

SUMMARY
Translational control of gene expression has recently been recognized as an important mechanism controlling cell proliferation and oncogenesis and it mainly occurs in the initiation step of protein synthesis that involves multiple eukaryotic initiation factors (eIFs). Many eIFs have been found to have aberrant expression in human tumors and the aberrant expression may contribute to oncogenesis. However, how these previously considered house-keeping proteins are potentially oncogenic remains elusive. In this study, we investigated the expression of eIF3i in human colon cancers, tested its contribution to colon oncogenesis, and determined the mechanism of eIF3i action in colon oncogenesis. We found that eIF3i expression was up-regulated in both human colon adenocarcinoma and adenoma polyps as well as in model inducible colon tumorigenic cell lines. Over-expression of ectopic eIF3i in intestinal epithelial cells causes oncogenesis by directly up-regulating synthesis of COX-2 protein and activates the β-catenin/TCF4 signaling pathway that mediates the oncogenic function of eIF3i. Together, we conclude that eIF3i is a proto-oncogene that drives colon oncogenesis by translationally up-regulating COX-2 and activating β-catenin signaling pathway. These findings imply that protooncogenic eIFs likely exert their tumorigenic function by regulating/altering the synthesis level of down-stream tumor suppressor or oncogenes.
Keywords: eIF3i, COX-2, β-catenin, translational control, colon cancer, RNA-binding
INTRODUCTION
Translational control is one of the primary regulations of gene expression and it mainly takes place in the initiation step of protein synthesis, which in eukaryotes involves more than ten eukaryotic initiation factor (eIF) complexes 1. Many of these eIFs, in particular eIF4E, have been shown to regulate protein synthesis, participate in signal transduction, and associate with human cancers 2.
eIF3, the largest eIF complex consisting of 13 putative subunits (eIF3a-eIF3m) is an important factor in maintaining free 40S ribosomes and in forming the 43S pre-initiation complex 3-5. Several eIF3 subunits have been found to have altered expression in human cancers and possibly associate with the disease while some of them have been shown to have oncogenic properties. However, how a eIF3 subunit contributes to oncogenesis remains elusive.
In this study, we took eIF3i as a model protein and investigated its mechanism of action in colon oncogenesis. In both human colon adenocarcinoma and adenoma polyps, eIF3i expression is up-regulated. It is also up-regulated in inducible colon tumorigenic cell lines. Over-expression of ectopic eIF3i causes colon oncogenesis as determined in vitro and in a xenograft animal model. Further analyses revealed that eIF3i over-expression directly up-regulates synthesis of functionally active COX-2, which, in turn, activates β-catenin/TCF4 signaling pathway that mediates the oncogenic function of eIF3i. Together, we conclude that eIF3i is a proto-oncogene that drives colon oncogenesis by up-regulating COX-2 synthesis and activating the β-catenin signaling pathway.
RESULTS
Association of eIF3i expression with colon cancers
To determine the expression status of eIF3i in colon cancer, we collected and performed Western blot analysis of 22 matched pairs of fresh frozen human colon normal and cancer tissues. As shown in Fig. 1A, 16 of the 22 cancer tissues (~73%) have increased eIF3i level than their respective matched normal tissues. This increase is statistically significant (Fig. 1B). Fig. 1C shows immunofluorescence staining of a representative pair of human colon normal and cancer tissues, confirming the elevated eIF3i expression in cancer tissues. eIF3i expression also appears to be increased in benign adenoma polyps (supplemental Fig. S1). We also tested the expression of the known eIF3i-binding proteins, eIF3b and eIF3g 6, in these tissues. While eIF3b expression remained the same between normal and cancer tissues, the expression of eIF3g is decreased in most cancer tissues (data not shown). Thus, the eIF3i-binding proteins, eIF3b and eIF3g, do not increase in concert with eIF3i in human colon cancers.
Figure 1. Increased expression of eIF3i in human colon cancer tissues.

A. Western blot analysis of eIF3i in matched normal (N) and cancer (C) human colon tissues. GAPDH was used as a loading control. B. Scatter plots of relative eIF3i level in normal and cancer colon tissues as determined from panel A. The relative mean eIF3i level is shown by the horizontal bar. C. Immunofluorescence staining of eIF3i in a pair of representative normal and cancer human colon tissues.
We next examined the expression pattern of eIF3i in two rat intestinal epithelial cell lines, IEC-iK Ras and RIE-iH Ras, which can be transformed by inducing expression of the mutant Ras using IPTG. As expected, both cell lines have significant increases in foci formation (Fig. 2A), proliferation (Fig. 2B), and colony formation (Fig. 2C) following induction of Ras expression (Fig. 2D). Interestingly, eIF3i expression is also increased in these cells following Ras induction (Fig. 2D), suggesting that eIF3i expression also associates with inducible cell transformations and it may be a downstream target gene of Ras.
Figure 2. Association of eIF3i up-regulation with inducible transformation of rat intestinal epithelial RIE-iH Ras and IEC-iK Ras cells.

RIE-iH Ras and IEC-iK Ras cells were treated with or without IPTG for 72 hrs followed by analysis of foci formation (A), proliferation (B), anchorage-independent growth (C), and eIF3i and Ras expression (D). Actin was used as a loading control for Western blot. (n=3; *p<0.05; **p<0.01).
We further examined the association of eIF3i expression with colon cancer using CaCo-2 cell line that can differentiate into intestine like cells and lose cancer phenotype upon confluency as indicated by expression of brush-border enzymes such as alkaline phosphatase (AP), a differentiation marker of colon epithelial cells 7-10. We first determined eIF3i expression in response to confluency using Western blot. Fig. 3A-B show that eIF3i expression decreases when cells reach confluency and begin to differentiate as indicated by the increased AP activity (Fig. 3C), Together, these findings suggest that eIF3i expression positively associates with colon cancer phenotype.
Figure 3. eIF3i expression and differentiation of CaCo-2 cells.

