

Abstract
Immune complexes formed between monoclonal antibodies (mAbs) and toxins can neutralize toxicity in vivo by multiple mechanisms. Toxin sequestration and clearance by mAbs may be improved by enhancing their ability to bind to red blood cells (RBCs) through immune adherence. This can be achieved by converting the mAbs to heteropolymers (HPs), which are antigen-specific mAbs cross-linked to mAbs targeting the complement receptor (CR1), a protein that is expressed on the surface of RBCs in primates and mediates delivery of complement C3b-containing immune complexes to tissue macrophages. Conversion of mAbs to HPs has been shown to enhance clearance of multivalent antigens from the blood circulation, but the interaction of HPs with monovalent toxins has not been examined. Using botulinum neurotoxin (BoNT) as a model system, we studied the effect of conversion of a pair of BoNT-specific mAbs into HPs on toxin neutralization and handling in vivo. Two HPs given in combination had 166-fold greater potency than un-modified mAbs, neutralizing 5,000 LD50 BoNT, when tested in transgenic mice expressing human CR1 on RBC membranes. Improvement required adherence of BoNT to the RBC in vivo and 2 HPs, rather than an HP + mAb pair. The HP pair bound BoNT to RBCs in the circulation for 2 hours, in comparison to BoNT-neutralizing anti-serum, which induced no detectable RBC binding. HP pairs exhibited enhanced uptake by peritoneal macrophages in vitro, compared to pairs of mAbs or mAb + HP pairs. In a post-exposure therapeutic model, HPs gave complete protection from a lethal BoNT dose up to 3 hours after toxin exposure. In a pre-exposure prophylaxis model, mice given HP up to 5 days prior to BoNT administration were fully protected from a lethal BoNT dose. These studies elucidate general mechanisms for the neutralization of toxins by HP pairs and demonstrate the potential utility of HPs as BoNT therapeutics.
Keywords: botulinum neurotoxin, monoclonal antibody, immune adherence, Type 1 complement receptor, heteropolymer, peritoneal macrophage, toxin clearance
1. Introduction
Botulinum neurotoxins (BoNT) are a serologically diverse family of molecules produced by organisms of the genus Clostridium. BoNTs are the most potent biological toxins known and have been designated as category A select bioterror agents (Arnon et al., 2001). BoNTs induce peripheral neuromuscular and autonomic paralysis by inhibiting cholinergic function. The process of intoxication proceeds by a number of steps, generally beginning with either oral or inhalational exposure. BoNT crosses the intestinal or respiratory epithelium and then transits through the blood circulation to reach its target sites, cholinergic nerve endings at neuromuscular junctions (NMJ) (Simpson, 2013). At the NMJ, BoNT is internalized by the presynaptic neuron through endocytosis. Within the neuron, the BoNT catalytic light chain domain exits the endocytic vesicle and enters the cytoplasm, where it cleaves proteins that are required for the release of acetylcholine in response to neuronal stimulation. Once BoNT has been internalized by a nerve ending and has cleaved its substrate, the nerve ending is no longer functional. Therefore, BoNT countermeasures need to prevent interaction of the toxin with cholinergic nerve endings. Methods that use monoclonal antibodies (mAbs) to sequester BoNT in the blood circulation and enhance clearance can contribute to BoNT neutralization by interfering with a key step in BoNT intoxication.
Because BoNT exists in 7 known serotypes and multiple sub-serotypes that can differ dramatically in mAb binding and sensitivity, a comprehensive biodefense preparedness strategy for BoNT exposure may require dozens of different mAbs (Hill et al., 2007; Smith et al., 2005). The primary motivation for the present study is that mAbs capable of binding to multiple BoNT serotypes appear to be less potent at neutralization than single serotype-specific mAbs, so optimizing BoNT sequestration and clearance may be important for creating a definitive, poly-specific BoNT therapeutic (Garcia-Rodriguez et al., 2011).
Antibody binding induces rapid clearance of BoNT from the bloodstream through sequestration of BoNT in the liver and spleen (Ravichandran et al., 2006). Clearance requires binding of polyclonal antiserum or at least three distinct antibodies (L. Simpson and F. Al-Saleem, unpublished observations) (Nowakowski et al., 2002; Ravichandran et al., 2006). The mechanism is extremely potent, with a capacity of neutralizing > 10,000 LD50 BoNT, and occurs within minutes of intravenous injection (Nowakowski et al., 2002; Ravichandran et al., 2006). This clearance can also be induced with polypeptide-tagged single-chain variable fragments (scFv) that form immune complexes when mixed with a mAb specific for the polypeptide tag (Sepulveda et al., 2010). The mechanism for clearance of BoNT in an immune complex likely involves capture by Fcγ receptor-bearing fixed tissue macrophages (Takai, 2005). Complement-mediated mechanisms may contribute to this process, as a study in humans showed that a proportion of antibody-containing immune complexes can incorporate complement C3b and adhere to red blood cells (RBCs) through complement receptor type 1 (CR1) (Davies et al., 1990).
The ability of mAbs to sequester antigens in the blood circulation and deliver them to fixed tissue macrophages can be enhanced by directly binding them to RBCs through CR1 binding. “Heteropolymers” (HPs) are cross-linked mAb complexes in which one of the mAbs is specific for CR1 and the other mAb binds to a specific antigen (Lindorfer et al., 2001a). HPs are superior to un-modified mAbs in promoting antigen clearance. HP + antigen complexes bound to RBCs are taken up and processed by macrophages using essentially the same mechanism by which C3b-opsonized antigens bound to RBCs are cleared (Mohamed et al., 2005). This increases the efficiency of clearance of antigen from the circulation. This process of immune adherence may contribute to the defense against bacteria and viral pathogens via sequestration, preventing interaction with susceptible tissues.
In a previous study, we induced RBC immune adherence of BoNT + mAb complexes using a fusion protein (FP) that comprised a streptavidin molecule fused to an scFv specific for the RBC membrane protein glycophorin (Adekar et al., 2011). The FP enhanced BoNT neutralization of a pair of mAbs 166-fold by molar ratio. Compared to targeting glycophorin, which primarily plays a structural role on the RBC surface, targeting of CR1 may differ in its mechanism of neutralization because it may replicate aspects of complement-mediated immune complex clearance. HPs may also improve clearance through better interaction with Fcγ receptor-bearing fixed tissue macrophages, because they each contain two Fcγ domains, double that of IgG + FP complexes. We were also interested in studying the interaction of HPs with heterodimeric toxins, such as BoNT, which may behave differently from previously studied HPs that target multivalent antigens, such as phage, bacteria, and IgM (Lindorfer et al., 2001a; Lindorfer et al., 2001b; Mohamed et al., 2005).
