

Abstract
Hyperactivation of the calcium-dependent cysteine protease, calpain-1 (Cal1), is implicated as a primary or secondary pathological event in a wide range of illnesses, and in neurodegenerative states, including Alzheimer’s disease (AD). E-64 is an epoxide-containing natural product identified as a potent non-selective, calpain inhibitor, with demonstrated efficacy in animal models of AD. Using E-64 as a lead, three successive generations of calpain inhibitors were developed using computationally assisted design to increase selectivity for Cal1. First generation analogs were potent inhibitors, effecting covalent modification of recombinant Cal1 catalytic domain (Cal1cat), demonstrated using LC-MS/MS. Refinement yielded 2nd generation inhibitors with improved selectivity. Further library expansion and ligand refinement gave three Cal1 inhibitors, one of which was designed as an activity-based protein profiling probe. These were determined to be irreversible and selective inhibitors by kinetic studies comparing full length Cal1 with the general cysteine protease, papain.
Keywords: calpain, epoxide, cysteine protease inhibitor, calpain inhibitors, Alzheimer’s disease, selective calpain inhibitors, peptidomimetics, epoxysuccinate
INTRODUCTION
Calpains are a class of ubiquitously expressed calcium-dependent cysteine proteases that regulate numerous intracellular signaling cascades and are fundamentally involved in regulating protein kinases responsible for cytoskeletal dynamics and remodeling.1-4 Under physiological conditions, transient and localized Ca2+ fluxes from extracellular or intracellular calcium stores result in controlled activation of local calpain populations. While regional calpain proteolytic activity is essential for native Ca2+signaling and cytoskeletal remodeling, pathological conditions may produce excessive levels of Ca2+, resulting in widespread calpain activation and unregulated proteolysis.5, 6 Calpain proteolytic activity contributes to secondary degeneration in situations of acute cellular stress following myocardial ischemia, cerebral ischemia, and traumatic brain injury.7-10 Enhanced calpain activity in platelets is a contributor to atherothromobosis and diabetes pathology.11, 12 Calpain hyperactivation associated with altered Ca2+ homeostasis, contributes to pathogenesis of: Huntington’s disease, Parkinson’s disease, cataract formation, glaucoma, multiple sclerosis, and Alzheimer’s disease (AD).5, 13-18 In this context, an appropriate selective calpain inhibitor may hold therapeutic potential in selected disease states.
Attempts to develop calpain inhibitors have been cataloged previously.19, 20 The structural nomenclature of cysteine protease substrates and inhibitors is introduced in Figure 1. The majority of reported calpain inhibitors rely upon the ability of an electrophilic reactive group, or “warhead”, to either reversibly or irreversibly modify the active site cysteine of calpain. A natural product, E-64, L-trans-epoxysuccinyl-leucylamido(4-guanidino)butane (Figure 1), was an early identified cysteine protease inhibitor, utilizing an epoxide for active site modification.21, 22 While being non-reactive towards other protease super-families (i.e. aspartic, serine, etc.), E-64 serves as a benchmark, high affinity, non-selective, irreversible calpain inhibitor.23, 24 In a transgenic model of AD, E-64 demonstrated excellent in vivo efficacy.25 Therefore, using E-64 as a lead and benchmark, the object of the present study was to maintain potency whilst increasing Cal selectivity and druggability.
Figure 1. Overview of nomenclature, mechanism, and design motif of epoxysuccinate peptidomimetic cysteine protease inhibitors.

Protease substrates are designated according to their amino acid residues extending from the scissile bond. “Unprimed” and “primed” substrate residues are designated P1, P2, etc. and P1’, P2’, etc., respectively. The design rationale is illustrated using E-64 as a lead.
CA clan cysteine proteases (i.e. papain, calpains, and lysosomal cathepsins) have similar P1-P3 substrate binding pockets, which results in a common preference for hydrophobic residues at the S2 subsite (i.e. Leu, Ile, Val, Phe, Tyr).26, 27 It has been suggested that inhibitors containing S,S epoxide stereochemistry bind preferentially into the P1-P3 pocket of CA clan proteases.28, 29 Accordingly, E-64 and related S,S epoxides containing hydrophobic residues at the P2 position have shown poor selectivity and good potency for these proteases. Recent efforts examined an array of P4-P3-P2-epoxysuccinate peptides for inhibitory activity against CA clan cysteine proteases, including: Cal1, Cal1cat (recombinant calpain-1 catalytic domain), lysosomal cysteine proteases (cathepsins, Cath).30 While these peptides do not have optimal drug-like properties, their activity profiles give valuable insight into the design and development of novel selective calpain inhibitors using peptidomimetic epoxides. Three successive generations of inhibitors were synthesized using computationally assisted design and Cal1 inhibition data. Modification of the active site cysteine was confirmed using LC-MS/MS and the relative selectivity assessed against papain using an enzyme kinetics analysis. The study presents novel, selective Cal1 inhibitors that due to the presence of the electrophilic epoxide warhead also provide an ideal activity-based protein profiling (ABPP) probe for future mechanistic investigation.
RESULTS AND DISCUSSION
Design & Synthesis
The epoxysuccinate moiety of E-64 is considered essential for potent cysteine protease inhibition. Alternatives to the epoxide warhead, such as alkene and aziridine analogs, possess weak inhibitory activity.21, 31 Despite concerns regarding potential ADMET complications stemming from incorporation of the epoxysuccinate moiety, E-64 and derivatives have been approved for clinical studies,32-34 and there are multiple reports of in vivo efficacy and safety by E-64 and related epoxysuccinate analogs in mice.35-38 Retention of the epoxysuccinate group also facilitates the design of ABPP probes to identify off-target proteins that may contribute to efficacy or toxicity. Furthermore, we observed epoxysuccinate containing peptidomimetics show negligible reactivity after 24 hr incubation in the presence of excess GSH at physiological pH and temperature (PBS, 50 mM, pH 7.4, 37 °C; [Figure 2]). A study describing the low inherent reactivity of the epoxysuccinate moiety with thiols has been reported previously.39 Given these considerations, the epoxysuccinate moiety was retained and design focused on modifying and evaluating two main portions of the peptidomimetic scaffold: 1) the P3/P4 cap group, and 2) the P2 site that is occupied by a L-leucine in E-64.
Figure 2. Reaction between an epoxysuccinate containing peptidomimetic and GSH.

HPLC-UV-Vis (λ = 280 nm) chromatogram from incubation of 31 (100 μM) with excess GSH (5 mM) in PBS (50mM, pH 7.4) at 37 °C for 24 hr. Product identity was confirmed using LC-MS/MS and comparison of retention time of authentic sample in the absence of GSH.
Synthesis was directed at generation of a library of peptidomimetic epoxides possessing either natural or non-natural peptidomimetic residues at the P2 position, designated the R1 substituent, and varying the P3/P4 cap groups, designated as R2 in Scheme 1. Initially, the R2-amines were coupled to the commercially available Boc-protected amino acids following typical peptide coupling procedures using either, HOBT and EDCI, or in the presence of CDI to give the corresponding Boc-protected peptidomimetic scaffolds (9-13). The epoxysuccinate moiety was synthesized in 3 steps starting from either L-DET or D-DET following published procedures to yield the unambiguous epoxysuccinate R,R (7) and S,S (8), respectively.40 Following TFA deprotection, the appropriate peptidomimetic scaffold was coupled to 7 or 8 using EDCI and HOBT in the presence of DIPEA to give the corresponding epoxide esters (14-19). Ester saponification using LiOH at 0 °C afforded the analogous epoxy acids 20-34 (Table 1). In four instances (28a, b and 29a, b), histidine containing analogs were found to be diastereomeric mixtures (L/D, S, S) and (L/D, R, R) resulting from racemization of the peptidyl α-carbon during the initial peptide coupling using CDI.
Scheme 1a.

Table 1.
Epoxysuccinate peptidomimetic structures and inhibition data for full length Cal1
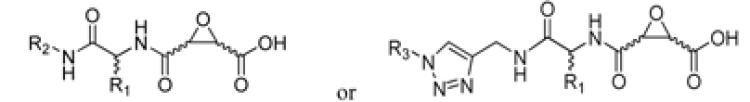
| ||||
|---|---|---|---|---|
|
| ||||
| R1 | R2 or R3 | epoxide stereochem |
Cal1 IC50a | |
|
| ||||
| E-64 | L-Leucine | R2 = Guanidinyl-1-pentyl | S,S | 100 nM |
| 20 | L-Leucine | R2= Phenyl | S,S | 100 nM |
| 21a | L-Leucine | R2= 2,6-Difluorophenyl | S,S | 100 nM |
| 21b | L-Leucine | R2= 2,6-Difluorophenyl | R,R | >1 μM |
| 22a | L-Leucine | R2 = 4-(4-Fluorophenyl)thiazol-2-amino | S,S | 50 nM |
| 22b | L-Leucine | R2 = 4-(4-Fluorophenyl)thiazol-2-amino | R,R | 250 nM |
| 23 | L-Leucine | R2 = 4-F-Phenyl-SO2-NH-(CH2)4-NH | S,S | 150 nM |
| 24 | L-Leucine | R2 = Lipoyl-NH-(CH2)4-NH | S,S | 100 nM |
| 25 | L-Leucine | R2 = D-Biotin-NH-(CH2)4-NH | S,S | 100 nM |
| 26 | L-Histidine | R2 = Phenyl | S,S | 5 μM |
| 27 | L-Histidine | R2 = 1,3,5-Trimethylanilino | S,S | 5 μM |
| 28a | L/D-Histidine | R2 = 4-(4-Fluorophenyl)thiazol-2-amino | S,S | 100 nM |
| 28b | L/D-Histidine | R2 = 4-(4-Fluorophenyl)thiazol-2-amino | R,R | >1 μMb |
| 29a | L/D-Histidine | R2 = 6-Fluorobenzo[d]thiazol-2-amino | S,S | 530 nM |
| 29b | L/D-Histidine | R2 = 6-Fluorobenzo[d]thiazol-2-amino | R,R | >1 μMb |
| 30 | L-Ala(4-thiazyl) | R2 = Phenyl | S,S | 2.5 μM |
| 31 | L-Ala(4-thiazyl) | R2 = 4-(4-Fluorophenyl)thiazol-2-amino | S,S | 100 nM |
| 32 | L-Ala(4-thiazyl) | R2 = 4-(4-Ethynylphenyl)thiazol-2-amino | S,S | 100 nM |
| 33 | L-His(2-Me) | R2 = 4-(4-Fluorophenyl)thiazol-2-amino | S,S | 5 μM |
| 34 | L-His(4-Me) | R2 = 4-(4-Fluorophenyl)thiazol-2-amino | S,S | 225 nM |
|
| ||||
| 35 | L-Ala(4-thiazyl) | R3 = Phenyl | S,S | 1 μM |
| 36 | L-Ala(4-thiazyl) | R3 = 4-Fluorophenyl | S,S | 100 nM |
| 37 | L-Ala(4-thiazyl) | R3 = 4-(Piperidin-l-ylsulfonyl)phenyl | S,S | 1.5 μM |
| 38 | L-Ala(4-thiazyl) | R3 = Benzo[d][l,3]dioxol-5-yl | S,S | 400 nM |
| 39 | L-Ala(4-thiazyl) | R3 = 4-Sulfamoylphenyl | S,S | >1 μMb |
| 40 | L-Ala(4-thiazyl) | R3 = 3,5-Bis(trifluoromethyl)phenyl | S,S | >1 μMb |
| 41 | L-Ala(4-thiazyl) | R3 = 4-Bromophenyl | S,S | 40 nM |
| 42 | L-Ala(4-thiazyl) | R3 = 4-Nitrophenyl | S,S | 300 nM |
| 43 | L-Ala(4-thiazyl) | R3 = 2,6-Bis(fiuoro)phenyl | S,S | >1 μMb |
| 44 | L-Ala(4-thiazyl) | R3 = 2,4,6-Tris(methyl)phenyl | S,S | >1 μMb |
Inhibition was measured by monitoring fluorescence emission at 480 nm after 20 min incubations with varying concentration of inhibitor in the presence of the FRET substrate: % inhibition normalized to control incubations in the absence of inhibitor. Data represents the mean ± S.D. of triplicate experiments.
Inhibition not quantifiable at1 μM.
A limitation in the synthesis of epoxide incorporating inhibitors is the propensity of the epoxide to undergo ring-opening. Hence, convergent synthesis is commonly utilized and the epoxide functionality is incorporated after derivatization has taken place, although a divergent synthetic approach is preferred for library development during lead optimization (Scheme 2).
Scheme 2.

Copper catalyzed Huisgen cycloadditions, a.k.a. “click chemistry”, has received much attention due to low cross-reactivity with other functional groups, making it the ideal tool for the generation of a library of epoxide incorporating inhibitors. Computational guidance was employed, vide infra, along with a novel click chemistry route to generate a library of triazole incorporating calpain inhibitors. The key alkynyl intermediate, 19, was functionalized with aryl-azides using a Cu[II] catalyzed Huisgen cycloaddition, with the aid of TBTA to give the corresponding triazole-aryl incorporating epoxyester peptidomimetics (35E-44E). Ester saponification yielded the desired epoxyacids (35-44) in good yield over 2 steps (85-100 %).
Calpain inhibition by 1st generation inhibitors: P3/P4 cap group selection
Calpain activity was measured using full-length human Cal1 in the presence of a FRET substrate after 20 min. IC50 values were approximated by co-incubation of varying concentrations (10-1000 nM) in the presence of the FRET substrate (DABCYL)TPLK-SPPSPR-(EDANS) and % inhibition calculated by normalizing to control experiments containing no inhibitor. A peptide inhibitor, Z-LLY-FMK, was used as a positive control.
1st generation inhibitors 20-25 (Table 1) were designed to mimic E-64 at the P2 position while varying the P3 cap group to increase druggability compared to the ionizable guanidino group of E-64. The majority of 1st generation inhibitors were found to be approximately equipotent to E-64, and one inhibitor, 22a, displayed improved potency with an IC50 value of 50 nM (± 25). These results are consistent with the understanding that hydrophobic amino acid residues at the P2 position deliver high affinity for Cal1. In two instances, 21a-b and 22a-b, the importance of the S,S epoxide stereochemistry was confirmed.28, 29
Active site modification by novel calpain inhibitors
The recombinant Cal1 catalytic domain (Cal1cat) is composed of the proteolytic domain of the full-length enzyme and has been used as a surrogate to study calpain activity.41-44 Cal1cat provides an advantage, because upon Ca2+-induced activation, full length Cal1 engages in autocatalytic, self-proteolysis complicating analysis of activity and inhibition. Cal1cat is devoid of the autocatalytic activity observed for full length Cal1. Recombinant rat Cal1cat was expressed and purified from E. coli, to examine aspects of inhibition in more detail.
Inhibition kinetics and x-ray crystallography support a mechanism of calpain inhibition by E-64, resulting from covalent modification of the active site Cys.45 LC-MS/MS supports a similar mechanism for 22a. The experimental methodology is outlined in Figure 3A. The peptide fragment containing the active site Cys (TDICQGALGDC*WLLAAIASLTNETILHR) was identified after covalent modification by IAA, 22a or E-64. A peptide fragment containing a labeled non-active site cysteine was used as an internal standard for semi-quantitative analysis of the degree of active site modification observed (Figure 3B). It is noteworthy that although Cal1cat was employed in these studies, analysis of full length Cal1 gave an identical active site peptide fragment (TIC of control digest from Cal1cat and full length Cal1 are compared in the Supporting Materials).
