

Abstract
Herein, a combination of microcontact printing of functionalized alkanethiols and site-specific modification of proteins is utilized to chemoselectively immobilize proteins onto gold surfaces either by oxime or copper catalyzed alkyne-azide click chemistry. Two molecules capable of click reactions, an aminooxy-functionalized alkanethiol and an azide-functionalized alkanethiol, were synthesized, and self-assembled monolayer (SAM) formation on gold was confirmed by IR spectroscopy. The alkanethiols were then individually patterned onto gold surfaces by microcontact printing. Site-specifically modified proteins, horse heart myoglobin (HHMb) containing an N-terminal α-oxoamide and a red-fluorescent protein (mCherry-CVIA) with a C-terminal alkyne, respectively were immobilized by incubation onto the stamped functionalized alkanethiol patterns. Pattern formation was confirmed by fluorescence microscopy.
Keywords: chemoselectivity, click chemistry, self-assembly, protein immobilization, microcontact printing
Introduction
Chemoselective immobilization of molecules, functional nanoparticles, macromolecules, and biomolecules on surfaces with high spatial control is important in array technologies and for micro- and nanofabrication.[1] An elegant approach for covalent immobilization of biomolecules with minimal loss of activity is to utilize click reactions such as thiol-maleimide[2], sulfonamide[3], and Staudinger ligation[4]. Recently, bioorthogonal click reactions such as the Cu(I) catalyzed Huisgen 1,3-dipolar cycloaddition[5], oxime bond formation[6], are of significant interest in these applications owing to their typically high conversions and mild reaction conditions.[7-11]
Oxime bonds form by condensation of an aminooxy moiety with a ketone or aldehyde. This approach has several advantages. First, molecules can be easily modified with one of the reactive partners. For example, biomolecules can be modified specifically at the N-terminus with an α-oxoamide[12, 13] or synthesized with N-levulinyl lysine residues.[14] Second, alkoxyamines react with aldehydes and ketones in the absence of other reagents and under mild conditions.[15, 16] In an earlier study, we demonstrated that α-ketoamide-modified proteins could be immobilized on micron-sized aminooxy surfaces produced by photoacid generator (PAG)-based photolithography or electron beam lithography.[12, 17, 18] Meijer and coworkers found that aniline-catalyzed oxime formation when used to immobilize pyridoxal 5’-phosphate (PLP) transaminated proteins on surfaces resulted in superior bioactivities compared to non-site-selective immobilization.[19] In addition, aldehyde and aminooxy groups have been site-specifically installed onto proteins by enzymatic C-terminal modification.[20, 21] Herein, we describe the generation of aminooxy-functionalized alkanethiol micropatterns by microcontact printing for the conjugation of an N-terminally transaminated horse-heart myoglobin.
Introduction of azides and alkynes into small molecules, macromolecules, and proteins has received increasing interest due to the mild reaction conditions required for copper-catalyzed Huisgen 1,3-dipolar cycloaddition.[11] The reactions proceed with high yield to form stable triazoles with high regioselectivity.[22] Furthermore, site-specific incorporation of these functional groups into proteins has been successfully demonstrated using protein engineering techniques or chemical techniques.[22-27] This approach has been employed to immobilize biomolecules on surfaces.[28-34] In the work described here, we demonstrated the ability to click red fluorescent protein where the alkyne functionality was introduced into the C-terminus by enzymatic prenylation onto azide functionalized alkanethiol patterned surfaces.
Biomolecule patterns have been made utilizing using various techniques[35] including microcontact printing,[36] dip-pen lithography (DPN),[37, 38] nanografting,[39] imprint lithography,[40] photolithography,[41] and electron beam lithography.[17, 42] Microcontact printing, developed by Whitesides and coworkers, has been widely exploited to pattern reactive surfaces at the micrometer scale.[43] In this technique, a poly(dimethylsiloxane) (PDMS) stamp is “inked” with the molecule and then brought into conformal contact with the substrate of choice. The “ink” molecule transfers from the stamp onto the surface forming the desired features. This system is amenable to a wide variety of “inks” and different surface substrates.[44] It is also straightforward to carry out, relatively inexpensive, and does not require clean room technologies. Both covalent and non-covalent immobilization strategies have been employed to immobilize biomolecules by microcontact printing.[45-51] While covalent immobilization by reactive microcontact printing results in relatively stable biomolecule patterns, earlier efforts have relied mainly on non-chemoselective strategies targeting free amine groups on the biomolecules[48-51] that could lead to loss of biomolecule functionality.[19] Covalent immobilization of site-selectively modified proteins onto surfaces by microcontact printing and bioorthogonal click reactions is relatively rare.[45] In this report, we describe the use of site selective chemical or biological protein modification with microcontact printing and two bioorthogonal click reactions, either oxime ligation or the copper catalized Click reaction, for oriented and chemoselective immobilization of proteins (Scheme 1)
Scheme 1.

Microcontact printing of the alkanethiols and chemoselective immobilization of the proteins via oxime and Huisgens cycloaddition.
Results and Discussion
Alkanethiol synthesis
To begin, alkanethiols functionalized with moieties suitable for click reactions were synthesized. We targeted an azide-containing alkanethiol for use in the Huisgen 1,3-dipolar cycloaddition along with an aminooxy containing alkanethiol for oxime bond formation. Both alkanethiols were designed to contain a triethylene glycol moiety to reduce non-specific adsorption.[52-56] As such, both alkanethiols were synthesized from the same precursor molecule. Schemes 2 and 3 outline the synthesis of these alkanethiols, respectively, starting from 2-(2-(2-(undec-10-enyloxy)ethoxy)ethoxy)ethanol.
Scheme 2.
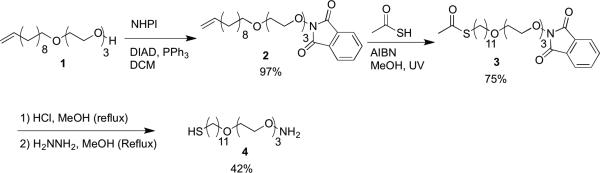
Aminooxy alkanethiol synthesis.
Scheme 3.
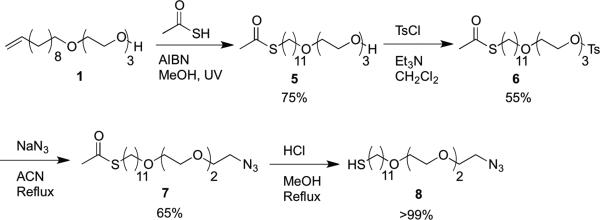
Azide alkanethiol synthesis.
