkit, version 1.5. Heme parameters and partial charges were taken from the Hemoglobin model included with Amber 8, except with an additional +1 charge added to the iron to create Fe(III), as in ref. 57. Additional parameters were added to create the Fe-Nε bond between the Heme and His175, and the four Heme N-Fe-Nε angles, using force constants taken from ref. 57. The equilibrium bond length and angles were generated based on the average of existing crystal structures of CCP W191G.
4.4 Protein protonation
Protonation states of titratable residues were selected by running MCCE 2.258,59 on PDB structure 1KXN7 with the Heme removed at pH 4.5. When the Heme was added back in with Fe[III] and both propionates ionized, this created a model of CCP with a net charge of −5. Because the pI of CCP is between 4.9 and 5.2535, we also created a positively charged model of CCP (net charge of +9) to more accurately model the true receptor protonation state. We generated this model by protonating the highest pKa Asp and Glu residues (according to MCCE) which were not already predicted to be protonated at pH 4.5. All calculations were performed using the initial net charge −5 model before the experimental protein pI was considered. However, to avoid errors resulting from an incorrect protein net charge state, we recalculated all charge insertion free energies (SI Methods section 5a step 4) using the more appropriate net charge +9 model, and used these calculations to determine ΔGP → PR. We did not recalculate the other alchemical steps in the net charge +9 model, as these steps are unlikely to depend on the receptor net charge state because no charges are inserted or removed.
4.5 Convergence
We assessed the convergence of all free energy estimates by examining the change in the estimate with additional simulation time. To control for un-equilibrated data at the start of our trajectories, we also examined the convergence of our free energy estimates when data was added in reverse, i.e. starting with the end of all trajectories and incrementally adding data from earlier in the trajectory. Further details are given in SI Results. Convergence plots for all alchemical steps are provided in SI Dataset 1; see also SI Results Figs. S7 and S8.
4.6 Long range electrostatics
Long range electrostatics were treated using the Particle-mesh Ewald method. All free energies involving changes in net charge (both in solution and in the protein) were corrected for artifacts resulting from the artificial periodicity of the cell and non-coulombic electrostatics, as in ref. 36. Full details of these corrections are given in SI Results section 10 and SI Methods section 8. The total correction for these effects amounted to +1 kcal/mol to all binding affinities.
4.7 QM Calculations
All calculations were performed using Jaguar 7.8, release 109, accessed using Maestro 9.2.109. Ligands structures from OEChem were optimized using Hartree-Fock theory and the 6–31G** basis set. Following optimization, a single point calculation was performed using LMP2 theory and the cc-PVTZ basis set with 5 D functions. This calculation was used in Jaguar to generate ESP-fit atom-centered partial charges, based on a spherical grid. Both QM calculations used the SCF accuracy setting ‘Accurate’ within Jaguar. All calculations were performed on the ligand alone in vacuum. To determine the binding affinities of ligands using the ESP charges, the ligand partial charges were perturbed from their original AM1-BCC charges to the ESP charges, both in the protein and in solution. Only ligand poses which were originally found to be within 1 kcal/mol of the most favorable ligand pose in the original AM1-BCC simulations were considered; we did not attempt to identify new poses which might be more favorable with ESP charges.
4.8 Miniature Site Simulations
The miniature binding site contains CCP residues 175–180, 189–191, 228–230, 233, and the Heme. We selected the reference structure and force constants using a procedure described in SI Methods section 9, and verified that the model site reproduced the atomic fluctuations seen in the full protein (Figure S6 b) and the expansion and contraction of the binding site with different bound ligands (Figure S6 c). To determine binding free energies to the miniature site, we used only the ligand poses originally found to be within 1 kcal/mol of the most favorable ligand pose in the original simulations in the full complex; we did not attempt to identify new poses which might be more favorable in the miniature site. The absolute binding free energy cycle used is described in SI Methods section 5e.
4.9 Protein Preparation
The plasmid for the open cavity mutant protein was provided by the Goodin lab and protein expression and purification was performed as in ref. 16.
4.10 Titration of the UV-Vis Soret Band
Ligand stocks were made up to 1M in DMSO, solubility permitting. Titration of the heme Soret band was performed as in ref. 15. Ligand binding was measured by endpoint titration in 100 mM citric buffer at pH 4.5 or 500mM MES buffer pH 6.0. To avoid competition in ligand binding with small cations like potassium the pH of both buffer conditions was adjusted with Bis-Tris16. The compounds stocks were made in DMSO. Binding was monitored by the red shift and increase of absorbance of the heme Soret band, except for the neutral ligands where a blue shift was observed16.