2. Materials and Methods
2.1. Monoclonal antibodies and conversion into heteropolymers
We used human mAbs specific for either the BoNT serotype A (BoNT/A) heavy chain or light chain A, referred to as 6A and 4LCA, respectively; the anti-CR1 mouse IgGs mAbs 7G9 and HB8592, and the isotype control 7B7 (anti-ΦX174), which have all been described previously (Adekar et al., 2008a; Adekar et al., 2008b; Lindorfer et al., 2001a). The HPs were constructed by chemical cross-linking as previously described (Lindorfer et al., 2001b). The final products were subjected to gel filtration in borate saline buffer on Superose 6 (GE Healthcare Life Sciences, Piscataway, NJ), which was calibrated with monomeric IgG, in order to separate cross-linked from monomeric IgG. Cross-linked HP products were pooled and stored at 4°C. The specific HPs are noted by the conventions we have previously described (Lindorfer et al., 2001a). For example, the anti-botulinum neurotoxin heavy chain A mAb (6A), cross-linked with anti-CR1 mAb (7G9), is 6A X 7G9. Here, these names have been abbreviated, with the suffixes HP, HP-HB, and HP-CTRL denoting HPs containing the 7G9, HB8592, or 7B7 mAbs, respectively (e.g. 6A-HP, 6A-HP-HB, 6A-HP-CTRL, 4LCA-HP, 4LCA-HP-HB, and 4LCA-HP-CTRL).
2.2. Tg-hCR1 transgenic mouse colony breeding and genotyping
Tg-hCR1 transgenic mice (courtesy of Dr. Robert W. Finberg) express the human complement receptor (hCR1) gene under the control of the RBC-specific GATA1 promoter (Repik et al., 2005). Tg-hCR1 heterozygous breeders were mated with C57BL/6 mice (Taconic, Hudson, NY). hCR1 positive animals were detected using PCR of tail DNA extracted using DNeasy Blood and Tissue Kit (Qiagen, Valencia, CA). Amplification of DNA was performed using GoTaq Flexi DNA polymerase (Promega, Madison, WI). CR1 forward and reverse primers reported previously, were (5′-ACCCTTTCTGTCC-TCACA-3′) and (5′-TTTCTCCCTCCGCTTCCAGTTG-3′) (Repik et al., 2005). Thermal cycling consisted of 35 cycles of 94°C, 30 sec, 60°C 30 sec, 72°C 60 sec. RBC hCR1 expression was verified by flow cytometry with the anti-mouse TER-119 FITC (eBiosciences, San Diego, CA) and anti-CR1-PE (Southern Biotechnology, Birmingham, AL) on a BD FACSCantoII (Becton Dickson, Franklin Lakes, NJ), using FlowJo 8.8.6. software (Tree Star, Ashland, OR).
2.3. Analysis in vitro of HP and HP complexes binding to RBCs
Blood from Tg-hCR1 mice was collected in heparinized tubes and RBCs were isolated. The RBCs were washed with 200 μl PBS/1% BSA (PBSA) and centrifuged at 326 × g in a microfuge. HC50A, the 50 kD C-terminal domain of BoNT serotype A (13), was biotinylated using a FluoReporter Mini-biotin-XX protein labeling kit (Invitrogen, Carlsbad, CA). Biotinylated HC50A (BIOT-A) was incubated with 1:100 diluted PE-Streptavidin (PE-SA; Jackson ImmunoResearch, West Grove, PA), rotating for 30 min at 4° C. BIOT-A with PE-SA was then added to RBCs with 20 ng HP and anti-human IgG APC (Jackson Immunoresearch), incubated at RT for 30 min, washed twice in PBSA, resuspended in a final volume of 1 ml PBSA, and analyzed by flow cytometry for RBCs that were “double positive”, thus indicating that both HP and biotinylated HC50A were bound to the RBCs.
2.4. BoNT proteins
Serotype A1 BoNT (BoNT/A) was obtained from Metabiologics, Inc. (Madison, WI). The recombinant 50 kD C-terminal domain (HC50A) and a recombinant inactive BoNT/A (RI-BoNT) were produced in E. coli following published methods (Pier et al., 2008; Ravichandran et al., 2007).
2.5. Analysis of RBC binding by HPs in vivo
50 ng of biotinylated RI-BoNT was mixed with 6 μg each of 6A-HP and 4LCA-HP or 25 μl rabbit anti-BoNT serum and injected intravenously (i.v.) into Tg-hCR1 mice. RBCs were collected at 5, 30, 90, 120 minutes and 24 hours following the injection, stained with PE-SA, and assessed by flow cytometry.
2.6. Macrophage uptake of BoNT HP complexes
Tg-hCR1 transgenic mice received intraperitoneal (i.p.) injections with sterile 3% Thioglycollate Medium, Brewer Modified solution (Thermo Scientific) (Zhang et al., 2008). After 4 days, elicited peritoneal macrophages were collected using cold PBS, centrifuged at 1000 rpm for 10 min at 4° C and washed with DMEM containing 20% FBS, 100 U/ml penicillin and 100 μg/ml streptomycin. 106 cells were plated on cover slips in 1 ml DMEM in 24 well tissue culture plates and incubated at 37° C (5% CO2). After 2 hours, non-adherent cells were removed by 3 washes with warm DMEM. RI-BoNT was labeled using the Alexa Fluor 488 Microscale Protein Labeling Kit (Invitrogen). 15 ng labeled BoNT was incubated with antibody and HP reagents as follows: no mAb or HP (negative control), 15 μg purified polyclonal rabbit IgG against BoNT, 8 μg each 6A and 4LCA, 8 μg 6A and 4 μg 4LCA-HP, 8 μg 6A-HP and 4 μg 4LCA, 4 μg each 6A-HP-CTRL and 4LCA-HP-CTRL, or 4 μg each 6A-HP and 4LCA-HP, all diluted in a total of 100 μl volume of DMEM and incubated at 20° C for 1 hour. Each mixture was added to a cover slip and incubated at 4° C for 30 min and then another 30 min at 37° C. Cover slips were washed with serum free medium 3 times and fixed with 4% paraformaldehyde solution for 30 min at 4° C and washed 3 times with PBS. The cover slips were then mounted on microscopic slides using Prolong Gold antifade reagent with 4′,6-diamidino-2-phenylindole (DAPI, Life Technologies). Images were acquired using a Carl Zeiss LSM 510 UV META inverted confocal microscope with a Plan-Apo 40X oil immersion lens at room temperature and Zeiss AIM 4.2 SP1 software (Zeiss Microimaging, Thornwood, NY).
2.7 Mouse protection assay
We incubated mixtures of the HPs and BoNT at room temperature for 1 hour prior to injection in the tail veins of mice. Mice were sedated with isoflurane prior to injection and monitored twice daily for seven days. Mice exhibiting signs of BoNT intoxication, such as paralysis, cachexia, hunched backs, eye secretions, rapid breathing, or hypokinesis were euthanized by CO2 inhalation.