A-C. eIF3i expression and confluency-induced CaCo-2 cell differentiation. CaCo-2 cells were harvested at different times followed by Western blot analysis of eIF3i expression (A), quantitation of eIF3i level in panel A using Image J software (B), and analysis of alkaline phosphatase (AP) activity (C). Panel A shows a representative experiment and panels B and C show summary of three independent experiments. GAPDH was used as a loading control and for normalization during quantification. D-E. Effect of eIF3i expression on CaCo-2 cell differentiation. Pre- (Pre-C) and post-confluent (Post-C) cells were transfected with eIF3i cDNA (eIF3i) and siRNA (Si) to enforce and reduce eIF3i expression, respectively, followed by analysis of eIF3i level (D) and alkaline phosphatase activity (E). Vector (Vec)- and scrambled siRNA (Scr)-transfected cells were used as controls. Untransfected pre- and post-confluent cells were also analyzed as controls. (n=3; *p<0.05; **p<0.01).
Contribution of eIF3i over-expression to colon oncogenesis
The finding that eIF3i expression is up-regulated in colon benign polyps suggests that eIF3i up-regulation may occur before oncogenesis. To determine if eIF3i over-expression possibly contributes to colon oncogenesis, we first examined if reducing eIF3i expression in CaCo-2 cells would reduce oncogenic property and contributes to confluency-induced differentiation. For this purpose, CaCo-2 cells were allowed to grow to confluency and then transiently transfected with eIF3i for ectopic over-expression (Fig. 3D) followed by analysis of AP. Fig. 3D-3E show that ectopic over-expression of eIF3i suppressed confluency-induced differentiation as indicated by the reduced AP activity, suggesting that eIF3i over-expression inhibits confluency-induced-loss of cancer phenotype of CaCo-2 cells. Conversely, eIF3i knockdown in pre-confluent cells effectively induced differentiation as indicated by increased AP activity (Fig. 3D-3E), suggesting that eIF3i knockdown can potentially reverse the cancer phenotype of CaCo-2 cells.
To further determine the role of eIF3i in colon oncogenesis, we used the normal rat intestinal epithelial cells (IEC) and determined if eIF3i over-expression could transform these cells. This cell was chosen because colon cancer arises from intestinal epithelial cells and the rat IEC cell is well characterized and widely used for oncogenic research. Two stable clones with eIF3i over-expression (eIF3i-1 and eIF3i-2) along with a vector-transfected control clone (Vec) were established (Fig. 4A) and tested for their proliferation rate, colony formation efficiency, and anchorage-independent growth. Fig. 4B shows that both eIF3i-1 and eIF3i-2 stable clones proliferate much faster than the Vec control cells. Both eIF3i-1 and eIF3i-2 clones also have significant increases in colony formation efficiency (Fig. 4C) and in anchorage-independent growth in both colony size and numbers (Fig. 4D). Together, these observations suggest that ectopic eIF3i over-expression can effectively transform normal IEC cells.
Figure 4. Contribution of eIF3i to colon oncogenesis.

A-D. Characterization of eIF3i-1 and eIF3i-2 stable clones. The stable eIF3i-1 and eIF3i-2 clones along with the vector-transfected control cells (Vec) were analyzed for their expression of eIF3i (A), proliferation rate (B), colony-formation efficiency (C), and anchorage-independent growth (D). (n=3), **p<0.01). E. Growth of eIF3i-1 and eIF3i-2 xenograft tumors. Equal numbers of the stable eIF3i-1 and eIF3i-2 clones as well as the Vec control cells were injected subcutaneously into NOD/SCID mice and the growth of xenograft tumors were monitored weekly (E). F. Final dissected tumors at 9 weeks after implantation with histology of representative eIF3i-1 and eIF3i-2 xenograft tumor sections. G. Western blot analysis of eIF3i in xenograft tumors. Lysates from the tumors were separated by SDS-PAGE followed by Western blot analysis with lysate from Vec cells as a control.
We next examined if these two stable IEC clones could form xenograft tumors. Equal numbers of eIF3i-1, eIF3i-2, and control Vec cells were injected subcutaneously into different groups of NOD/SCID mice and followed by observation for 9 weeks. As shown in Fig. 4E, tumors were formed and palpable 2 weeks after implantation and grow exponentially from eIF3i-1 and eIF3i-2 clones in all animals (3 mice for each clone). The average size of these tumors reached about 250 mm3 at 9 weeks after implantation. The average weight of these tumors at 9 weeks is 0.16±0.09 g. H&E staining of paraffin-embedded sections of xenograft tumors showed typical Adenocarcinoma characteristics (Fig. 4F). Xenograft tumors continue to express high level of eIF3i (Fig. 4G). However, no tumor was formed from any of the 3 animals that were injected with the Vec control cells. Taken together, we conclude that eIF3i over-expression contributes to colon oncogenesis.
eIF3i activates β-catenin/TCF4 signaling
The finding that eIF3i over-expression increases cell proliferation indicates that eIF3i may affect cell cycle progression. To test this possibility, we analyzed cell cycle distributions and found that eIF3i-1 and eIF3i-2 clones have increased S (~47%) and decreased G0/G1 (~41%) phase populations compared to the Vec control cells which have 35% S and 51% G0/G1 phase cells (Fig. 5A). This observation is consistent with the increased proliferation of eIF3i-1 and eIF3i-2 stable cells (Fig. 4B). To verify that the increase in S phase population was due to eIF3i over-expression, we knocked down eIF3i level in both eIF3i-1 and eIF3i-2 clones using siRNA (supplemental Fig. S2A) and tested its effect on S phase population. Fig. S2B shows that the S phase population was effectively decreased in both eIF3i-1 and eIF3i-2 clones following eIF3i knockdown. Thus, eIF3i may play an important role in regulating cell proliferation by regulating S phase entry.
Figure 5. eIF3i regulates S phase entry by activating β-catenin.

A-C. Effect of eIF3i on cell cycle distribution and cyclin D1 expression. Stable eIF3i-1 and eIF3i-2 clones and the vector-transfected control (Vec) cells were subjected to cell cycle analysis (A), Western blot analysis of cyclin D1, cyclin E, and c-Myc protein level (B), and real-time RT-PCR analysis of cyclin D1 mRNA level (C). D-F. Effect of eIF3i on β-catenin activity. Stable eIF3i-1 and eIF3i-2 clones and Vec control cells were transiently transfected with reporter constructs driven by β-catenin followed by luciferase assay for β-catenin activity (D) or first transfected with eIF3i siRNA to knockdown eIF3i expression as confirmed by Western blot analysis (E) before transfection with reporter construct for luciferase activity assay (F). (n=3; *p<0.05; **p<0.01).
