

Abstract
Human papillomavirus (HPV) 58 accounts for a notable proportion of cervical cancers in East Asia and parts of Latin America, but it is uncommon elsewhere. The reason for such ethnogeographical predilection is unknown. In our study, nucleotide sequences of E6 and E7 genes of 401 HPV58 isolates collected from 15 countries/cities across four continents were examined. Phylogenetic relationship, geographical distribution and risk association of nucleotide sequence variations were analyzed. We found that the E6 genes of HPV58 variants were more conserved than E7. Thus, E6 is a more appropriate target for type-specific detection, whereas E7 is more appropriate for strain differentiation. The frequency of sequence variation varied geographically. Africa had significantly more isolates with E6-367A (D86E) but significantly less isolates with E6-203G, -245G, -367C (prototype-like) than other regions (p ≤ 0.003). E7-632T, -760A (T20I, G63S) was more frequently found in Asia, and E7-793G (T74A) was more frequent in Africa (p < 0.001). Variants with T20I and G63S substitutions at E7 conferred a significantly higher risk for cervical intraepithelial neoplasia grade III and invasive cervical cancer compared to other HPV58 variants (odds ratio = 4.44, p = 0.007). In conclusion, T20I and/or G63S substitution(s) at E7 of HPV58 is/are associated with a higher risk for cervical neoplasia. These substitutions are more commonly found in Asia and the Americas, which may account for the higher disease attribution of HPV58 in these areas.
Keywords: HPV, variant, cervical cancer, phylogeny, oncogenic risk
High-risk human papillomavirus (HPV) plays a necessary role in the development of cervical cancer, which is the third most common cancer in women worldwide.1 Among the 15 HPV types classified as high-risk, HPV16 and HPV18 are the two most important types accounting for about 70% of cervical cancers across the world. The remaining cancers are caused by HPV31, 33, 35, 39, 45, 51, 52, 56, 58 and 59. The distribution of these less common types shows some degree of ethnogeographical variation.2,3 HPV58 is an uncommon type with skewed ethnogeographical distribution. The prevalence of HPV58 is remarkably high among cervical squamous cell carcinomas in East Asia (33% in Shaanxi, 26% in Shanghai, 16% in South Korea, 10% in Hong Kong and Taiwan and 8% in Japan),4–9 whereas it only accounts for about 2% of cervical cancers in other regions.2,3 In addition to East Asia, a higher prevalence of HPV58 has also been observed in a few places in Latin America. HPV58 was found in 12% of cervical cancers in Costa Rica,10 21% in Southeastern Mexico,11 9.5% in Northeastern Brazil12 and was the second most common type (about 13%) found in precancerous lesions in Central and Southern Brazil.13,14
The reason for an ethnogeographical predilection in the attribution of HPV58 to cervical cancer is not fully understood. HPV58 variants are classified into four lineages. These lineages exhibit a distribution pattern that varies across the world.15,16 Studies comparing the risk association of these variant lineages are very limited and a conclusion has not yet been reached.17,18
The E6 and E7 proteins encoded by high-risk HPV play a key role in cellular transformation. Previous studies from Eastern19 and Southern Chinese populations20 suggested that sequence variations within these regions may be associated with a higher risk for cervical neoplasia. At present, information on sequence variation of the E6 and E7 open reading frames (ORF) of HPV58 have been derived mainly from Chinese populations.18–20 Our study examined the sequence polymorphisms of HPV58 E6 and E7 ORF and assessed the geographical distribution and risk association with cervical neoplasia from a global perspective.
Material and Methods
Study samples
A total of 401 samples including 295 cervical scrapes or swabs, 69 vaginal swabs and 37 anal samples collected from 15 countries and cities were examined in our study (Table 1). These samples have been used to establish a lineage classification system for HPV58 variants.16
Table 1. Source and cervical pathology status of study samples.
| Region | Country/city | Total no. of specimen | No. of specimen per cervical pathology status | |||||
|---|---|---|---|---|---|---|---|---|
| Normal | ASCUS1 | LGSIL2 | HGSIL3 | Carcinoma | Unknown | |||
| Africa | Zimbabwe4 | 69 | 0 | 0 | 0 | 0 | 0 | 69 |
| Americas | 63 | 6 | 1 | 6 | 10 | 2 | 1 | |
| Canada | 10 | 2 | 0 | 4 | 4 | 0 | 0 | |
| USA5 | 37 | |||||||
| Mexico | 2 | 0 | 0 | 0 | 1 | 1 | 0 | |
| Argentina | 6 | 3 | 1 | 0 | 0 | 1 | 1 | |
| Brazil | 3 | 1 | 0 | 1 | 1 | 0 | 0 | |
| Honduras | 5 | 0 | 0 | 1 | 4 | 0 | 0 | |
| Asia | 238 | 57 | 23 | 57 | 66 | 31 | 4 | |
| Mainland China | 3 | 0 | 0 | 0 | 0 | 0 | 3 | |
| Hong Kong | 90 | 17 | 0 | 15 | 41 | 17 | 0 | |
| Taiwan | 6 | 0 | 0 | 0 | 0 | 6 | 0 | |
| Korea | 119 | 30 | 23 | 40 | 22 | 4 | 0 | |
| Japan | 14 | 9 | 0 | 2 | 1 | 1 | 1 | |
| Thailand | 6 | 1 | 0 | 0 | 2 | 3 | 0 | |
| Europe | 31 | 16 | 1 | 9 | 3 | 0 | 2 | |
| Great Britain | 14 | 6 | 0 | 6 | 2 | 0 | 0 | |
| Italy | 17 | 10 | 1 | 3 | 1 | 0 | 2 | |
ASCUS, atypical squamous cells of undetermined significance.