Figure 3. Examination of modified Cal1cat active site using LC-MS/MS.

Confirmation of covalent modification of Cal1 active site Cys by epoxysuccinate inhibitors. (A) After in gel digest and capping of free cysteines with iodoacetamide (IAA), peptide fragments were analyzed by LC-MS/MS. (B) Total ion chromatograms (TIC) for the incubation of recombinant Cal1cat (5 μM) in the absence or presence of inhibitors (10 μM). A non-active site and IAA-capped peptide fragment (Rt = 41.3 min) was used as an internal standard for EIC quantitation. (C) Active site Cys modification of Cal1cat was concentration dependent. (D) Co-incubation of Cal1cat (1 μM) with an inhibitor mixture (5 μM) showed competitive modification of the active site by 22a and E-64.
Incubations of Cal1cat (1 μM) with either E-64 or 22a showed a concentration dependent modification of the active site cysteine (Figure 3C). Co-incubation of Cal1cat with E-64 and 22a (both 5 μM) demonstrated relatively greater modification by 22a (Figure 3D). This outcome, combined with the potency of inhibition by 22a, supported retention of the 4-F-phenylthiazole P3/P4 cap group for subsequent ligand development.
Incorporation of selectivity in 2nd generation calpain inhibitors
CA clan cysteine proteases have similar “unprimed” substrate binding pockets, with a common preference for hydrophobic residues at the S2 binding site (i.e. Leu, Ile, Val, Phe). 26, 27, 29 Previous work on peptides appended with an epoxysuccinate ester warhead has indicated that a P2 histidine might confer Cal1 selectivity. The rationale given being the presence of a stabilizing hydrogen bond occurring within the S2 pocket with a highly conserved water molecule chelated by Glu-349 and Thr-210.30 A focused series of L-histidine incorporating analogs was synthesized and evaluated (26-29, 33-34) to probe the S2 pocket.
Facile protonation of the histidine imidazole ring is anticipated to potentially hinder bioavailability, therefore, thiazolyl analogues were also developed (30-32).46 Screening of the 2nd generation compounds revealed a general loss of activity on replacement of L-leucine (Table 1), although the P2 analogues, 28a and 31, were approximately equipotent to E-64 (IC50 ~ 100 nM). The crystal structures used to rationalize the selectivity and active site interactions of L-histidine at P2 contained ethyl epoxysuccinate esters, yet in our hands, simple ethyl esters of 1st and 2nd generation epoxysuccinates did not give measurable inhibition of Cal1 (data not shown).
Target selectivity is the primary goal in mechanism-based drug design, however, for AD pharmacotherapy targeting Cal1, the selection of an off-target enzyme screen needs consideration. Many other cysteine proteases, including pro-apoptotic caspases have frequently been proposed as targets for inhibition in AD, dementia, and neurodegenerative disorders.47, 48 Notably, CathB has variously been proposed as a drug target for inhibition in AD therapy.49, 50,51 The introduction of P2 histidine into epoxusuccinate Cal1 inhibitors by Cuerrier et al. led to selectivity for Cal1 over Cal2, and cathepsins, including CathB. We initially compared E-64, 22a, and the P2-histidine (28a) and thiazolyl (31) analogues, using an activity directed profiling assay, to confirm the predicted selectivity for Cal1 over CathB (Supporting Materials), however, we chose papain for the quantitative counter-screen, as a promiscuous cysteine protease, generally representative of the family of typically lysosomal or secreted cysteine proteases, including the human cathepsins (B, C, F, H, K, L1-2, O, S, W, Z).
For papain, the P1 peptide residue is cationic and P2 is hydrophobic and includes Leu, therefore papain represents a useful counter-screen for Cal1. Several methods have been reported in the literature for detailed kinetic analysis of the irreversible inhibition of cysteine proteases, including Cal1. The FRET assay used for initial screening proved to have insufficient signal over noise for detailed kinetic analysis, therefore conditions were optimized for use of the aminomethylcoumarin substrate, SucLLVYAMC. Furthermore, the truncated rat Cal1cat, is catalytically inferior to the full length enzyme,41, 42, 44 and therefore full length Cal1 was used for kinetic analysis.
Using SucLLVYAMC as substrate, kinetic parameters were derived using the method of Davies and co-workers, whereby “progress curves” were initially obtained measuring product formation as a function of time and fitted to y=P∞*(1-e(−k*x)) (Figure 4).30 Plots of P∞ versus 1/[I] gave excellent linear correlations supporting the validity of the approach (not shown). Kinetic parameters (ki, Ki, ki/Ki) were obtained from double reciprocal plots (Table 2). Using ki/Ki to assess relative selectivity for inhibition of Cal1 over papain, the data presented in Table 2 show that 22a is less selective for Cal1 than is the lead compound E-64, whereas 31, 32, and 36 all show Cal1 selectivity. Reference to Table 2 and Figure 4 clearly shows that the cause of this selectivity is not enhanced inhibition of Cal1, but attenuated inhibition of papain caused by the P2-thiazole. Thus introduction of the thiazolyl side chain leads to diminished inhibitory activity against general cysteine proteases as represented by papain, whilst maintaining efficiency for inhibition of Cal1. The P3 cap group of 36 also contributes to the higher selectivity of this derivative by reducing binding affinity to papain, but not Cal1.
Figure 4. Kinetic analysis of papain and Cal1 enzyme inhibition by selected inhibitors.

Plots of secondary and primary kinetic data for inhibition of papain (A) and Cal1 (B) used to derive detailed kinetic parameters reported in Table 2: E-64 (diamonds – solid green); 22a (closed circles – dashed red); 31 (squares – solid orange); 36 (open circles – dashed blue). The observed rate constants (kobs) shown in the double reciprocal plots were measured from non-linear fitting of “progress curves” for product formation from substrate. Exemplar progress curves are shown for Cal1 inhibition by E-64.
Table 2.
Detailed inhibition constants for Cal1 and papain and relative inhibitor selectivity.a
| Calpain1 | Papain | 102 × ki/Ki Call ki/Ki papain |
Relative selectivity
compared to E-64c |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
|
||||||||||
| Ki
(μM) |
ki
(s−1) |
ki/Kia
×l0−4 s−1M−1 |
re1b | Ki
(μM) |
ki
(s−1) |
ki/Kia
×l0−4s−1M−1 |
re1b | |||
| E-64 | 3.96 | 0.12 | 3.02±0.15 | 1 | 5.08 | 28.6 | 563 ± 52 | 1 | 0.54 | 1 |
| 22a | 6.02 | 0.16 | 2.74±0.19 | 0.91 | 1.62 | 17.1 | 1,050 ±41 | 1.9 | 0.26 | 0.5 |
| 31 | 2.59 | 0.08 | 2.96±0.14 | 0.98 | 5.31 | 14.8 | 279 ± 47 | 0.50 | 1.06 | 2.0 |
| 32 | 3.53 | 0.14 | 3.89±0.41 | 1.3 | 9.70 | 21.6 | 223 ± 2.4 | 0.40 | 1.74 | 3.3 |
| 36 | 2.89 | 0.10 | 3.31±0.32 | 1.1 | 19.2 | 12.7 | 66.2 ±7.4 | 0.12 | 5.0 | 9.3 |
Calculated inhibition rate and apparent equilibrium constants versus full length Call and papain.
ki/Ki calculated relative to E-64.
Selectivity calculated using ki/Ki relative to E-64 set at 1.0. Inhibition constants were calculated as described in the text, varying both enzyme and inhibitor concentrations. Data represent the mean ± S.D. of triplicate experiments.
Structural overlay of docking results for 22a, 28a, and 31 predicts a common binding motif, with the P3 cap group extended into the solvent exposed S4-S3 region and the P2 peptidomimetic moiety situated within the S2 pocket (Figure 5). Docking poses are compatible with the potential of 28a and 31 to interact with the conserved water molecule at the S2 site. It is important to note that incorporation of imidazolyl or thiazolyl groups did not increase affinity for the Cal1 binding site relative to the leucine-based inhibitors, as indicated by IC50 values.
Figure 5. Representative proposed binding modes of selected inhibitors.

Selected inhibitors (compounds 22a, 28a, and 31) were docked within the WR-18 x-ray structure of Cal1cat [PDB: 2NQG]. (A) Structural overlay of putative docking poses; (B) Magnified S2 pocket illustrating proposed H-bond formed between the conserved H2O molecule and the P2 moiety. Docking was carried out using GOLD docking platform. Docking poses were rendered using UCSF Chimera molecular modeling software.
In silico guidance towards P3/P4 refinement
Computer aided molecular design, based upon crystal structures of cysteine proteases modified by epoxide inhibitors, is expected to be problematic, since nucleophilic attack by the active site Cys thiol leads to ring-opening, bond-rotations, and relaxation of the resulting ligand-protein adduct. We attempted a variety of docking approaches and programs and found that docking to the X-ray models of the Cal1 that contained a co-crystallized ligand resulted in no correlation between the docking scores and the IC50 values. A common assumption for accurate prediction of binding energies of covalently linked ligands is that the binding site and the ligands should undergo minimal conformational changes.52 This is unlikely to be the case for Cal1 and the epoxide-based ligands. As depicted in Figure 6, reaction at the active site requires close interaction (d2) of the cysteine nucleophile with the electrophilic C2 of the epoxide, and probably interaction of the histidine acting as a general acid towards the ring oxygen of the epoxide. In crystal structures of Cal1 adducted with ring-opened inhibitors, gross structural relaxation leads to loss of active site interactions essential for reaction. In addition to these large structural changes expected for unbound and bound epoxide-based ligands, conformational changes to the protein upon ligand binding are expected to be pronounced based on the comparison of X-ray models of Cal1. The conformations of the gating loops43 in domain I and especially in domain II, which are both part of the binding site and directly interact with the ligands, vary quite dramatically depending on the presence and the structure of the ligand. For example, the gating loop in domain II could be considered “open” in the 2ARY (no ligand), 2G8J, 2NQG, 2NQI, 2R9C, 2R9F (with ligand) and “closed” in 1KXR (no ligand), 1TL9, 1TLO, 1ZCM, 2G8E (with ligand) X-ray structures found in the PDB.
Figure 6. Model of active site describing the bond distances and angles associated with active site modification by epoxysuccinate containing peptidomimetics.
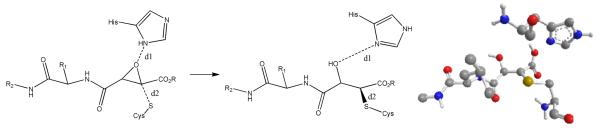
Crystal structures of epoxide inhibitors bound at the active site of Cal1 show a relaxed, ring-opened structure, whereas a transition state for epoxide ring-opening requires the Cys nucleophile and His general acid to be aligned for SN2 substitution. Triazole-based inhibitors were designed based upon docking scores biased towards d2 = ~2Å. For comparison, in the crystal structures: 2NQG d1= 4.606 and d2= 1.810; and 1TLO (shown with E-64, His, and Cys) d1= 4.775, d2= 1.833.
To make use of the structural information, we hypothesized that IC50 observed for inhibition of calpain by a covalently bound ligand is proportional to the probability of reaction that in turn is proportional to the number of protein-ligand conformations where such reaction can occur. A conceptually similar approach was successfully used for fast computational prediction of P450 drug metabolism.53-55 First, we docked a 1,2-substituted ethylene oxide portion general for all the ligands in Table 1 to 2ARY and 1KXR (both are ligand free). The “substrate” was then positioned manually with the assumption that the epoxide should adopt a conformation optimal for nucleophile attack of thiol and its likely interaction with the oxyanion hole formed by the side chain of Gln109. The placement of the non-hydrogen atoms of the oxirane ring was then used to generate RMSD with the corresponding atoms in the docked ligands. To evaluate the probability of reaction using docking, we decided to approximate it as the number of docking poses (NSig) within the RMSD threshold at or below 2 Å. We hypothesized that the higher the number of docking poses complying with this criterion, the better would be the IC50. To test this computational approach and to explore P3/P4 modifications further, a click chemistry route was used to generate a virtual library of analogs of compound 31, using a divergent synthetic strategy (Scheme 2). Thirty two synthetically accessible analogs of compound 31 were screened in silico to select ten for synthesis and assay (Scheme 3). We found that compounds with low NSig values, and therefore predicted to be poor inhibitors (39, 40, 43, and 44), yielded relatively poor IC50 values, whereas compounds with high NSig, and therefore predicted to be effective inhibitors, displayed higher activity against calpain (36, 38, 41, and 42). These promising results require further validation with a larger set of ligands, and suggest that this approach originally developed for phase I metabolic enzymes may have broader application.
Scheme 3a.

CONCLUSION
Calpain inhibition has been proposed as a promising therapeutic intervention in many disease states associated with disrupted Ca-signaling and hyperactivated Cal1, including neuronal stress leading to neurodegeneration. E-64 and simple analogs have been reported to display in vivo efficacy in murine models of muscular dystrophy and AD, however, the non-selective nature of these compounds is seen as unfavorable for clinical use. Exploiting E-64 as a lead compound, novel peptidomimetic epoxides were designed and generated as Cal1 inhibitors. Active site modification was confirmed by LC-MS/MS. The negligible reactivity towards GSH of epoxysuccinate containing peptidomimetics contrasted with the observed covalent modification of the active site cysteine of Cal1. Potency was maintained via incorporation of the correct epoxide stereochemistry and a 4-F-phenylthiazole P3 cap group (22a), allowing selectivity to be achieved by incorporating a P2 thiazolyl group (31). Diversity was achieved by developing a divergent synthetic route leading to 36. Increased selectivity for Cal1 was an objective of this study, however, several cysteine proteases, including caspases and Cath B have been proposed as targets for AD, dementia, and neurodegenerative disorders,47-51 and would not be appropriate counter screens predictive of adverse effects, therefore papain was used as a counter screen for selectivity. Retention of the electrophilic epoxide in the present work was deemed as important for the preparation of a Cal1 inhibitor (32) that could be used in future as an ABPP chemical probe for identification of on-target and off-target proteins to aid in further refinement of both drug targets and drug optimization. Several inhibitors reported herein have shown activity in AD mouse models and these results in addition to toxicity data will be presented in subsequent manuscripts.
EXPERIMENTAL
Calpain Inhibition FRET Assay
The Calbiochem InnoZyme activity kit was used for measuring the inhibition effect inhibitors on human erythrocyte calpain 1 activity. A calpain FRET substrate, (DABCYL)-TPLKSPPPSPR-(EDANS), was used to detect the activity of Cal1. 20 μM of the FRET substrate, 10 nM of native Cal1, and 20 μM TCEP (reducing agent) were added to the reaction mixture containing assay buffer (Tris [10 mM], NaCl [100 mM], pH 7.4). Cal1 was activated by the addition of 10 μM calcium. Cleavage of the scissile bond amide bond, K-S, releases the fluorophore (EDANS) from the internal quenching molecule (DABCYL), resulting in an increase in fluorescence measured at 320 nm excitation and 480 nm emission wave lengths for 20 min. Each inhibitor was added to the reaction mixture at varying concentrations (10 nM, 100 nM, 1 μM, and 10 μM) to detect inhibition of Cal1 and reduction in fluorescence was measured by 96 well-plate reader. Approximate IC50 values of each compound were generated using linear regression within Graphpad Prism Software.