4.11 Low C-value Isothermal Titration Calorimetry
Experiments were performed as in ref. 15 using a Microcal VP-ITC model calorimeter60 in 100 mM pH 4.5 citrate buffer at 10° C. Ligand stocks were prepared in buffer from overnight dialysis of the protein to prevent buffer mismatch. The protein concentration was determined by UV-Vis spectroscopy measuring the absorbance of the heme Soret band with Ext coeff412nMv= 101 M−1cm−1.
4.12 Crystallography
Ligands were soaked into crystals grown under previously published conditions at concentrations from 50mM up to 100mM in the crystallization buffer (MES pH 6.0) solubility permitting7 or 100mM acetic acid–bis-tris propane pH 4.5. If solubility was an issue, soaks were instead performed directly in the cryoprotectant, 25% MPD. Compound 3 was co-crystallized as previously described16. Multiple conformations of the engineered loop were frequently observed but in most cases electron density quality only allowed the modeling of the major conformation.
Supplementary Material
Free energy calculations are the state of the art for binding affinity prediction.
We blindly predicted affinities for charged compounds to a charged protein site.
New experiments measured 19 affinities and defined 13 ligand binding poses.
All predicted affinities were systematically too strong, based on ligand polarity.
Results show directions for improvement in modeling charges and affinity prediction.
Acknowledgements
This work supported by: the National Science Foundation (NSF) and Department of Defense pre-doctoral fellowships (to G.J.R.); National Institutes of Health (NIH) grant GM59957 (to BKS) GM096257, NSF EPSCoR Cooperative Agreement No. EPS-1003897, Louisiana Board of Regents Research Competitiveness and Research Enhancement Subprograms and additional support from the Louisiana Board of Regents (to D.L.M.); XSEDE allocation MCB100142 (to B.K.S.); and NIH grant GM063592 and GM090205 (to K.A.D.). We thank Jens Carlsson, Ryan Coleman, Chris Fennell, and Justin McCallum for suggestions on the manuscript, and Allison Doak for protein preparations. We also thank Michael Shirts for providing and supporting the FEP branch of GROMACS 4.5, and John Chodera for helpful discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mobley DL, Dill KA. Binding of small-molecule ligands to proteins: “what you see” is not always “what you get.”. Structure. 2009;17:489–498. doi: 10.1016/j.str.2009.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gilson MK, Given JA, Bush BL, McCammon JA. The statistical-thermodynamic basis for computation of binding affinities: a critical review. Biophys J. 1997;72:1047–1069. doi: 10.1016/S0006-3495(97)78756-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gumbart JC, Roux B, Chipot C. Standard Binding Free Energies from Computer Simulations: What Is the Best Strategy? J. Chem. Theory Comput. 2013;9:794–802. doi: 10.1021/ct3008099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beauchamp KA, Lin Y-S, Das R, Pande VS. Are Protein Force Fields Getting Better? A Systematic Benchmark on 524 Diverse NMR Measurements. J. Chem. Theory Comput. 2012;8:1409–1414. doi: 10.1021/ct2007814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lindorff-Larsen K, Maragakis P, Piana S, Eastwood MP, Dror RO, Shaw DE. Systematic Validation of Protein Force Fields against Experimental Data. PLoS ONE. 2012;7:e32131. doi: 10.1371/journal.pone.0032131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mobley DL, Graves AP, Chodera JD, McReynolds AC, Shoichet BK, Dill KA. Predicting absolute ligand binding free energies to a simple model site RID C-3885-2009. J. Mol. Biol. 2007;371:1118–1134. doi: 10.1016/j.jmb.2007.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rosenfeld R, Hays A, Musah R, Goodin D. Excision of a proposed electron transfer pathway in cytochrome c peroxidase and its replacement by a ligand-binding channel. Protein Sci. 2002;11:1251–1259. doi: 10.1110/ps.4870102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Banba S, Brooks CL. Free energy screening of small ligands binding to an artificial protein cavity. The Journal of Chemical Physics. 2000;113:3423–3433. [Google Scholar]