LGSIL, low-grade squamous intraepithelial lesions, including CIN1, and mild dysplasia.
HGSIL, high-grade squamous intraepithelial lesions, including CIN2, CIN3, and severe dysplasia.
Cervical or vaginal swabs.
Anal swab samples.
Nucleotide sequencing
The full length of E6 and E7 ORF was amplified by polymerase chain reaction (PCR) using primers 5′-CGA AAA CGG TCT GACCGA AA-3′ and 5′-TAT CGT CTG CTG TTT CGT CC-3′, followed by a second PCR using inner primers (5′-GAC CGA AAC CGG TGC ATA TA-3′ and 5′-ACC GCT TCT ACC TCA AAC CA-3′) when necessary. Briefly, the PCR was conducted in a 50-μL reaction mix containing 4 μL of extracted DNA, 200 μM of deoxynucleotide triphosphates, forward and reverse primers at 0.25 μM each, 1.25 unit of HotStarTaq Plus polymerase (Qiagen, Hilden, Germany). The cycling conditions were activation of polymerase at 95°C for 5 min, 40 cycles of denaturation at 94°C for 50 sec, annealing at 57°C (59°C for nested PCR) for 1 min and extension at 72°C for 1 min, followed by a final extension at 72°C for 8 min. Ten microliters of the purified PCR products were mixed with 2 μL of BigDye Terminator v.3.1 sequencing reaction mix (Applied Biosystems, Foster City, USA), 3 μL of 5× sequencing buffer and 3.2 pmol of the sequencing primer which was the corresponding PCR primer as described above and made up to a final volume of 20 μL. The cycling conditions for the labeling PCR were 25 cycles at 95°C for 15 sec, 50°C for 15 sec and 65°C for 75 sec. Fluorescence-labeled PCR products were purified with DyeEx (Qiagen) and run on an ABI 3130 automated sequence analyzer (Applied Biosystems). Sequence data were obtained from both directions and analyzed with SeqScape v2.5 (Applied Biosystems). Variations that occurred only in one isolate were confirmed by repeating PCR sequencing from the original sample.
Phylogenetic analysis
The program MEGA5 was applied to identify the best evolutionary model and to construct the maximum-likelihood trees using the close-neighbor-interchange (CNI) search approach.21 The data were bootstrap resampled 1,000 times for tree topology evaluation. HPV33 was used as an outgroup sequence. The program Datamonkey (http://www.datamonkey.org) was used to detect positive or negative selection.22,23
Statistical analysis
All data analyses were performed by the Statistical Package for Social Sciences (version 18.0.0, IBM Corp., NY). The rate of detection of E6 and E7 sequence patterns was compared between the four regions by χ2 test and using the Bonferroni method to account for multiple comparisons.24 For the purpose of risk association analysis, women with normal cytology or absence of lesion at colposcopy or biopsy were grouped as “normal cervices.” Using cervical intraepithelial neoplasia grade III (CIN3) and cervical cancer as an outcome variable and normal cervices as control, the risk association of nonsynonymous substitutions was first assessed by univariate analysis using χ2 test or Fisher's exact test. Those mutations found to be significant for the association (p < 0.05) were tested for interactions, followed by unconditional binary logistic regression analyses controlling for patients' age and their country of origin. All p values <0.05 were regarded as statistically significant.
This study has included 37 anal samples collected from men in USA. In our previous study on HPV58 variant lineage based on the same cohort of samples, no differences in the distribution of variants between these anal samples and cervical samples collected from the same region was observed.16 In our study also, there were no differences in the distribution of E6 and E7 variants among these two sources of samples. For these reasons, anal samples were pooled together with cervical samples for the purpose of analysis in our study.
Results
E6 sequence variation
Altogether, 19 variants showing nucleotide sequence variations in the E6 ORF were identified (Fig. 1). The maximum variation between any two variants was nine nucleotides, a variability of 2.0% over the 450-bp E6 ORF. The mean dN/dS was 0.916, and no site with significant positive or negative selection was found. Altogether, 16 nucleotide positions showing sequence polymorphism were identified, with 11 positions had nonsynonymous substitutions leading to amino acid changes. Based on the phylogenetic relationship, the E6 sequence patterns could be classified into four groups designated as E6-A, -B, -C, and -D, and each group was associated with a signature nucleotide sequence variation as shown in Figure 2a. The maximum intergroup nucleotide sequence variability range from 0.9% (E6-A and -B) to 2.0% (E6-B and -D), whereas the maximum intragroup variability range from 0.7% (E6-A and -C) to 1.3% (E6-B).