The aminooxy functionalized alkanethiol 4 (Scheme 2) was prepared by first coupling the triethylene glycol-undecene (1) to N-hydroxyphthalimide (NHPl) under Mitsunobu conditions to give the alkene-phthalimide (2) in 97% yield. The protected thiol was next synthesized in 75% yield by AIBN-catalyzed thiol-ene reaction between the alkene and thioacetic acid under UV irradiation to give the N-hydroxyphthalimide-thioacetate (3). Compound 3 was then deprotected by subsequent reaction with concentrated HCl in refluxing methanol and exposure to hydrazine under reflux. This produced the final aminooxyalkanethiol 4 in 42% yield over two steps. The deprotection was performed without isolation between steps due to the partial deprotection of the phthalimide upon exposure to HCl. Deprotecting the phthalimide first was also attempted; however simultaneous removal of the acetate resulted in acetate capping of the aminooxy moiety.
The azide-functionalized alkanethiol 8 (Scheme 3) was synthesized by first coupling a thioacetate via an AIBN-catalyzed thiol-ene reaction between the 1 and thioacetic acid under UV irradiation to give the alcohol-thioacetate 5 in 75% yield. The alcohol was activated with p-toluenesulfonylchloride (TsCl) using triethylamine in methylene chloride to give the tosylate 6 in 55% yield. The azide was then incorporated via a substitution reaction providing the thioacetate-azide in 65% yield. Deprotection of 7 with concentrated HCl in refluxing methanol to 8 proceeded quantitatively.
Protein Modification
Protein modification was accomplished by two approaches. In the first, HHMb was chemically modified by the PLP reaction to install a group that can react with aminooxy surfaces (Scheme 4). Specifically, pyridoxal 5’-phosphate was added to HHMb in slightly acidic buffer. PLP transforms the end terminus of proteins to oxo-amide groups.[57] In this case, the end terminus of HHMb is a glycine so the transformation results in a glyoxyl amide. The alkyne protein for reaction with azide surfaces was prepared by enzymatic transformation (Scheme 5). Specifically mCherry-CVIA was modified a by post-translational lipid modification called protein prenylation. Protein farnesyl transferase (PFTase)[58] or geranyl-geranyl transferase[59] are known to transfer non-natural substrates. Thus, when alkyne modified farnesyl diphosphate and either enzyme was added to mCherry-CVIA protein in reducing buffer, the resulting protein contained an alkyne group at the C-terminus.
Scheme 4.
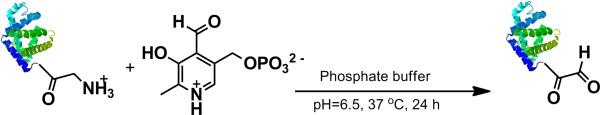
Transamination of horse heart myoglobin.
Scheme 5.
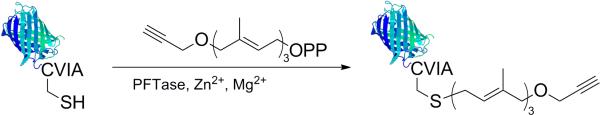
Prenylation of mCherry-CVIA.
SAM Formation
Prior to stamping, SAM formation of each individual molecule was assessed. SAMs of either 4 or 8 were formed by incubating gold plated silicon in 1 mM solutions of the alkanethiols in ethanol for 1 h. SAM formation was confirmed by surface IR (Figure 1). SAM 4 showed absorbances at 2920, 2850 (CH stretches), and 1130 (CO stretches) cm−1, similar to our previously reported aminooxy SAM.[60] The broad –NH2 stretch of the aminooxy moiety was also visible at 1410 cm−1 and above 3000 cm−1. The azide functionality of 8 (Figure 1b) was clearly visible at 2107 cm−1. SAM 8 contained a large, broad peak above 3000 cm−1; this was attributed to residual ethanol from the SAM formation and rinsing steps.
Figure 1.
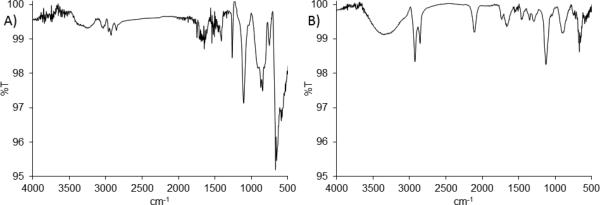
Surface FTIR spectra of A) aminooxy alkanethiol (4) SAM and B) azide alkanethiol (8) SAM on gold substrates.
Microcontact Printing and Protein Immobilization
Compounds 4 and 8 were printed onto separate gold surfaces in a 1:3 ratio along with a triethylene glycol-functionalized alkanethiol using a PDMS stamp containing circular features 200 μm in diameter. The 1:3 ratio was determined from a dilution study to be the optimal ratio for maximal signal-to-noise. Following printing, the gold surfaces were backfilled with the triethylene glycol-functionalized alkanethiol and incubated with the appropriate coupling partner. Horse heart myoglobin that had been modified by PLP transamination was exposed to SAMs of 4 and a red-fluorescent protein (mCherry-CVIA) containing an N-terminal alkyne was exposed to SAMs of 8.
Transaminated HHMb was incubated on the aminooxy surface for 1 h. This SAM was then treated with a goat-anti horse myoglobin antibody followed by an AlexaFluor® 488 modified mouse-anti goat antibody for 1 h each. The surface was then examined via fluorescence microscopy (Figure 2). There was a green signal where 4 had been printed, indicating immobilization of the myoglobin. When either the triethylene glycol alkanethiol or 8 were stamped and incubated with the transaminated myoglobin and antibodies in the same manner as the aminooxy SAMs, protein immobilization was not observed. This indicated that the aminooxy moiety was required for immobilization of transaminated myoglobin. In separate controls, the aminooxy surface was incubated with non-modified protein and both antibodies or modified protein with the secondary antibody only (without primary antibody). No protein patterning was observed in any of these samples, indicating that oxoamide modification must be present and that modification of the surface proceeds by oxime bond formation.
Figure 2.

Fluorescence micrograph showing the covalent immobilization of transaminated horse heart myoglobin on aminooxy alkanethiol SAM patterns after antibody staining.