- 9.Banba S, Guo Z, Brooks CL. Efficient Sampling of Ligand Orientations and Conformations in Free Energy Calculations Using the λ-Dynamics Method. J. Phys. Chem. B. 2000;104:6903–6910. [Google Scholar]
- 10.Boyce SE, Mobley DL, Rocklin GJ, Graves AP, Dill KA, Shoichet BK. Predicting Ligand Binding Affinity with Alchemical Free Energy Methods in a Polar Model Binding Site RID C-3885-2009. J. Mol. Biol. 2009;394:747–763. doi: 10.1016/j.jmb.2009.09.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang J, Deng Y, Roux B. Absolute Binding Free Energy Calculations Using Molecular Dynamics Simulations with Restraining Potentials. Biophysical Journal. 2006;91:2798–2814. doi: 10.1529/biophysj.106.084301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gallicchio E, Lapelosa M, Levy RM. Binding Energy Distribution Analysis Method (BEDAM) for Estimation of Protein-Ligand Binding Affinities. J. Chem. Theory Comput. 2010;6:2961–2977. doi: 10.1021/ct1002913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hermans J, Wang LU. Inclusion of loss of translational and rotational freedom in theoretical estimates of free energies of binding. Application to a complex of benzene and mutant T4 lysozyme. Journal of the American Chemical Society. 1997;119:2707–2714. [Google Scholar]
- 14.Merski M, Shoichet BK. The impact of introducing a histidine into an apolar cavity site on docking and ligand recognition. J. Med. Chem. 2013;56:2874–2884. doi: 10.1021/jm301823g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Musah R, Jensen G, Bunte S, Rosenfeld R, Goodin D. Artificial protein cavities as specific ligand-binding templates: Characterization of an engineered heterocyclic cation-binding site that preserves the evolved specificity of the parent protein. J. Mol. Biol. 2002;315:845–857. doi: 10.1006/jmbi.2001.5287. [DOI] [PubMed] [Google Scholar]
- 16.Brenk R, Vetter SW, Boyce SE, Goodin DB, Shoichet BK. Probing molecular docking in a charged model binding site. J. Mol. Biol. 2006;357:1449–1470. doi: 10.1016/j.jmb.2006.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Warshel A, Sharma PK, Kato M, Parson WW. Modeling electrostatic effects in proteins. Biochimica et Biophysica Acta (BBA) - Proteins & Proteomics. 2006;1764:1647–1676. doi: 10.1016/j.bbapap.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 18.Pliego JR, Jr, Riveros JM. Gibbs energy of solvation of organic ions in aqueous and dimethyl sulfoxide solutions. Phys. Chem. Chem. Phys. 2002;4:1622–1627. [Google Scholar]
- 19.Davis A, Teague S. Hydrogen bonding, hydrophobic interactions, and failure of the rigid receptor hypothesis. Angew. Chem.-Int. Edit. 1999;38:737–749. doi: 10.1002/(SICI)1521-3773(19990315)38:6<736::AID-ANIE736>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 20.Gao J, Bosco DA, Powers ET, Kelly JW. Localized thermodynamic coupling between hydrogen bonding and microenvironment polarity substantially stabilizes proteins. Nat. Struct. Mol. Biol. 2009;16:684–690. doi: 10.1038/nsmb.1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.HyunJoong Joh N, Min A, Faham S, Whitelegge JP, Yang D, Woods VL, Bowie JU. Modest stabilization by most hydrogen-bonded side-chain interactions in membrane proteins. Nature. 2008;453:1266–1270. doi: 10.1038/nature06977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schutz CN, Warshel A. What are the dielectric “constants” of proteins and how to validate electrostatic models? Proteins. 2001;44:400–417. doi: 10.1002/prot.1106. [DOI] [PubMed] [Google Scholar]
- 23.Yang T, Wu JC, Yan C, Wang Y, Luo R, Gonzales MB, Dalby KN, Ren P. Virtual Screening Using Molecular Simulations. Proteins. 2011;79:1940–1951. doi: 10.1002/prot.23018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Simonson T, Carlsson J, Case D. Proton binding to proteins: pK(a) calculations with explicit and implicit solvent models RID A-5733-2008 RID A-8019-2010. J. Am. Chem. Soc. 2004;126:4167–4180. doi: 10.1021/ja039788m. [DOI] [PubMed] [Google Scholar]