Figure 1.
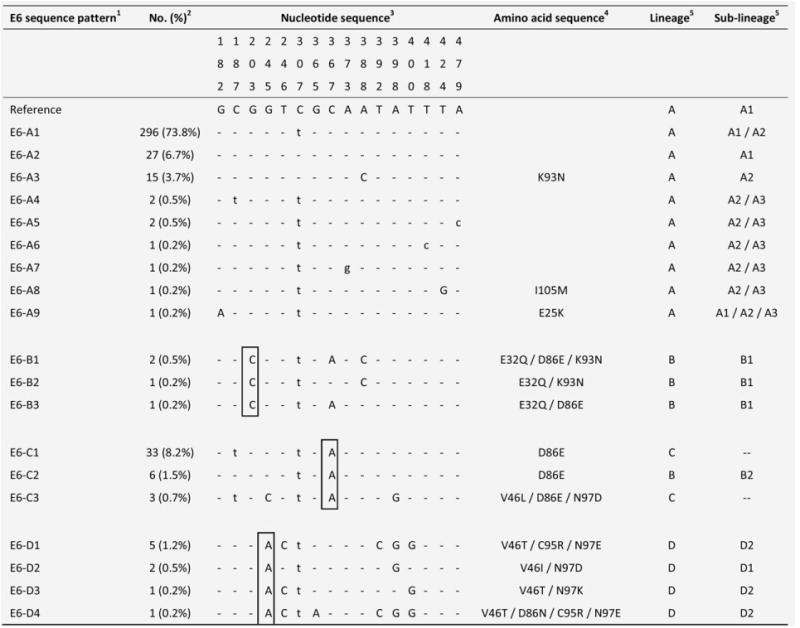
Nucleotide sequence variations at E6 of 19 HPV58 isolates.1 The group containing the prototype was assigned as E6-A. The HPV58 prototype (http://www.ncbi.nlm.nih.gov/genbank accession no. NC_001443) was used as reference.2No. (%) of each sequence pattern found among the 401 samples examined.3 Nucleotide positions where variations were detected are written vertically across the top. Sites with no changes are marked with dashes. Capital letters indicate alternations result in an amino acid change. Lower case letters indicate synonymous mutations.4 The one letter amino acid code is used. The position of amino acid change is stated numerically. The letter preceding this number refers to the reference amino acid, and the letter following refers to its substitution.5 Lineage and sublineage classification based on full genome or concatenated E6-E7-E2-E5-L1-PCR sequences described in previous studies.15,16 Some E6 sequence patterns are not sublineage-specific. –, no sublineage assigned. Single nucleotide substitutions diagnostic for E6 variants are boxed. GenBank accession numbers for E6 sequences are KC190273 - KC190291.
Figure 2.
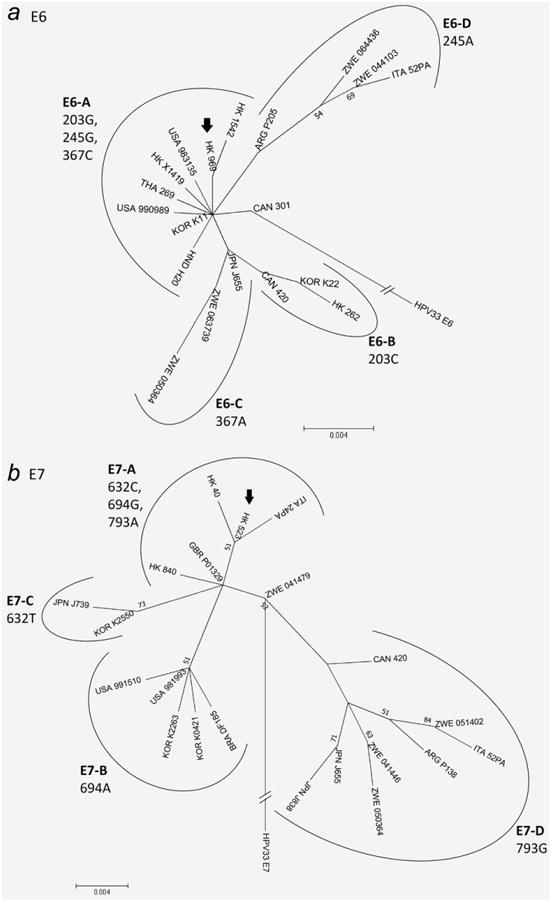
Phylogenetic trees of E6 and E7 of HPV58. The maximum-likelihood trees were constructed by MEGA5 using the CNI search approach. Bootstrap values of ≥50% generated by 1,000 resamplings are shown. (a) Constructed from 450-bp E6 nucleotide sequences of 19 HPV58 isolates. The length of scale bar represents 0.004 substitutions per nucleotide position. The sample HK969 with E6 nucleotide sequence identical to the prototype (GenBank accession no. NC_001443) is indicated by an arrow. The group containing the prototype sequence was assigned as E6-A. Sample CAN301 was grouped into sequence pattern E6-A. (b) Constructed from 297-bp E7 nucleotide sequences of 21 HPV58 isolates. The length of scale bar represents 0.004 substitutions per nucleotide position. The sample HK523 with nucleotide sequence identical to the prototype (GenBank accession no. NC_001443) is indicated by an arrow. The group containing the prototype sequence was assigned as E7-A.