Since cyclin D1 is an important cyclin for S phase entry, we then determined cyclin D1 expression using Western blot analysis and found that it was drastically increased in both eIF3i-1 and eIF3i-2 clones (Fig. 5B). Real time RT-PCR analyses showed that the mRNA level of cyclin D1 was also increased in both eIF3i-1 and eIF3i-2 stable clones (Fig. 5C). However, the expression of cyclin E was not changed. Therefore, cyclin D1, not cyclin E, is likely a downstream mediator of eIF3i in cell proliferation and cell cycle regulation.
To investigate how eIF3i regulates cyclin D1 expression, we hypothesized that β-catenin, which promotes cyclin D1 transcription and also a known proto-oncogene and major mediator of adenomatous polyposis coli in colon oncogenesis 11, might be activated by eIF3i. To test this hypothesis, we first determined if another well known β-catenin downstream target gene, c-Myc, is up-regulated similarly as cyclin D1 by eIF3i. As shown in Fig. 5B, c-Myc expression is increased in both eIF3i stable clones. Next, we used luciferase reporter assay, where the luciferase gene is driven by a promoter containing a β-catenin/TCF4 element, to monitor β-catenin activity. As shown in Fig. 5D, eIF3i over-expression increases β-catenin activity by 1.7-2.8 folds. Immunofluorescence staining also shows the increase in β-catenin level in both cytoplasm and nucleus in the two eIF3i over-expression clones than the Vec control cells (supplemental Fig. S3). To further determine if the increase in β-catenin activity in eIF3i-1 and eIF3i-2 clones was due to eIF3i over-expression, we transiently knocked down eIF3i expression using siRNA followed by testing β-catenin activity using luciferase reporter assay. Fig. 5E shows the knockdown of eIF3i in these cells and Fig. 5F shows that the β-catenin activity is effectively reduced in eIF3i knockdown cells compared with the scrambled siRNA-transfected control cells.
To verify these findings and to eliminate cell line specificity, we investigated the effect of eIF3i on β-catenin activity in RIE cells and CaCo-2 cells. As shown in supplemental Fig. S4A, eIF3i over-expression significantly increases β-catenin activity in RIE cells. Interestingly, following confluency and differentiation of CaCo-2 cells, the β-catenin activity is dramatically decreased (Supplemental Fig. S4B), which coincides with the reduced eIF3i expression following confluency (see Fig. 3). Furthermore, transient eIF3i over-expression in the post-confluent CaCo-2 cells increased while transient eIF3i knockdown in pre-confluent CaCo-2 cells decreased β-catenin activity (Supplemental Fig. S4B). These observations strongly support the possible role of eIF3i in regulating β-catenin activity.
Cyclooxygenase-2 is the direct effector of eIF3i in β-catenin activation
Prostaglandin E2 (PGE2), a product of cyclooxygenase-2 (COX-2), promotes colon cancer cell growth by activating and stabilizing β-catenin 12. To determine if eIF3i possibly increases COX-2 expression and, thus, PGE-2 production, which in turn activates β-catenin, we first performed a Western blot analysis of COX-2 in the stable IEC clones. As shown in Fig. 6A, both eIF3i-1 and eIF3i-2 clones have much higher level of COX-2 than the control cells. Also, transient eIF3i over-expression in post-confluent or knockdown in pre-confluent CaCo-2 cells increased and decreased COX-2 level, respectively (supplemental Fig. S5). Furthermore, the increased COX-2 level is accompanied with an increased PGE2 production in eIF3i-1 and eIF3i-2 clones (Fig. 6B). Thus, eIF3i over-expression likely increases the level of the functionally active COX-2.
Figure 6. eIF3i regulates β-catenin and oncogenesis via COX-2.

A and B. Effect of eIF3i on COX-2 expression and PGE2 production. Stable eIF3i-1 and eIF3i-2 clones and the vector-transfected control (Vec) cells were analyzed for COX-2 expression using Western blot (A) and culture media were collected for EIA assay of PGE2 (B). C and D. COX-2 mediates eIF3i effect on β-catenin activation. Stable eIF3i-1 and eIF3i-2 clones were transiently transfected with siRNAs targeting COX-2 [Si(Cox)] and β-catenin [Si(Cat)] or control scrambled siRNAs (Scr) followed by Western blot analysis of COX-2 and β-catenin (C) or luciferase assay of β-catenin activity (D). E. COX-2 and β-catenin mediate the oncogenic function of eIF3i. Stable IEC clones with eIF3i over-expression (eIF3i-1 and eIF3i-2) were transiently transfected with siRNAs against COX-2 [Si(Cox)] or β-catenin [Si(Cat)] followed by colony formation assay. (n=3; *p<0.05; **p<0.01).
To determine if COX-2 is responsible for eIF3i-mediated up-regulation of β-catenin activity, we next knocked down COX-2 expression in eIF3i-1 and eIF3i-2 clones and monitored the change in β-catenin activity using reporter assay. Fig. 6C-6D show that reducing COX-2 expression using siRNA significantly reduced β-catenin activity. We also found that COX-2 over-expression in RIE cells dramatically increased β-catenin activity (supplemental Fig. S6A) and knocking down COX-2 in RIE cells that have transient eIF3i over-expression reduced β-catenin activity (supplemental Fig. S6B). Similarly, knocking down COX-2 in CaCo-2 cells that have transient eIF3i over-expression also reduced β-catenin activity (supplemental Fig. S7). Furthermore, knocking down COX-2 downstream effectors, β-catenin and TCF4, in these cells with ectopic eIF3i over-expression all substantially reduced β-catenin activity (Supplemental Fig. S6B and S7), confirming the above observation and validating the β-catenin activity assay. Thus, we conclude that COX-2 likely mediates eIF3i-induced up-regulation of β-catenin activity.
Finally, to determine if COX-2 and β-catenin mediates the oncogenic function of eIF3i, we performed a colony formation assay of eIF3i-1 and eIF3i-2 clones following transient knockdown of COX-2 or β-catenin (Fig. 6C). As shown in Fig. 6E, the colony formation potential of both eIF3i-1 and eIF3i-2 clones were reduced about 50% by COX-2 or β-catenin knockdown. Likely, COX-2 and β-catenin partly mediate the oncogenic function of eIF3i.