LC-MS/MS examination of inhibitor modified Cal1cat active site
Expression from E. coli and purification protocols can be found in Supporting Materials. Method A refers to the data shown in Fig. 3A-B, and Method B describes the semi-quantitative approach shown in Fig.3C-D. The recombinant Cal1cat (Method A: 5 μM; Method B: 1 μM) was activated via addition of CaCl2 (10 mM) and incubated with either; Method A: E-64 or 22a (10 μM) for 30 min, or Method B: E-64 and/or 22a (1 μM, 5 μM, 10 μM) for 20 min. The competition experiments from Fig.2D were only examined at 5 μM of E-64 and 22a. The reaction was quenched with EDTA (10 μM) and reaction mixture ran on a SDS PAGE gel. The Cal1cat containing band was cut from the gel and submitted to in-gel alkylation with IAA (100 mM) for 60 min, followed by trypsin digestion. LC-MS/MS was carried out on an Agilent 6300 Ion-Trap LC/MS. HPLC separation employed a phenomenex Jupiter reverse phase HPLC column (5 micron, 150 mm, 2.00 mm); mobile phase ACN [0.1% formic acid])/water ([0.1% formic acid]). The resulting TIC and EIC were analyzed for anticipated m/z modified peptide fragments. Chromatograms are shown in Figure 3 and m/z peak assignments are included in the Supporting Materials.
Calpain and papain inhibition kinetics studies
Full length porcine calpain (156 nM), or papain (236 pM) was added to a solution of 100 mM NaCl, 50 mM HEPES, pH 7.6, 1 mM TCEP, 30 μM Suc-LLVY-AMC substrate, and inhibitor (0.5 to 50 μM). Calpain reactions also contained CaCl2 (1 mM and 100 mM for porcine and rat respectively). Both substrate and inhibitors were dissolved in acetonitrile/DMSO (1:1) with the exception of E-64, dissolved in water. Organic solvent remained < 2 % in all reactions, and most often < 1 %. Reactions were carried out in microtiter 96-well plates, with 150 μL per well, 30 °C, and product formation was monitored over time by fluorescence (Ex/Em 346/444 nm, with 420 nm cutoff filter). Kinetic values of kobs were determined via non-linear regression using one-phase association analysis and linear plots of 1/ kobs vs. 1/[I] provided kinetic constants ki and Ki. Representative primary data are supplied in Supporting Materials.
Synthesis
General Methods
Unless stated otherwise, all reactions were carried out under an atmosphere of dry argon in oven-dried glassware. Indicated reaction temperatures refer to those of the reaction bath, while room temperature (rt) is noted as 25 °C. Dichloromethane (CH2Cl2) was distilled over CaH2, and THF distilled over Na(s). All other solvents were of anhydrous quality purchased from Aldrich Chemical Co. and used as received. Pure reaction products were typically dried under high vacuum in the presence of phosphorus pentoxide. Commercially available starting materials and reagents were purchased from Aldrich, TCI and Fisher Scientific and were used as received unless specified otherwise. Analytical thin layer chromatography (TLC) was performed with (5 × 20 cm, 60 Å, 250 μm). Visualization was accomplished using a 254 nm UV lamp. 1H and 13C NMR spectra were recorded on either a Bruker Avance 400 MHz spectrometer or Bruker DPX 400 MHz spectrophotometer. Chemical shifts are reported in ppm with the solvent resonance as internal standard ([CDCl3 7.27 ppm, 77.23 ppm] [DMSO-d6 2.5 ppm, 39.51 ppm] and [MeOD-d4 4.78, 49.0] for 1H, 13C respectively). Data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, dd = doublet of doublet, t = triplet, q = quartet, br = broad, m = multiplet, abq = ab quartet), number of protons, and coupling constants. Low-resolution mass spectra (LRMS) were acquired on an Agilent 6300 Ion-Trap LC/MS. High resolution mass spectral data was collected in-house using a Shimadzu QTOF 6500. All reported compounds were fully characterized by 1H NMR, and 13C NMR. The structures of all novel final compounds were supported by HRMS. All compounds submitted for biological testing were confirmed to be > 95 % pure by analytical HPLC. Synthetic procedures, tabulated spectral data, HRMS, and purity analysis for final compounds and novel intermediates are described below. Consult the Supporting Materials for extended synthetic procedures and characterization data.
HPLC analysis of stability/reactivity
31 (100 uM) was incubated with reduced glutathione (5 mM) in PBS (50 mM, pH 7.4, [2 % DMSO v/v]) in a temperature controlled HPLC autosampler (37 °C) and reaction progress monitored via 20 uL injections every hour for 24 hours. Product identity was confirmed using LC-MS/MS and comparison of retention time of authentic sample in the absence of GSH.
Optimized procedure for the coupling of Arylamines to N-protected peptidomimetics
A round bottom was charged with the appropriate Boc-protected carboxylic acid (1.0 eq) dissolved in minimal DMF (~3 ml/mmol) under argon and maintained at 0 °C. EDCI (1.2 eq) was then added in one portion and the suspension stirred until homogenous (typically < 5 min), followed by the addition of HOBt (1.5 eq). After stirring for an additional 15 min, the amine (1.0 eq) in DMF (~2 ml/mmol) was added dropwise, and the reaction allowed to warm to r.t., monitored by TLC. In instances when the amine remained on TLC after 8 h, reaction was again brought to 0 °C, and additional EDCI (0.5 eq) was added, followed by stirring for an additional 4 h. Reaction was acidified to pH ~ 4 with 1 N HCl, extracted CH2Cl2 (3x). Combined organic extracts were washed with 1 N HCl (2x), and brine (1x), dried over Na2SO4, concentrated in vacuo, and purified by column chromatography to give the desired peptidomimetic scaffolds.
General procedure for coupling of the epoxide monoester with the peptidomimetic amine
TFA (10 eq) was added to a suspension of compound the Boc protected peptidomimetic (1 eq) in CH2Cl2 (20 ml/ 1 mmol peptidomimetic) at 0 °C and the reaction mixture stirred at the same temperature for 2 h. Excess TFA and CH2Cl2 were removed under vacuum and the residual TFA-salt was either dried under high vacuum and subjected to next reaction without further purification, or dissolved in MeOH/H2O and brought to pH of ~ 7 using a saturated NaHCO3 solution, followed by extraction with CH2Cl2 (3 × 50 ml) and removal of solvent in vacuo to afford the pure free amine, which was used without further purification. The intermediate deprotected amine (1 eq), the epoxide monoester (1 eq), and HOBt (1.1 eq) were dissolved in minimal DMF and then DIPEA (1 mL, 5.6 mmol) and EDCI (326 mg, 1.7 mmol) were added. After stirring at ambient temperature for 12 h H2O was added to the reaction mixture and diluted with 200 mL of CH2Cl2. The organic layer was separated and washed with 1 N HCl (20 mL), saturated NaHCO3 solution (10 mL), water (30 mL), brine (10 mL), and dried over anhydrous Na2SO4 and concentrated in vacuo. Chromatographic purification of the crude mixture gave the title compounds.
Optimized procedure for coupling of the epoxide monoester with the peptidomimetic amine Boc-deprotection
TFA (10 eq) was added to a suspension of the appropriate Boc-protected peptidomimetic (1 eq) in freshly distilled CH2Cl2 (10 ml/ 1 mmol peptidomimetic) at 0 °C and the reaction mixture stirred at the same temperature for 6 h. If needed, additional TFA (5 eq) was added at 0 °C every 30 min until no starting material remained on TLC. The reaction was then concentrated in vacuo and the residual TFA-salt was dissolved in MeOH/H2O and brought to pH ~ 7 using a sat. NaHCO3 solution, followed by extraction with CH2Cl2 (3 × 50 ml) and removal of solvent in vacuo to afford the pure free amine, which was dried under high vacuum and used in the next reaction without further purification.
Coupling
A round bottom was charged with the appropriate epoxide monoacid (1.0 eq) dissolved in minimal DMF (~2 ml/mmol) with DIPEA (1.1 eq) under argon and maintained at 0 °C. EDCI (1.0 eq) was then added in one portion and the suspension stirred until homogenous (typically < 5 min), followed by the addition of HOBt (1.2 eq). After stirring for an additional 15 min, the free amine (1.0 eq) in DMF (~3 ml/mmol) was added dropwise, and the reaction allowed to warm to room temperature, monitored by TLC. After 12 h, the reaction was quenched acidified to pH ~ 4 with 1 N HCl, extracted with CH2Cl2 (3x). Combined organic extracts were washed with 1 N HCl (2x), and brine (1x), dried over Na2SO4, concentrated in vacuo, and purified by column chromatography to give the desired peptidomimetic scaffolds.
General procedure for peptidomimetic-epoxide ester saponification
The peptidomimetic epoxide ester (1 eq) was dissolved in THF/MeOH/H2O (3:1:1) and cooled to 0 °C and LiOH (1 eq) was added and reaction allowed to warm to r.t. Additional LiOH (0.2 eq) was added at 0 °C every 3 h as needed until no starting material remained on TLC. The resulting solution was acidified with cold 1 N HCl and poured into CH2Cl2. In some cases the acid precipitated out and was filtered through sintered funnel and washed consecutively with CH2Cl2 and H2O and isolated. In instances when the compound stayed in solution, the aqueous phase was extracted with CH2Cl2 (3 × 50 ml), combined organic extracts dried over Na2SO4 and concentrated to give the desired product. Typically, the purity of the desired product reflects the purity of the ester starting material. It was found that having sufficiently pure ester starting material is essential to afford a pure acid upon hydrolysis.
(2S,3S)-3-((S)-4-methyl-1-oxo-1-(phenylamino)pentan-2-ylcarbamoyl)oxirane-2-carboxylic acid (20)
Followed general procedure using: the corresponding peptidomimetic epoxide ethyl ester 14a (70 mg, 0.2 mmol); LiOH (4.8 mg, 0.2 mmol); after extraction afforded the desired product as a white solid (54 mg, 83.9 %). 1H NMR (MeOD-d4, 400 MHz): δ 7.57-7.55 (d, 2H); 7.33-7.29 (t, 2H); 7.13-7.09 (t, 1H); 4.64-4.61 (m, 1H); 3.71 (s, 1H); 3.57 (s, 1H); 1.76-1.65 (m, 3H); 1.02-0.98 (t, 6H). 13C NMR (MeOD-d4, 100 MHz): 171.22, 169.07, 167.14, 137.98, 128.41, 124.12, 120.14, 52.90, 52.58, 51.69, 40.70, 29.49, 24.64, 22.04, 20.59. ESI-HRMS (m/z): [M-H+] calcd. for C16H20N2O5: 319.3404, observed: 319.1332. HPLC Method 1: Rt = 21.0 min, purity = 96.1 %.
(2S,3S)-3-((S)-1-(2,6-difluorophenylamino)-4-methyl-1-oxopentan-2-ylcarbamoyl)oxirane-2-carboxylic acid (21a)
Followed general procedure using: the corresponding peptidomimetic epoxide ethyl ester 14b (28 mg, 0.07 mmol); LiOH (1.7 mg, 0.07 mmol); after extraction afforded the desired product as a white solid (20 mg, 77.0 %). 1H NMR (MeOD-d4, 400 MHz,): δ 7.91 (s, 1H); 7.36-7.31 (m, 1H); 7.07-7.02 (t, 2H); 4.72 (m, 1H); 3.65 (s, 1H); 3.52 (s, 1H); 1.74-1.68 (m, 3H); 1.01-0.99 (d, 6H). 13C NMR (DMSO-d6, 100 MHz): 171.26, 169.20, 165.81, 159.49 (d, J = 5.2 Hz), 157.00 (d, J = 5.2 Hz), 128.98 (t, J = 19.7 Hz); 115.34 (t, J = 34.0 Hz); 113.10 (d, J = 5.2 Hz), 107.43, 106.22, 52.96, 51.51, 51.32, 41.30, 24.71, 23.81. ESI-HRMS (m/z): [M-H]+ calcd. for C16H20N2O5: 355.3216, observed: 355.3101. HPLC Method 1: Rt = 19.7 min, purity = 95.0 %.
(2R,3R)-3-((S)-1-(2,6-difluorophenylamino)-4-methyl-1-oxopentan-2-ylcarbamoyl)oxirane-2-carboxylic acid (21b)
Followed general procedure using: the corresponding peptidomimetic epoxide ethyl ester 14c (85 mg, 0.22 mmol); LiOH (5.2 mg, 0.22 mmol); after extraction afforded the desired product as a white solid (59 mg, 74.8 %). 1H NMR (DMSO-d6, 400 MHz,): δ 9.88 (s, 1H); 8.81-8.79 (d, 1H, J = 8.21 Hz); 7.38-7.33 (m, 1H); 7.17-7.13 (t, 2H); 4.61-4.56 (q, 1H); 3.70-3.69 (d, 1H, J = 1.79 Hz); 3.52-3.51 (d, 1H, J = 1.79 Hz); 1.66-1.58 (m, 3H); 0.93-0.88 (dd, 6H). 13C NMR (DMSO-d6, 100 MHz): 171.35, 169.20, 165.79, 159.48 (d, J = 5.16 Hz); 157.01 (d, J = 5.21Hz); 128.58 (t, 19.7 Hz); 114.63 (t, J = 34.0 Hz); 112.39 (d, J = 22.6 Hz); 53.05, 51.71, 51.62, 41.32, 24.71, 21.96. ESI-HRMS (m/z): [M-H+] calcd. for C16H20N2O5: 355.3216, observed: 355.3201. HPLC Method 1: Rt = 18.9 min, purity = 96.5 %.
(2S,3S)-3-((S)-1-(4-(4-fluorophenyl)thiazol-2-ylamino)-4-methyl-1-oxopentan-2-ylcarbamoyl)oxirane-2-carboxylic acid (22a)
Followed general procedure using: the corresponding peptidomimetic epoxide ethyl ester 14d (203 mg, 0.45 mmol); LiOH (10.8 mg, 0.45 mmol); after extraction afforded the desired product as a white solid (150 mg, 78.8 %). 1H NMR (DMSO-d6, 400 MHz): δ 12.51 (bs, 1H); 8.64-8.61 (d, 1H, J = 12.0 Hz); 7.95-7.92 (t, 2H); 7.62 (s, 1H); 7.28-7.24 (t, 2H); 4.63-4.58 (m, 1H); 3.51-3.50 (d, 1H, J = 1.60 Hz); 3.34-3.33 (d, 1H, J = 1.60 Hz); 165-1.54 (m, 3H); 0.91-0.88 (t, 6H). 13C NMR (MeOD-d4, 100 MHz): 172.44, 165.32, 162.87, 159.37, 150.45, 132.60, 132.57, 129.14, 129.06, 116.56, 116.34, 108.69, 54.41, 53.55, 53.19, 41.79, 26.21, 23.54, 22.02. ESI-HRMS (m/z): [M+H+] calcd. for C19H20FN3O5S: 422.1180, observed: 422.1188. HPLC Method 1: Rt = 24.6 min, purity = 95.7 %.