The azide SAM was incubated with the mCherry alkyne for 2 h in the presence of ligand (tris-(triazolyl)benzylamine), CuSO4, and TCEP. In Figure 3 is shown the resulting fluorescence image. Patterning was visible indicating that the mCherry-CVIA had been immobilized as desired. Control experiments with mCherry lacking the alkyne modification and alkyne-modified mCherry incubated without copper did not show patterning. This indicated that copper-catalyzed click chemistry was required to immobilize the protein.
Figure 3.

Fluorescence micrograph showing the covalent immobilization of mCherry-CVIA-alkyne on azide alkanethiol SAM patterns.
These data together demonstrate that the combination of chemoselective modification of proteins either at the N-terminus or C-terminus and bioorthogonal click reactions is highly effective for the immobilization of proteins on surfaces. Patterned substrates are easily made by microcontact printing of alkanethiols. Proteins bound to the surfaces chemoselectively under mild reaction conditions via bioorthogonal oxime and cycloaddition click reactions as designed. This is important because harsh conditions and non-specific immobilizations can considerably reduce protein activity. Thus, it is anticipated that this approach will be useful for protein arrays, cell surface substrates and other applications were oriented proteins are desired
Conclusion
The synthesized alkanethiols having azide or aminooxy functionality have been demonstrated to be suitable for reactive SAM formation by use of microcontact printing. Immobilization of oriented proteins on these reactive surfaces has been shown using both oxime chemistry and the copper catalyzed Huisgen 1,3-dipolar cycloadditions. The proteins were generated either chemically to introduce a oxo-amide group at the N-terminus or by protein prenylation to the C-terminus. This platform can facilitate the oriented immobilization of proteins and hence should be useful for a wide range of biomedical and biotechnology applications.
Experimental Section
Materials
All chemicals were purchased from Sigma-Aldrich or Acros and used as received, unless otherwise specified. Antibodies, FluoSpheres® aldehyde−sulfate spheres (20 nm, yellow-green), and Click-iT® Alexa Fluor® 594 DIBO alkyne were purchased from Invitrogen. Deuterated solvents were obtained from Cambridge Isotope Laboratories. A 450 Watt Ace Glass Incorporated water-cooled mercury vapor U.V. lamp (λmax = 366 nm) was used for photochemical reactions.
Analytical Techniques
Column chromatography was performed using Sorbent Technologies silica gel 60 (200–400 mesh). Images of stained samples were acquired with an AxioCam MRm camera mounted on an Axiovert 200 inverted microscope equipped with a FluoArc mercury lamp (Carl Zeiss MicroImaging GmbH, Germany), using Axio Vision 4.8 software. Infrared spectroscopy of small molecules was recorded using a PerkinElmer Spectrum 100 FT-IR equipped with a universal ATR sample accessory. Infrared spectroscopy of the SAMs was performed using a Jasco FT/IR-670 plus equipped with a Harrick Seagull grazing-angle ATR attachment with a hemispherical germanium crystal. An FTA 135 Version 2.0 Dynamic contact angle analyzer was employed for contact angle measurements.1H and 13C NMR spectra were obtained on either a Bruker ARX 500 MHz or AVANCE 500 DRX MHz spectrometer. Mass spectra were obtained on either an Applied Biosystems Voyager-DE-STR MALDI-TOF or a high resolution ESI Applied BioSystems Q-Star Elite supported by Grant Number S10RR024605 from the National Center for Research Resources. The spectra are solely the responsibility of the authors and do not necessarily represent the official views of the National Center for Research Resources or the National Institutes of Health.
Methods
Synthesis of 1
1 was synthesized according to a literature procedure[61] with some modification. A mixture of 50% aqueous sodium hydroxide (0.97 mL, 12.3 mmol) and triethylene glycol (TEG) (8 mL, 60 mmol) was stirred at 100 °C under argon atmosphere. After 30 min, 11-bromoundec-1-ene (2.7 mL, 12.3 mmol) was added. After 24 h, the reaction mixture was cooled to room temperature and extracted six times with hexanes. The hexanes layers were consolidated and the solvent was evaporated by rotary evaporation. The product was purified by silica gel column chromatography with 4:1 hexanes:ethyl acetate (EtOAc) to afford 1.4 g of 1 as a light yellow, viscous oil in 38% yield. 1H NMR (CDCl3): δ = 5.80 (m, 1H), 4.92 (m, 2H), 3.64 (m, 14H), 3.44 (t, J= 6.85 Hz, 2H), 2.52 (br s, 1H), 2.04 (m, 2H), 1.57 (m, 2H), 1.30 (m, 10H). 13C NMR (CDCl3): δ = 139.22, 114.15, 72.62, 71.60, 70.61, 70.37, 70.04, 61.71, 33.83, 29.59, 28.56, 29.46, 29.14, 28.95, 26.09. IR νmax 3484, 3065, 2853, 1640, 1463, 1350, 1244 1100, 993, 908, 722 cm−1. MS (MALDI) calculated for C17H34O4Na [M + Na]+ 325.23, found 325.22.
Synthesis of 2
2 was synthesized according to our literature procedure.[60] Alkene-PEG 1 (1.4 g, 4.6 mmol) was dissolved in 20 mL of dichloromethane (DCM); triphenylphosphine (1.5 g, 5.6 mmol) and N-hydroxyphthalimide (0.9 g, 5.6 mmol) were added. Diisopropyl azidodicarboxylate (DIAD) (1.0 mL, 5.1 mmol) was then added drop wise over the course of one min. The reaction mixture was stirred at 25 °C for 16 h. The solvent was evaporated and the triphenylphosphine oxide byproduct was removed by filtration. The filtrate was collected, and the solvent evaporated. The product was purified by silica gel column chromatography by eluting with 4:1 hexanes:EtOAc to afford 2 g of 2 as a white solid in 97% yield. 1H NMR (CDCl3): δ = 7.85–7.79 (m, 2H), 7.76–7.71 (m, 2H), 5.85–5.77 (m, 1H), 4.97–4.87 (m, 2H,), 3.86–3.80 (m, 2H), 3.66–3.59 (m, 2H), 3.57–3.44(m, 6H), 3.41 (t, J = 7.0 Hz, 2H), 2.80 (bs, 1H, OH) 2.02–1.97 (m, 2 H) 1.57–1.46 (m, 2H), 1.38–1.16 (m, 12H). 13C NMR (CDCl3): δ = 163.6, 139.3, 134.5, 129.1, 123.6, 114.2, 76.9, 71.6, 70.9, 70.7, 70.6, 70.1, 69.4, 33.9, 29.7, 29.7, 29.6, 29.2, 29.0, 28.7, 25.9, 21.5. IR νmax 2924, 2854, 1790, 1731, 1639, 1611, 1524, 1467, 1374,1325, 1292, 1257, 1186, 1109, 1083, 1033, 996, 978, 953, 908,877, 787, 699. MS (ESI) calculated for C25H37NO6Na[M + Na]+ 470.25; found 470.25.
