

Abstract
We report the case of a 22-year-old woman who presented with self-poisoning by cyanide ingestion. We have elected to pay particular attention to describing the neuropsychological sequelae of cyanide poisoning, and the evolution of these deficits over a 6-month period. Prominent deficits in episodic memory were noted from an early stage, which were consistent with the findings noted on structural neuroimaging. These deficits remained persistent, although improving in severity over the follow-up period. No focal neurological deficits or abnormal involuntary movements emerged, and the patient's overall functional status remained satisfactory. The patient's psychiatric presentation and background history are briefly discussed.
Background
Cyanide poisoning has been noted to occur in different contexts, and can be secondary to ingestion, inhalation or skin contact. Poisoning through ingestion is a frequently fatal event, and literature offering systematic data on follow-up of survivors is scant. Published reports have, in turn, focused on the neurological aspects of the clinical presentation, in addition to emergent findings on structural neuroimaging.1–4 The latter reports have emphasised the involvement of key memory areas of the brain, including the thalamus and the basal ganglia (BG).1 2 4 As such, a thorough neuropsychological investigation is warranted, as we have chosen to present in our case report. Also, the evolution of cognitive impairments over time delineated through repeat assessment has, to our knowledge, not been attended to in the literature, and we attempt to address this gap. In doing so, it is our intention to highlight the potential for cognitive recovery in patients such as ours. We also provide a brief update on the putative pathomechanisms of brain injury in cyanide ingestion.
Case presentation
A 22-year-old woman of mixed Caucasian–Chinese ethnicity presented to the emergency department (ED) at Prince of Wales Hospital in Sydney, Australia. She had reported intentional self-poisoning by cyanide ingestion to attending ambulance officers, and had a Glasgow Coma Scale (GCS) of 3 at the time of presentation to ED. She required intubation and received intravenous (IV) fluids, atropine and metaraminol for worsening hypotension and bradycardia with partial response. Sodium thiosulfate infusion was started as specific antidotal therapy for cyanide poisoning, with rapid resolution of hypotension over 20–30 min. Metabolic acidosis, and her overall clinical state, improved over the next 12–24 h in the intensive care unit (ICU) setting. A second dose of sodium thiosulfate was administered 12 h after admission, and she was extubated on day 2.
She was seen by the consultation-liaison psychiatry team on day 3, and cooperated with a full assessment. She gave a history of mild depressive symptoms in the months preceding the overdose, superimposed on a background of deficient and maladaptive coping with interpersonal stressors. Her depressive syndrome appeared mild, and had not precluded stable sociovocational functioning. The overdose appeared to be impulsive in part, though she had accessed the cyanide through work a few days prior to ingesting it. Her recall of events in the hours leading up to the overdose was patchy. The patient was transferred, with her agreement, to the psychiatric unit after being medically cleared by the ICU team on day 5. Her clinical presentation appeared stable, and there was no evidence of an acute confusional state, with generally intact attention and orientation on bedside assessment. Deficits in episodic memory, particularly for day-to-day events, were apparent from this early stage on. Neurological examination at this time was otherwise unremarkable. Specifically, there was no evidence of parkinsonism; nor were any dyskinetic movements noted.
Following transfer to the mental health unit, the patient maintained stability in her mood and behaviour overall. She was occasionally low in mood and teary, and she presented with symptoms of a mild depressive disorder, in part reactive to her cognitive difficulties and their projected impact on her future functioning. Retrograde amnesia for events on the day of the overdose was noted and remained a consistent finding. The most prominent feature of her presentation, however, was noted to be marked impairment in episodic memory. Less pronounced deficits were noted in her ability to sustain and divide attention. She would frequently require staff to remind her about planned appointments and activities and forget having had visitors earlier in the day. She reported difficulty with concentrating while reading, and could not recall content she had read previously in the day. These issues persisted throughout her admission, and were formally evaluated with detailed neuropsychological testing at 3 weeks and 6 months post self-poisoning.