(2R,3R)-3-((S)-1-(4-(4-fluorophenyl)thiazol-2-ylamino)-4-methyl-1-oxopentan-2-ylcarbamoyl)oxirane-2-carboxylic acid (22b)
Followed general procedure using: the corresponding peptidomimetic epoxide ethyl ester 14e (76.4 mg, 0.16 mmol); LiOH (8 mg, 0.34 mmol); after extraction afforded the desired product as a white solid (51 mg, 71.5 %). 1H NMR (DMSO-d6, 400 MHz): δ 12.52 (s, 1H); 8.78-8.76 (d, 1H, J = 8.0 Hz); 7.94-7.92 (t, 2H); 7.63 (s, 1H); 7.29-7.24 (t, 2H); 4.58-4.57 (m, 1H); 3.62-3.61 (d, 1H, J = 1.80 Hz); 3.41-3.40 (d, 1H, J = 1.80 Hz); 1.64-1.51 (m, 3H); 0.91-0.88 (dd, 6H). 13C NMR (DMSO-d6, 100 MHz): 171.02, 166.26, 162.88, 160.46, 157.72, 147.81, 130.80, 130.77, 127.62, 127.54, 115.59 (d, J = 21.6 Hz); 107.97, 52.36, 52.08, 51.37, 28.89, 24.25, 22.82, 21.19. ESI-HRMS (m/z): [M+H+] calcd. for C19H20FN3O5S: 422.1180, observed: 422.1186. HPLC Method 1: Rt = 23.8 min, purity = 95.1 %.
(2S,3S)-3-((S)-1-(4-(4-fluorophenylsulfonamido)butylamino)-4-methyl-1-oxopentan-2-ylcarbamoyl)oxirane-2-carboxylic acid (23)
Followed general procedure using: the corresponding peptidomimetic epoxide ethyl ester 14f (213 mg, 0.42 mmol); LiOH (21 mg, 0.85 mmol); after extraction afforded the desired product as a white solid (110 mg, 54.7 %). 1H NMR (DMSO-d6, 400 MHz): δ 8.15 (s, 1H); 7.89-7.85 (m, 2H); 7.38 (s, 1H); 7.21-7.17 (t, 2H); 6.14 (s, 1H); 3.68-3.60 (d, 2H); 3.38-3.37 (d, 1H, J = 1.60 Hz); 3.10-3.09 (d, 1H, J = 1.60 Hz); 2.97 (s, 1H); 2.85 (s, 1H); 1.65-1.56 (m, 7H); 0.91-0.88 (m, 6H). 13C NMR (DMSO-d6, 100 MHz): 173.41, 170.47, 166.90, 166.52, 163.99, 135.75, 135.72, 130.03, 129.93, 116.71, 116.48, 53.83, 52.32, 43.08, 41.34, 39.65, 26.76, 26.22, 24.95, 22.94, 22.06. ESI-HRMS (m/z): [M+H+] calcd. for C20H28FN3O7S: 474.1705, observed: 474.1707. HPLC Method 1: Rt = 20.9 min, purity = 99.0 %.
(2S,3S)-3-((2S)-1-(4-(5-(1,2-dithiolan-3-yl)pentanamido)butylamino)-4-methyl-1-oxopentan-2-ylcarbamoyl)oxirane-2-carboxylic acid (24)
14g (53 mg, 0.1 mmol) was dissolved in 2 mL of 3:1:1 mixture THF, methanol and water and cooled to 0 °C and LiOH (5 mg, 0.2 mmol) was added. After stirring at the same temperature for 15 min the resulting solution was acidified with 1N HCl and extracted with CH2Cl2. The organic layer was washed with water, brine, and dried over anhydrous Na2SO4 and concentrated in vacuo. Column purification of the crude mixture (SiO2, 15 % MeOH/CHCl3) gave the title compound as a white solid. 1H NMR (CDCl3, 400 MHz): δ 8.01-7.99 (d, 1H, J = 10.8 Hz); 7.26 (s, 1H); 6.25 (bs, 1H); 4.57-4.53 (q, 1H); 3.69 (s, 1H); 3.59 (s, 1H); 3.21-3.31 (m, 5H); 2.45-2.42 (m, 1H); 2.24-2.20 (t, 2H); 1.94-1.92 (m, 1H); 1.66-1.51 (m, 12H); 1.25-1.21 (m, 2H); 0.94-0.91 (m, 6H). 13C NMR (CDCl3, 100 MHz): 174.12, 172.13, 168.92, 166.64, 56.52, 53.57, 52.57, 51.62, 41.19, 40.31, 39.09, 38.93, 38.50, 36.24, 34.63, 28.93, 26.56, 26.42, 25.55, 24.85, 22.80, 21.87. ESI-HRMS (m/z): [M+H+] calcd. for C22H37N3O6S2: 504.2197, observed: 504.2192. HPLC Method 1: Rt = 21.7 min, purity = 95.4 %.
(2S,3S)-3-((S)-4-methyl-1-oxo-1-(4-(5-((3aS,4S,6aR)-2-oxohexahydro-1H-thieno[3,4-d]imidazol-4-yl)pentanamido)butylamino)pentan-2-ylcarbamoyl)oxirane-2-carboxylic acid (25)
Synthesized following general saponification procedure using the following quantities: the corresponding peptidomimetic epoxide ethyl ester 14h (155 mg, 0.3 mmol); LiOH (14 mg, 0.58 mmol); MeOH/H2O (1.5 mL:0.5 mL: 0.5 mL); yielded 29 as a white solid (40 mg, 27.1 %). 1H NMR (DMSO-d6, 400 MHz): 8.56 (d, 1H); 8.06 (t, 1H); 7.76 (t, 1H); 6.41 (s, 1H), 6.35 (s, 1H); 4.57 (m, 2H); 4.31 (m, 1 H); 3.66 (d, 1H); 3.31 (d, 1H); 3.08 (m, 1H); 3.01 (m, 3H); 2.80 (dd, 1H); 2.56 (d, 1H); 2.04 (t, 2H); 1.53 (m, 6H); 1.43 (m, 3H); 1.36 (m, 2H); 0.89 (d, 3H); 0.84 (d, 3H).13C NMR (DMSO-d6, 100 MHz): 171.80, 171.06, 168.78, 164.85, 162.68, 61.00, 59.16, 55.36, 52.65, 51.19, 41.14, 35.17, 28.17, 27.98, 26.59, 26.45, 25.28, 24.24, 22.87, 21.61. ESI-HRMS (m/z): no ionization, mass not seen. HPLC Method 1: Rt = 14.8 min, purity = 95.6 %.
(2S,3S)-3-((S)-3-(1H-imidazol-4-yl)-1-oxo-1-(phenylamino)propan-2-ylcarbamoyl)oxirane-2-carboxylic acid (26)
Synthesized following general saponification procedure using the following quantities: the corresponding peptidomimetic epoxide ethyl ester 15a (155 mg, 0.3 mmol); LiOH (14 mg, 0.58 mmol); MeOH/H2O (1.5 mL:0.5 mL: 0.5 mL); yielded 26 as a white solid (40 mg, 27.1 %). 1H NMR (MeOD-d4, 400 MHz): δ 8.66 (s, 1H); 7.57-7.55 (d, 2H, J = 7.86 Hz ); 7.29-7.27 (t, 3H); 7.10-7.07 (t, 1H); 4.97-4.94 (q, 1H); 3.64-3.63 (d, 1H, J = 1.66 Hz); 3.529-3.525 (d, 1H, J =1.66 Hz); 3.36-3.15 (m, 2H). 13C NMR (MeOD-d4, 100 MHz): 169.79, 167.26, 166.95, 137.90, 134.99, 128.37, 124.10, 120.11, 54.13, 52.94, 51.67. ESI-HRMS (m/z): [M+H+] calcd. for C16H16N4O5: 345.1194, observed: 345.1185. HPLC Method 1: Rt = 18.2 min, purity = 95.2 %
(2S,3S)-3-((S)-3-(1H-imidazol-5-yl)-1-(mesitylamino)-1-oxopropan-2-ylcarbamoyl)oxirane-2-carboxylic acid (27)
Synthesized following general saponification procedure using the following quantities: the corresponding peptidomimetic epoxide ethyl ester 15b (51.6 mg, 0.1 mmol); LiOH (5 mg, 0.2 mmol); MeOH/H2O (1.5 mL:0.5 mL: 0.5 mL); yielded 27 as a white solid (26.2 mg, 54.2 %). 1H NMR (MeOD-d4, 400 MHz): δ 7.62 (s. 1H); 6.93 (s, 1H); 6.87 (s, 2H); 4.84-4.82 (m, 1H); 3.53-3.49 (d, 1H); 3.38-3.35 (d, 1H); 3.25-3.22 (m, 2H); 2.25 (s, 3H); 2.07 (s, 6H). 13C NMR (MeOD-d4, 100 MHz): 174.40, 172.62, 170.80, 168.89, 136.54, 135.74, 135.11, 133.43, 131.25, 128.18, 117.26, 54.20, 53.82, 53.69, 52.80, 29.34, 19.55, 16.86. ESI-HRMS (m/z): [M-H+] calcd. for C19H22N4O5: 385.1517, observed: 385.1516. HPLC Method 2: Rt = 16.4 min, purity = 95.4 %
(2S,3S)-3-(1-(4-(4-fluorophenyl)thiazol-2-ylamino)-3-(1H-imidazol-4-yl)-1-oxopropan-2-ylcarbamoyl)oxirane-2-carboxylic acid (28a)
Synthesized following general saponification procedure using the following quantities: the corresponding peptidomimetic epoxide ethyl ester 15c (184 mg, 0.39 mmol); LiOH (14 mg, 0.58 mmol); MeOH/H2O (1.5 mL:0.5 mL: 0.5 mL); yielded 28a as a white solid (40 mg, 27.1 %).1H NMR (DMSO-d6, 400 MHz): δ 12.50 (bs, 1H); 8.84-8.73 (dd, 1H, J = 7.3 Hz, 7.3 Hz); 7.95-7.91 (q, 2H); 7.73 (s, 1H); 7.63 (s, 1H); 7.29-7.24 (t, 2H); 6.90 (s, 1H); 4.81-4.74 (m, 1H); 3.65-3.64 (dd, 2H, J = 1.68 Hz, 1.68 Hz); 3.46-3.42 (dd, 2H, J = 1.65 Hz, 1.65 Hz); 3.09-3.01 (m, 2H). 13C NMR (DMSO-d6, 100 MHz): 169.81, 165.89, 163.95, 157.92, 147.96, 134.99, 132.93, 132.91, 127.89, 127.72, 115.77, 115.56, 108.17, 99.59, 52.69, 51.85, 51.77. ESI-HRMS (m/z): [M+H+] calcd. for C19H16FN5O5S: 446.0929, observed: 446.0922. HPLC method 2: Rt = 16.2 min (S-isomer) & 12.8 min (R-isomer), purity = 98.2 %. For characterization purposes, isomeric identity was determined by resynthesis with the use of HOBT in the initial peptide coupling. This led to isolation of the enantiomerically pure S-isomer, which elutes at Rt = 16.2 min, corresponding to the S-isomer in the enantiomeric mixture. Only the racemic mixture was tested for potency and selectivity.
(2R,3R)-3-((S)-1-(4-(4-fluorophenyl)thiazol-2-ylamino)-3-(1H-imidazol-4-yl)-1-oxopropan-2-ylcarbamoyl)oxirane-2-carboxylic acid (28b)
Synthesized following general saponification procedure using the following quantities: the corresponding peptidomimetic epoxide ethyl ester 15d (200 mg, 0.42 mmol); LiOH (12 mg, 0.50 mmol); THF/MeOH/H2O (2.5 mL:1.5 mL: 1.5 mL); yielded 28b as a white solid (85 mg, 45.2 %). 1H NMR (DMSO-d6, 400 MHz,): δ 12.52 (bs, 1H); 8.85-8.75 (dd, 2H, J = 6.9 Hz; J = 43.1 Hz); 7.94-7.91 (t, 2H); 7.80 (s, 1H); 7.62 (s, 1H); 7.28-7.24 (t, 2H); 6.93 (s, 1H); 4.80-4.76 (q, 1H); 3.65-3.64 (d, 1H, J = 4.40 Hz); 3.46-3.42 (d, 1H, J = 14.0 Hz); 3.09-2.96 (m, 2H). 13C NMR (DMSO-d6, 100 MHz): 169.5, 168.8, 165.9, 162.9, 160.5, 157.6, 147.8, 134.6, 132.1, 130.7, 127.6, 116.4, 115.5 (d, J = 21.4 Hz), 108.0, 52.9, 52.7, 52.6, 51.9, 28.6. ESI-HRMS (m/z): [M+H+] calcd. for C19H16FN5O5S: 446.0928, observed: 446.0939; HPLC Method 2: Rt = 15.8 min, purity = 97.2 %.
(2S,3S)-3-((S)-1-(6-fluorobenzo[d]thiazol-2-ylamino)-3-(1H-imidazol-4-yl)-1-oxopropan-2-ylcarbamoyl)oxirane-2-carboxylic acid (29a)
Synthesized following general saponification procedure using the following quantities: the corresponding peptidomimetic epoxide ethyl ester 15e (153 mg, 0.32 mmol); LiOH (7.7 mg, 0.32 mmol); THF/MeOH/H2O (3.5 mL:1.5 mL: 1.5 mL); yielded 29a as a white solid (103 mg, 71.5 %). 1H NMR (DMSO-d6, 400 MHz): δ 14.55 (bs, 1H); 14.36 (bs, 1H); 9.18-9.02 (m, 2H); 7.93-7.91 (m, 1H); 7.79-7.75 (m, 1H); 7.44 (s, 1H); 7.31-7.29 (t, 1H); 4.96-4.91 (q, 1H); 3.72-3.71 (d, 1H, J = 1.62 Hz); 3.51-3.50 (d, 1H, J = 1.62 Hz); 3.29-3.16 (m, 2H). %). 13C NMR (DMSO-d6, 100 MHz): 169.50, 168.51, 165.84, 165.84, 159.82, 157.53, 157.44, 145.02, 133.67, 132.68, 132.57, 128.56, 121.63, 117.01, 114.40, 114.16, 108.27, 108.00, 52.67, 52.19, 51.34, 26.20. ESI-HRMS (m/z): [M+H+] calcd. for C17H14FN5O5S: 420.0773, observed: 420.0777.
(2R,3R)-3-((S)-1-(6-fluorobenzo[d]thiazol-2-ylamino)-3-(1H-imidazol-4-yl)-1-oxopropan-2-ylcarbamoyl)oxirane-2-carboxylic acid (29b)
Synthesized following general saponification procedure using the following quantities: the corresponding peptidomimetic epoxide ethyl ester 15f (125 mg, 0.26 mmol); LiOH (6.2 mg, 0.26 mmol); THF/MeOH/H2O (1.5 mL:1.0 mL: 0.5 mL); yielded 29b as a white solid (46 mg, 39.1 %).1H NMR (400 MHz, MeOD-d4): δ 11.88 (bs, 1H), 8.89-8.83 (dd, 1H, J = 7.4 Hz, J = 7.4 Hz); 7.91-7.89 (d, 1H, J = 2.5 Hz); 7.78-7.72 (q, 1H); 7.56 (s, 1H); 7.30 (td, 1H, J = 2.6 Hz); 6.85 (s, 1H); 4.81-4.76 (m, 1H); 3.76-3.74 (dd, 1H, J = 1.68 Hz); 3.45-3.43 (dd, 1H, J = 1.65 Hz); 3.11-3.00 (m, 2H). 13C NMR (DMSO-d6, 100 MHz,): 171.13, 167.48, 165.83, 160.32, 158.21, 145.64, 135.40, 133.23, 122.17, 114.83, 108.74, 53.86, 53.40, 51.86. ESI-HRMS (m/z): [M+H+] calcd. for C17H14FN5O5S: 420.0773, observed: 420.0769.