Translational regulation of COX-2 by eIF3i
In order to elucidate the potential mechanisms of eIF3i regulation of COX-2, we determined the potential effect of eIF3i on COX-2 protein synthesis with consideration of the potential role of eIF3i in translational control. Fig. 7A shows that the synthesis of endogenous COX-2 in the eIF3i-over-expressing IEC cells is much faster than that in the Vec control cells as determined using pulse labeling in combination with immunoprecipitation and autoradiography. To determine if eIF3i regulation of COX-2 synthesis is a direct effect not indirectly by regulating the expression of another gene, we used the in-vitro transcription/translation system to program translation of COX-2 in the absence or presence of purified eIF3i. In this system, only COX-2 is newly expressed by programing its translation with purified COX-2 cRNA. Supplementation of eIF3i will not affect the level of any other proteins in this system. Fig. 7B-7C show that supplementation of purified eIF3i significantly up-regulates the synthesis of COX-2 in a dose-dependent manner. However, supplementation of purified 14-3-3σ, an irrelevant protein of similar size as eIF3i as a control, did not affect the synthesis of COX-2. To eliminate the possibility that eIF3i potentially stimulates translation of all transcripts in the in-vitro system, we also tested translation of 14-3-3σ cRNA. As shown in Fig. 7D, addition of purified eIF3i to the in-vitro system increased translation of COX-2 but not that of 14-3-3σ cRNAs.
Figure 7. eIF3i regulates COX-2 protein synthesis and binds to COX-2 mRNA.

A. Effect of eIF3i on the synthesis of endogenous COX-2. Stable eIF3i-1 clone was pulse labeled with [35S]methionine and COX-2 was immunoprecipitated for separation by SDS-PAGE and autoradiography (ARG) analysis. B. Effect of eIF3i on in-vitro translation of COX-2 cRNA. COX-2 cRNA was used to program cell-free translation in rabbit reticulocyte lysate supplemented with [35S]methionine in the absence or presence of purified eIF3i or 14-3-3σ recombinant proteins followed by SDS-PAGE separation and autoradiography (ARG) or Western blot (IB) analyses of newly synthesized COX-2 proteins. C. Quantitative analysis of [35S]-labeled COX-2 protein produced by in vitro transcription/translation system as shown in panel B using scintillation of counting of the excised COX-2 band (n=3; **p<0.01). D. Effect of eIF3i on in-vitro translation of 14-3-3σ cRNA. 14-3-3-σ and COX-2 cRNAs were use to program cell-free translation in the absence or presence of purified eIF3i and the newly synthesized 14-3-3σ and COX-2 were separated by SDS-PAGE and detected using autoradiograph as described for panel B.
To further demonstrate the direct effect of eIF3i on COX-2 synthesis, we performed a pull-down assay of purified eIF3i using biotin-labeled COX-2 cRNA. As shown in Fig. 8A, dramatically more purified eIF3i is pulled down by the biotin-labeled COX-2 cRNA probe than the unlabeled control COX-2 cRNA probe. The binding of eIF3i to the biotin-labeled COX-2 cRNA probe could be completely inhibited by the unlabeled probe (Fig. 8B), suggesting that the binding is specific. However, the control biotin-labeled 14-3-3σ cRNA probe did not pull down any purified eIF3i, indicating that it cannot bind to eIF3i (Fig. 8B) and that eIF3i regulation of COX-2 synthesis may be specific. Similarly, using RNA-EMSA we found that purified eIF3i could bind directly to [32P]-labeled COX-2 but not 14-3-3σ cRNA probes and that the un-labeled COX-2 competitor cRNA probe could completely block the binding between eIF3i and the [32P]-labeled COX-2 cRNA probe (Fig. 8C). Thus, we conclude that eIF3i likely up-regulates the COX-2 synthesis by directly binding to COX-2 mRNA.
Figure. 8. Binding of eIF3i to COX-2 cRNA.
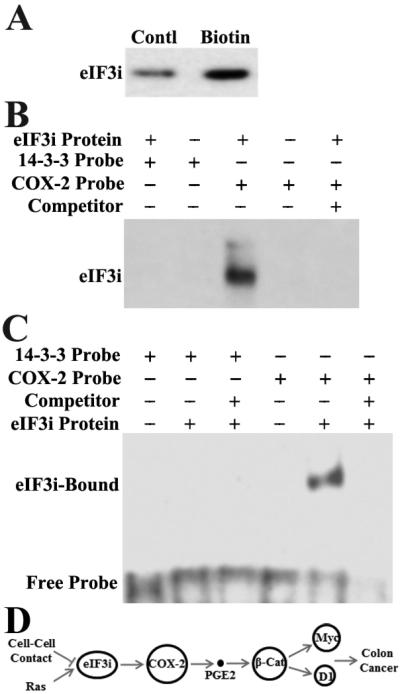
A and B. Pull-down assay. Purified recombinant eIF3i was incubated with biotin-labeled or control unlabeled COX-2 cRNA (A) or pre-incubated with unlabeled probe competitor prior to incubation with labeled probe (B) followed by pull down with Streptavidin-beads and Western blot analysis of eIF3i. C. Electrophoretic Mobility Shift Assay (EMSA). Purified recombinant eIF3i was pre-incubated without or with unlabeled cRNA probe competitors followed by incubation with [32P]-labeled COX-2 cRNA and 14-3-3-σ cRNA. The unbound probes were digested with RNase T1 before separation of eIF3i-bound probe on non-denaturing PAGE and detection by autoradiograph. D. Schematic model of eIF3i function and regulation in colon oncogenesis. It is noteworthy that β-catenin is a known activator of COX-2 expression and, thus, there may be a feed-forward loop that involves eIF3i.
We also tested if eIF3i up-regulation in colon cancer tissues correlates with COX-2 expression. Supplemental Fig. S8 shows that the COX-2 level is increased in 11 of 12 (~92%) cancer colon tissues with increased eIF3i expression. COX-2 remains unchanged in two cancer tissues that do not have increased eIF3i. Together with above observations, these findings suggest that eIF3i may up-regulate COX-2 expression in human colon cancer tissues.
DISCUSSION
The findings that eIF3i is over-expressed in human colon cancer tissues and drives colon oncogenesis along with previous findings on eIF3i over-expression in head and neck cancer 13, breast cancer 14, hepatocellular carcinoma 15, neuroblastoma and melanoma 16, suggest that eIF3i over-expression may be a oncogenic factor. In supporting this conclusion, it has been shown previously that over-expression of ectopic eIF3i causes transformation of mouse fibroblast NIH3T3 and human endothelial ECV304 cells 17. Further studies using in vivo animal models on other cancers such as breast cancer are needed to demonstrate the oncogenic function of eIF3i for these cancers.