3. Results
3.1. Creation and binding activities of HPs that bind BoNT
We established a model to study the effect of HPs on toxin neutralization and clearance, based on use of the BoNT-neutralizing mAb pair, 6A and 4LCA (Adekar et al., 2008b). 6A is specific for the BoNT serotype A (BoNT/A) heavy chain (HC) and 4LCA is specific for the BoNT/A light chain (LC) (Adekar et al., 2008a; Adekar et al., 2008b). These two mAbs were ideal for the present study because we have fully characterized their activity in vivo as unmodified mAbs and in studies of immune adherence induced by the FP (Adekar et al., 2011; Adekar et al., 2008b). Both mAbs were converted into HPs by cross-linking with murine mAbs, 7G9 or HB8592 or 7B7. 7G9 and HB8592 are specific for the hCR1, but bind different CR1 epitopes; 7B7 is an isotype control mAb that does not bind CR1. Following cross-linking, the HPs were separated from monomeric IgG by chromatography using a Superose 6 column (M.A. Lindorfer and R. P. Taylor, data not shown). HPs incorporating the 7G9 were named 6A-HP and 4LCA-HP, those with the HB8592 mAb were named 6A-HP-HB and 4LCA-HP-HB, and those with the control mAb 7B7 were named 6A-HP-CTRL and 4LCA-HP-CTRL.
To test the binding and activity of the HPs, we used the transgenic mouse Tg-hCR1, which expresses the human CR1 protein (hCR1) on the surface of its RBCs (Repik et al., 2005). Murine RBCs do not express a CR1 receptor that can bind complement-opsonized immune complexes, rather, their platelets perform this function using platelet-associated factor H (Alexander et al., 2001). We tested the ability of the HPs to adhere BoNT to the Tg-hCR1 RBC surface by mixing the HPs and biotinylated RI-BoNT holotoxin with RBCs and detecting the bound complexes with PE:SA and an APC anti-human Fcγ secondary (Figure 1). A double positive population of RBCs was only seen with the CR1-specific HPs 6A-HP (75.5%), 6A-HP-HB (76.4%), 4LCA-HP (75.4%), 4LCA-HP-HB (73.3%). Substantially less binding was observed with the two non-binding HPs, 6A-HP-CTRL (12.8%) and 4LCA-HP-CTRL (17.6%).
Figure 1.
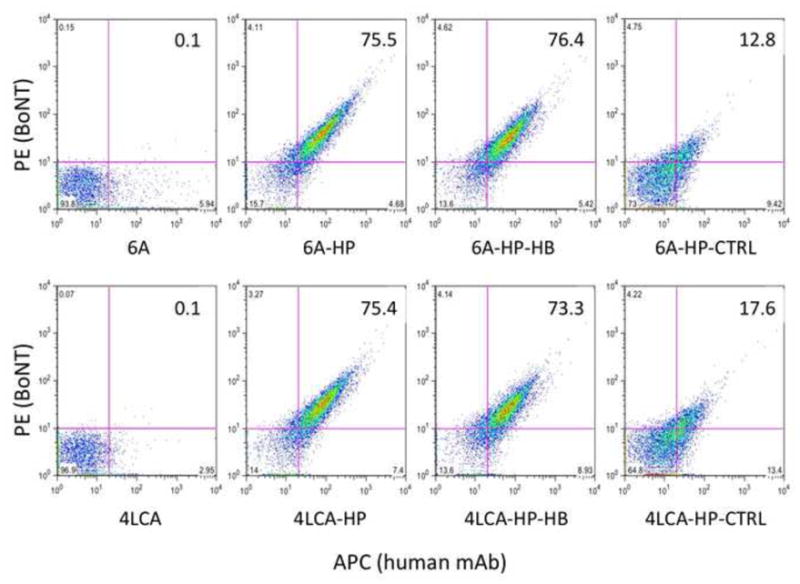
Binding of HP + BoNT complexes to Tg-hCR1 RBCs. Tg-hCR1 RBCs were incubated with biotinylated RI-BoNT/A, PE-SA, anti-human IgG APC, along with the following mAbs or HPs: 6A mAb, 6A-HP, 6A-HP-HB, 6A-HP-CTRL, 4LCA mAb, 4LCA-HP, 4LCA-HP-HB, or 4LCA-HP-CTRL. Percent double positive RBC values are given in the upper right quadrant of each diagram.
3.2. Protection conferred by HPs
We first tested whether conversion of the mAbs to HPs improved their ability to neutralize toxin in vivo. We tested the HPs in the Tg-hCR1 mouse strain (Table 1) using the standard mouse protection assay (MPA) (Pearce et al., 1994). We began with 6 μg each of the HPs injected intravenously, mixed with BoNT prior to injection. In two separate experiments with a total of 8 mice, 1/8 survived at 100 LD50 with the 6A-HP and 7/8 survived with the 4LCA-HP. This is superior to our previous results with un-modified 6A and 4LCA mAbs, which neutralized 2.5 and 25 LD50 BoNT, respectively (Adekar et al., 2008b). Challenge with 1,000 LD50 and a higher dose of 4LCA-HP (50 μg) gave no survival, with 0/5 mice surviving. When combined, the HP combination of 6A-HP + 4LCA-HP gave 93% survival at 5000 LD50s when administered at 6 μg each HP (14/15 mice surviving among four different experiments) (Table 2). An additional 5 mice survived 5,000 LD50 when given the 6A-HP-HB + 4LCA-HP-HB combination (6 μg each).
Table 1.
Neutralization of BoNT by heteropolymers (HPs)
| Antibody | mAb μg | BONT/A LD50 | Mice Alive/total | % Survival |
|---|---|---|---|---|
| 6A-HP | 12 | 100 | 1/8 | 12.5 |
| 4LCA-HP | 12 | 100 | 7/8 | 87.5 |
| 4LCA-HP | 50 | 1,000 | 0/5 | 0 |
Table 2.
Neutralization of BoNT by immune complexes containing HPs.
| Group | HP/mAb μg | BoNT/A LD50 | Mice alive/total | % survival |
|---|---|---|---|---|
| 6A-HP + 4LCA-HP | 6 + 6 | 5000 | 14/15 | 93 |
| 6A-HP + 4LCA-HP | * | 10,000 | 0/21 | 0 |
| 6A-HP + 4LCA-HP-CTRL | 6 + 6 | 5000 | 8/8 | 100 |
| 6A-HP-CTRL + 4LCA-HP | 6 + 6 | 5000 | 8/8 | 100 |
| 6A-HP-CTRL + 4LCA-HP-CTRL | 6 + 6 | 5000 | 0/8 | 0 |
| 6A-HP-HB + 4LCA-HP-HB | 6 + 6 | 5000 | 5/5 | 100 |
| 6A + 4LCA | 6 + 6 | 5000 | 0/9 | 0 |
| 6A-HP + 4LCA | 6 + 6 | 5000 | 1/6 | 17 |
| 6A + 4LCA-HP | 6 + 6 | 5000 | 0/6 | 0 |
various doses: 6 + 6, 12 + 12, or 50 + 50 μg each HP
We repeatedly attempted to neutralize 10,000 LD50, testing a total of 21 mice with the 6A-HP + 4LCA-HP combination at either 6 + 6, 12 + 12, or 50 + 50 μg each HP (Table 2). Likewise, an additional 15 mice that received the HPs containing the HB8592 mAb did not survive 10,000 LD50, tested in groups of 5 with 6A-HP + 4LCA-HP-HB, 6A-HP-HB + 4LCA-HP or 6A-HP-HB + 4LCA-HP-HB (data not shown). Successful neutralization of 5,000 LD50 with 12 μg HP total is 166-fold greater than neutralization achieved with naked 4LCA + 6A by molar ratio (1000 LD50 with 100 μg each mAb) (Adekar et al., 2008b) and is equivalent to what was achieved with the FP + mAb combination (Adekar et al., 2011).