E7 sequence variation
Twenty-one variants showing sequence variations in the E7 ORF were identified (Fig. 3). The maximum nucleotide sequence variation between any two variants was nine, a variability of 3.0% over the 297-bp ORF. The mean dN/dS of E7 was 0.748 indicating a positive selection. Overall, 18 nucleotide positions showing sequence polymorphisms were identified, and 11 had nonsynonymous substitutions (Fig. 3). Four groups of E7 sequence patterns designated as E7-A with signature sequence variation of 632C, 694G, 793A; E7-B (694A); E7-C (632T) and E7-D (793G) were identified. The phylogenetic relationship and their associated signature nucleotide sequence variations are shown in Figure 2b.
Figure 3.
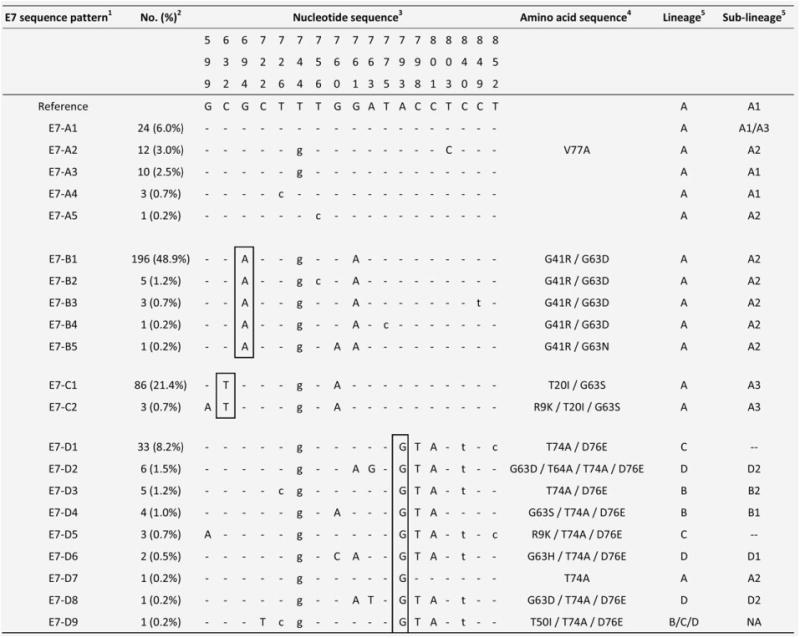
Nucleotide sequence variations at E7 of 21 HPV58 isolates.1 The group containing the prototype was assigned as E7-A. The HPV58 prototype (http://www.ncbi.nlm.nih.gov/genbank accession no. NC_001443) was used as reference.2 No. (%) of each sequence pattern found among the 401 samples examined.3 Nucleotide positions where variations were detected are written vertically across the top. Sites with no changes are marked with dashes. Capital letters indicate alternations result in an amino acid change. Lower case letters indicate synonymous mutations.4 The one letter amino acid code is used. The position of amino acid change is stated numerically. The letter preceding this number refers to the reference amino acid, and the letter following refers to its substitution.5 Lineage and sublineage classification based on full genomes or concatenated E6-E7-E2-E5-L1-PCR sequences described in previous studies.15,16 Some E7 sequence patterns are not lineage/sublineage-specific. –, no sublineage assigned. NA, not applicable. Single nucleotide substitutions diagnostic for E7 variants are boxed. GenBank accession numbers for E7 sequences are JX896403 - JX896423.
Geographical distribution
The frequencies of different E6 and E7 sequence variation patterns among isolates collected from different parts of the world are shown in Figure 4. E6-A was the most common pattern found overall (Fig. 4a). In Asia, Americas and Europe, E6-A predominated and accounted for 86–96% of isolates. In Africa, E6-A (51%) cocirculated with E6-C (40%). E6-A was significantly less frequent in Africa compared to the other three regions (p = 0.003 for Americas, p < 0.001 for Asia and Europe); whereas E6-C was significantly more frequent in Africa than in other regions (p < 0.001 for all three regions).
Figure 4.