Although it was previously thought that the eIF3 complex behaves as a house-keeping initiation factor, accumulating evidences suggest that subunits of this complex may have additional functions in regulating protein synthesis and cell proliferation 3, 5. One such good example is eIF3a, which has been extensively demonstrated to regulate protein synthesis and cell proliferation 17-21. It has been shown that eIF3a up-regulation causes transformation of fibroblast 17 and its down-regulation reverses malignant phenotype of cancer cells 18. Interestingly, eIF3a up-regulation does not up-regulate the synthesis of all proteins. In fact, the synthesis of some proteins including potential tumor suppressors such as p27, XPA, and RPA are inhibited by eIF3a over-expression 20, 22, 23.
Similarly, eIF3i may also play an important role in regulating protein synthesis. The finding in this study that eIF3i over-expression up-regulates the synthesis of COX-2 supports this argument. Recently, we found that, similar to eIF3a, eIF3i up-regulation inhibits the synthesis of p27 (unpublished observation), further supporting the regulatory role of eIF3i in protein synthesis. Although eIF3i does not have any known RNA-binding motif, the fact that purified eIF3i can directly bind to COX-2 but not 14-3-3σ cRNAs argues that eIF3i alone may be able to bind and stimulate COX-2 synthesis. eIF3a can also bind to mRNAs without any known RNA-binding motif 24, 25. It is, thus, tempting to speculate that these proteins may have an unknown RNA-binding sequence. The possibility that by forming inactive subcomplexes with other eIF3 subunits 4, 26-28, eIF3i over-expression suppresses general translation, which leads to up-regulation of translation of specific mRNAs such as COX-2 as suggested previously for eIF3a 17, cannot be ruled out. However, increased cellular proliferation due to eIF3i over-expression is inconsistent with suppression of general protein synthesis. The finding that the eIF3i-binding partners do not increase in concert with eIF3i in cancer tissues also argues against this possibility.
COX-2 mRNA has been shown to contain an AU-rich element (ARE) in its 3’-UTR that inhibits COX-2 mRNA translation 29, 30 and accelerate COX-2 mRNA degradation. However, we found that the increased eIF3i expression does not appear to affect the degradation of COX-2 mRNA in IEC cells (unpublished observation) and, thus, we believe that eIF3i does not affect COX-2 mRNA stability. Nevertheless, it will be of interest to determine if eIF3i possibly binds to the inhibitory ARE in the 3’-UTR of COX-2 mRNA to potentially relieve the inhibitory effect of ARE on translation of COX-2 mRNAs. We are currently testing this possibility.
COX-2 and its product PGE2 have been shown to play an important role in oncogenesis and disease progression possibly by activating β-catenin in the Wnt/β-catenin signaling pathway 31-33. PGE2 has been shown to promote β-catenin stabilization and activation in colon cancer cell lines by displacing GKS-3β from the β-catenin complex and, thus, preventing its phosphorylation and degradation 12, 34. Indeed, eIF3i over-expression up-regulates COX-2 synthesis, increases production of PGE2, and increases β-catenin activity. Knocking down COX-2 or β-catenin could partially reverse malignant phenotype induced by eIF3i over-expression. Hence, it is not surprising to find that eIF3i has oncogenic functions.
COX-2 and its product PGE2 may be one of many eIF3i downstream targets that mediate the oncogenic function of eIF3i. This possibility is consistent with the finding that knocking down COX-2 expression in the eIF3i-over-expressing cells only partially reversed the eIF3i-over-expression-induced malignant phenotype (Fig. 6) and that the expression of a tumor suppressor gene p27 is inhibited by eIF3i over-expression (unpublished observation). Currently, what other genes possibly mediate the oncogenic function of eIF3i is unknown. However, it has been found that the C-terminus of eIF3i interacts with Akt, which is required for eIF3i-mediated Akt activation 35 and that mTOR inhibition reduces eIF3i phosphorylation and eIF3i oncogenic activity 36. Since Akt is known to mediate the effect of K-ras on COX-2 expression 37, eIF3i may participate in the intricate signaling network involving Akt, K-ras, and COX-2 in colon oncogenesis.
Equally important but unknown issues regarding eIF3i is its upstream regulator in colon oncogenesis. Our findings using the Ras inducible cell lines suggest that eIF3i may be under the control of Ras. Considering that 30-50% of colon cancers have K-Ras mutation, the finding that eIF3i may be under K-Ras control and contribute to colon oncogenesis by regulating the translation of its downstream target genes is important and may have significant impact on future studies of colon cancer. However, it is noteworthy that all colon cancer tissues examined in this study appear to have increased eIF3i expression. This is consistent with genomic data (www.oncomine.org), where increased eIF3i expression was observed in adenoma and adenocarcinoma compared with normal tissues. With the average rate of K-ras mutation in colon cancers as 30-50%, it is not clear if K-ras regulates eIF3i expression in human tissues and there may be other mechanisms that up-regulate eIF3i expression.
One of the other potential regulators of eIF3i expression is signaling from cell-cell contact. It appears that eIF3i expression decreases as CaCo-2 cells reaches confluency. Further studies are needed to investigate what signaling pathways are possibly involved in suppressing eIF3i expression in response to confluency. Nevertheless, eIF3i reduction in response to confluency may be responsible for the confluency-induced CaCo-2 cell differentiation. This is consistent with our previous finding that eIF3a may play a role in differentiation and that the decreased eIF3a expression could be a pre-requisite of intestinal epithelial cell differentiation 21.
In summary, we showed that eIF3i is a proto-oncogene, functioning by up-regulating the COX-2 synthesis and PGE2 production, which in turn activates β-catenin (Fig. 8D). The regulation of COX-2 synthesis appears to be via direct binding of eIF3i to its mRNA. These findings suggest that eIF3i unlikely participates in protein synthesis only as a subunit of the house-keeping eIF3 complex and it may have additional non-canonic functions and play an important regulatory role in protein synthesis, cell proliferation and oncogenesis.