(2S,3S)-3-((S)-1-oxo-1-(phenylamino)-3-(thiazol-4-yl)propan-2-ylcarbamoyl)oxirane-2-carboxylic acid (30)
Synthesized following general saponification procedure using the following quantities: the corresponding peptidomimetic epoxide ethyl ester 16a (112 mg, 0.28 mmol); LiOH (6.9 mg, 0.29 mmol); THF/MeOH/H2O (2.5 mL:1.0 mL: 1.0 mL); yielded 30 as a white solid (45 mg, 43.3 %).1H NMR (MeOD-d4, 400 MHz,): δ 8.97 (s, 1H); 7.52-7.50 (d, 2H); 7.32 (s, 1H); 7.30-7.26 (t, 3H); 4.94-4.92 (t, 1H); 3.62 (s, 1H); 3.52-3.45 (q, 2H); 3.43 (s, 1H). 13C NMR (DMSO-d6, 100 MHz): 172.48, 169.65, 168.87, 153.83, 152.41, 138.16, 128.33, 124.04, 120.73, 115.89, 54.49, 53.60, 52.89 32.87. ESI-HRMS (m/z): [M+H+]calcd. for C16H15N3O5S: 360.0660, observed: 360.0673; HPLC Method 2: Rt = 16.4 min; Purity: 95.2 %.
3-((S)-1-(4-(4-fluorophenyl)thiazol-2-ylamino)-1-oxo-3-(thiazol-4-yl)propan-2-ylcarbamoyl)oxirane-2-carboxylic acid (31)
Synthesized following general saponification procedure using the following quantities: the corresponding peptidomimetic epoxide ethyl ester 16b (974 mg, 2.0 mmol); LiOH (47.5 mg, 2.0 mmol); THF/MeOH/H2O (25 mL:5 mL: 2 mL); yielded 31 as a white solid (685 mg, 74.6 %). 1H NMR (MeOD-d4, 400 MHz,): δ 8.99-8.98(d, 1H, J = 1.67 Hz); 7.94-7.91 (q, 2H); 7.37 (s, 1H); 7.36 (s, 1H); 7.15-7.10 (t, 2H); 5.06-5.02 (q, 1H); 3.64-3.63 (d, 1H, J = 1.57 Hz); 3.49-3.45 (m, 3H). 13C NMR (MeOD-d4, 100 MHz): 170.05, 169.01, 166.19, 163.43, 158.19, 154.25, 152.66, 148.36, 131.30, 128.18, 116.63, 116.15, 115.93, 108.62, 53.12, 53.05, 52.21, 33.09. ESI-HRMS (m/z): [M+H+] calcd. for C19H15FN4O5S2: 463.0541, observed: 463.0545; HPLC Method 2: Rt = 13.7 min; purity = 96.3 %.
(2S,3S)-3-((S)-1-(4-(4-ethynylphenyl)thiazol-2-ylamino)-1-oxo-3-(thiazol-4-yl)propan-2-ylcarbamoyl)oxirane-2-carboxylic acid (32)
Synthesized following general saponification procedure using the following quantities: 16c (115 mg, 0.23 mmol); LiOH (6.7 mg, 0.28 mmol); THF/MeOH/H2O (4.5 ml:1.5 ml: 1.5 ml); yielded 32 as a white solid (48 mg, 44.2 %). 1H NMR (DMSO-d6, 400 MHz) : δ 12.57 (s, 1H); 9.06-9.05 (d, 1H, J = 1.92 Hz); 8.81-8.79 (d, 1H, J = 7.8 Hz); 7.93-7.91 (d, 2H, J = 8.4 Hz); 7.76 (s, 1H); 7.55-7.53 (d, 2H, J = 8.4 Hz); 7.45-7.44 (d, 1H, 1.8 Hz); 4.98-4.93 (q, 1H); 4.25 (s, 1H); 3.66-3.65 (d, 1H, J = 1.8 Hz); 3.42-3.41 (d, 1H, J = 1.8 Hz); 3.35-3.22 (m, 2H). 13C NMR (DMSO-d6, 100 MHz): 170.08, 169.03, 165.98, 158.25, 154.33, 152.54, 148.47, 134.95, 132.61, 126.26, 121.32, 116.75, 110.20, 83.87, 81.99, 53.15, 53.08, 51.88, 33.05. ESI-HRMS (m/z): [M+H+] calcd. for C21H16N4O5S2: 469.0635; observed, 469.0627; HPLC Method 1: Rt = 12.5 min, Purity = 95.7 %.
(2S,3S)-3-((S)-1-(4-(4-fluorophenyl)thiazol-2-ylamino)-3-(1-methyl-1H-imidazol-5-yl)-1-oxopropan-2-ylcarbamoyl)oxirane-2-carboxylic acid (33)
Synthesized following general saponification procedure using the following quantities: the corresponding peptidomimetic epoxide ethyl ester 17 (129 mg, 0.26 mmol); LiOH (6.3 mg, 0.26 mmol); THF/MeOH/H2O (2.5 mL:1.5 mL: 1.5 mL); yielded 33 as a white solid (56 mg, 46.1 %). 1H NMR (DMSO-d6, 400 MHz): 8.92-8.90 (d, 1H); 7.96-7.91 (m, 2H); 7.68 (s, 1H); 7.35-7.24 (m, 3H); 4.94-4.87 (m, 1H); 4.64-4.56 (m, 2H); 3.82-3.50 (m, 5H). 13C NMR (400 MHz, DMSO-d6): 171.52, 167.50, 165.57, 162.45, 161.02, 148.42, 138.62, 131.27, 128.18, 128.01, 127.83, 127.05, 116.17, 115.99, 106.70, 53.24, 52.58, 51.90, 31.29, 29.52. ESI-HRMS (m/z): [M+H+] calcd. for C20H18FN5O5S: 460.1085; observed, 460.1092; HPLC Method 1: Rt = 17.8 min, purity = 93.6 %.
(2S,3S)-3-((S)-1-(4-(4-fluorophenyl)thiazol-2-ylamino)-3-(1-methyl-1H-imidazol-4-yl)-1-oxopropan-2-ylcarbamoyl)oxirane-2-carboxylic acid (34)
Synthesized following general saponification procedure using the following quantities: the corresponding peptidomimetic epoxide ethyl ester 18 (93 mg, 0.19 mmol); LiOH (4.5 mg, 0.19 mmol); THF/MeOH/H2O (1.5 mL:0.5 mL: 0.5 mL); yielded 34 as a white solid (29 mg, 33.1 %). 1H NMR (400 MHz, DMSO-d6): 12.50 (bs, 1H), 8.82-8.80 (d, 1H); 7.93-7.91 (m, 2H); 7.76 (s, 1H); 7.63 (s, 1H); 7.46-7.24 (t, 2H); 6.99 (s, 1H); 4.79-4.74 (m, 1H); 3.67-3.66 (d, 1H); 3.62 (s, 3H); 3.49-3.48 (d, 1H); 3.12-2.90 (m, 2H). 13C NMR (400 MHz, DMSO-d6): 169.72, 168.60, 165.56, 162.97, 160.54, 157.78, 147.88, 137.17, 130.84, 127.71, 127.63, 118.35, 115.68, 115.47, 108.08, 53.07, 52.98, 51.56, 33.11, 29.60. ESI-HRMS (m/z): [M+H+] calcd. for C20H18FNO5S: 460.1085, observed: 460.1090. HPLC Method 1: Rt = 18.1 min, purity = 95.3 %.
Preparation of the alkynyl epoxide-ester for click derivatization, 19
19 was generated using the optimized peptide coupling procedure using: Boc-protected peptidomimetic (740 mg, 3.5 mmol); (2S, 3S)-epoxysuccinic acid monoester (564 mg, 3.52 mmol), DIPEA (1.53 mL, 8.8 mmol); EDCI (740 mg, 3.9 mmol); HOBT (520 mg, 3.9 mmol) in DMF (10 ml); afforded the title compound (960 mg, 77.6 %) as white solid.
(2S,3S)-ethyl-3-((S)-1-oxo-1-(prop-2-ynylamino)-3-(thiazol-4-yl)propan-2-ylcarbamoyl)oxirane-2-carboxylate (19)
1H NMR (CDCl3, 400 MHz): δ 8.78 (s, 1H); 7.65-7.63 (d, 1H, J = 1.00 Hz); 7.14 (s, 1H); 7.04 (bs, 1H); 4.80-4.75 (q, 1H); 4.29-4.24 (m, 2H); 3.99-3.97 (q, 2H); 3.72-3.71 (d, 1H, J = 1.6 Hz); 3.53-3.52 (d, 1H, J = 1.6 Hz); 3.34-3.15 (m, 2H); 2.19 (bs, 1H); 1.33-1.30 (t, 3H). 13C NMR (CDCl3, 100 MHz): 169.59, 166.49, 166.45, 153.22, 152.42, 116.20, 78.98, 71.59, 62.31, 53.77, 52.81, 52.44, 32.65, 29.20, 14.02.
General procedure for Huisgen cycloaddition to give the intermediate esters (35E-44E)
To the appropriate aryl-azide (1.2 eq) and 19 (1.0 eq) were dissolved in t-BuOH/EtOH/H2O (1:1:0.5). CuSO4 (0.2 eq), sodium ascorbate (0.4 eq), and a catalytic amount of TBTA (0.01 eq) were added sequentially and the reaction stirred for 12 h. The resulting precipitate was filtered off, dissolved in CH2Cl2, filtered through celite and concentrated in vacuo to afford the desired product in yields and quantities as follows.
(2S,3S)-ethyl-3-((S)-1-oxo-1-(1-phenyl-1H-1,2,3-triazol-4-yl)methylamino)-3-(thiazol-4-yl)propan-2-ylcarbamoyl)oxirane-2-carboxylate (35E)
The general click procedure was used substituting the following quantities: 19 (25.0 mg, 0.08 mmol); phenylazide (9.5 mg, 0.07 mmol); CuSO4 (2.0 mg, 0.01 mmol); NaAsc (6.0 mg, 0.03 mmol); TBTA (5.0 mg, 0.01 mmol); in t-BuOH/EtOH/H2O (2:1:0.5); afforded the 35E as a white solid (32 mg, 92.6 %). 1H NMR (CDCl3, 400 MHz): δ 8.74-7.73 (d, 1H, J = 1.90 Hz), 7.93 (s, 1H); 7.73-7.71 (d, 3H, J = 8.68 Hz); 7.55-7.52 (t, 2H); 7.47-7.39 (m, 2H); 7.07-7.06 (d, 1H, J = 1.71 Hz); 4.84-4.79 (q, 1H); 4.56-4.54 (d, 2H, J = 5.87 Hz); 4.32-4.21 (m, 2H); 3.71-3.70 (d, 1H, J = 1.78 Hz); 3.57-3.56 (d, 1H, J = 1.78 Hz); 3.39-3.18 (m, 2H); 1.33-1.31 (t, 3H). 13C NMR (CDCl3, 100 MHz): 170.06, 166.52, 166.39, 153.25, 152.36, 145.11, 136.91, 129.75, 128.81, 120.57, 120.46, 115.97, 62.27, 53.80, 52.79, 52.58, 34.98, 32.76, 14.01.
(2S,3S)-ethyl-3-((S)-1-((1-(4-fluorophenyl)-1H-1,2,3-triazol-4-yl)methylamino)-1-oxo-3-(thiazol-4-yl)propan-2-ylcarbamoyl)oxirane-2-carboxylate (36E)
The general click procedure was used substituting the following quantities: 19 (27.3 mg, 0.08 mmol); 1-azido-4-fluorobenzene (10.4 mg, 0.08 mmol); CuSO4 (2.0 mg, 0.01 mmol); NaAsc (6.0 mg, 0.03 mmol); TBTA (5.0 mg, 0.01 mmol); in t-BuOH/EtOH/H2O (2:1:0.5); afforded the 36E as a white solid (35 mg, 92.2 %). 1H NMR (CDCl3, 400 MHz): δ 8.73-8.72 (d, 1H, J = 1.91 Hz); 7.90 (s, 1H); 7.74-7.67 (m, 3H); 7.58-7.55 (t, 1H); 7.24-7.18 (t, 2H); 7.07-7.06 (d, 1H, J = 1.71 Hz); 4.84-4.79 (q, 1H); 4.54-4.50 (d, 2H, J = 5.90 Hz); 4.29-4.21 (m, 2H); 3.68-3.67 (d, 1H, J = 1.78 Hz); 3.53-3.52 (d, 1H, J = 1.78 Hz); 3.37-3.18 (m, 2H); 1.30-1.26 (t, 3H). 13C NMR (CDCl3, 100 MHz): 169.82, 166.17, 166.03, 163.28, 160.81, 152.86, 152.00, 144.94, 132.78, 122.10, 122.01, 120.48, 116.45, 116.22, 115.59, 61.91, 53.43, 52.40, 52.24, 34.55, 32.42, 13.63.
(2S,3S)-ethyl-3-((S)-1-oxo-1-((1-(4-(piperidin-1-ylsulfonyl)phenyl)-1H-1,2,3-triazol-4-yl)methylamino)-3-(thiazol-4-yl)propan-2-ylcarbamoyl)oxirane-2-carboxylate (37E)
The general click procedure was used substituting the following quantities using: 19 (28.1 mg, 0.08 mmol); 4-piperidinophenylazide (10.7 mg, 0.08 mmol); CuSO4 (3.0 mg, 0.02 mmol); NaAsc (6.0 mg, 0.03 mmol); TBTA (5.0 mg, 0.01 mmol); in t-BuOH/EtOH/H2O (2:1:0.5); afforded the 37E as a white solid (31 mg, 79.8 %). 1H NMR (CDCl3, 400 MHz):δ 8.76-8.75 (d, 1H, J = 1.84 Hz); 8.05 (s, 1H); 7.93 (s, 4H); 7.76-7.74 (d, 1H, J = 7.04 Hz); 7.38-7.36 (t, 1H); 7.10-7.09 (d, 1H, J = 1.70 Hz); 4.79-4.76 (q, 1H); 4.58-4.55 (q, 2H); 4.30-4.25 (m, 2H); 3.72-3.71 (d, 1H, J = 1.78 Hz); 3.55-3.54 (d, 1H, J = 1.78 Hz); 3.38-3.17 (abq, 2H); 3.07-304 (t, 3H); 1.75-1.60 (m, 1H); 1.31-1.26 (t, 3H). 13C NMR (CDCl3, 100 MHz): 170.31, 169.65, 166.52, 166.44, 153.30, 152.40, 152.34, 145.93, 139.64, 136.68, 129.40, 120.61, 120.41, 116.20, 116.02, 79.01, 71.57, 62.31, 53.77, 52.80, 52.45, 46.94, 32.70, 29.19, 25.11, 23.42, 14.02.
(2S,3S)-ethyl-3-((S)-1-(1-(benzo[d][1,3]dioxol-5-yl)-1H-1,2,3-triazol-4-yl)methylamino)-1-oxo-3-(thiazol-4-yl)propan-2-ylcarbamoyl)oxirane-2-carboxylate (38E)
The general click procedure was used substituting the following quantities using: 19 (25.0 mg, 0.07 mmol); 5-azidobenzo[d][1,3]dioxole (9.5 mg, 0.07 mmol); CuSO4 (2.0 mg, 0.01 mmol); NaAsc (6.0 mg, 0.03 mmol); TBTA (5.0 mg, 0.01 mmol); in t-BuOH/EtOH/H2O (2:1:0.5); afforded the 38E as a white solid (31 mg, 89.7 %). 1H NMR (CDCl3, 400 MHz): δ 8.76-8.75 (d, 1H, J = 1.86 Hz); 7.79 (s, 1H); 7.73-7.71 (d, 1H, J = 7.16 Hz); 7.24-7.21 (dd, 2H); 7.14-7.11 (dd, 1H); 7.08-7.07 (d, 1H, J = 1.63 Hz); 6.93-6.91 (d, 1H, J = 8.32 Hz); 6.09 (s, 2H); 4.79-4.76 (q, 1H); 4.54-4.53 (d, 2H, J = 5.90 Hz); 4.31-4.25 (m, 2H); 3.72-3.71 (d, 1H, J = 1.76 Hz); 3.55-3.54 (d, 1H, J = 1.76 Hz); 3.38-3.15 (ab, 2H, J = 92.8 Hz, 57.57 Hz); 1.35-1.30 (t, 3H). 13C NMR (CDCl3, 100 MHz): 169.49, 169.05, 164.22, 155.01, 153.30, 148.84, 147.92, 148.27, 130.98, 121.72, 116.26, 114.17, 109.12, 102.60, 102.33, 62.14, 52.95, 52.54, 51.90, 34.78, 33.49, 14.23.