Having established 5,000 LD50 as a dose that could be routinely survived with HP treatment, and failing to see a significant difference between 6, 12 and 50 μg HP at the 10,000 LD50 dose, we used 5,000 LD50 BoNT and 6 μg HP for testing factors that contribute to neutralizing activity. We tested HP combinations in which only one of the HPs was able to bind hCR1, but both of the HPs included the BoNT-specific mAb. We tested groups of 4 mice in 2 separate experiments (Table 2). At 5000 LD50 BoNT, either 6A-HP (CR1 binding) + 4LCA-HP-CTRL (non-CR1 binding) or 6A-HP-CTRL (non-CR1 binding) + 4LCA-HP (CR1 binding) gave full protection. The combination of the non-CR1 binding HPs provided no protection (6A-HP-CTRL + 4LCA-HP-CTRL). Moreover, pairing an RBC-binding HP with an un-modified mAb gave either 17% (6A-HP + 4LCA) or 0% survival (6A + 4LCA-HP), in 2 separate experiments testing 6 mice total for each combination (Table 2). Thus, two HPs were more potent than HP + mAb combinations and maximal neutralization required that at least one of the HPs in a pair could bind to hCR1.
3.3. Macrophage uptake by HP + mAb complexes
The finding that pairs of HPs provided better neutralization than HP + mAb combinations suggests that the macrophages may be preferentially recognizing the larger complexes, which contain 4 Fc domains. Both of the human mAbs are IgG1 subtype, which binds to macrophage Fcγ Rla (CD64) with approximately the same affinity as murine IgG2a (Takai, 2005). We tested uptake of opsonized BoNT using thioglycollate-elicited murine peritoneal macrophages from the Tg-hCR1 mice and different combinations of HPs and/or mAbs. Alexa Fluor 488-labeled BoNT holotoxin (15 ng) was mixed with either rabbit anti-BoNT/A heavy chain serum (15 μg), 6A + 4 LCA, 6A + 4LCA-HP, 6A-HP + 4LCA, 6A-HP-CTRL + 4LCA-HP-CTRL or 6A-HP + 4LCA-HP. We used 4 μg each mAb or 8 μg each HP (Figure 2). Virtually no uptake was seen with the 6A + 4LCA pair. Rare, cytoplasmic, vesicular uptake was seen with 6A mAb + 4LCA-HP and 6A-HP + 4LCA mAb pairs. Widespread cytoplasmic uptake was observed with both of the HP pairs (note the substantial green intensity associated with individual cells in Figures 2 g, h), which was somewhat greater than the uptake observed with the BoNT antiserum (Figure 2 b).
Figure 2.
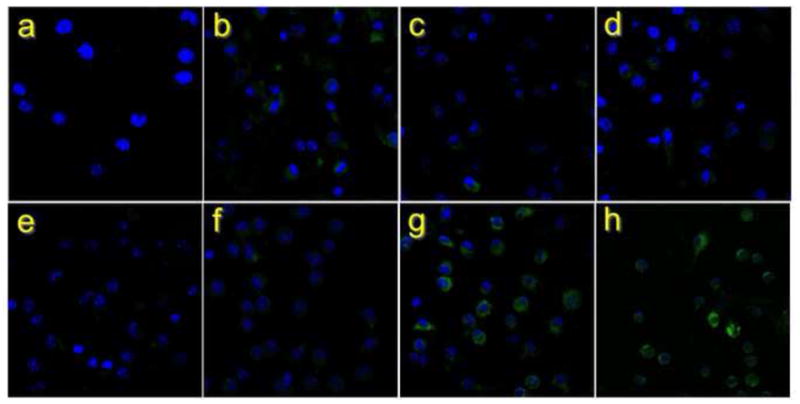
Macrophage uptake of BoNT in HP and antibody complexes. BoNT labeled with Alexa Fluor 488 was bound to the HP or BoNT-specific mAbs indicated and applied in vitro to activated peritoneal macrophages from Tg-hCR1 mice. Nuclei were stained with DAPI. Preparations include: a) Cells only; b) BoNT antiserum; c) 6A + 4LCA-HP; d) 6A-HP + 4LCA; e) BoNT alone (no HPs or mAbs); f) 6A + 4LCA mAb; g) 6A-HP-CTRL + 4LCA-HP-CTRL ; h) 6A-HP + 4LCA-HP. 80X.
We quantitated these results by measuring the Alexa-fluor corrected total cell fluorescence (CTCF) for each image using IMAGEj software (http://imagej.nih.gov/ij/) (Figure 3). Compared to 6A + 4LCA, the cells treated with 2 HPs (6A-HP and 4LCA-HP, 6A-HP-CTRL and 4LCA-HP-CTRL) or the anti-serum had significantly increased mean CTCF. BoNT uptake for the 6A + 4LCA-HP and 6A-HP + 4LCA combinations was also elevated, but to a lesser extent. Thus, conversion of the 6A and 4LCA mAbs to HPs enhanced their ability to induce BoNT uptake by macrophages. Optimum uptake required pairs of HPs (4 Fc domains, rather than 3), and this effect was independent of whether the HP contained a mAb specific for CR1 or a control mAb.
Figure 3.