Synthesis of 3
A solution of 2 (500 mg, 1.1 mmol), thioacetic acid (0.24 mL, 3.4 mmol), and AIBN (16.4 mg, 0.10 mmol) were stirred in 10 mL of methanol (MeOH) under argon. The reaction mixture was exposed to 366 nm light for 8 h. Then, the solvent was evaporated and the product was purified by silica gel column chromatography by eluting with 2:1 hexanes:EtOAc to afford 0.44 g of 3 as a white solid in 75% yield. 1H NMR (CDCl3): δ = 7.82 (m, 2H), 7.74 (m, 2H), 4.38 (m, 2H), 3.86 (m, 2H), 3.67 (m, 2H), 3.55 (m, 6H), 3.41 (t, J = 11.5 Hz, 2H), 2.85 (t, J =12 Hz, 2H), 2.32 (s, 3H), 1.56 (m, 4H), 1.28 (br s, 14H). 13C NMR (CDCl3): δ = 163.51, 134.51, 129.08, 123.57, 71.59, 70.89, 70.66, 70.59, 70.07, 69.39, 30.73, 29.71, 29.63, 29.58, 29.55, 29.24. IR νmax 2929, 2849, 1787, 1750, 1684, 1466, 1377, 1318, 1222, 1187, 1118, 1081, 1033, 975, 877, 791, 697 cm−1. MS (MALDI) calculated for C27H41NO7SNa [M + Na]+ 546.24, found 546.15.
Synthesis of 4
Compound 3 (50 mg, 0.1 mmol) and 1 mL of 12N HCL in 5 mL of MeOH was refluxed at 70 °C for 6 h. Hydrazine hydrate (0.7 mL, 14 mmol) was then added and the solution was stirred for 4h under reflux. After cooling to room temperature, MeOH was removed by rotary evaporation. 20 mL of DCM was then added and the solution was washed with saturated sodium bicarbonate. The organic layer was dried over MgSO4 and the solvent removed by rotary evaporation. to yield 14 mg of 4 as a yellow oil in 42% yield; 1H NMR (CDCl3): δ = 5.5 (broad s 2H), 3.8 (t, J = 4.6 Hz, 2H), 3.6 (m, 10H), 3.4 (t, J = 6.8 Hz, 2H), 2.5 (q, 2H), 1.6 (m, 4H), 1.3 (m, 14H). 13C NMR (CDCl3): δ = 75.01, 71.70, 70.79, 70.74, 70.71, 70.22, 69.76, 39.35, 29,79, 29.72, 29.68, 29.65, 29.38, 29.21, 28.68, 26.24. IR νmax 2960, 2925, 1460, 1377, 1261, 1125, 750cm−1; MS (MALDI) calculated for C17H37NO4S [M + 1]+ 352.24, found 352.24.
Synthesis of 5
A solution of 1 (800 mg, 2.7 mmol), thioacetic acid (0.58 mL, 8.1 mmol), and AIBN (44 mg, 0.27 mmol) were stirred in 10 mL of MeOH under argon. The reaction was exposed to 366 nm light for 8 h. The reactants were removed from the light source, and the solvent was evaporated. The product was purified by silica gel column chromatography by eluting with 2:1 followed hexanes:EtOAc to afford 0.77 g of 5 as a clear oil in 75% yield. 1H NMR (CDCl3): δ = 4.38 (m, 2H), 3.86 (m, 2H), 3.67 (m, 2H), 3.55 (m, 6H), 3.41 (t, J = 11.5 Hz, 2H), 2.85 (t, J = 12 Hz, 2H), 2.32 (s, 3H), 1.56 (m, 4H), 1.28 (br s, 14H). 13C NMR (CDCl3): δ = 163.51, 71.59, 70.89, 70.66, 70.59, 70.07, 69.39, 30.73, 29.71, 29.63, 29.58, 29.55, 29.24. IR νmax 3484, 2929, 2849, 1787, 1750, 1684, 1466, 1377, 1318, 1222, 1187, 1118, 1081, 1033, 975, 877, 791, 697 cm−1. MS (MALDI) calculated for C19H38O5SNa [M + Na]+ 401.23, found 401.25.
Synthesis of 6
5 (100 mg, 0.255 mmol) was stirred with triethylamine (TEA, 55 μL, 0.383 mmol) under argon atmosphere in dry DCM (5 mL). The mixture was cooled to 0 °C in an ice bath and p-toluenesulfonylchloride (97 mg, 0.510 mmol) dissolved in DCM (5 mL) was added. The reaction was allowed to warm to 23 °C slowly over 24 h before removal of the solvent under vacuum. Purification of the residue by column chromatography (1:2 EtOAc:hexanes) yielded 6 as a white, crystalline solid in 55% yield (74 mg). 1H NMR (CDCl3): δ = 7.79 (d, J = 8.0, 2H), 7.33 (d, J = 8.0, 2H), 4.15 (t, J = 5.6, 2H), 3.72-3.36 (m, 12H), 2.85 (t, J = 7.3, 2H), 2.44 (s, 3H), 2.31 (s, 3H), 1.73-1.15 (m, 18H). 13C NMR (CDCl3): δ = 195.92, 144.71, 133.00, 129.66, 127.90, 73.75, 70.63, 70.53, 70.40, 69.89, 69.11, 68.55, 30.50, 29.49, 29.41, 29.36, 29.33, 29.31, 29.01, 28.96, 28.67, 25.94, 21.50. IR νmax 2923, 2854, 1687, 1598, 1456, 1354, 1290, 1188, 1096, 1017, 917, 814, 772 cm−1. MS (MALDI): calculated for C26H44O7S2Na [M + Na]+ 555.24, found 555.15.