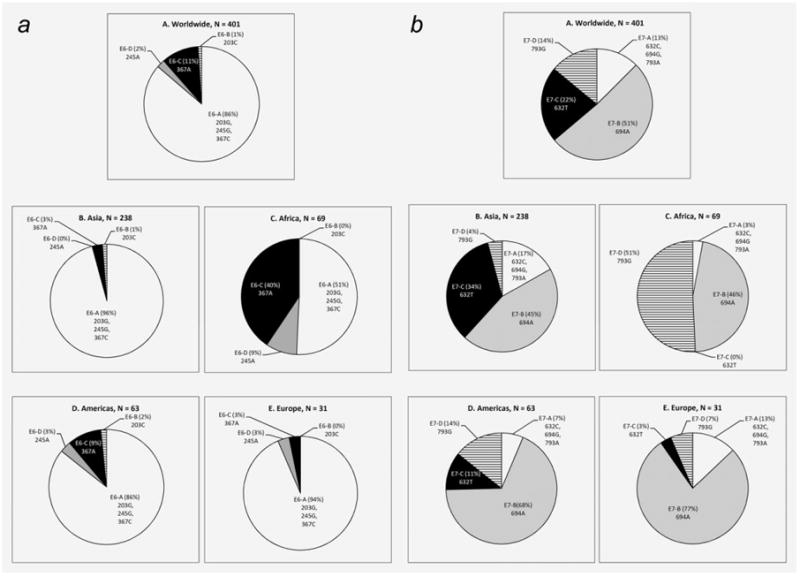
Geographical distribution of HPV58 isolates according to E6 and E7 sequence variation patterns. (a) E6; (b) E7.
The geographical distribution of E7 sequence patterns was more diverse (Fig. 4b). In Asia, both E7-B and E7-C patterns were commonly found, whereas E7-B and E7-D patterns codominated in Africa. E7-C was significantly more frequent in Asia (p < 0.001 for all three regions), whereas E7-D was significantly more frequent in Africa (p < 0.001 for all three regions).
Risk association with cervical neoplasia
Among the 401 samples examined, 33 were histology-confirmed invasive cervical cancer, 31 were CIN3 and 79 were normal cervices as defined by normal cytology or no lesion detected at colposcopy or biopsy. These cases were used to analyze the risk association of sequence variations. Two E7 substitutions (G41R and G63D) showed a significantly lower risk, and the other two E7 substitutions (T20I and G63S) showed a significantly higher risk based on univariate analysis (Table 2). All isolates with G41R also carried G63D, and a significant interaction was found (p = 0.023). Similarly, all isolates with T20I also carried G63S, and a significant interaction was found (p = 0.002). We therefore selected G41R and T20I into two separate final regression models, respectively, controlling for age and country of origin. It was found that T20I and/or G63S conferred an independent increase in risk for CIN3 and invasive cervical cancer (adjusted odds ratio 4.44, 95% CI. 1.51–13.05, p = 0.007). The risk association observed for G41R and/or G63D was no longer significant after multivariate analysis.
Table 2. Univariate analysis on risk association of HPV58 E6 and E7 amino acid substitutions.
| Cervical pathology1 | ||||
|---|---|---|---|---|
|
|
||||
| Amino acid substitution | Normal (N = 79) | CIN3/ICC (N = 64) | p-value2 | Odds ratio (95% CI)3 |
| E6 | ||||
|
| ||||
| E25K | 0 | 1 | 0.448 | – |
|
| ||||
| E32Q | 3 | 0 | 0.253 | – |
|
| ||||
| V46I | 1 | 1 | 1.000 | 1.24 (0.00–46.34) |
|
| ||||
| V46T | 1 | 0 | 1.000 | – |
|
| ||||
| D86E | 4 | 1 | 0.380 | 0.30 (0.01–2.94) |
|
| ||||
| D86N | 1 | 0 | 1.000 | – |
|
| ||||
| K93N | 6 | 4 | 0.987 | 0.81 (0.18–3.45) |
|
| ||||
| C95R | 1 | 0 | 1.000 | – |
|
| ||||
| N97D | 1 | 1 | 1.000 | 1.24 (0.00–46.34) |
|
| ||||
| N97E | 1 | 0 | 1.000 | – |
|
| ||||
| E7 | ||||
|
| ||||
| R9K | 0 | 2 | 0.199 | – |
|
| ||||
| T20I | 15 | 27 | 0.004 | 3.11 (1.38–7.07) |
|
| ||||
| G41R | 46 | 25 | 0.035 | 0.46 (0.22–0.95) |
|
| ||||
| T50I | 1 | 0 | 1.000 | – |
|
| ||||
| G63D | 47 | 25 | 0.024 | 0.44 (0.21–0.90) |
|
| ||||
| G63H | 1 | 1 | 1.000 | 1.24 (0.00–46.34) |
|
| ||||
| G63S | 18 | 27 | 0.021 | 2.47 (1.13–5.44) |
|
| ||||
| T74A | 7 | 2 | 0.188 | 0.33 (0.05–1.84) |
|
| ||||
| D76E | 7 | 2 | 0.188 | 0.33 (0.05–1.84) |
|
| ||||
| V77A | 1 | 6 | 0.045 | 8.07 (0.92–182.76) |
Histology-confirmed CIN3 or invasive cervical cancer. Normal, no abnormalities detected by cytology or colposcopy or biopsy.
2 × 2 comparison between samples with and without the indicated amino acid substitution using χ2 test or Fisher's exact test. Significant associations are bolded.
–, odds ratio undefined; CI, confidence interval.