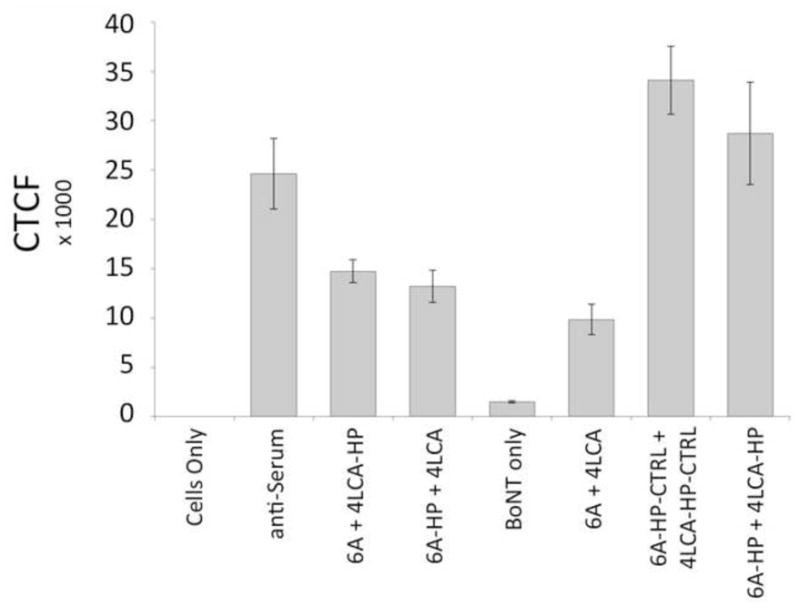
Quantification of the fluorescent signal from the experiment displayed in Figure 2, indicating uptake of Alexa-Fluor labeled BoNT by peritoneal macrophages. Each cell was measured individually using IMAGEj software and the mean corrected total cell intensity (CTCF) is shown. Error bars indicate the S.E.M.
3.4. Adherence of HP complexes to RBCs in vitro
Effective macrophage uptake suggested that the HP immune complexes should be effectively recognized by macrophages, however, the pairs of HPs that did not bind RBCs were ineffective at 5,000 LD50. Thus, some of the neutralization effect may result from improved BoNT sequestration. We tested the time course of HP-mediated adherence of BoNT to RBCs in vivo. We biotinylated the RI-BoNT and injected 6 μg along with the 6A-HP and 4LCA-HP (6 μg each) into groups of 3 mice. As a positive control for clearance, we injected three additional mice with biotinylated RI-BoNT and 25 μl undiluted rabbit anti-BoNT/A heavy chain serum. RBC-bound BoNT was assessed by incubation with PE-SA and flow cytometry at 5 min, 30 min, 90 min, 2 hours, and 24 hrs (Figure 4). BoNT was detectable on the RBCs 5 minutes after injection, peaking at 30 minutes and still evident at 2 hours (Figure 4 a, c). In contrast, mice that received BoNT and the anti-BoNT serum showed no binding at any time point (Figure 4 b, c). These results indicate that the HPs capture BoNT in the plasma by immune adherence to the surface of RBCs. But, the residence time of BoNT in the circulation is substantially longer than multivalent antigens bound to HPs or BoNT bound to anti-serum (< 20 min) (Lindorfer et al., 2001b; Ravichandran et al., 2006; Taylor et al., 1997a).
Figure 4.
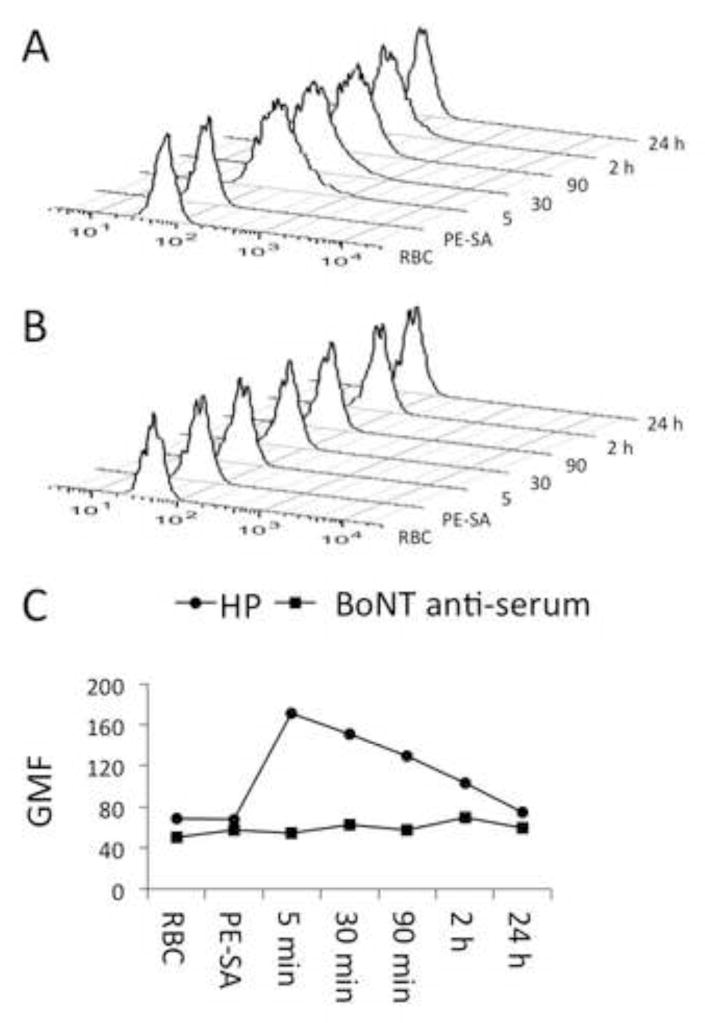
HP-mediated adherence of BoNT/A to Tg-hCR1 RBCs in vivo. Groups of three Tg-hCR1 mice received intravenous biotinylated recombinant inactive BoNT/A and either the 6A-HP + 4LCA-HP combination (Panel A) or rabbit BoNT anti-serum (Panel B). RBCs were collected at 5, 30, 90 minutes, and 2 and 24 hours following the injection and stained with PE-streptavidin (PE-SA). Histograms show the RBC PE signal over time. Panel C plots the histogram data as geometric mean fluorescence intensity (GMF) (HP RBCs, circles; anti-serum RBCs, squares).
3.5. Pre- and post-exposure protection with the HP combination
We next assessed the HPs in post-exposure and pre-exposure models, in which the HPs were administered separately from 10 LD50 BoNT. This dose corresponds to recently reported human outbreaks of BoNT/A, in which 7 subjects with severe disease had serum toxin titers of 4–16 mouse LD50/ml serum (Mazuet et al., 2012), and results in death of the mice at approximately 12 hours after injection. BoNT was delivered by i.p. injection and HP complexes were given i.v. 1, 2, 3, or 4 hours later. Six μg each of 6A-HP + 4LCA-HP were tested in groups of 5 mice monitored for survival for 5 days. In the post-exposure model, complete survival was provided by the 6A-HP + 4LCA-HP combination given up to 3 hours following BoNT injection, with 80% survival at 4 hours post-BoNT (4/5) (Figure 5). In the pre-exposure model, groups of 5 mice received the HP combination i.v., followed by i.p. 10 LD50 BoNT. When given up to 5 days (120 hours) before BoNT administration, the 6A-HP + 4LCA-HP combination completely protected the mice. Partial protection (4/5) was observed with HPs given 6 days prior to BoNT (144 hours), and none of the mice survived when given HPs given 7 days (168 hours) prior to BoNT administration.
Figure 5.