Synthesis of 7
Sodium azide (72.2 mg, 1.11 mmol) was added to 6 (198.1 mg, 0.37 mmol) in acetonitrile (15 mL). The reaction was heated to reflux for 24 h before removal of solvent under vacuum. Purification of the residue by column chromatography (1:2 EtOAc:hexanes) yielded 7 as a clear oil in 65% yield (97 mg). 1H NMR (CDCl3): δ = 3.75-3.35 (m, 14H), 2.85 (t, J = 6.7, 2H), 2.34 (s, 3H) 1.70-1.18 (m, 18H,). 13C NMR (CDCl3): δ = 195.6, 71.42, 70.61, 70.54, 69.94, 69.91, 50.57, 39.06, 33.91, 29.51, 29.44, 29.40, 29.36, 29.10, 28.93, 28.40, 28.24, 25.96. IR νmax 2924, 2853, 2098, 1689, 1456, 1352, 1284, 1105, 952, 852, 721 cm−1. Mass spec (MALDI): calcd for C19H37N3O4SK [M + K]+ 442.21, found 442.02.
Synthesis of 8
7 (50.4 mg, 0.12 mmol) was stirred in 5 mL of MeOH. Concentrated HCl (1 mL) was added, and the mixture was heated to reflux for 4 h before removal of solvent under vacuum. The resulting residue was dissolved in hexanes (10 mL) and washed with saturated sodium bicarbonate (3 × 10 mL). The organic layer was removed under reduced pressure and the resulting oil was freeze-dried from benzene to yield 8 as clear oil in quantitative yield. The product was a mixture of thiol and the disulfide. 1H NMR (CDCl3): δ 3.72-3.33 (m, 14H), 2.69-2.48 (m, 2H), 1.70-1.18 (m, 18H). 13C NMR (CDCl3): δ = 71.42, 70.61, 70.54, 69.94, 69.91, 50.57, 39.06, 33.91, 29.51, 29.44, 29.40, 29.36, 29.10, 28.93, 28.40, 28.24, 25.96. IR νmax 2923, 2853, 2103, 1457, 1349, 1260, 1103, 934, 801, 734 cm−1. Mass spec (MALDI): calcd for C17H35SN3Na [M + Na]+ 384.23, found 384.21.
Preparation of gold-coated substrates
Briefly, 34 mm diameter glass coverslides (Fisher) were cleaned with Piranha solution (70% H2SO4, 30% H2O2, Caution: Piranha reacts violently with organic materials) at 100°C for 20 min followed by thorough rinsing in deionized (DI) water and drying under a stream of nitrogen. Immediately following this cleaning procedure, a 10 nm layer of gold with a 5 nm titanium adhesion layer was deposited onto the cleaned substrate with a CHA e-beam evaporator.
SAM Formation
A gold-coated wafer was immersed in hot piranha solution (1:3 v/v H2O2:H2SO4) for 5 min (Caution: Piranha reacts violently with organic materials), rinsed with Milli-Q water and ethanol, and dried under a stream of filtered air. The wafer was then exposed to a 1 mM ethanolic solution of either 4 or 8 at 23 °C for 1 h. The surface was rinsed with ethanol (5 mL) and dried with filtered air before analysis via contact angle (SAM 4 62 ± 3°, SAM 8 68 ± 4°) and IR spectroscopy to confirm the formation of SAMs 4 and 8.
Preparation of stamps for microcontact printing
Photolithographically prepared silicon masters were used as templates to cast poly(dimethylsiloxane) (PDMS) stamps for microcontact printing. Masters were fabricated using conventional photolithography techniques. Four-inch Si (100) test wafers (Tech Gopher Inc.) were first cleaned in a Piranha solution (Caution: Piranha reacts violently with organic materials) at 100°C for 20 min. The wafers were then thoroughly rinsed in deionized (DI) water and spun dry at 2000 rpm for 10 min in a wafer drier. Next, a film of the negative photoresist SU-8-50 (MicroChem, Bedford, MA) with an approximate thickness of 50 μm was applied to the surface of a cleaned wafer via spin coating. The SU-8 film was then soft baked on a hotplate using annealing parameters provided by the photoresist supplier. Subsequently, the films were exposed to UV irradiation through a photomask containing the desired pattern with a Karl Suss contact aligner. Photomasks were designed in Adobe Illustrator and printed onto transparencies at a resolution of 24,000 dpi with a high-resolution printer (CAD Art Sciences). Next, the non-exposed photoresist was selectively removed with SU-8 developer solution to reveal the patterned features. Upon pattern development, the final designs were rinsed with isopropyl alcohol, gently dried with N2, and hard baked for 20 min at 150 °C per instructions from the photoresist supplier. A Veeco Dektak 150 profiler was used to confirm the step heights of the patterned SU-8 features. Prior to replica molding of PDMS stamps, the masters were exposed to tridecafluoro-1,1,2,2-tetrahydrooctyl-1-trichlorosilane vapor under vacuum for 1 h, rendering the surface of the master hydrophobic and non-sticking.
PDMS stamps were cast by pouring a mixture of Sylgard 184 (Dow Corning, Midland, MI), prepared in the standard 10:1 prepolymer-tocrosslinker ratio, onto the desired master. Samples were held under vacuum overnight at room temperature and subsequently cured at 60°C for one hour. Upon curing, PDMS stamps were then removed from the master and sectioned into roughly 34 mm diameter circular stamps for patterning SAMs onto gold coated glass substrates.
Microcontact Printing of 4
A PDMS stamp containing circular features 200 μm in diameter was inked with a 3:1 solution of tri(ethylene glycol)undecyl alkanethiol: 4 (total concentration of 1 mM in ethanol) and the solvent was allowed to evaporate. The stamp was then dried under a stream of filtered air and pressed onto a gold wafer that had been immersed in hot piranha solution, rinsed with Milli-Q water and ethanol, and dried under a stream of filtered air. The wafer was then exposed to a 1 mM ethanolic solution of tri(ethylene glycol)undecyl alkanethiol for 1 h. The surface was rinsed with ethanol (5 mL) and dried.
Preparation of Transaminated Horse Heart Myoglobin
Horse heart myoglobin (HHMb) was transaminated according to a literature procedure.[13] Briefly, myoglobin (50 μM) and pyridoxal 5’-phosphate (PLP, 10 mM) were dissolved in 25 mM pH=6.5 phosphate buffer. The solution was kept at 37 °C and gently mixed for 24 h. The transaminated protein was purified by centrifugal filtration (Centriprep® centrifugal filter, 3000 MWCO, Millipore, MA).
SAM 4 Visualization
Patterned SAM 4 was incubated with PLP transaminated horse heart myoglobin (50 μM, 25 mM phosphate buffer, pH = 6.5) for 1h and rinsed with MilliQ H2O (5 mL). This surface was then treated with a goat-anti horse myoglobin antibody (1:1000 in H2O) for 1 h, rinsed with MilliQ H2O (5 mL) followed by an AlexaFluor® 488 modified mouse-anti goat antibody (1:1000 in H2O) for 1 h and rinsed with MilliQ H2O (5 mL). The surface was dried with filtered air and examined via fluorescence microscopy. Signal/ Noise: 12/1.