Discussion
Our study has delineated the sequence variations of two onco-protein-encoding genes of HPV58. This dataset provides essential information for further in depth studies to elucidate the epidemiology of this characteristic HPV type that exhibits a higher disease attribution in certain ethnogeographical groups. While our study has examined a large series of samples covering a wide range of geographical regions, one should note that the number of samples available from certain areas was small and might not be representative of those areas.
The degree of nucleotide sequence variation among HPV58 isolates was found to be small (maximum 2% for E6, 3% for E7), as expected for HPV in general.15,25–27 Phylogenetically related E6 and E7 sequence patterns could be identified by distinct signatures of nonsynonymous sequence variations. These molecular signatures not only provide convenient markers for large-scale epidemiological studies, but the substitutions themselves may carry biological implications.
Overexpression of E6 and E7 proteins is required to maintain the transformed phenotype of keratinocytes, and thus they are constitutively retained in cancer cells and not affected by viral genome disruption resulting from integration. Therefore, E6 and E7 are good diagnostic targets, particularly for type-specific detection as E6 and E7 genes are diverse among HPV types. Our results show that the sequences of E7 were more variable than those of E6 and more heterogenously distributed across the world. This observation of HPV58 is in contrast to those reported for HPV16, 31, 33, 35 and 52 where the E7 ORF is less variable, and therefore E7 is often chosen as the target for diagnostic detection of HPV16.28,29 Based on the current findings, E6 rather than E7 is a better target for type-specific detection of HPV58.
The first clone of HPV58 was obtained from a Japanese patient with invasive cervical cancer.30 This strain is regarded as the prototype and commonly used for primer design. The E6-A2 and E7-A1 sequence pattern, as assigned by our study, corresponds, respectively, to the E6 and E7 sequence of the prototype. Our data showed that these “prototype-like” sequences were uncommon (10%) in Asia and rare (<3%) in other regions. Primers and probes designed based on the prototype sequence may have the problem of mismatch with the more commonly found strains.
Geographical predilection in the distribution of E6 and E7 sequence patterns was observed. E6-A predominated in most parts of the world except Africa, where it codominated with E6-C that was uncommon elsewhere. Two characteristics in the distribution of E7 sequence variation patterns were observed. First, E7-D with a nucleotide signature of 793G was a common sequence pattern found in 51% of isolates from Africa but uncommon (14%) in Americas and rare in Asia (4%) and Europe (7%). The second observation was that E7-C with an amino acid substitution signature of T20I and G63S was commonly found in Asia (34%) and Americas (11%) but rare elsewhere. Geographical variation in the distribution of HPV58 E6 and E7 sequence patterns should be considered in designing diagnostic assays.
HPV58 is rare worldwide but attributes to a higher proportion of cervical cancers in East Asia and some parts of Latin America. Our study attempted to identify HPV58 E6 and E7 sequence variations that might carry a higher oncogenic risk and examined whether the distribution of these variations could account for the ethnogeographical predilection in disease burden. Isolates carrying G63S and/or T20I were found to have an independent increase in risk for CIN3 and invasive cervical cancer. Only one variant harbored G63S alone without T20I. This variant was only found in four samples precluding analysis on these two amino acid variations separately.
The high-risk mutations T20I and G63S were found in 33% of isolates in Asia and 10% in Americas but rare in Europe (3%) and Africa (0%). This distribution is reminiscent of the geographical predilection in cervical cancers associated with HPV58. A previous study from southern China (Hong Kong) also revealed that substitutions T20I and/or G63S were associated with a 6.9-fold increase in odds ratio for developing invasive cervical cancer, and the age of patients were significantly younger than those infected with other variants of HPV58.20 Another study from eastern China (Zhejiang) also reported a significant positive trend of association between these substitutions and the severity of cervical lesions.19
It has been shown that HPV58 variants can be divided into four lineages, which is mainly based on the sequence variations within the long control region (LCR).15,16 Although, naturally occurring LCR sequence variations have been shown to carry different promoter activity in vitro,31 two previous studies that analyzed epidemiological risk association based on the LCR-lineage classification system did not reveal any significant differences among the lineages or sublineages.16,18 This is special for HPV58 as an association between LCR polymorphisms and lesion has been reported for other types. Our study suggested that the nucleotide sequence variations at E7 are more useful for predicting the risk of HPV58 variants. In vitro study to compare the transforming activities among E7 proteins carrying these amino acid substitutions is ongoing.
In conclusion, this study provides a comprehensive analysis on the sequence variation of E6 and E7 of HPV58 from a worldwide perspective. E6 is less variable and more appropriate as a target for type-specific diagnostic detection. Geographical variation in the distribution of HPV58 E6 and E7 sequence variations exists, which should be considered when designing diagnostic assays and therapeutic vaccines. Amino acid substitution(s) T20I and/or G63S at E7 is(are) associated with a higher risk for CIN3 and invasive cervical cancer. This high-risk sequence signature is more commonly found in isolates from Asia and the Americas than elsewhere, resembling the geographical predilection of cervical cancers attributed to HPV58.