By way of background, the patient had no history suggestive of neurodevelopmental vulnerability or of acquired brain injury. Her socioeducational functioning had been at a high level, and she was a few months short of completing a university graduate degree with honours. Her medical history was unremarkable. There was no history of a substance abuse disorder. In the recent past, she had been diagnosed with a major depressive episode and been on treatment with venlafaxine at the time of self-poisoning. Other psychiatric history of relevance included a brief period of self-harming by cutting a few years earlier, and intermittent disordered eating behaviours with mild food restriction and purging.
The patient remained an inpatient for a period of 10 weeks postinjury. The anterograde memory deficits described above remained prominent but stable throughout this period. She was initiated on escitalopram as an antidepressant therapy. In addition, she was engaged in brief supportive psychotherapy with the team clinical psychologist, and had input from the broader allied health team. Functional assessment revealed well-preserved overall functional levels, commensurate with high premorbid functioning, though the need for prompting with complex, multistep tasks was identified. The patient's mood improved and the family was supported through regular meetings with the team.
Investigations
Structural neuroimaging
MRI of the brain at 1 week postinjury showed extensive long repetition time (TR) high signal and restricted diffusion in the hippocampal formation and globus pallidus bilaterally (figure 1). Subtle long TR signal changes in the cingulate gyri and heads of the caudate nuclei, as well as in the ventroanterior thalami and posterior putamen bilaterally, were noted. These appearances were considered consistent with those expected in anoxic brain injury secondary, as in our patient, to cyanide poisoning.
Figure 1.
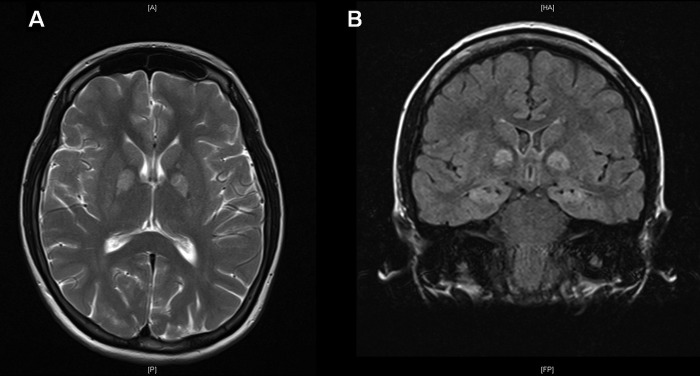
Images showing extensive high signal in the globus pallidi (A—axial T2 image) and hippocampi (B—coronal FLAIR (fluid attenuated inversion recovery) image) at 1 week postinjury.
Repeat neuroimaging by MRI at 5 months postinjury allowed for comparison with prior radiological findings. Previously demonstrated areas of signal change within the BG and thalami were noted to be smaller in size with residual T2-weighted/FLAIR (fluid attenuated inversion recovery) hyperintensity and areas of serpiginous increased surrounding T1-weighted hyperintensity peripherally in the globus pallidi (figure 2A). There were new linear areas of T1-weighted hyperintensity within both cerebellar hemispheres at the grey–white matter interface, affecting mostly the semilunar and gracile lobules. These areas demonstrated no restricted diffusion or blooming artefact.
Figure 2.
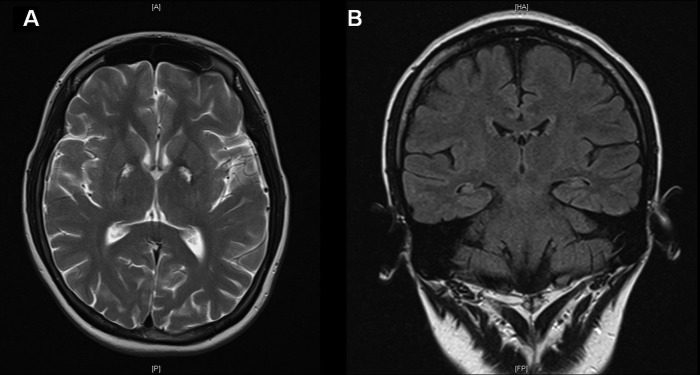
Images showing reduced size of previously demonstrated signal change in globus pallidi bilaterally (A—axial T2 image), in addition to the mildly atrophic appearance of both hippocampi (B—coronal FLAIR (fluid attenuated inversion recovery) image).