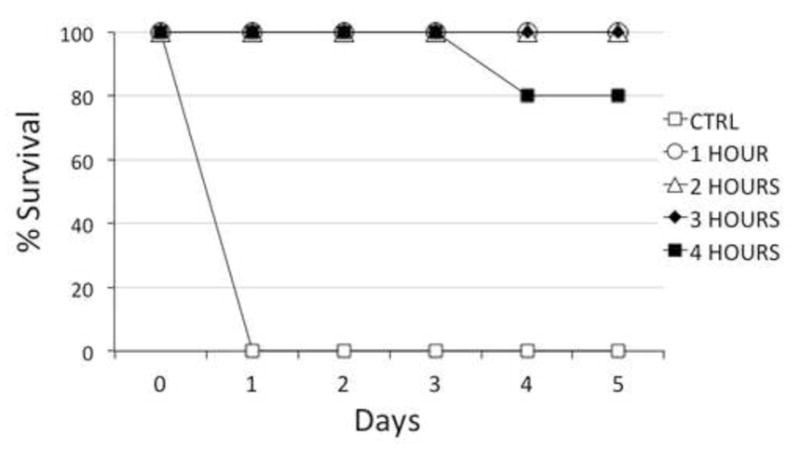
Post-exposure treatment of BoNT using the 6A-HP + 4LCA-HP combination. Groups of 5 mice received 10 LD50 BoNT i.p. This was followed by 6 μg of each HP administered i.v. at either 1, 2, 3 or 4 hours after the BoNT.
4. Discussion
The ability of mAbs to neutralize a toxin transiting through the bloodstream can be significantly enhanced through immune adherence, in which the mAb-toxin immune complex is tethered to the RBC surface. Immune adherence can potentially contribute two advantages in neutralization: toxin sequestration and improved clearance. In this study, we explored these phenomena using BoNT as a model system, converting two BoNT neutralizing mAbs into HPs capable of adhering BoNT to the RBC surface through interaction with hCR1. The HPs had 166-fold improved neutralization potency in vivo, compared to un-modified mAbs, which resulted from a combination of sequestration and improved clearance effects.
Adherence of BoNT to RBCs can limit access of the toxin into the NMJ. We observed that the HPs bound BoNT to RBCs in vitro and in vivo. RBC adherent complexes circulated in the bloodstream for at least 2 hours but were not detectable at 24 hours. BoNT neutralization at 5,000 LD50 occurred only when an HP was included that could bind RBCs; the pair of HPs that did not bind CR1 mAbs was not effective. This indicates that immune adherence-mediated sequestration contributed to BoNT neutralization. In our previous study with the FP, RBC adherence was also essential to enhanced neutralization ability (Adekar et al., 2011). Thus, RBC sequestration through immune adherence is a general mechanism for improving BoNT neutralization by mAbs in vivo.
The immune complexes formed with an HP and an un-modified mAb were less potent than those formed with two HPs. Consistent with this result, peritoneal macrophages internalized BoNT better when it was bound to two HPs rather than to an HP + mAb or mAb + mAb combination. This was independent of whether the HP pair contained a CR1-binding or non-binding mAb, indicating that the productive interaction with macrophages was based on the structure of the HP complexes, rather than any RBC binding and/or delivery effects. These data suggest that that improved BoNT clearance from the blood circulation by fixed tissue macrophages contributed to the effectiveness of the HPs via opsonization of multiple Fc domains in the HP complexes.
Our findings are in good agreement with previous reports, which examined how the degree of opsonization of antigens with IgG mAbs can influence their potential interaction with acceptor cells as well as their clearance from the bloodstream. Montero-Julian et al. reported, in a mouse model, that binding of 1 or 2 IgG mAbs to IL-6 actually increased its residence time in the circulation (Montero-Julian et al., 1995). However, when the IL-6 was chelated by 3 different IgG mAbs, clearance of the resulting immune complex from the circulation was increased substantially, with rapid uptake by the liver. They suggested that this finding reflected multivalent interaction of the IL-6 immune complex with Fc receptors on liver Kupffer cells. Similarly, optimal neutralization of BoNT requires at least three independent mAbs to induce rapid clearance from the circulation (L. Simpson and F. Al-Saleem, unpublished observations) (Nowakowski et al., 2002; Ravichandran et al., 2006).
Taylor et al. reported, in a non-human primate model, that HP constructed only with Fab′ mAb fragments could effectively mediate stable binding of ΦX174 to RBCs in the circulation (Taylor et al., 1997b). However, the bound ΦX174 was not removed from the RBCs or cleared from the bloodstream unless a second, intact anti-ΦX174 IgG mAb was infused. Reinagel et al. reported that transfer of HP-ΦX174 complexes from RBCs to macrophages was increased considerably when a second mAb (not used to construct the HP) was used to additionally opsonize the ΦX174 (Reinagel and Taylor, 2000). These results support the concept that opsonization with more IgGs allows for better recognition and uptake of substrates promoted by Fc receptors on acceptor macrophages.
An important aspect of the antigens previously studied with HPs, such as ΦX174, is that they are multivalent, capable of binding multiple copies of a single HP. In contrast, BoNT exists as a heterodimer that contains only one binding site for each HP, so the BoNT immune complexes we tested consisted of a single BoNT molecule with 2 HPs. In terms of macrophage uptake, there was a clear improvement with the HPs, compared to un-modified mAbs, but it is notable that our double HP combination was not able to neutralize the > = 10,000 LD50 achieved by some triplet BoNT-specific mAb combinations (Smith et al., 2005). The most likely explanation is that the BoNT + HP complexes were less efficient in interaction with Fc receptors than multivalent antigens bound to HPs. For example, multivalent antigens bound to HPs are completely cleared from RBCs in 10–20 minutes, rather than the ~ 2 hours we observed for BoNT + HP clearance (Lindorfer et al., 2001b; Taylor et al., 1997a). HP complexes bound to RBCs during that time could transiently release BoNT, enabling lethal intoxication.
The lack of efficient uptake of the HP + mAb complexes suggests that the Fc domains in those complexes are not ideally positioned for Fc receptor interaction. Little is known about the determinants of efficient Fc receptor recognition and uptake of immune complexes, and it is clear that simply binding three mAbs to BoNT is not sufficient to give maximal (> 10,000 LD50) neutralization (R. Sharma, F. Al-Saleem, S.K. Dessain, and L.L. Simpson, data not shown). In our case, the HC and LC binding sites on the BoNT molecule targeted by the two mAbs may be separated by as much as 130 Å, which may reduce the potential for close Fc receptor clustering on the acceptor macrophage surface (Lacy et al., 1998).
In our earlier study, the glycophorin-binding FP gave approximately the same neutralization potency as the HP tested here (5,000 LD50 with 3 μg each mAb). Maximum neutralization with the FP required that both the 6A and 4LCA mAbs be associated with an FP, so that the complex was bound to the RBCs at 2 sites. The antibodies were mixed with the tetrameric FPs in a 1:1 ratio (antibody:tetramer) so that the average number of Fc domains per BoNT molecule was 2. Thus, the enhancement of neutralization provided by the FP may differ from the HP in that it depended more on efficient sequestration on RBCs than on improved macrophage uptake.