What's new?
For unknown reasons, human papillomavirus (HPV) 58 accounts for a significant proportion of cervical cancers in East Asia and parts of Latin America, but it is uncommon elsewhere. In this study, the authors analyzed the E6 and E7 genes from HPV58 samples from around the world. They found that patients with an HPV58 variant containing T20I and G63S substitutions in E7 had a greater risk of developing cancer. This variant was also more prevalent in Asia than in other regions, and may therefore help to explain the higher disease burden observed in this region.
Acknowledgments
The authors are grateful to the contribution of Dr. Sergio Andres Tonon and Dr. Cláudia Renata F. Martins who unfortunately have passed away before the completion of this study. Dr. Francois Coutlée's research on HPV variants is supported by the Cancer Society of Canada. Dr. Federico De Marco is partly supported by the Italian Ministry of Foreign Affairs, DGPC Uff V and by the Italian Ministry of Health. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Dr. Francois Coutlée has received financial support for research projects and honoraria for oral presentations on HPV from Roche
Diagnostics and Merck. Dr. Karen Smith McCune is a member of the Scientific Advisory Board and owner of shares of stock in OncoHealth Inc., a startup company developing diagnostic assays for cervical cancer screening.
Grant sponsors: International Centre for Genetic Engineering and Biotechnology (ICGEB) (project no. CRP/CHN08-03)
References
- 1.Ferlay J, Shin HR, Bray F, et al. Estimates of worldwide burden of cancer in 2008 : GLOBOCAN 2008. Int J Cancer. 2010;127:2893–917. doi: 10.1002/ijc.25516. [DOI] [PubMed] [Google Scholar]
- 2.de Sanjose S, Quint WG, Alemany L, et al. Human papillomavirus genotype attribution in invasive cervical cancer: a retrospective cross-sectional worldwide study. Lancet Oncol. 2010;11:1048–56. doi: 10.1016/S1470-2045(10)70230-8. [DOI] [PubMed] [Google Scholar]
- 3.Li N, Franceschi S, Howell-Jones R, et al. Human papillomavirus type distribution in 30,848 invasive cervical cancers worldwide: variation by geographical region, histological type and year of publication. Int J Cancer. 2011;128:927–35. doi: 10.1002/ijc.25396. [DOI] [PubMed] [Google Scholar]
- 4.Shah W, Hongwei C, Jin Z, et al. The prevalence of human papillomavirus type 58 in Chinese patients with cervical carcinoma and its influence on survival. Clin Oncol (R Coll Radiol) 2009;21:768–74. doi: 10.1016/j.clon.2009.09.003. [DOI] [PubMed] [Google Scholar]
- 5.Huang S, Afonina I, Miller BA, et al. Human papillomavirus types 52 and 58 are prevalent in cervical cancers from Chinese women. Int J Cancer. 1997;70:408–11. doi: 10.1002/(sici)1097-0215(19970207)70:4<408::aid-ijc6>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 6.Hwang T. Detection and typing of human papillomavirus DNA by PCR using consensus primers in various cervical lesions of Korean women. J Korean Med Sci. 1999;14:593–9. doi: 10.3346/jkms.1999.14.6.593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chan PK, Ho WC, Yu MY, et al. Distribution of human papillomavirus types in cervical cancers in Hong Kong: current situation and changes over the last decades. Int J Cancer. 2009;125:1671–7. doi: 10.1002/ijc.24495. [DOI] [PubMed] [Google Scholar]
- 8.Ding DC, Hsu HC, Huang RL, et al. Type-specific distribution of HPV along the full spectrum of cervical carcinogenesis in Taiwan: an indication of viral oncogenic potential. Eur J Obstet Gynecol Reprod Biol. 2008;140:245–51. doi: 10.1016/j.ejogrb.2008.03.014. [DOI] [PubMed] [Google Scholar]
- 9.Asato T, Maehama T, Nagai Y, et al. A large case-control study of cervical cancer risk associated with human papillomavirus infection in Japan, by nucleotide sequencing-based genotyping. J Infect Dis. 2004;189:1829–32. doi: 10.1086/382896. [DOI] [PubMed] [Google Scholar]
- 10.Herrero R, Hildesheim A, Bratti C, et al. Population-based study of human papillomavirus infection and cervical neoplasia in rural Costa Rica. J Natl Cancer Inst. 2000;92:464–74. doi: 10.1093/jnci/92.6.464. [DOI] [PubMed] [Google Scholar]
- 11.Canche JC, López IR, Suárez NG, et al. High prevalence and low E6 genetic variability of human papillomavirus 58 in women with cervical cancer and precursor lesions in Southeast Mexico. Mem Inst Oswaldo Cruz. 2010;105:144–8. doi: 10.1590/s0074-02762010000200006. [DOI] [PubMed] [Google Scholar]