Protein Controls of SAM 4
SAMs of tri(ethylene glycol)undecyl alkanethiol or patterns of SAM 8 incubated with transaminated horse heart myoglobin and visualized by controls showed no patterns. Patterned SAM 4 was incubated with either non-transaminated horse heart myoglobin or PLP transaminated horse heart myoglobin (50 μM, 25 mM phosphate buffer, pH = 6.5) for 1h and both were rinsed with 5 mL of MilliQ H2O. The surface incubated with non-transaminated myoglobin was then treated with a goat-anti horse myoglobin antibody (1:1000 in H2O) for 1 h, rinsed with MilliQ H2O (5 mL) followed by an AlexaFluor® 488 modified mouse-anti goat antibody (1:1000 in H2O) for 1 h and rinsed with MilliQ H2O (5 mL). The surface incubated with the modified myoglobin was treated with an AlexaFluor® 488 modified mouse-anti goat antibody (1:1000 in H2O) for 1 h (without prior incubation with the primary antibody) and rinsed with MilliQ H2O (5 mL). The surfaces were dried with filtered air and examined via fluorescence microscopy. No protein patterning was observed in either case.
Microcontact Printing of 8
A PDMS stamp containing circular features (200 μm in diameter) was inked with a 3:1 solution of tri(ethylene glycol)undecyl alkanethiol: 8 (total concentration of 1 mM in ethanol) and the solvent was allowed to evaporate. The stamp was dried under a stream of filtered air and pressed onto a gold wafer that had been immersed in hot piranha solution, rinsed with Milli-Q water and ethanol, and dried under a stream of filtered air. The wafer was then exposed to a 1 mM ethanolic solution of tri(ethylene glycol)undecyl alkanethiol for 1 h. The surface was rinsed with ethanol (5 mL).
Preparation of mCherry-alkyne
mCherry-CVIA protein was purified as previously described.[62] Enzymatic prenylation reactions of the resulting protein were conducted on a 10 mL scale in buffer containing 50 mM Tris-HCl (pH 7.5), 10 mM MgCl2, 50 μM ZnCl2, 15 mM dithiothreitol, 20 mM KCl, and 2.4 μM mCherry-CVIA. The mixtures were incubated for 1 h at 25 °C to reduce any disulfide-linked proteins before performing the prenylation reaction. Alkyne modified farnesyl diphosphate[63] and PFTase[58] or PGGTase[59] were then added to final concentrations of 30 μM (isoprenoid substrate) and 170 nM (enzyme), respectively. The enzymatic reaction mixtures were incubated at 30 °C for 2 h followed by concentration to 500 μL by centrifugal dialysis (Amicon Ultra-15, molecular weight cutoff 10.0 kDa, Millipore). That material was then loaded onto an Illustra NAP-5 column (GE Healthcare, cat. #17-0853-02) and eluted with 50 mM phosphate buffer (pH=7.5) in a final volume of 500 μL. The concentrations of the alkyne modified mCherry-CVIA protein solutions were 25 μM (from the PFTase-catalyzed reaction) and 21 μM (from the PGGTase-catalyzed reaction); those values were determined by UV (ε492 = 72,000 M−1cm−1).[64] The resulting mCherry-CVIA protein that contains an alkyne-modified farnesyl group is referred to herein as mCherry-alkyne.
SAM 8 Visualization
Patterned SAM 8 was clicked using the conditions described by Distefano and coworkers.[65] Briefly, mCherry-alkyne (19 μL, 160 μM in 50 mM NaH2PO4, pH 7.3), tris-(triazolyl)benzylamine (1.8 μL, 1.67 mM in DMSO:t-BuOH 1:4,[66] CuSO4 (1.2 μL, 25 mM), TCEP (1.2 μL, 25 mM), and NaH2PO4 (6.8 μL, 50 mM, pH 7.3) for 2 h. The wafer was rinsed with 5 mL of MilliQ H2O, dried with filtered air, and examined via fluorescence microscopy. Signal/ Noise: 3.7/1.
Protein Controls of SAM 8
SAMs of 8 were exposed to mCherry-CVIA lacking the alkyne modification using the click conditions described above or alkyne-modified mCherry-CVIA using the click conditions described above but with no CuSO4. The wafers was rinsed with MilliQ H2O (5 mL), dried with filtered air, and examined via fluorescence microscopy. No protein patterning was observed.
Dye Control of SAM 4
FluoSpheres aldehyde−sulfate spheres (1 mL, 1:100, 20mg/ mL: 50 mM phosphate buffer pH = 5.5) were incubated with 1 M hydroxylamine (1 mL) for 1 h at 23 °C to ensure oxime formation. SAM 4 was subsequently incubated in this solution for 1 h. The wafer was rinsed with MilliQ H2O (5 mL), dried with filtered air, and examined via fluorescence microscopy. No FluoSphere pattern was observed.
Dye Control of SAM 8
Click-iT® Alexa Fluor® 594 DIBO alkyne (10 μL, 1 mM in DMSO) was incubated (1-azidoethyl)benzene (10 μL, 10 mM in DMSO) for 4 h at 23 °C to ensure consumption of the cyclooctyne. The solution was diluted with H2O (480 μL) and SAM 8 was incubated in this solution for 4 h at 23 °C. The wafer was rinsed with MilliQ H2O (5 mL), DMSO (5 mL), and MilliQ H2O (5 mL), dried with filtered air, and examined via fluorescence microscopy. No pattern was observed.
Acknowledgements
This work was financially supported by the NSF (CHE-0416359) and an Alfred P. Sloan Research Fellowship (HDM). ZPT thanks the NIH-sponsored Biotechnology Training in Biomedical Sciences and Engineering Program for funding. Partial funding was also provided by the National Institutes of Health through Grant P01 GM081621 (WR, BD), Grant R01 GM084152 (MD), and by a training grant (T1-0005) for WR from the California Institute of Regenerative Medicine (CIRM). Additional support for WR was provided by a training award from the Eli and Edythe Broad Center of Regenerative Medicine and Stem Cell Research at UCLA. EB thanks the Netherlands Organization for Scientific Research and Marie Curie Cofund Action for the financial support (Rubicon Grant 680-50-1101).