This study extends previous work with HPs by demonstrating that they have therapeutic utility as anti-toxins. The BoNT HPs were capable of protection in vivo in the post-exposure and pre-exposure models. In the post-exposure model, protection was complete for up to three hours, which is comparable to what was demonstrated with FP complexes and other polyclonal antibody mixtures (Al-Saleem et al., 2011; Cheng et al., 2009; Sepulveda et al., 2010). This supports the concept that there is a threshold of intoxication beyond which additional antigen clearance or binding cannot be effective, so that the effectiveness of a BoNT anti-toxin will depend on the dose of BoNT received and the time elapsed between exposure and the antidote.
The pre-exposure model is relevant for passive immunization of individuals facing potential BoNT exposure, such as first responders to a BoNT contaminated site. The pair of HPs provided protection from a 10 LD50 dose of BoNT when administered up to 6 days prior to the BoNT injection. This is 2 days longer than afforded by the FP and indicates that the HP complexes have sufficient stability in vivo for prolonged protection. TThe maintenance of our HPs in the circulation may have been limited by generation of an anti-human IgG humoral immune response in the mice.
In conclusion, we have demonstrated that conversion of mAbs to HPs consisting of a toxin-specific mAb conjugated to a mAb specific for CR1 can improve toxin neutralization in vivo through a mechanism that involves RBC sequestration and improved macrophage uptake.
Sharma Highlights.
Bloodstream clearance is essential for neutralization of botulinum neurotoxin (BoNT).
Antibodies against BoNT and human complement receptor (hCR1) were linked to make HPs.
Improved BoNT neutralization required HP-mediated binding of BoNT to hCR1 on RBCs.
HP pairs induced better BoNT uptake by peritoneal macrophages than mAbs.
HP pairs were effective in pre- and post-exposure protection from BoNT.
Acknowledgments
This work was supported in part by Public Health Service grants R43AI079999 (S.P.A.) and R01AI06596 (S.K.D.) from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services. We are grateful to Robert W. Finberg of the University of Massachusetts Medical School for the Tg-hCR1 mouse strain. We thank Sarang Puranik, Cindy Chen, and Chandana Devi for technical assistance, Lisa Laury-Kleintop and Paul Simon and Minzhou Huang for technical advice and critical reading of the manuscript. Maria Yolanda Covarrubias provided assistance with microscopy at the Bioimaging Facility of the Kimmel Cancer Center (NIH Cancer Center Core grant 5 P30 CA-56036).
Abbreviations
HP names have been abbreviated: with the suffixes HP, HP-HB, and HP-CTRL denoting HPs containing the 7G9, HB8592, or 7B7 mAbs, respectively (e.g. 6A-HP, 6A-HP-HB, 6A-HP-CTRL, 4LCA-HP, 4LCA-HP-HB, and 4LCA-HP-CTRL)
- BoNT
botulinum neurotoxin
- BoNT/A
serotype A botulinum neurotoxin
- CR1
complement receptor
- Fab′
mAb antigen binding domain
- HC50A
BoNT/A recombinant 50 kD C-terminal domain
- FP
a fusion protein consisting of a streptavidin molecule and an scFv specific for glycophorin
- hCR1
human complement receptor
- HP
heteropolymer
- HRP
horseradish peroxidase
- i.p
intra-peritoneal
- i.v
intravenous
- mAb
monoclonal antibody
- mAb
monoclonal antibody
- NMJ
neuromuscular junction
- OPD
o-phenylenediamine dihydrochloride
- PBS
phosphate buffered saline
- RBCs
red blood cells
- RI-BoNT
recombinant inactive BoNT
- scFv
single-chain variable fragment
Footnotes
The content of this report is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Allergy and Infectious Diseases or the National Institutes of Health.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adekar SP, Jones RM, Elias MD, Al-Saleem FH, Root MJ, Simpson LL, Dessain SK. Hybridoma populations enriched for affinity-matured human IgGs yield high-affinity antibodies specific for botulinum neurotoxins. J Immunol Methods. 2008a;333:156–166. doi: 10.1016/j.jim.2008.01.015. [DOI] [PubMed] [Google Scholar]
- Adekar SP, Segan AT, Chen C, Bermudez R, Elias MD, Selling BH, Kapadnis BP, Simpson LL, Simon PM, Dessain SK. Enhanced neutralization potency of botulinum neurotoxin antibodies using a red blood cell-targeting fusion protein. PLoS ONE. 2011;6:e17491. doi: 10.1371/journal.pone.0017491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adekar SP, Takahashi T, Jones RM, Al-Saleem FH, Ancharski DM, Root MJ, Kapadnis BP, Simpson LL, Dessain SK. Neutralization of botulinum neurotoxin by a human monoclonal antibody specific for the catalytic light chain. PLoS ONE. 2008b;3:e3023. doi: 10.1371/journal.pone.0003023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Saleem FH, Nasser Z, Olson RM, Cao L, Simpson LL. Identification of the factors that govern the ability of therapeutic antibodies to provide postchallenge protection against botulinum toxin: a model for assessing postchallenge efficacy of medical countermeasures against agents of bioterrorism and biological warfare. J Pharmacol Exp Ther. 2011;338:503–17. doi: 10.1124/jpet.111.180653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander JJ, Hack BK, Cunningham PN, Quigg RJ. A protein with characteristics of factor H is present on rodent platelets and functions as the immune adherence receptor. J Biol Chem. 2001;276:32129–35. doi: 10.1074/jbc.M101299200. [DOI] [PubMed] [Google Scholar]
- Arnon SS, Schechter R, Inglesby TV, Henderson DA, Bartlett JG, Ascher MS, Eitzen E, Fine AD, Hauer J, Layton M, Lillibridge S, Osterholm MT, O’Toole T, Parker G, Perl TM, Russell PK, Swerdlow DL, Tonat K. Botulinum toxin as a biological weapon: medical and public health management. Jama. 2001;285:1059–70. doi: 10.1001/jama.285.8.1059. [DOI] [PubMed] [Google Scholar]
- Cheng LW, Stanker LH, Henderson TD, 2nd, Lou J, Marks JD. Antibody protection against botulinum neurotoxin intoxication in mice. Infect Immun. 2009;77:4305–13. doi: 10.1128/IAI.00405-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies KA, Hird V, Stewart S, Sivolapenko GB, Jose P, Epenetos AA, Walport MJ. A study of in vivo immune complex formation and clearance in man. J Immunol. 1990;144:4613–20. [PubMed] [Google Scholar]