- 25.Woo H-J, Roux B. Calculation of absolute protein-ligand binding free energy from computer simulations. Proc. Natl. Acad. Sci. U.S.A. 2005;102:6825–6830. doi: 10.1073/pnas.0409005102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiao D, Golubkov PA, Darden TA, Ren P. Calculation of protein-ligand binding free energy by using a polarizable potential RID E-5814-2011. Proc. Natl. Acad. Sci. U. S. A. 2008;105:6290–6295. doi: 10.1073/pnas.0711686105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jiao D, Zhang J, Duke RE, Li G, Schnieders MJ, Ren P. Trypsin-ligand binding free energies from explicit and implicit solvent simulations with polarizable potential. Journal of Computational Chemistry. 2009;30:1701–1711. doi: 10.1002/jcc.21268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lau AY, Roux B. The Hidden Energetics of Ligand-Binding and Activation in a Glutamate Receptor. Nat Struct Mol Biol. 2011;18:283–287. doi: 10.1038/nsmb.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Irwin JJ, Sterling T, Mysinger MM, Bolstad ES, Coleman RG. ZINC: A Free Tool to Discover Chemistry for Biology. J. Chem. Inf. Model. 2012;52:1757–1768. doi: 10.1021/ci3001277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jorgensen WL. Efficient drug lead discovery and optimization. Acc. Chem. Res. 2009;42:724–733. doi: 10.1021/ar800236t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Michel J, Essex JW. Prediction of protein–ligand binding affinity by free energy simulations: assumptions, pitfalls and expectations. J Comput Aided Mol Des. 2010;24:639–658. doi: 10.1007/s10822-010-9363-3. [DOI] [PubMed] [Google Scholar]
- 32.Chodera JD, Mobley DL, Shirts MR, Dixon RW, Branson K, Pande VS. Alchemical free energy methods for drug discovery: progress and challenges. Curr. Opin. Struct. Biol. 2011;21:150–160. doi: 10.1016/j.sbi.2011.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Deng Y, Roux B. Computation of binding free energy with molecular dynamics and grand canonical Monte Carlo simulations. The Journal of Chemical Physics. 2008;128:115103. doi: 10.1063/1.2842080. [DOI] [PubMed] [Google Scholar]
- 34.Caravella JA, Carbeck JD, Duffy DC, Whitesides GM, Tidor B. Long-Range Electrostatic Contributions to Protein-Ligand Binding Estimated Using Protein Charge Ladders, Affinity Capillary Electrophoresis, and Continuum Electrostatic Theory. J. Am. Chem. Soc. 1999;121:4340–4347. [Google Scholar]
- 35.Erman JE, Vitello LB. Yeast cytochrome c peroxidase: mechanistic studies via protein engineering. Biochimica et Biophysica Acta (BBA) - Protein Structure and Molecular Enzymology. 2002;1597:193–220. doi: 10.1016/s0167-4838(02)00317-5. [DOI] [PubMed] [Google Scholar]
- 36.Kastenholz MA, Hunenberger PH. Computation of methodology-independent ionic solvation free energies from molecular simulations. II. The hydration free energy of the sodium cation. J. Chem. Phys. 2006;124 doi: 10.1063/1.2201698. [DOI] [PubMed] [Google Scholar]
- 37.Kastenholz M, Hunenberger P. Computation of methodology-independent ionic solvation free energies from molecular simulations. I. The electrostatic potential in molecular liquids. J. Chem. Phys. 2006;124 doi: 10.1063/1.2172593. [DOI] [PubMed] [Google Scholar]
- 38.Leontyev IV, Stuchebrukhov AA. Electronic Continuum Model for Molecular Dynamics Simulations of Biological Molecules. J. Chem. Theory Comput. 2010;6:1498–1508. doi: 10.1021/ct9005807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Leontyev I, Stuchebrukhov A. Accounting for electronic polarization in non-polarizable force fields. Phys. Chem. Chem. Phys. 2011;13:2613–2626. doi: 10.1039/c0cp01971b. [DOI] [PubMed] [Google Scholar]
- 40.Youngs TGA, Hardacre C. Application of Static Charge Transfer within an Ionic-Liquid Force Field and Its Effect on Structure and Dynamics. ChemPhysChem. 2008;9:1548–1558. doi: 10.1002/cphc.200800200. [DOI] [PubMed] [Google Scholar]
- 41.Schmidt J, Krekeler C, Dommert F, Zhao Y, Berger R, Site LD, Holm C. Ionic Charge Reduction and Atomic Partial Charges from First-Principles Calculations of 1,3-Dimethylimidazolium Chloride. J. Phys. Chem. B. 2010;114:6150–6155. doi: 10.1021/jp910771q. [DOI] [PubMed] [Google Scholar]