References
- 1.Hawker CJ, Wooley KL. Science. 2005;309:1200. doi: 10.1126/science.1109778. [DOI] [PubMed] [Google Scholar]
- 2.Hong HG, Jiang M, Sligar SG, Bohn PW. Langmuir. 1994;10:153. [Google Scholar]
- 3.Govindaraju T, Jonkheijm P, Gogolin L, Schroeder H, Becker CFW, Niemeyer CM, Waldmann H. Chem. Commun. 2008:3723. doi: 10.1039/b806764c. [DOI] [PubMed] [Google Scholar]
- 4.Kalia J, Abbott NL, Raines RT. Bioconjugate Chem. 2007;18:1064. doi: 10.1021/bc0603034. [DOI] [PubMed] [Google Scholar]
- 5.Huisgen R. Angew. Chem. Int. Ed. 1963;2:633. [Google Scholar]
- 6.Prescher JA, Bertozzi CR. Nat. Chem. Biol. 2005;1:13. doi: 10.1038/nchembio0605-13. [DOI] [PubMed] [Google Scholar]
- 7.Heredia KL, Tolstyka ZP, Maynard HD. Macromolecules. 2007;40:4772. [Google Scholar]
- 8.Kolb HC, Finn MG, Sharpless KB. Angew. Chem. Int. Ed. 2001;40:2004. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 9.Saxon E, Bertozzi CR. Science. 2000;287:2007. doi: 10.1126/science.287.5460.2007. [DOI] [PubMed] [Google Scholar]
- 10.Le Droumaguet B, Velonia K. Macromol. Rapid Commun. 2008;29:1073. [Google Scholar]
- 11.Nandivada H, Jiang XW, Lahann J. Adv. Mater. 2007;19:2197. [Google Scholar]
- 12.Christman KL, Broyer RM, Tolstyka ZP, Maynard HD. J. Mater. Chem. 2007;17:2021. [Google Scholar]
- 13.Gilmore JM, Scheck RA, Esser-Kahn AP, Joshi NS, Francis MB. Angew. Chem. Int. Ed. 2006;45:5307. doi: 10.1002/anie.200600368. [DOI] [PubMed] [Google Scholar]
- 14.Kochendoerfer GG, Chen SY, Mao F, Cressman S, Traviglia S, Shao HY, Hunter CL, Low DW, Cagle EN, Carnevali M, Gueriguian V, Keogh PJ, Porter H, Stratton SM, Wiedeke MC, Wilken J, Tang J, Levy JJ, Miranda LP, Crnogorac MM, et al. Science. 2003;299:884. doi: 10.1126/science.1079085. [DOI] [PubMed] [Google Scholar]
- 15.Amyes TL, Jencks WP. J. Am. Chem. Soc. 1988;110:3677. [Google Scholar]
- 16.Lemieux GA, Bertozzi CR. Trends Biotechnol. 1998;16:506. doi: 10.1016/s0167-7799(98)01230-x. [DOI] [PubMed] [Google Scholar]
- 17.Christman KL, Schopf E, Broyer RM, Li RC, Chen Y, Maynard HD. J. Am. Chem. Soc. 2009;131:521. doi: 10.1021/ja804767j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Christman KL, Broyer RM, Schopf E, Kolodziej CM, Chen Y, Maynard HD. Langmuir. 2011;27:1415. doi: 10.1021/la103978x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lempens EHM, Helms BA, Merkx M, Meijer EW. ChemBioChem. 2009;10:658. doi: 10.1002/cbic.200900028. [DOI] [PubMed] [Google Scholar]
- 20.Rashidian M, Song JM, Pricer RE, Distefano MD. J. Am. Chem. Soc. 2012;134:8455. doi: 10.1021/ja211308s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yi L, Sun HY, Wu YW, Triola G, Waldmann H, Goody RS. Angew. Chem. Int. Ed. 2010;49:9417. doi: 10.1002/anie.201003834. [DOI] [PubMed] [Google Scholar]
- 22.van Dongen SFM, Teeuwen RLM, Nallani M, van Berkel SS, Cornelissen JJLM, Nolte RJM, van Hest JCM. Bioconjugate Chem. 2009;20:20. doi: 10.1021/bc8004304. [DOI] [PubMed] [Google Scholar]
- 23.Schoffelen S, Lambermon MHL, van Eldijk MB, van Hest JCM. Bioconjugate Chem. 2008;19:1127. doi: 10.1021/bc800019v. [DOI] [PubMed] [Google Scholar]
- 24.Wang L, Xie J, Schultz PG. Annu. Rev. Biophys. 2006;35:225. doi: 10.1146/annurev.biophys.35.101105.121507. [DOI] [PubMed] [Google Scholar]
- 25.Lin PC, Ueng SH, Tseng MC, Ko JL, Huang KT, Yu SC, Adak AK, Chen YJ, Lin CC. Angew. Chem. Int. Ed. 2006;45:4286. doi: 10.1002/anie.200600756. [DOI] [PubMed] [Google Scholar]
- 26.Soundrarajan N, Sokalingam S, Raghunathan G, Budisa N, Paik HJ, Yoo TH, Lee SG. Plos One. 2012:7. doi: 10.1371/journal.pone.0046741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Viswanathan R, Labadie GR, Poulter CD. Bioconjugate Chem. 2013;24:571. doi: 10.1021/bc300462j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Devaraj NK, Miller GP, Ebina W, Kakaradov B, Collman JP, Kool ET, Chidsey CED. J. Am. Chem. Soc. 2005;127:8600. doi: 10.1021/ja051462l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sun X-L, Stabler CL, Cazalis CS, Chaikof EL. Bioconjugate Chem. 2005;17:52. doi: 10.1021/bc0502311. [DOI] [PubMed] [Google Scholar]
- 30.Zhang Y, Luo S, Tang Y, Yu L, Hou K-Y, Cheng J-P, Zeng X, Wang PG. Anal. Chem. 2006;78:2001. doi: 10.1021/ac051919+. [DOI] [PubMed] [Google Scholar]
- 31.Kleinert M, Winkler T, Terfort A, Lindhorst TK. Org. Biomol. Chem. 2008;6:2118. doi: 10.1039/b801595c. [DOI] [PubMed] [Google Scholar]
- 32.Rozkiewicz DI, Gierlich J, Burley GA, Gutsmiedl K, Carell T, Ravoo BJ, Reinhoudt DN. ChemBioChem. 2007;8:1997. doi: 10.1002/cbic.200700402. [DOI] [PubMed] [Google Scholar]