MATERIALS AND METHODS
Materials
Antibodies against eIF3i, cyclinD1, COX-2, and siRNAs targeting human TCF4, β-catenin, and COX-2 were from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies against Cyclin E, β-catenin, and TCF4 were from Cell Signaling Technology (Beverly, MA). TOP-FLASH and FOP-FLASH reporter plasmids and anti-pan-Ras antibody were from EMD Millipore (Temecula, CA). Anti-GAPDH and β-Actin antibodies were from Abcam Inc. (Cambridge, MA). Anti-His-Tag antibody, HRP- and FITC-labeled secondary antibodies were from Sigma-Aldrich (St Louis, MO). siRNAs targeting rat β-catenin and COX-2, and human eIF3i were from Dharmacon (Lafayette, CO). RNeasy Mini Kit, SYBR Green qPCR Master Mixes, [35S]methionine, and enhanced chemiluminenscence (ECL) were from Qiagen (Valencia, CA), Applied Biosystems (Foster City, CA), Perkin Elmer (Boston, MA), and GE Healthcare (Pittsburgh, PA), respectively. Culture media, reagents, and pCMV-β-gal were from Invitrogen (Grand Island, NY). Luciferase Reporter Assay Kit and Streptavidin-magnetic beads were from Promega (Madison, WI). Bradford reagents and iScript cDNA Synthesis Kit were from BioRad Laboratories (Berkeley, CA). All other reagents of molecular biology grade were from Sigma or Fisher Scientific (Chicago, IL).
Human Tissues
Frozen and paraffin embedded surgical specimens of matched human colon cancer and adjacent normal tissues were from IUSCC Tissue Procurement and Distribution Core at Indiana University School of Medicine, Indianapolis, IN. These tissues were verified by certified pathologist at IUSCC.
Construct Engineering
The mammalian eIF3i expression construct was engineered by releasing the eIF3i cDNA from pOTB7/eIF3i (Open Biosystems, AL) and cloning it into pCβA to generate pCβA/eIF3i. To generate the plasmid for production of recombinant protein, eIF3i cDNA in pOTB7/eIF3i was amplified by PCR using primers 5’-GGAATTGATGAAGCCGATCCTACTGCAGGGC-3’ (forward) and 5’-CCTCGAGTTAAGCCTCAAACTCAAATTCGAAGTA-3’ (reverse). The PCR product was then cloned into pET28a to generate pET28a/eIF3i. The COX-2 expression plasmid was engineered by releasing human COX-2 cDNA from COX2/SPORT6 plasmid (Open Biosystems, AL) and cloning it into pCβA, resulting in pCβA/COX-2. All plasmids were confirmed by DNA sequencing.
Cell Lines and Transfection
IEC-6, IEC-iK Ras 30 and RIE-iH Ras 37 cell lines that harbor inducible K-RasVal12 and Ha-RasVal12 gene, respectively, were gifts from Dr. Hongmiao Sheng (Indiana University School of Medicine). CaCo-2 was obtained from ATCC (Manassas, VA). RIE-iH Ras and IEC-iK Ras cells were cultured in selective DMEM medium containing 400 mg/ml G418 and 150 mg/ml hygromycin B. For confluency studies, CaCo-2 cells were seeded into 60 mm dishes at 1×106 cell per dish and cultured for various time with medium changed every two days.
Transient transfections were performed using Lipofectamine Plus reagent or Lipofectin 2000 (Invitrogen) as we previously described 18-20. Cells were harvested for analysis at 48 hours following transfection. For co-transfections, the secondary construct or siRNAs were introduced into cells at 24 hrs following the first transfection. To establish stable transfectants, IEC cells were transfected with pCβA/eIF3i construct or pCβA vector. Positive clones were selected using G418 and identified using Western blot.
RNA purification and real-time RT-PCR
RNA purification and real-time RT-PCR was performed as previously described 38. The primers used were 5’-AAGGACTCATGACCACAGTCCAT-3’ (forward) and 5’-CCATCACGCCACAGTTTCC-3’ (reverse) for GAPDH and 5’-CCGTCCATGCGGAAGATC-3’ (forward) and 5’-GAAGACCTCCTCCTCGCACT-3’ (reverse) for cyclin D1. The threshold cycle (Ct) of cyclin D1 was determined and normalized against that of internal control, GAPDH.
Alkaline phosphatase (AP) and luciferase reporter assay
AP assay was performed as previously described 21 using a kit from Sigma-Aldrich. Luciferase reporter assay was performed using a kit from Promega according to manufacturer's instructions. The luciferase activity was normalized by β-galactosidase activity and reported as a ratio of TOP-FLASH/FOP-FLASH as previously described 39.
Determination of COX-2 synthesis and PGE2 production
Synthesis of COX-2 was analyzed by pulse labeling using [35S]methionine as previously described 18, 20. Briefly, cells were incubated in methionine-free medium supplemented with [35S]methionine at 300 μCi/ml for 2 hrs, followed by lysis of cells and immunoprecipitation of COX-2. The precipitants were separated by 12% SDS-PAGE and the COX-2 was detected by autoradiography. Production of PGE2 was determined using PGE2 EIA Kit (Cayman, Ann Arbor, Michigan) according to manufacture's instructions.
Colony formation, anchorage-independent growth, and xenograft tumor formation assays
Colony formation and anchorage-independent growth in soft agar were performed as previously described 18. Briefly, cells were seeded in 6-well plates and cultured for 9 days with media changed every two days. For growth in soft agar, a base layer of 0.7% Noble agar (Difco) in DMEM in 60-mm dish was generated followed by addition of top layer containing 1000 cells in DMEM, and 0.4% agar. Cells were cultured for 14-21 days. Colonies were stained using 0.005% crystal violet, and counted manually.
For xenograft tumor formation, ~1×107cells were injected subcutaneously into 7-week old female NOD/SCID mice (one injection/mouse). Tumor growth was measured by a caliper once a week for a total of 9 weeks. The tumor volume was calculated from two perpendicular diameters using the formula: volume = (length/2) × (width2). Tumors were removed, measured, fixed in 10% formalin buffer and stained with Hematoxylin & eosin (H&E) using standard histology procedures. This study was approved by Indiana University ICAUC.
RNA/protein pull-down assay
Pull-down of proteins using biotinylated RNA was performed as previously described 25. Briefly, Streptavidin-magnetic beads was blocked with 3% BSA and then incubated with 1μg biotinylated or naked cRNA probes for 1 hr, followed by incubation with 1 μg purified eIF3i for 1 hr. The pull-down materials were then washed extensively and separated by SDS-PAGE followed by Western blot analysis of eIF3i.