(2S,3S)-ethyl-3-((S)-1-oxo-1-((1-(4-sulfamoylphenyl)-1H-1,2,3-triazol-4-yl)methylamino)-3-(thiazol-4-yl)propan-2-ylcarbamoyl)oxirane-2-carboxylate (39E)
The general click procedure was used substituting the following quantities using: 19 (109.0 mg, 0.31 mmol); 4-azidobenzenesulfonamide (61.5 mg, 0.31 mmol); CuSO4 (10.0 mg, 0.06 mmol); NaAsc (20.0 mg, 0.03 mmol); TBTA (10.0 mg, 0.02 mmol); in t-BuOH/EtOH/H2O (4:4:2); afforded the 39E as a white solid (119 mg, 69.8 %). 1H NMR (CDCl3, 400 MHz): δ 8.93 (s, 1H); 8.45 (s, 1H); 8.19-8.05 (q, 4H); 7.33 (s, 1H); 4.81-4.77 (t, 1H); 4.57-4.48 (q, 2H); 4.28-4.23 (q, 2H); 3.64 (s, 1H); 3.50 (s, 1H); 3.40-3.21 (m, 2H); 1.28-1.23 (t, 3H). 13C NMR (MeOD-d4, 100 MHz): 170.94, 166.80, 166.72, 153.45, 151.92, 145.70, 143.49, 138.85, 127.31, 120.74, 119.76, 115.56, 61.42, 52.72, 52.67, 51.47, 33.93, 31.91, 12.52.
(2S,3S)-ethyl-3-((S)-1-((1-(3,5-bis(trifluoromethyl)phenyl)-1H-1,2,3-triazol-4-yl)methylamino)-1-oxo-3-(thiazol-4-yl)propan-2-ylcarbamoyl)oxirane-2-carboxylate (40E)
The general click procedure was used substituting the following quantities using: 19 (93.7 mg, 0.27 mmol); 1-azido-3,5-bis(trifluoromethyl)benzene (82.5 mg, 0.32 mmol); CuSO4 (0.6 mg, 0.01 mmol); NaAsc (3.5 mg, 0.02 mmol); TBTA (15.0 mg, 0.03 mmol); in t-BuOH/EtOH/H2O (4:4:2) with an additional 3 drops of DMF; afforded the 40E as a white solid (142 mg, 87.8 %). 1H NMR (CDCl3, 400 MHz): δ 8.77 (s, 1H); 8.25 (s, 2H); 8.09 (s, 1H); 7.96 (s, 1H); 7.77-7.75 (d, 1H, J = 7.09 Hz); 7.34-7.32 (t, 1H); 7.11 (s, 1H); 4.81-4.76 (q, 1H); 4.59-4.57 (d, 2H, J = 5.90 Hz); 4.31-4.25 (m, 2H); 3.72-3.711 (d, 1H, J = 1.64 Hz); 3.55-3.54 (d, 1H, J = 1.64 Hz); 3.41-3.17 (m, 2H); 1.01-0.99 (t, 3H). 13C NMR (DMSO-d6, 100 MHz): 170.62, 167.49, 165.37, 154.06, 153.24, 146.92, 138.31, 124.57, 122.37, 121.85, 121.09, 116.21, 61.97, 53.44, 52.97, 51.74, 34.64, 33.49, 14.29.
(2S,3S)-ethyl-3-((S)-1-((1-(4-bromophenyl)-1H-1,2,3-triazol-4-yl)methylamino)-1-oxo-3-(thiazol-4-yl)propan-2-ylcarbamoyl)oxirane-2-carboxylate (41E)
The general click procedure was used substituting the following quantities using: 19 (60 mg, 0.17 mmol); 1-azido-4-bromobenzene (51 mg, 0.26 mmol); CuSO4 (4.4 mg, 0.03 mmol); NaAsc (22 mg, 0.11 mmol); TBTA (20.0 mg, 0.04 mmol); in t-BuOH/EtOH/H2O (2:2:1); afforded the 41E as a white solid (40 mg, 42.6 %). 1H NMR (DMSO-d6, 400 MHz): δ 9.00-8.99 (d, 1H, J = 1.66 Hz); 8.74-8.71 (t, 1H); 8.63-8.61 (d, 1H, J = 8.22 Hz); 8.54 (s, 1H); 7.60-7.56 (d, 2H, J = 14.85 Hz); 7.345-7.341 (d, 1H, J = 1.63 Hz); 7.11-7.07 (d, 2H, J = 14.85 Hz); 4.69-4.66 (q, 1H); 4.40-4.38 (d, 2H, J = 5.59 Hz); 4.20-4.13 (m, 2H); 3.66-3.66 (d, 1H, J = 1.73 Hz); 3.52-3.51 (d,1H, J = 1.73 Hz); 3.33-3.12 (m, 2H); 1.23-1.20 (t, 3H). 13C NMR (100 MHz, DMSO-d6): 170.57, 167.51, 165.36, 154.07, 153.27, 146.56, 139.39, 136.25, 122.30, 121.54, 117.51, 116.23, 61.97, 53.46, 52.99, 51.76, 34.75, 33.50, 14.32.
(2S,3S)-ethyl-3-((S)-1-((1-(4-nitrophenyl)-1H-1,2,3-triazol-4-yl)methylamino)-1-oxo-3-(thiazol-4-yl)propan-2-ylcarbamoyl)oxirane-2-carboxylate (42E)
The general click procedure was used substituting the following quantities using: 19 (60 mg, 0.17 mmol); 1-azido-4-nitrobenzene (42 mg, 0.26 mmol); CuSO4 (4.4 mg, 0.03 mmol); NaAsc (22 mg, 0.11 mmol); TBTA (20.0 mg, 0.04 mmol); in t-BuOH/EtOH/H2O (2:2:1); afforded the 42E as an orange solid (40 mg, 45.6 %). 1H NMR (DMSO-d6, 400 MHz): δ 9.00-8.99 (d, 1H, J = 1.8 Hz); 8.77-8.74 (t, 1H); 8.73 (s, 1H); 8.63-8.61 (d, 1H, J = 8.17 Hz); 8.47-8.44 (d, 2H, J = 9.13 Hz); 8.21-8.19 (d, 2H, J = 9.13 Hz); 7.36 (d, 1H, J = 1.78 Hz); 4.71-4.65 (q, 1H); 4.42-4.40 (d, 2H, J = 5.50 Hz); 4.20-4.09 (m, 2H); 3.66-3.65 (d, 1H, J = 1.75 Hz); 3.52-3.51 (d, 1H, J = 1.75 Hz); 3.28-3.06 (m, 2H); 1.22-1.17 (t, 3H). 13C NMR (CDCl3, 100 MHz): 170.25, 167.14, 165.00, 153.70, 152.87, 146.77, 146.68, 140.89, 125.70, 121.59, 120.42, 115.86, 61.60, 53.09, 52.61, 51.40, 34.32, 33.12, 13.94.
(2S,3S)-ethyl-3-((S)-1-((1-(2,6-difluorophenyl)-1H-1,2,3-triazol-4-yl)methylamino)-1-oxo-3-(thiazol-4-yl)propan-2-ylcarbamoyl)oxirane-2-carboxylate (43E)
The general click procedure was used substituting the following quantities using: 19 (48 mg, 0.14 mmol); 2-azido-1,3-difluorobenzene (24 mg, 0.15 mmol); CuSO4 (3.5 mg, 0.02 mmol); NaAsc (18 mg, 0.09 mmol); TBTA (12 mg, 0.01 mmol); in t-BuOH/EtOH/H2O (1:1:0.5); afforded 43E as a white solid (20 mg, 31.6 %). 1H NMR (DMSO-d6, 400 MHz): δ 8.74-8.73 (d, 1H, J = 1.79 Hz); 7.74-7.72 (m, 2H); 7.55-7.45 (m, 1H); 7.34-7.32 (m, 1H); 7.17-7.13 (t, 2H); 7.04-7.03 (d, 1H, J = 1.72 Hz); 4.82-4.80 (q, 1H); 4.57-4.56 (d, 2H, J = 5.97 Hz); 4.29-4.24 (m, 2H); 3.71-3.70 (d, 1H, J = 1.78 Hz); 3.55-3.54 (d, 1H, J = 1.78 Hz); 3.35-3.18 (m, 2H); 1.33-1.30 (t, 3H). 13C NMR (CDCl3, 100 MHz): 170.16, 166.65, 166.30, 158.01, 155.46, 153.29, 152.24, 144.36, 131.48, 129.14, 125.10, 116.01, 112.64, 62.24, 53.81, 52.74, 52.62, 34.74, 32.91, 13.99.
(2S,3S)-ethyl-3-((S)-1-((1-mesityl-1H-1,2,3-triazol-4-yl)methylamino)-1-oxo-3-(thiazol-4-yl)propan-2-ylcarbamoyl)oxirane-2-carboxylate (44E)
The general click procedure was used substituting the following quantities using: 19 (48 mg, 0.14 mmol); 2-azido-1,3-difluorobenzene (24 mg, 0.15 mmol); CuSO4 (3.5 mg, 0.02 mmol); NaAsc (18 mg, 0.09 mmol); TBTA (12 mg, 0.01 mmol); in t-BuOH/EtOH/H2O (1:1:0.5); afforded 44E as a white solid (30 mg, 42.8 %). 1H NMR (400 MHz, DMSO-d6): δ 8.79-8.74 (d, 1H, J =1.79 Hz); 7.68-7.64 (m, 2H); 7.50 (s, 1H); 7.36-7.34 (d, 2H); 7.28-7.26 (d, 2H), 7.15-7.10 (m, 2H), 6.98 (s, 1H); 5.50 (s, 1H); 4.83-4.78 (m, 1H); 4.59-4.58 (d, 1H); 4.29-4.25 (m, 2H); 3.99-3.97 (q, 1H); 3.81 (s, 3H); 3.69-3.68 (m, 3H); 3.55-3.54 (d, 1H); 3.37-3.19 (abq, 2H); 2.35 (s, 3H); 1.93 (s, 3H) 1.33-1.29 (t, 3H). 13C NMR (DMSO-d6, 100 MHz): 169.25, 165.62, 153.65, 152.98, 144.93, 140.11, 134.51, 134.50, 128.12, 124.69, 115.88, 63.51, 52.69, 52.78, 51.98, 48.66, 34.54, 33.10, 20.69, 16.89, 13.52.
(2S,3S)-3-((S)-1-oxo-1-((1-phenyl-1H-1,2,3-triazol-4-yl)methylamino)-3-(thiazol-4-yl)propan-2-ylcarbamoyl)oxirane-2-carboxylic acid (35)
Followed general procedure using: the corresponding peptidomimetic epoxide ethyl ester 35E (23 mg, 0.47 mmol); LiOH (1.2 mg, 1.5 mmol); after extraction afforded the desired product as a white solid (18 mg, 84.9 %). 1H NMR (DMSO-d6, 400 MHz): δ 9.00-8.99 (d, 1H, J = 1.76 Hz); 8.73-8.70 (t, 1H); 8.58-8.56 (d, 1H, J = 8.24 Hz); 8.50 (s, 1H); 7.87-7.85 (d, 2H, J = 7.76 Hz); 7.61-7.49 (m, 3H); 7.344-7.341 (d, 1H, J = 1.21 Hz); 4.69-4.66 (q, 1H); 4.41-4.39 (d, 2H, J = 5.49 Hz); 3.59-3.59 (d, 1H, J = 1.58 Hz); 3.35-3.10 (m, 3H). 13C NMR (DMSO-d6, 100 MHz): 170.62, 169.06, 165.75, 154.05, 153.31, 146.36, 137.07, 130.38, 129.06, 131.48, 120.41, 116.19, 53.25, 52.96, 51.90, 34.80, 29.42. HRMS-ESI: m/z [M+H+] calcd. for C18H16N6O5S: 443.1137, observed: m/z 443.1135 (M+H+);. HPLC Method 2: Rt = 16.7 min; purity = 97.9 %.
(2S,3S)-3-((S)-1-((1-(4-fluorophenyl)-1H-1,2,3-triazol-4-yl)methylamino)-1-oxo-3-(thiazol-4-yl)propan-2-ylcarbamoyl)oxirane-2-carboxylic acid (36)
Followed general procedure using: the corresponding peptidomimetic epoxide ethyl ester 36E (25 mg, 0.51 mmol); LiOH (1.3 mg, 0.06 mmol); after extraction afforded the desired product as a white solid (18 mg, 76.2 %). 1H NMR (DMSO-d6, 400 MHz): δ 8.98 (s, 1H); 8.72-8.70 (t, 1H); 8.57-8.55 (d, 1H, J = 8.06); 8.48 (s, 1H); 7.93-7.90 (q, 2H); 7.48-7.44 (t, 2H); 7.34 (s, 1H); 4.70-4.65 (q, 1H); 4.40-4.38 (d, 2H; J = 5.52); 3.66 (s, 1H); 3.54-3.06 (m, 3H). 13C NMR (DMSO-d6, 100 MHz): 170.62, 165.75, 163.24, 160.80, 154.06, 153.32, 146.40, 133.62, 122.81, 122.72, 121.76, 117.33, 117.10, 53.24, 53.01, 52.94, 34.77, 33.50. HRMS-ESI: m/z [M+H+]calculated for C19H17FN6O5S: 461.1043, observed: m/z 461.1049 (M+H+); HPLC Method 2: Rt = 17.9 min; purity = 97.2 %.
(2S,3S)-3-((S)-1-oxo-1-((1-(4-(piperidin-1-ylsulfonyl)phenyl)-1H-1,2,3-triazol-4-yl)methylamino)-3-(thiazol-4-yl)propan-2-ylcarbamoyl)oxirane-2-carboxylic acid (37)
Followed general procedure using: the corresponding peptidomimetic epoxide ethyl ester 37E (26 mg, 0.42 mmol); LiOH (1.0 mg, 0.04 mmol); after extraction afforded the desired product as a white solid (12 mg, 48.3 %). 1H NMR (Acetone-d6, 400 MHz): δ 8.98 (bs, 1H); 8.48 (s, 1H); 8.16-8.12 (d, 2H, 8.48); 8.02-8.00 (d, 2H, J = 8.48 Hz); 7.37 (bs, 1H); 4.81 (bs, 1H); 4.52 (s, 2H); 3.85-3.79 (m, 4H); 3.61 (s, 1H); 3.55 (s, 1H); 1.82-1.60 (m, 6H). 13C NMR (Acetone-d6, 100 MHz): 170.07, 169.01, 165.12, 153.52, 139.36, 136.19, 129.47, 120.84, 120.21, 107.39, 107.25, 66.62, 53.44, 52.80, 52.77, 46.83, 32.52, 30.07, 25.02, 23.13. HRMS-ESI: m/z [M+H+] calcd. for C24H27N7O7S2: 590.6440, observed: m/z 590.1050 (M+H+); HPLC Method 2: Rt = 21.7 min; purity = 96.3 %.