- 33.Michel O, Ravoo BJ. Langmuir. 2008;24:12116. doi: 10.1021/la802304w. [DOI] [PubMed] [Google Scholar]
- 34.Godula K, Rabuka D, Nam KT, Bertozzi CR. Angew. Chem. Int. Ed. 2009;48:4973. doi: 10.1002/anie.200805756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Christman KL, Enriquez-Rios VD, Maynard HD. Soft Matter. 2006;2:928. doi: 10.1039/b611000b. [DOI] [PubMed] [Google Scholar]
- 36.Renault JP, Bernard A, Bietsch A, Michel B, Bosshard HR, Delamarche E, Kreiter M, Hecht B, Wild UP. J. Phys. Chem. B. 2003;107:703. [Google Scholar]
- 37.Hyun J, Ahn SJ, Lee WK, Chilkoti A, Zauscher S. Nano Lett. 2002;2:1203. [Google Scholar]
- 38.Lee KB, Park SJ, Mirkin CA, Smith JC, Mrksich M. Science. 2002;295:1702. doi: 10.1126/science.1067172. [DOI] [PubMed] [Google Scholar]
- 39.Wadu-Mesthrige K, Xu S, Amro NA, Liu GY. Langmuir. 1999;15:8580. [Google Scholar]
- 40.Hoff JD, Cheng LJ, Meyhofer E, Guo LJ, Hunt AJ. Nano Lett. 2004;4:853. [Google Scholar]
- 41.Reynolds NP, Tucker JD, Davison PA, Timney JA, Hunter CN, Leggett GJ. J. Am. Chem. Soc. 2009;131:896. doi: 10.1021/ja8079252. [DOI] [PubMed] [Google Scholar]
- 42.Hong Y, Krsko P, Libera M. Langmuir. 2004;20:11123. doi: 10.1021/la048651m. [DOI] [PubMed] [Google Scholar]
- 43.Kumar A, Whitesides GM. Appl. Phys. Lett. 1993;63:2002. [Google Scholar]
- 44.Kaufmann T, Ravoo BJ. Polym. Chem. 2010;1:371. [Google Scholar]
- 45.Wasserberg D, Nicosia C, Tromp EE, Subramaniam V, Huskens J, Jonkheijm P. J. Am. Chem. Soc. 2013;135:3104. doi: 10.1021/ja3102133. [DOI] [PubMed] [Google Scholar]
- 46.Ludden MJW, Li X, Greve J, van Amerongen A, Escalante M, Subramaniam V, Reinhoudt DN, Huskens J. J. Am. Chem. Soc. 2008;130:6964. doi: 10.1021/ja078109v. [DOI] [PubMed] [Google Scholar]
- 47.Vega RA, Maspoch D, Shen CKF, Kakkassery JJ, Chen BJ, Lamb RA, Mirkin CA. ChemBioChem. 2006;7:1653. doi: 10.1002/cbic.200600271. [DOI] [PubMed] [Google Scholar]
- 48.Coq N, van Bommel T, Hikmet RA, Stapert HR, Dittmer WU. Langmuir. 2007;23:5154. doi: 10.1021/la0700321. [DOI] [PubMed] [Google Scholar]
- 49.Lahiri J, Ostuni E, Whitesides GM. Langmuir. 1999;15:2055. [Google Scholar]
- 50.Rozkiewicz DI, Kraan Y, Werten MWT, de Wolf FA, Subramaniam V, Ravoo BJ, Reinhoudt DN. Chem. Eur. J. 2006;12:6290. doi: 10.1002/chem.200501554. [DOI] [PubMed] [Google Scholar]
- 51.Feng CL, Vancso GJ, Schonherr H. Adv. Funct. Mater. 2006;16:1306. [Google Scholar]
- 52.Prime KL, Whitesides GM. J. Am. Chem. Soc. 1993;115:10714. [Google Scholar]
- 53.Harder P, Grunze M, Dahint R, Whitesides GM, Laibinis PE. J. Phys. Chem. B. 1998;102:426. [Google Scholar]
- 54.Chapman RG, Ostuni E, Yan L, Whitesides GM. Langmuir. 2000;16:6927. [Google Scholar]
- 55.Benesch J, Svedhem S, Svensson SCT, Valiokas R, Liedberg B, Tengvall P. J. Biomater. Sci. Polym. Ed. 2001;12:581. doi: 10.1163/156856201316883421. [DOI] [PubMed] [Google Scholar]
- 56.Pale-Grosdemange C, Simon ES, Prime KL, Whitesides GM. J. Am. Chem. Soc. 1991;113:12. [Google Scholar]
- 57.Gilmore JM, Scheck RA, Esser-Kahn AP, Joshi NS, Francis MB. Angew. Chem. Int. Ed. 2006;45:5307. doi: 10.1002/anie.200600368. [DOI] [PubMed] [Google Scholar]
- 58.Wollack JW, Silverman JM, Petzold CJ, Mougous JD, Distefano MD. ChemBioChem. 2009;10:2934. doi: 10.1002/cbic.200900566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Clausen VA, Edelstein RL, Distefano MD. Biochemistry. 2001;40:3920. doi: 10.1021/bi002011a. [DOI] [PubMed] [Google Scholar]
- 60.Mancini RJ, Li RC, Tolstyka ZP, Maynard HD. Org. Biomol. Chem. 2009;7:4954. doi: 10.1039/b904195h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chirakul P, Perez-Luna VH, Owen H, Lopez GP, Hampton PD. Langmuir. 2002;18:4324. [Google Scholar]
- 62.Chung JA, Wollack JW, Hovlid ML, Okesli A, Chen Y, Mueller JD, Distefano MD, Taton TA. Anal. Biochem. 2009;386:1. doi: 10.1016/j.ab.2008.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hosokawa A, Wollack JW, Zhang ZY, Chen L, Barany G, Distefano MD. Int. J. Pept. Res. Ther. 2007;13:345. [Google Scholar]
- 64.Shaner NC, Campbell RE, Steinbach PA, Giepmans BNG, Palmer AE, Tsien RY. Nat. Biotechnol. 2004;22:1567. doi: 10.1038/nbt1037. [DOI] [PubMed] [Google Scholar]
- 65.Duckworth BP, Xu JH, Taton TA, Guo A, Distefano MD. Bioconjugate Chem. 2006;17:967. doi: 10.1021/bc060125e. [DOI] [PubMed] [Google Scholar]
- 66.Chan TR, Hilgraf R, Sharpless KB, Fokin VV. Org. Lett. 2004;6:2853. doi: 10.1021/ol0493094. [DOI] [PubMed] [Google Scholar]