- Garcia-Rodriguez C, Geren IN, Lou J, Conrad F, Forsyth C, Wen W, Chakraborti S, Zao H, Manzanarez G, Smith TJ, Brown J, Tepp WH, Liu N, Wijesuriya S, Tomic MT, Johnson EA, Smith LA, Marks JD. Neutralizing human monoclonal antibodies binding multiple serotypes of botulinum neurotoxin. Protein Eng Des Sel. 2011;24:321–31. doi: 10.1093/protein/gzq111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill KK, Smith TJ, Helma CH, Ticknor LO, Foley BT, Svensson RT, Brown JL, Johnson EA, Smith LA, Okinaka RT, Jackson PJ, Marks JD. Genetic diversity among Botulinum Neurotoxin-producing clostridial strains. J Bacteriol. 2007;189:818–32. doi: 10.1128/JB.01180-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacy DB, Tepp W, Cohen AC, DasGupta BR, Stevens RC. Crystal structure of botulinum neurotoxin type A and implications for toxicity. Nat Struct Biol. 1998;5:898–902. doi: 10.1038/2338. [DOI] [PubMed] [Google Scholar]
- Lindorfer MA, Hahn CS, Foley PL, Taylor RP. Heteropolymer-mediated clearance of immune complexes via erythrocyte CR1: mechanisms and applications. Immunol Rev. 2001a;183:10–24. doi: 10.1034/j.1600-065x.2001.1830102.x. [DOI] [PubMed] [Google Scholar]
- Lindorfer MA, Nardin A, Foley PL, Solga MD, Bankovich AJ, Martin EN, Henderson AL, Price CW, Gyimesi E, Wozencraft CP, Goldberg JB, Sutherland WM, Taylor RP. Targeting of Pseudomonas aeruginosa in the bloodstream with bispecific monoclonal antibodies. J Immunol. 2001b;167:2240–9. doi: 10.4049/jimmunol.167.4.2240. [DOI] [PubMed] [Google Scholar]
- Mazuet C, Ezan E, Volland H, Popoff MR, Becher F. Toxin detection in patients’ sera by mass spectrometry during two outbreaks of type A Botulism in France. J Clin Microbiol. 2012;50:4091–4. doi: 10.1128/JCM.02392-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohamed N, Jones SM, Casey LS, Pincus SE, Spitalny GL. Heteropolymers: a novel technology against blood-borne infections. Curr Opin Mol Ther. 2005;7:144–50. [PubMed] [Google Scholar]
- Montero-Julian FA, Klein B, Gautherot E, Brailly H. Pharmacokinetic study of anti-interleukin-6 (IL-6) therapy with monoclonal antibodies: enhancement of IL-6 clearance by cocktails of anti-IL-6 antibodies. Blood. 1995;85:917–24. [PubMed] [Google Scholar]
- Nowakowski A, Wang C, Powers DB, Amersdorfer P, Smith TJ, Montgomery VA, Sheridan R, Blake R, Smith LA, Marks JD. Potent neutralization of botulinum neurotoxin by recombinant oligoclonal antibody. Proc Natl Acad Sci U S A. 2002;99:11346–50. doi: 10.1073/pnas.172229899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce LB, Borodic GE, First ER, MacCallum RD. Measurement of botulinum toxin activity: evaluation of the lethality assay. Toxicol Appl Pharmacol. 1994;128:69–77. doi: 10.1006/taap.1994.1181. [DOI] [PubMed] [Google Scholar]
- Pier CL, Tepp WH, Bradshaw M, Johnson EA, Barbieri JT, Baldwin MR. Recombinant holotoxoid vaccine against botulism. Infect Immun. 2008;76:437–42. doi: 10.1128/IAI.00843-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravichandran E, Al-Saleem FH, Ancharski DM, Elias MD, Singh AK, Shamim M, Gong Y, Simpson LL. Trivalent vaccine against botulinum toxin serotypes A, B, and E that can be administered by the mucosal route. Infect Immun. 2007;75:3043–54. doi: 10.1128/IAI.01893-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravichandran E, Gong Y, Al Saleem FH, Ancharski DM, Joshi SG, Simpson LL. An initial assessment of the systemic pharmacokinetics of botulinum toxin. J Pharmacol Exp Ther. 2006;318:1343–51. doi: 10.1124/jpet.106.104661. [DOI] [PubMed] [Google Scholar]
- Reinagel ML, Taylor RP. Transfer of immune complexes from erythrocyte CR1 to mouse macrophages. J Immunol. 2000;164:1977–85. doi: 10.4049/jimmunol.164.4.1977. [DOI] [PubMed] [Google Scholar]
- Repik A, Pincus SE, Ghiran I, Nicholson-Weller A, Asher DR, Cerny AM, Casey LS, Jones SM, Jones SN, Mohamed N, Klickstein LB, Spitalny G, Finberg RW. A transgenic mouse model for studying the clearance of blood-borne pathogens via human complement receptor 1 (CR1) Clin Exp Immunol. 2005;140:230–40. doi: 10.1111/j.1365-2249.2005.02764.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sepulveda J, Mukherjee J, Tzipori S, Simpson LL, Shoemaker CB. Efficient serum clearance of botulinum neurotoxin achieved using a pool of small antitoxin binding agents. Infect Immun. 2010;78:756–63. doi: 10.1128/IAI.01084-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson L. The life history of a botulinum toxin molecule. Toxicon. 2013;68:40–59. doi: 10.1016/j.toxicon.2013.02.014. [DOI] [PubMed] [Google Scholar]
- Smith TJ, Lou J, Geren IN, Forsyth CM, Tsai R, Laporte SL, Tepp WH, Bradshaw M, Johnson EA, Smith LA, Marks JD. Sequence variation within botulinum neurotoxin serotypes impacts antibody binding and neutralization. Infect Immun. 2005;73:5450–7. doi: 10.1128/IAI.73.9.5450-5457.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai T. Fc receptors and their role in immune regulation and autoimmunity. J Clin Immunol. 2005;25:1–18. doi: 10.1007/s10875-005-0353-8. [DOI] [PubMed] [Google Scholar]
- Taylor RP, Ferguson PJ, Martin EN, Cooke J, Greene KL, Grinspun K, Guttman M, Kuhn S. Immune complexes bound to the primate erythrocyte complement receptor (CR1) via anti-CR1 mAbs are cleared simultaneously with loss of CR1 in a concerted reaction in a rhesus monkey model. Clin Immunol Immunopathol. 1997a;82:49–59. doi: 10.1006/clin.1996.4286. [DOI] [PubMed] [Google Scholar]
- Taylor RP, Martin EN, Reinagel ML, Nardin A, Craig M, Choice Q, Schlimgen R, Greenbaum S, Incardona NL, Ochs HD. Bispecific monoclonal antibody complexes facilitate erythrocyte binding and liver clearance of a prototype particulate pathogen in a monkey model. J Immunol. 1997b;159:4035–44. [PubMed] [Google Scholar]
- Zhang X, Goncalves R, Mosser DM. The isolation and characterization of murine macrophages. In: Coligan John E, et al., editors. Current protocols in immunology. Unit 14. Chapter 14. 2008. p. 1. [DOI] [PMC free article] [PubMed] [Google Scholar]