Electrophoretic Mobility Shift Assay (EMSA)
EMSA was performed by mixing 1 μg purified His-tagged eIF3i and 150,000 cpm [32P]-labeled COX-2 or 14-3-3σ cRNA transcript in a buffer consisting of 15 mM HEPES (pH 7.9), 50 mM KCl, 10% glycerol, 0.2 mM dithiothreitol, 5 mM MgCl2, 200 μg/ml tRNA with or without 1 μg un-labeled COX-2 or 14-3-3σ cRNA as cold probe competitors followed by incubation for 30 min at room temperature. Unbound probes were digested by 100 units RNase T1 for 15 min at 30°C. The reaction mixtures were then separated by non-denaturing PAGE and detection of eIF3i-bound probe by autoradiography.
Supplementary Material
ACKNOWLEDGEMENT
The authors wish to thank Dr. Hongmiao Sheng for the generous gift of the rat intestinal epithelial cell lines. This work was supported in part by a NIH grant R01 CA94961 (JTZ).
Footnotes
Conflict of Interest: The authors declare no conflict of interest.
REFERENCES
- 1.Mathews MB, Sonenberg N, Hershey JW. Translational Control in Biology and Medicine. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schneider RJ, Sonenberg N. Translational control in cancer development and progression. In: Mathews MB, Sonenberg N, Hershey JW, editors. Translational control in biology and medicine. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 2007. pp. 401–431. [Google Scholar]
- 3.Dong Z, Zhang JT. Initiation factor eIF3 and regulation of mRNA translation, cell growth, and cancer. Crit Rev Oncol Hematol. 2006;59:169–180. doi: 10.1016/j.critrevonc.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 4.Masutani M, Sonenberg N, Yokoyama S, Imataka H. Reconstitution reveals the functional core of mammalian eIF3. EMBO J. 2007;26:3373–3383. doi: 10.1038/sj.emboj.7601765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yin JY, Dong Z, Liu ZQ, Zhang JT. Translational control gone awry: a new mechanism of tumorigenesis and novel targets of cancer treatments. Biosci Rep. 2011;31:1–15. doi: 10.1042/BSR20100077. [DOI] [PubMed] [Google Scholar]
- 6.Zhou M, Sandercock AM, Fraser CS, Ridlova G, Stephens E, Schenauer MR, et al. Mass spectrometry reveals modularity and a complete subunit interaction map of the eukaryotic translation factor eIF3. Proc Natl Acad Sci U S A. 2008;105:18139–18144. doi: 10.1073/pnas.0801313105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Markowitz AJ, Wu GD, Bader A, Cui Z, Chen L, Traber PG. Regulation of lineage-specific transcription of the sucrase-isomaltase gene in transgenic mice and cell lines. Am J Physiol. 1995;269:G925–939. doi: 10.1152/ajpgi.1995.269.6.G925. [DOI] [PubMed] [Google Scholar]
- 8.Pinto M, Robine-Leon S, Appay MD, Kedinger M, Triadou N, Dussaulx E, et al. Enterocyte-like differentiation and polarization of the human colon carcinoma cell line Caco-2 in culture. Biol Cell. 1983;47:323–330. [Google Scholar]
- 9.Zweibaum A, Chantret I. Human colon carcinoma cell lines as in vitro models for the study of intestinal cell differentiation. In: Smith MW, Sepulveda FV, editors. Adaptation and Development of Gastrointestinal Function. Manchester University Press; Manchester, UK: 1989. pp. 103–112. [Google Scholar]
- 10.Ding QM, Ko TC, Evers BM. Caco-2 intestinal cell differentiation is associated with G1 arrest and suppression of CDK2 and CDK4. Am J Physiol. 1998;275:C1193–1200. doi: 10.1152/ajpcell.1998.275.5.C1193. [DOI] [PubMed] [Google Scholar]
- 11.Markowitz SD, Bertagnolli MM. Molecular origins of cancer: Molecular basis of colorectal cancer. N Engl J Med. 2009;361:2449–2460. doi: 10.1056/NEJMra0804588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Castellone MD, Teramoto H, Williams BO, Druey KM, Gutkind JS. Prostaglandin E2 promotes colon cancer cell growth through a Gs-axin-beta-catenin signaling axis. Science. 2005;310:1504–1510. doi: 10.1126/science.1116221. [DOI] [PubMed] [Google Scholar]
- 13.Rauch J, Ahlemann M, Schaffrik M, Mack B, Ertongur S, Andratschke M, et al. Allogenic antibody-mediated identification of head and neck cancer antigens. Biochem Biophys Res Commun. 2004;323:156–162. doi: 10.1016/j.bbrc.2004.08.071. [DOI] [PubMed] [Google Scholar]
- 14.Matsuda S, Katsumata R, Okuda T, Yamamoto T, Miyazaki K, Senga T, et al. Molecular cloning and characterization of human MAWD, a novel protein containing WD-40 repeats frequently overexpressed in breast cancer. Cancer Res. 2000;60:13–17. [PubMed] [Google Scholar]
- 15.Gray SG, Kytola S, Lui WO, Larsson C, Ekstrom TJ. Modulating IGFBP-3 expression by trichostatin A: potential therapeutic role in the treatment of hepatocellular carcinoma. Int J Mol Med. 2000;5:33–41. doi: 10.3892/ijmm.5.1.33. [DOI] [PubMed] [Google Scholar]
- 16.Bernardini S, Melino G, Saura F, Annicchiarico-Petruzzelli M, Motti C, Cortese C, et al. Expression of co-factors (SMRT and Trip-1) for retinoic acid receptors in human neuroectodermal cell lines. Biochem Biophys Res Commun. 1997;234:278–282. doi: 10.1006/bbrc.1997.6626. [DOI] [PubMed] [Google Scholar]
- 17.Zhang L, Pan X, Hershey JW. Individual overexpression of five subunits of human translation initiation factor eIF3 promotes malignant transformation of immortal fibroblast cells. J Biol Chem. 2007;282:5790–5800. doi: 10.1074/jbc.M606284200. [DOI] [PubMed] [Google Scholar]
- 18.Dong Z, Liu LH, Han B, Pincheira R, Zhang JT. Role of eIF3 p170 in controlling synthesis of ribonucleotide reductase M2 and cell growth. Oncogene. 2004;23:3790–3801. doi: 10.1038/sj.onc.1207465. [DOI] [PubMed] [Google Scholar]