(2S,3S)-3-((S)-1-((1-(benzo[d][1,3]dioxol-5-yl)-1H-1,2,3-triazol-4-yl)methylamino)-1-oxo-3-(thiazol-4-yl)propan-2-ylcarbamoyl)oxirane-2-carboxylic acid (38)
Followed general procedure using: the corresponding peptidomimetic epoxide ethyl ester 38E (10 mg, 0.16 mmol); LiOH (0.35 mg, 0.02 mmol); after extraction afforded the desired product as a white solid (7 mg, 73.1 %). 1H NMR (DMSO-d6, 400 MHz): δ 9.00-8.99 (d, 1H, J = 1.88 Hz); 8.71-8.68 (t, 1H); 8.57-8.55 (d, 1H, J = 8.28 Hz); 8.38 (s, 1H); 7.46-7.45 (d, 1H, J = 2.15 Hz); 7.33-7.31 (m, 2H); 7.11 (s, 1H); 7.09 (s,1H); 6.15 (s, 2H); 4.67-4.65 (m, 1H); 4.38-4.36 (d, 2H); 3.60-3.58 (d, 1H, J = 1.72 Hz); 3.50-3.17 (m, 3H). 13C NMR (DMSO-d6, 100 MHz): 170.59, 169.06, 165.75, 154.05, 153.31, 148.65, 147.81, 146.07, 131.53, 121.69, 116.19, 114.15, 109.11, 102.58, 102.33, 53.24, 52.94, 51.90, 34.78, 33.49. HRMS-ESI: m/z [M+H+] calcd. for C20H18N6O7S: 487.4643, observed: m/z 487.1050 (M+H+); HPLC Method 2: Rt = 18.1 min; purity = 97.7 %.
(2S,3S)-3-((S)-1-oxo-1-((1-(4-sulfamoylphenyl)-1H-1,2,3-triazol-4-yl)methylamino)-3-(thiazol-4-yl)propan-2-ylcarbamoyl)oxirane-2-carboxylic acid (39)
Followed general procedure using: the corresponding peptidomimetic epoxide ethyl ester 39E (30 mg, 0.55 mmol); LiOH (1.3 mg, 0.055 mmol); product goes into aqueous layer during work up. Aqueous layer washed with CH2Cl2 and lyophilized to give the desired product as a white solid (8 mg, 45.1 %). 1H NMR (DMSO-d6, 400 MHz): δ 9.00-8.99 (d, 1H, J = 1.92 Hz); 8.79-8.76 (t, 1H); 8.70-8.68 (d, 1H, J = 8.0 Hz); 8.66 (s, 1H); 8.12-8.01 (m, 4H); 7.55 (s, 2H); 7.36 (s, 1H); 4.70-4.65 (m, 1H); 4.41-4.40 (d, 2H, J = 5.20 Hz); 3.61 (s, 1H); 3.44-3.10 (m, 3H). 13C NMR (DMSO-d6, 100 MHz): 172.25, 170.66, 169.06, 165.74, 154.10, 152.52, 138.29, 146.80, 144.19, 128.01, 121.75, 120.59, 116.29, 53.25, 52.98, 51.84, 49.01, 33.43.HRMS-ESI: m/z [M+H+] calcd. for C19H19N7O7S2: 522.0865, observed: m/z 522.0858 (M+H+); HPLC Method 2: Rt = 14.8 min; purity = 95.1 %.
(2S,3S)-3-((S)-1-((1-(3,5-bis(trifluoromethyl)phenyl)-1H-1,2,3-triazol-4-yl)methylamino)-1-oxo-3-(thiazol-4-yl)propan-2-ylcarbamoyl)oxirane-2-carboxylic acid (40)
Followed general procedure using: the corresponding peptidomimetic epoxide ethyl ester 40E (94 mg, 0.15 mmol); LiOH (3.7 mg, 0.15 mmol); after extraction afforded the desired product as a white solid (72 mg, 80.3 %). 1H NMR (DMSO-d6, 400 MHz): δ 8.99-8.98 (d, 1H, J = 1.92 Hz); 8.92 (s, 1H); 8.79-8.77 (t, 1H); 8.63 (s, 2H); 8.57-8.55 (d, 1H, J = 8.27 Hz); 8.27 (s, 1H); 7.34-7.33 (d, 1H, J = 1.85 Hz); 4.71-4.66 (m, 1H); 4.43-4.42 (d, 2H, J = 5.67 Hz); 3.58-3.57 (d, 1H, J = 1.73 Hz); 3.36-3.35 (d, 1H, J = 1.70 Hz); 3.28-3.10 (m, 2H). 13C NMR (DMSO-d6, 100 MHz): 170.67, 169.07, 165.80, 154.06, 153.29, 146.93, 138.32, 132.48, 132.14, 122.38, 121.11, 116.18, 53.22, 52.90, 51.96, 34.64, 33.51. HRMS-ESI: m/z [M+H+] calcd. for C21H16F6N6O5S: 579.4443, observed: m/z 579.0890 (M+H+) ; m/z 577.0775 (M-H+). HPLC Method 2: Rt = 24.5 min; purity = 95.3 %.
(2S,3S)-3-((S)-1-((1-(4-bromophenyl)-1H-1,2,3-triazol-4-yl)methylamino)-1-oxo-3-(thiazol-4-yl)propan-2-ylcarbamoyl)oxirane-2-carboxylic acid (41)
Followed general procedure using: the corresponding peptidomimetic epoxide ethyl ester 41E (25 mg, 0.04 mmol); LiOH (1.1 mg, 0.04 mmol); after extraction afforded the desired product as a white solid (18 mg, 75.9 %). 1H NMR (DMSO-d6, 400 MHz): δ 8.99-8.98 (d, 1H, J = 1.87 Hz); 8.74-8.71 (t, 1H); 8.58-8.56 (d, 1H, J = 8.27 Hz); 8.54 (s, 1H); 7.86-7.79 (q, 4H); 7.34-7.33 (d, 1H, J = 1.75 Hz); 4.70-4.65 (q, 1H); 4.40-4.38 (d, 2H, J = 5.59 Hz); 3.60-3.59 (d, 1H, J = 1.77 Hz); 3.38-3.37 (d, 1H, J = 1.77 Hz); 3.28-3.10 (m, 2H). 13C NMR (DMSO-d6, 100 MHz):170.62, 169.08, 165.74, 154.06, 153.29, 146.57, 136.25, 133.27, 122.31, 121.67, 121.54, 116.20, 53.25, 52.94, 51.89, 34.76, 33.49. HRMS-ESI: m/z [M+H+] calcd. for C19H17BrN6O5S: 521.34, observed: m/z 523.0229 (M+H+) ; m/z 519.0123 (M-H+); HPLC Method 2: Rt = 21.8 min; 97.2 %
(2S,3S)-3-((S)-1-((1-(4-nitrophenyl)-1H-1,2,3-triazol-4-yl)methylamino)-1-oxo-3-(thiazol-4-yl)propan-2-ylcarbamoyl)oxirane-2-carboxylic acid (42)
Followed general procedure using: the corresponding peptidomimetic epoxide ethyl ester 42E (27 mg, 0.05 mmol); LiOH (1.2 mg, 0.05 mmol); after extraction afforded the desired product as a white solid (20 mg, 78.3 %). 1H NMR (DMSO-d6, 400 MHz): δ 9.00-8.99 (d, 1H, J = 1.79 Hz); 8.74-8.71 (t, 1H); 8.59 (s, 1H); 8.48-8.47 (d, 1H, J = 8.42 Hz); 8.47-8.45 (d, 2H, J = 9.15 Hz); 8.21-8.19 (d, 2H, J = 9.15 Hz); 7.34-7.33 (d, 1H, J = 1.78 Hz); 4.69-4.66 (q, 1H); 4.42-4.41 (d, 2H, J = 5.60 Hz); 3.60-3.59 (d, 1H, J = 1.79 Hz); 3.50-3.10 (m, 3H). 13C NMR (DMSO-d6, 100 MHz):170.68, 169.07, 165.75, 154.10, 153.25, 147.11, 147.05, 141.26, 126.08, 121.95, 120.94, 116.23, 53.24, 52.95, 51.85, 34.69, 33.44. HRMS-ESI: m/z [M+H+] calcd. for C19H17N7O7S: 488.4543, observed: m/z 488.0986 (M+H+) ; m/z 486.0864 (M-H+). HPLC Method 2: Rt = 20.4 min; 93.4 %.
(2S,3S)-3-((S)-1-((1-(2,6-difluorophenyl)-1H-1,2,3-triazol-4-yl)methylamino)-1-oxo-3-(thiazol-4-yl)propan-2-ylcarbamoyl)oxirane-2-carboxylic acid (43)
Followed general procedure using: the corresponding peptidomimetic epoxide ethyl ester 43E (24 mg, 0.05 mmol); LiOH (1.1 mg, 0.05 mmol); after extraction afforded the desired product as a white solid (13 mg, 57.3 %). 1H NMR (DMSO-d6, 400 MHz): δ 9.00-8.99 (d, 1H, J = 1.90 Hz); 8.78-8.76 (t, 1H); 8.60-8.58 (d, 1H, J = 8.29 Hz); 8.27 (s, 1H); 7.77-7.69 (m, 1H); 7.49-7.45 (t, 2H); 7.36-7.27 (m, 2H); 4.71-4.65 (m, 1H); 4.44-4.42 (d, 2H, J = 5.63 Hz); 3.598-3.593 (d, 1H, J = 1.78); 3.360-3.355 (d, 1H, J = 1.78); 3.27-3.22 (m, 2H). 13C NMR (DMSO-d6, 100 MHz): 170.69, 169.07, 165.69, 158.07, 158.04, 155.56, 155.53, 154.05, 153.31, 145.67, 136.62, 129.17, 128.48, 126.17, 116.17, 53.23, , 52.93, 51.83, 34.67, 33.51. HRMS-ESI: m/z [M+H+] calcd. for C19H16F2N6O5S: 479.4343, observed: m/z 479.0960 (M+H+): HPLC Method 2: Rt = 17.32 min; 95.9 %
(2S,3S)-3-((S)-1-((1-mesityl-1H-1,2,3-triazol-4-yl)methylamino)-1-oxo-3-(thiazol-4-yl)propan-2-ylcarbamoyl)oxirane-2-carboxylic acid (44)
Followed general procedure using: the corresponding peptidomimetic epoxide ethyl ester 44E (24 mg, 0.05 mmol); LiOH (1.1 mg, 0.05 mmol); after extraction afforded the desired product as a white solid (15 mg, 66.1 %).1H NMR (DMSO-d6, 400 MHz): δ 8.98-8.97 (d, 1H, J = 1.87 Hz); 8.72-8.70 (t, 1H, J = 11.2 Hz); 8.55-8.53 (d, 1H, J = 8.23 Hz); 7.93 (s, 1H); 7.34 (s, 1H); 7.08 (s, 1H); 4.68-4.63 (m, 1H); 4.42-4.40 (d, 2H, J = 5.50 Hz); 3.61-3.50 (m, 4H); 2.32 (s, 3H); 1.86 (s, 6H). 13C NMR (DMSO-d6, 100 MHz):170.29, 165.60, 153.65, 152.98, 144.93, 139.46, 134.51, 133.50, 128.90, 124.69, 115.78, 52.79, 52.58, 48.66, 34.54, 33.10, 20.69, 16.89. HRMS-ESI: m/z [M+H+] calcd. for C22H24N6O5S: 485.5343, observed: m/z 485.1622 (M+H+): m/z 483.1600 (M-H+); HPLC Method 2: Rt = 23.6 min; 97.4 %.
Modeling
Coordinates of calpain protein structures were downloaded from the protein data bank (PDB).56 Visual inspection of the crystal structures 1KXR and 2ARY was performed using Chimera.57 Conserved water molecules near the binding site were included and allowed to be toggled on and off during docking. Ligands and the binding sites were prepared in MOE.58 GOLD59 was used for docking/scoring, employing default parameters and settings.
Supplementary Material
ACKNOWLEDGMENT
The work was supported by NIH grant: U01 AG028713. Molecular modeling was in part conducted using free academic license for the UCSF Chimera package from the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIH Grant P41 RR-01081).