- 12.Fernandes JV, Meissner RV, Carvalho MG, et al. Prevalence of human papillomavirus in archival samples obtained from patients with cervical pre-malignant and malignant lesions from Northeast Brazil. BMC Res Notes. 2010;3:96. doi: 10.1186/1756-0500-3-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Camara GN, Cerqueira DM, Oliveira AP, et al. Prevalence of human papillomavirus types in women with pre-neoplastic and neoplastic cervical lesions in the Federal District of Brazil. Mem Inst Oswaldo Cruz. 2003;98:879–83. doi: 10.1590/s0074-02762003000700003. [DOI] [PubMed] [Google Scholar]
- 14.Paesi S, Serafini EP, Barea F, et al. High prevalence of human papillomavirus type 58 in patients with cervical pre-malignant lesions in southern Brazil. J Med Virol. 2009;81:1270–5. doi: 10.1002/jmv.21410. [DOI] [PubMed] [Google Scholar]
- 15.Chen Z, Schiffman M, Herrero R, et al. Evolution and taxonomic classification of human papillomavirus 16 (HPV16)-related variant genomes: HPV31, HPV33, HPV35, HPV52, HPV58 and HPV67. PLoS One. 2011;6:e20183. doi: 10.1371/journal.pone.0020183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chan PK, Luk AC, Park JS, et al. Identification of human papillomavirus type 58 lineages and the distribution worldwide. J Infect Dis. 2011;203:1565–73. doi: 10.1093/infdis/jir157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schiffman M, Rodriguez AC, Chen Z, et al. A population-based prospective study of carcinogenic human papillomavirus variant lineages, viral persistence, and cervical neoplasia. Cancer Res. 2010;70:3159–69. doi: 10.1158/0008-5472.CAN-09-4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chang YJ, Chen HC, Lee BH, et al. CBCSP-HPV Study Group. Unique variants of human papillomavirus genotypes 52 and 58 and risk of cervical neoplasia. Int J Cancer. 2011;129:965–73. doi: 10.1002/ijc.25724. [DOI] [PubMed] [Google Scholar]
- 19.Ding T, Wang X, Ye F, et al. Distribution of human papillomavirus 58 and 52 E6/E7 variants in cervical neoplasia in Chinese women. Gynecol Oncol. 2010;119:436–43. doi: 10.1016/j.ygyno.2010.08.032. [DOI] [PubMed] [Google Scholar]
- 20.Chan PK, Lam CW, Cheung TH, et al. Association of human papillomavirus type 58 variant with the risk of cervical cancer. J Natl Cancer Inst. 2002;94:1249–53. doi: 10.1093/jnci/94.16.1249. [DOI] [PubMed] [Google Scholar]
- 21.Tamura K, Peterson D, Peterson N, et al. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–9. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Delport W, Poon AF, Frost SD, et al. Datamonkey 2010: a suite of phylogenetic analysis tools for evolutionary biology. Bioinformatics. 2010;26:2455–7. doi: 10.1093/bioinformatics/btq429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pond SL, Frost SD. Datamonkey: rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics. 2005;21:2531–3. doi: 10.1093/bioinformatics/bti320. [DOI] [PubMed] [Google Scholar]
- 24.Bland JM, Altman DG. Multiple significance tests: the Bonferroni method. BMJ. 1995;310:170. doi: 10.1136/bmj.310.6973.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stewart AC, Eriksson AM, Manos MM, et al. Intratype variation in 12 human papillomavirus types: a worldwide perspective. J Virol. 1996;70:3127–36. doi: 10.1128/jvi.70.5.3127-3136.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Prado JC, Calleja-Macias IE, Bernard HU, et al. Worldwide genomic diversity of the human papillomaviruses-53, 56, and 66, a group of high-risk HPVs unrelated to HPV-16 and HPV-18. Virology. 2005;340:95–104. doi: 10.1016/j.virol.2005.06.024. [DOI] [PubMed] [Google Scholar]
- 27.Calleja-Macias IE, Villa LL, Prado JC, et al. Worldwide genomic diversity of the high-risk human papillomavirus types 31, 35, 52, and 58, four close relatives of human papillomavirus type 16. J Virol. 2005;79:13630–40. doi: 10.1128/JVI.79.21.13630-13640.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen Z, Terai M, Fu L, et al. Diversifying selection in human papillomavirus type 16 lineages based on complete genome analyses. J Virol. 2005;79:7014–23. doi: 10.1128/JVI.79.11.7014-7023.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chan PK, Lam CW, Cheung TH, et al. Human papillomavirus type 16 intratypic variant infection and risk for cervical neoplasia in southern China. J Infect Dis. 2002;186:696–700. doi: 10.1086/342048. [DOI] [PubMed] [Google Scholar]
- 30.Matsukura T, Sugase M. Molecular cloning of a novel human papillomavirus (type 58) from an invasive cervical carcinoma. Virology. 1990;177:833–6. doi: 10.1016/0042-6822(90)90560-e. [DOI] [PubMed] [Google Scholar]
- 31.Raiol T, de Amorim RM, Galante P, et al. HPV-58 molecular variants exhibit different transcriptional activity. Intervirology. 2011;54:146–50. doi: 10.1159/000320199. [DOI] [PubMed] [Google Scholar]