- 42.Zhang Y, Maginn EJ. A Simple AIMD Approach to Derive Atomic Charges for Condensed Phase Simulation of Ionic Liquids. J. Phys. Chem. B. 2012;116:10036–10048. doi: 10.1021/jp3037999. [DOI] [PubMed] [Google Scholar]
- 43.Simonson T, Archontis G, Karplus M. Continuum Treatment of Long-Range Interactions in Free Energy Calculations. Application to Protein-Ligand Binding. J. Phys. Chem. B. 1997;101:8349–8362. [Google Scholar]
- 44.Mobley DL, Chodera JD, Dill KA. On the use of orientational restraints and symmetry corrections in alchemical free energy calculations RID C-3885-2009. J. Chem. Phys. 2006;125 doi: 10.1063/1.2221683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mobley DL, Bayly CI, Cooper MD, Shirts MR, Dill KA. Small Molecule Hydration Free Energies in Explicit Solvent: An Extensive Test of Fixed-Charge Atomistic Simulations RID C-3885-2009. J. Chem. Theory Comput. 2009;5:350–358. doi: 10.1021/ct800409d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mobley DL, Dumont É, Chodera JD, Dill KA. Comparison of Charge Models for Fixed-Charge Force Fields: Small-Molecule Hydration Free Energies in Explicit Solvent. J. Phys. Chem. B. 2007;111:2242–2254. doi: 10.1021/jp0667442. [DOI] [PubMed] [Google Scholar]
- 47.Rocklin GJ, Mobley DL, Dill KA. Separated topologies—A method for relative binding free energy calculations using orientational restraints. The Journal of Chemical Physics. 2013;138 doi: 10.1063/1.4792251. 085104–085104–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mobley DL, Chodera JD, Dill KA. Confine-and-release method: Obtaining correct binding free energies in the presence of protein conformational change RID C-3885-2009. J. Chem. Theory Comput. 2007;3:1231–1235. doi: 10.1021/ct700032n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bennett CH. Efficient estimation of free energy differences from Monte Carlo data. Journal of Computational Physics. 1976;22:245–268. [Google Scholar]
- 50.Shirts MR, Chodera JD. Statistically optimal analysis of samples from multiple equilibrium states. J Chem Phys. 2008;129 doi: 10.1063/1.2978177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hess B, Kutzner C, van der Spoel D, Lindahl E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 52.Pronk S, Páll S, Schulz R, Larsson P, Bjelkmar P, Apostolov R, Shirts MR, Smith JC, Kasson PM, Spoel D, van der Hess B, Lindahl E. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics. 2013 doi: 10.1093/bioinformatics/btt055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, Simmerling C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins. 2006;65:712–725. doi: 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang J, Wang W, Kollman PA, Case DA. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006;25:247–260. doi: 10.1016/j.jmgm.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 55.Jakalian A, Jack DB, Bayly CI. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J Comput Chem. 2002;23:1623–1641. doi: 10.1002/jcc.10128. [DOI] [PubMed] [Google Scholar]
- 56.Jakalian A, Bush BL, Jack DB, Bayly CI. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: I. Method. Journal of Computational Chemistry. 2000;21:132–146. doi: 10.1002/jcc.10128. [DOI] [PubMed] [Google Scholar]
- 57.Banci L, Carloni P, Savellini GG. Molecular dynamics studies on peroxidases: a structural model for horseradish peroxidase and a substrate adduct. Biochemistry. 1994;33:12356–12366. doi: 10.1021/bi00207a002. [DOI] [PubMed] [Google Scholar]
- 58.Alexov EG, Gunner MR. Incorporating protein conformational flexibility into the calculation of pH-dependent protein properties. Biophysical Journal. 1997;72:2075–2093. doi: 10.1016/S0006-3495(97)78851-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Georgescu RE, Alexov EG, Gunner MR. Combining Conformational Flexibility and Continuum Electrostatics for Calculating pKas in Proteins. Biophysical Journal. 2002;83:1731–1748. doi: 10.1016/S0006-3495(02)73940-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Plotnikov VV, Brandts JM, Lin LN, Brandts JF. A new ultrasensitive scanning calorimeter. Anal. Biochem. 1997;250:237–244. doi: 10.1006/abio.1997.2236. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.