ABBREVIATIONS
- AD
Alzheimer’s disease
- ADMET
absorption, distribution, metabolism, excretion, toxicity
- Cal1
Calpain
- Cal1cat
calpain 1 recombinant catalytic domain
- Cath
cathepsin
- CathB
Cathepsin B
- DET
diethyl tartrate
- EIC
extracted ion chromatogram
- EDCI
1-ethyl-3-(3-dimethylaminopropyl)carbodiimide)
- GSH
glutathione
- HOBT
hydroxybenzotriazole
- IAA
iodoacetamide
- S/N
signal-to-noise
- TIC
total ion chromatogram
- TBTA
Tris-[(1-benzyl-1H-1,2,3-triazol-4-yl) methyl]amine
Footnotes
Supporting Information Available: Further details of synthesis, characterization, protein expression; LC-MS; and selectivity assay. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Goll DE, Thompson VF, Li H, Wei WEI, Cong J. The Calpain System. Phys. Rev. 2003;83:731–801. doi: 10.1152/physrev.00029.2002. [DOI] [PubMed] [Google Scholar]
- 2.Perlmutter LS, Siman R, Gall C, Seubert P, Baudry M, Lynch G. The ultrastructural localization of calcium-activated protease "calpain" in rat brain. Synapse. 1988;2:79–88. doi: 10.1002/syn.890020111. [DOI] [PubMed] [Google Scholar]
- 3.Veeranna, Kaji T, Boland B, Odrljin T, Mohan P, Basavarajappa BS, Peterhoff C, Cataldo A, Rudnicki A, Amin N, Li BS, Pant HC, Hungund BL, Arancio O, Nixon RA. Calpain mediates calcium-induced activation of the erk1,2 MAPK pathway and cytoskeletal phosphorylation in neurons: relevance to Alzheimer's disease. Am. J. Pathol. 2004;165:795–805. doi: 10.1016/S0002-9440(10)63342-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shea TB. Restriction of microM-calcium-requiring calpain activation to the plasma membrane in human neuroblastoma cells: evidence for regionalized influence of a calpain activator protein. J. Neurosci. Res. 1997;48:543–550. [PubMed] [Google Scholar]
- 5.Di Rosa G, Odrijin T, Nixon RA, Arancio O. Calpain inhibitors: a treatment for Alzheimer's disease. J. Mol. Neurosci. 2002;19:135–141. doi: 10.1007/s12031-002-0024-4. [DOI] [PubMed] [Google Scholar]
- 6.Huang Y, Wang KKW. The calpain family and human disease. Trends in Mol. Med. 2001;7:355–362. doi: 10.1016/s1471-4914(01)02049-4. [DOI] [PubMed] [Google Scholar]
- 7.Saatman K, Creed J, Raghupathi R. Calpain as a therapeutic target in traumatic brain injury. Neurotherapeutics. 2010;7:31–42. doi: 10.1016/j.nurt.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hong SC, Goto Y, Lanzino G, Soleau S, Kassell NF, Lee KS. Neuroprotection with a calpain inhibitor in a model of focal cerebral ischemia. Stroke. 1994;25:663–669. doi: 10.1161/01.str.25.3.663. [DOI] [PubMed] [Google Scholar]
- 9.Yamashima T. Ca2+-dependent proteases in ischemic neuronal death: A conserved ‘calpain–cathepsin cascade’ from nematodes to primates. Cell Calcium. 2004;36:285–293. doi: 10.1016/j.ceca.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 10.Pike BR, Flint J, Dave JR, Lu XCM, Wang KKK, Tortella FC, Hayes RL. Accumulation of Calpain and Caspase-3 Proteolytic Fragments of Brain-Derived αII-Spectrin in Cerebral Spinal Fluid After Middle Cerebral Artery Occlusion in Rats. J. Cereb. Blood Flow Metab. 2004;24:98–106. doi: 10.1097/01.WCB.0000098520.11962.37. [DOI] [PubMed] [Google Scholar]
- 11.Randriamboavonjy V, Fleming I. All cut up! The consequences of calpain activation on platelet function. Vascul. Pharmacol. 2012;56:210–215. doi: 10.1016/j.vph.2012.02.009. [DOI] [PubMed] [Google Scholar]
- 12.Randriamboavonjy V, Isaak J, Elgheznawy A, Pistrosch F, Fromel T, Yin X, Badenhoop K, Heide H, Mayr M, Fleming I. Calpain inhibition stabilizes the platelet proteome and reactivity in diabetes. Blood. 2012;120:415–423. doi: 10.1182/blood-2011-12-399980. [DOI] [PubMed] [Google Scholar]
- 13.Shields DC, Schaecher KE, Saido TC, Banik NL. A putative mechanism of demyelination in multiple sclerosis by a proteolytic enzyme, calpain. Proc. Natl. Acad. Sci. USA. 1999;96:11486–11491. doi: 10.1073/pnas.96.20.11486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dufty BM, Warner LR, Hou ST, Jiang SX, Gomez-Isla T, Leenhouts KM, Oxford JT, Feany MB, Masliah E, Rohn TT. Calpain-Cleavage of α-Synuclein: Connecting Proteolytic Processing to Disease-Linked Aggregation. Am. J. Path. 2007;170:1725–1738. doi: 10.2353/ajpath.2007.061232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vosler P, Brennan C, Chen J. Calpain-Mediated Signaling Mechanisms in Neuronal Injury and Neurodegeneration. Mol. Neurobiol. 2008;38:78–100. doi: 10.1007/s12035-008-8036-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Robertson LJG, Morton JD, Yamaguchi M, Bickerstaffe R, Shearer TR, Azuma M. Calpain May Contribute to Hereditary Cataract Formation in Sheep. Invest. Ophth. Vis. Sci. 2005;46:4634–4640. doi: 10.1167/iovs.04-1291. [DOI] [PubMed] [Google Scholar]
- 17.Robertson LJG, Nakajima E, Morton JD, David LL, Shearer TR, Azuma M. Calpain-induced proteolysis in lens epithelial cell death during ovine inherited cataract. Invest. Ophthalmol. Vis. Sci. 2004;45:2653. [Google Scholar]
- 18.Govindarajan B, Laird J, Sherman R, Salomon RG, Bhattacharya SK. Neuroprotection in Glaucoma Using Calpain-1 Inhibitors: Regional Differences in Calpain-1 Activity in the Trabecular Meshwork, Optic Nerve and Implications for Therapeutics. CNS Neuro. Dis. 2008;7:295–304. doi: 10.2174/187152708784936644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Markus Pietsch KCHC. Calpains: Attractive Targets for the Development of Synthetic Inhibitors. Curr. Top. Med. Chem. 2010;10:270–293. doi: 10.2174/156802610790725489. A. D. A. [DOI] [PubMed] [Google Scholar]
- 20.Donkor IO. Calpain inhibitors: a survey of compounds reported in the patent and scientific literature. Expert Opinion on Therapeutic Patents. 2011;21:601–636. doi: 10.1517/13543776.2011.568480. [DOI] [PubMed] [Google Scholar]
- 21.Parkes C, Kembhavi AA, Barrett AJ. Calpain inhibition by peptide epoxides. Biochem. J. 1985;230:509–516. doi: 10.1042/bj2300509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sugita H, Ishiura S, Suzuki K, Imahori K. Inhibition of Epoxide Derivatives on Chicken Calcium-Activated Neutral Protease (CANP) In Vitro and In Vivo. J. Biochem. 1980;87:339–341. doi: 10.1093/oxfordjournals.jbchem.a132742. [DOI] [PubMed] [Google Scholar]
- 23.Hanada K, Yamagishi M, Ohmura S, Sawada J, Tanaka I. Isolation and Characterization of E-64, a New Thiol Protease Inhibitor. Agri. Biol. Chem. 1978;42:523–528. M. T. [Google Scholar]
- 24.Barrett AJ, Kembhavi AA, Brown MA, Kirschke H, Knight CG, Tamai M, Hanada K. L-trans-Epoxysuccinyl-leucylamido(4-guanidino)butane (E-64) and its analogues as inhibitors of cysteine proteinases including cathepsins B, H and L. Biochem. J. 1982;201:189–198. doi: 10.1042/bj2010189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Trinchese F, Fa M, Liu S, Zhang H, Hidalgo A, Schmidt SD, Yamaguchi H, Yoshii N, Mathews PM, Nixon RA, Arancio O. Inhibition of calpains improves memory and synaptic transmission in a mouse model of Alzheimer disease. J. Clin. Invest. 2008;118:2796–2807. doi: 10.1172/JCI34254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Greenbaum D, Medzihradszky KF, Burlingame A, Bogyo M. Epoxide electrophiles as activity-dependent cysteine protease profiling and discovery tools. Chem. Biol. 2000;7:569–581. doi: 10.1016/s1074-5521(00)00014-4. [DOI] [PubMed] [Google Scholar]
- 27.Greenbaum DC, Arnold WD, Lu F, Hayrapetian L, Baruch A, Krumrine J, Toba S, Chehade K, Brömme D, Kuntz ID, Bogyo M. Small Molecule Affinity Fingerprinting: a Tool for Enzyme Family Subclassification, Target Identification, and Inhibitor Design. Chem. Biol. 2002;9:1085–1094. doi: 10.1016/s1074-5521(02)00238-7. [DOI] [PubMed] [Google Scholar]
- 28.Mladenovic M, Ansorg K, Fink RF, Thiel W, Schirmeister T, Engels B. Atomistic Insights into the Inhibition of Cysteine Proteases: First QM/MM Calculations Clarifying the Stereoselectivity of Epoxide-Based Inhibitors. J. Phys. Chem. B. 2008;112:11798–11808. doi: 10.1021/jp803895f. [DOI] [PubMed] [Google Scholar]
- 29.Otto HH, Schirmeister T. Cysteine Proteases and Their Inhibitors. Chem. Rev. 1997;97:133–172. doi: 10.1021/cr950025u. [DOI] [PubMed] [Google Scholar]
- 30.Cuerrier D, Moldoveanu T, Campbell RL, Kelly J, Yoruk B, Verhelst SH, Greenbaum D, Bogyo M, Davies PL. Development of calpain-specific inactivators by screening of positional scanning epoxide libraries. J. Biol. Chem. 2007;282:9600–9611. doi: 10.1074/jbc.M610372200. [DOI] [PubMed] [Google Scholar]
- 31.Schirmeister T. New Peptidic Cysteine Protease Inhibitors Derived from the Electrophilic α-Amino Acid Aziridine-2,3-dicarboxylic Acid. J. Med. Chem. 1999;42:560–572. doi: 10.1021/jm981061z. [DOI] [PubMed] [Google Scholar]
- 32.Miyahara T, Shimojo S, Toyohara K, Imai T, Miyajima M, Honda H, Kamegai M, Ohzeki M, Kokatsu J. Phase I study of EST, a new thiol protease inhibitor—the 2nd report safety and pharmacokinetics in continuous administration. Rinsho Yakuri. 1985;16:537–546. [Google Scholar]
- 33.Satoyashi E. Therapeutic trials on progressive muscular dystrophy. Intern. Med. 1992;31:841–846. doi: 10.2169/internalmedicine.31.841. [DOI] [PubMed] [Google Scholar]
- 34.Scrip. 1992. p. 1765.
- 35.Tamai M, Omura S, Koyama I, Ozawa Y, Hanada K. In vitro and in vivo inhibition of cysteine proteases by EST, a new analog of E-64. J. Pharmacobio-dyn. 1986;9:672–677. doi: 10.1248/bpb1978.9.672. K. M. [DOI] [PubMed] [Google Scholar]
- 36.Towatari T, Nikawa T, Murata M, Yokoo C, Tamai M, Hanada K, Katunuma N. Novel epoxysuccinyl peptides. A selective inhibitor of cathepsin B, in vivo. FEBS Lett. 1991;280:311–315. doi: 10.1016/0014-5793(91)80319-x. [DOI] [PubMed] [Google Scholar]
- 37.Hook G, Hook VY, Kindy M. Cysteine protease inhibitors reduce brain beta-amyloid and beta-secretase activity in vivo and are potential Alzheimer's disease therapeutics. Biol. Chem. 2007;388:979–983. doi: 10.1515/BC.2007.117. [DOI] [PubMed] [Google Scholar]
- 38.Masaharu Tamai SO, Masaaki Kimura, Kazunori Hanada. Hideo Sugita Prolongation of Life Span of Dystrophic Hamster by Cysteine Proteinase Inhibitor, Loxistatin (EST) J. Pharmacobio-Dyn. 1987;10:678–681. doi: 10.1248/bpb1978.10.678. [DOI] [PubMed] [Google Scholar]
- 39.Bihovsky R. Reactions of .alpha.,.beta.-epoxy carbonyl compounds with methanethiolate: regioselectivity and rate. J. Org. Chem. 1992;57:1029–1031. [Google Scholar]
- 40.Saito S, Komada K, Moriwake T. Dietyhl (2S,2R)-2(N-tert-butoxycarbonyl)amino-3-hydroxysuccinate. Org. Syn. 1996;73:184. [Google Scholar]
- 41.Moldoveanu T, Hosfield CM, Lim D, Elce JS, Jia Z, Davies PL. A Ca2+ Switch Aligns the Active Site of Calpain. Cell. 2002;108:649–660. doi: 10.1016/s0092-8674(02)00659-1. [DOI] [PubMed] [Google Scholar]
- 42.Moldoveanu T, Campbell RL, Cuerrier D, Davies PL. Crystal Structures of Calpain-E64 and -Leupeptin Inhibitor Complexes Reveal Mobile Loops Gating the Active Site. J Mol Biol. 2004;343:1313–1326. doi: 10.1016/j.jmb.2004.09.016. [DOI] [PubMed] [Google Scholar]
- 43.Cuerrier D, Moldoveanu T, Inoue J, Davies PL, Campbell RL. Calpain inhibition by alpha-ketoamide and cyclic hemiacetal inhibitors revealed by X-ray crystallography. Biochemistry. 2006;45:7446–7452. doi: 10.1021/bi060425j. [DOI] [PubMed] [Google Scholar]
- 44.Cuerrier D, Moldoveanu T, Davies PL. Determination of Peptide Substrate Specificity for μ-Calpain by a Peptide Library-based Approach. J. Biol. Chem. 2005;280:40632–40641. doi: 10.1074/jbc.M506870200. [DOI] [PubMed] [Google Scholar]
- 45.Suzuki K, Hayashi H, Hayashi T, Iwai K. Amino acid sequence around the active site cysteine residue of calcium-activated neutral protease (CANP) FEBS Lett. 1983;152:67–70. doi: 10.1016/0014-5793(83)80483-9. [DOI] [PubMed] [Google Scholar]
- 46.Baumann M, Baxendale IR, Ley SV, Nikbin N. An overview of the key routes to the best selling 5-membered ring heterocyclic pharmaceuticals. Beilstein Journal of Organic Chemistry. 7:442–495. doi: 10.3762/bjoc.7.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tamayev R, Akpan N, Arancio O, Troy CM. Caspase-9 mediates synaptic plasticity and memory deficits of Danish dementia knock-in mice: caspase-9 inhibition provides therapeutic protection. Mol. Neurodegener. 2012;7:60. doi: 10.1186/1750-1326-7-60. L, D. A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.D'Amelio M, Cavallucci V, Middei S, Marchetti C, Pacioni S, Ferri A, Diamantini A, De Zio D, Carrara P, Battistini L, Moreno S, Bacci A, Ammassari-Teule M, Marie H, Cecconi F. Caspase-3 triggers early synaptic dysfunction in a mouse model of Alzheimer's disease. Nat. Neurosci. 2011;14:69–76. doi: 10.1038/nn.2709. [DOI] [PubMed] [Google Scholar]
- 49.Hook VY, Kindy M, Hook G. Inhibitors of cathepsin B improve memory and reduce beta-amyloid in transgenic Alzheimer disease mice expressing the wild-type, but not the Swedish mutant, beta-secretase site of the amyloid precursor protein. J. Biol. Chem. 2008;283:7745–7753. doi: 10.1074/jbc.M708362200. [DOI] [PubMed] [Google Scholar]
- 50.Hook G, Hook V, Kindy M. The cysteine protease inhibitor, E64d, reduces brain amyloid-beta and improves memory deficits in Alzheimer's disease animal models by inhibiting cathepsin B, but not BACE1, beta-secretase activity. J. Alzheimers Dis. 2011;26:387–408. doi: 10.3233/JAD-2011-110101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang C, Sun B, Zhou Y, Grubb A, Gan L. Cathepsin B degrades amyloid-beta in mice expressing wild-type human amyloid precursor protein. J. Biol. Chem. 2012;287:39834–39841. doi: 10.1074/jbc.M112.371641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Huo S, Wang J, Cieplak P, Kollman PA, Kuntz ID. Molecular dynamics and free energy analyses of cathepsin D-inhibitor interactions: insight into structure-based ligand design. J. Med. Chem. 2002;45:1412–1419. doi: 10.1021/jm010338j. [DOI] [PubMed] [Google Scholar]
- 53.Danielson ML, Desai PV, Mohutsky MA, Wrighton SA, Lill MA. Potentially increasing the metabolic stability of drug candidates via computational site of metabolism prediction by CYP2C9: The utility of incorporating protein flexibility via an ensemble of structures. Eur. J. Med. Chem. 2011;46:3953–3963. doi: 10.1016/j.ejmech.2011.05.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rydberg P, Vasanthanathan P, Oostenbrink C, Olsen L. Fast prediction of cytochrome P450 mediated drug metabolism. ChemMedChem. 2009;4:2070–2079. doi: 10.1002/cmdc.200900363. [DOI] [PubMed] [Google Scholar]
- 55.Moors SL, Vos AM, Cummings MD, Van Vlijmen H, Ceulemans A. Structure-based site of metabolism prediction for cytochrome P450 2D6. J. Med. Chem. 2011;54:6098–6105. doi: 10.1021/jm2006468. [DOI] [PubMed] [Google Scholar]
- 56.Kirchmair J, Markt P, Distinto S, Schuster D, Spitzer GM, Liedl KR, Langer T, Wolber G. The Protein Data Bank (PDB), Its Related Services and Software Tools as Key Components for In Silico Guided Drug Discovery. J. Med. Chem. 2008;51:7021–7040. doi: 10.1021/jm8005977. [DOI] [PubMed] [Google Scholar]
- 57.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comp. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 58.Molecular Operating Environment. http://www.chemcomp.com.
- 59.Jones G, Willett P, Glen RC. Molecular Recognition of Receptor-Sites Using a Genetic Algorithm with a Description of Desolvation. J. Mol. Biol. 1995;245:43–53. doi: 10.1016/s0022-2836(95)80037-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.