- 19.Dong Z, Liu Z, Cui P, Pincheira R, Yang Y, Liu J, et al. Role of eIF3a in regulating cell cycle progression. Exp Cell Res. 2009;315:1889–1894. doi: 10.1016/j.yexcr.2009.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dong Z, Zhang JT. EIF3 p170, a Mediator of Mimosine Effect on Protein Synthesis and Cell Cycle Progression. Mol Biol Cell. 2003;14:3942–3951. doi: 10.1091/mbc.E02-12-0784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu Z, Dong Z, Yang Z, Chen Q, Pan Y, Yang Y, et al. Role of eIF3a (eIF3 p170) in intestinal cell differentiation and its association with early development. Differentiation. 2007;75:652–661. doi: 10.1111/j.1432-0436.2007.00165.x. [DOI] [PubMed] [Google Scholar]
- 22.Liu RY, Dong Z, Liu J, Yin JY, Zhou L, Wu X, et al. Role of eIF3a in regulating cisplatin sensitivity and in translational control of nucleotide excision repair of nasopharyngeal carcinoma. Oncogene. 2011;30:4814–4823. doi: 10.1038/onc.2011.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yin JY, Shen J, Dong ZZ, Huang Q, Zhong MZ, Feng DY, et al. Effect of eIF3a on response of lung cancer patients to platinum-based chemotherapy by regulating DNA repair. Clin Cancer Res. 2011;17:4600–4609. doi: 10.1158/1078-0432.CCR-10-2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Block KL, Vornlocher HP, Hershey JW. Characterization of cDNAs encoding the p44 and p35 subunits of human translation initiation factor eIF3. Journal of Biological Chemistry. 1998;273:31901–31908. doi: 10.1074/jbc.273.48.31901. [DOI] [PubMed] [Google Scholar]
- 25.Yin JY, Dong ZZ, Liu RY, Chen J, Liu ZQ, Zhang JT. Translational regulation of RPA2 via internal ribosomal entry site and by eIF3a. Carcinogenesis. 2013;34:1224–1231. doi: 10.1093/carcin/bgt052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Asano K, Phan L, Anderson J, Hinnebusch AG. Complex formation by all five homologues of mammalian translation initiation factor 3 subunits from yeast Saccharomyces cerevisiae. J Biol Chem. 1998;273:18573–18585. doi: 10.1074/jbc.273.29.18573. [DOI] [PubMed] [Google Scholar]
- 27.Herrmannova A, Daujotyte D, Yang JC, Cuchalova L, Gorrec F, Wagner S, et al. Structural analysis of an eIF3 subcomplex reveals conserved interactions required for a stable and proper translation pre-initiation complex assembly. Nucleic Acids Res. 2012;40:2294–2311. doi: 10.1093/nar/gkr765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Phan L, Schoenfeld LW, Valasek L, Nielsen KH, Hinnebusch AG. A subcomplex of three eIF3 subunits binds eIF1 and eIF5 and stimulates ribosome binding of mRNA and tRNA(i)Met. Embo J. 2001;20:2954–2965. doi: 10.1093/emboj/20.11.2954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dixon DA, Kaplan CD, McIntyre TM, Zimmerman GA, Prescott SM. Post-transcriptional control of cyclooxygenase-2 gene expression. The role of the 3'-untranslated region. J Biol Chem. 2000;275:11750–11757. doi: 10.1074/jbc.275.16.11750. [DOI] [PubMed] [Google Scholar]
- 30.Sheng H, Shao J, Dixon DA, Williams CS, Prescott SM, DuBois RN, et al. Transforming growth factor-beta1 enhances Ha-ras-induced expression of cyclooxygenase-2 in intestinal epithelial cells via stabilization of mRNA. J Biol Chem. 2000;275:6628–6635. doi: 10.1074/jbc.275.9.6628. [DOI] [PubMed] [Google Scholar]
- 31.Greenhough A, Smartt HJ, Moore AE, Roberts HR, Williams AC, Paraskeva C, et al. The COX-2/PGE2 pathway: key roles in the hallmarks of cancer and adaptation to the tumour microenvironment. Carcinogenesis. 2009;30:377–386. doi: 10.1093/carcin/bgp014. [DOI] [PubMed] [Google Scholar]
- 32.Gupta RA, Dubois RN. Colorectal cancer prevention and treatment by inhibition of cyclooxygenase-2. Nat Rev Cancer. 2001;1:11–21. doi: 10.1038/35094017. [DOI] [PubMed] [Google Scholar]
- 33.Kikuchi A. Tumor formation by genetic mutations in the components of the Wnt signaling pathway. Cancer Sci. 2003;94:225–229. doi: 10.1111/j.1349-7006.2003.tb01424.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shao J, Jung C, Liu C, Sheng H. Prostaglandin E2 Stimulates the beta-catenin/T cell factor-dependent transcription in colon cancer. J Biol Chem. 2005;280:26565–26572. doi: 10.1074/jbc.M413056200. [DOI] [PubMed] [Google Scholar]
- 35.Wang YW, Lin KT, Chen SC, Gu DL, Chen CF, Tu PH, et al. Overexpressed-eIF3I interacted and activated oncogenic Akt1 is a theranostic target in human hepatocellular carcinoma. Hepatology. 2013;58:239–250. doi: 10.1002/hep.26352. [DOI] [PubMed] [Google Scholar]
- 36.Ahlemann M, Zeidler R, Lang S, Mack B, Munz M, Gires O. Carcinoma-associated eIF3i overexpression facilitates mTOR-dependent growth transformation. Mol Carcinog. 2006;45:957–967. doi: 10.1002/mc.20269. [DOI] [PubMed] [Google Scholar]
- 37.Sheng H, Shao J, Dubois RN. K-Ras-mediated increase in cyclooxygenase 2 mRNA stability involves activation of the protein kinase B1. Cancer Res. 2001;61:2670–2675. [PubMed] [Google Scholar]
- 38.Liu Y, Liu H, Han B, Zhang JT. Identification of 14-3-3sigma as a contributor to drug resistance in human breast cancer cells using functional proteomic analysis. Cancer Res. 2006;66:3248–3255. doi: 10.1158/0008-5472.CAN-05-3801. [DOI] [PubMed] [Google Scholar]
- 39.Kuroda T, Rabkin SD, Martuza RL. Effective treatment of tumors with strong beta-catenin/T-cell factor activity by transcriptionally targeted oncolytic herpes simplex virus vector. Cancer Res. 2006;66:10127–10135. doi: 10.1158/0008-5472.CAN-06-2744. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.