In discussion with the radiologist, decreased high signal in the hippocampi was noted, suggestive of resolution of acute oedema noted in the first scan. In the follow-up MRI, the hippocampi were noted to appear more atrophic (figure 2B), although formal volumetric analysis was not performed.
Neuropsychological assessment
Assessment of the patient's cognitive functioning was conducted 3 weeks postinjury. She appeared alert but flat in affect, and there was no spontaneous conversation on her part throughout the session. On specific questioning, she reported difficulty with recalling people's names and in recalling daily events. On assessment of intellectual functions, there was reduction (relative to her estimated above average intellectual capability) in her verbal abstract reasoning ability and visuospatial, problem-solving skills. Her verbal memory for contextual information (two stories), as well as her ability to assimilate and retain new information (a word list), was markedly impaired. Visual recall of simple and complex geometric designs was also significantly impaired. In contrast, her attention span and working memory span were intact. Her psychomotor and information processing speed were at a ‘superior level’, as shown in paper-and-pencil coding and tracking tasks. Frontal and executive functions such as verbal fluency, cognitive flexibility and visuospatial planning ability were relatively intact.
Repeat neuropsychological evaluation performed 5 months after initial assessment showed significant improvement in intellectual abilities. Significant improvements were also seen in her registration of verbally presented material (now low average), her ability to learn new verbal information, as well as her visual recall, although these remained at an impaired level. On this occasion, autobiographical memory and prospective memory were also assessed. She performed well with the former, with no difficulty in recalling and naming schools, providing addresses, names of teachers, and describing incidents relating to different stages in her life. She was also able to carry out all tasks involved in testing of prospective memory.
A comparison of the patient's neuropsychological profile with those previously reported in the literature revealed some differences. In a case of oral cyanide intoxication in a 22-year-old man reported by Rosenow et al,2 cognitive assessment at 5 weeks after injury showed ‘above average’ intellectual, verbal and visual memory functioning. However, the speed of information processing and motor reaction, and verbal fluency were reduced. The authors reported another case of a 43-year-old man with oral cyanide poisoning in whom assessment of cognitive functions at 4 months postinjury revealed attention and concentration deficits, but verbal and visual memory and intellectual functions were all within normal limits. In contrast, our patient had demonstrated reduction of intellectual abilities and impaired verbal and visual memory. A likely explanation of these inconsistencies in neuropsychological findings could be found in the discrepancy in MRI findings. Our patient's MRI (1 week postinjury) showed an ‘extensive long TR high signal and restricted diffusion in the hippocampal formation and globus pallidus bilaterally’. However, in the above two cases, the hippocampi were notably spared. In discussing changes in neuroimaging after acute cyanide intoxication, Rachinger et al1 stated that while cyanide particularly affects structures with a high oxygen dependency (including the hippocampus), the hippocampus was spared in all the cases that had been reported in the literature at the time. This is not the case with our patient in whom prospective memory and executive functions, as well as autobiographic memory, remained well preserved, consistent with the lack of structural changes in the frontal lobe and temporal cortex, respectively.
Discussion
Cyanide is a rapidly acting neurotoxic agent. Cyanide poisoning by oral ingestion, though infrequent, is frequently a fatal event. Consequently, there is a dearth of literature describing the pattern of clinical and neurocognitive features in survivors. In most of the patients reported, neurological sequelae including parkinsonism, dystonia and dyskinesia have been commonly noted, reflective of the involvement of structures within the BG.3 5 The striatum has been noted to be particularly vulnerable to cyanide-mediated neurotoxicity attributable to high glucose and oxygen utilisation within these structures, as well as region-specific considerations such as a higher local distribution of cytochrome oxidase.4
The cognitive salience of the BG has received much attention in the previous decade. Hitherto considered primarily motor, the critical role of the BG in the modulation of cognition and emotion has been emphasised in recent publications.6 The caudate, putamen and globus pallidi share rich connectivity with the cortex through a series of independent corticostriatal processing loops. Regional specificity within BG in relation to cortical interactions is suggested by contrasting the progression of cognitive deficits between prototypical disorders of the BG, namely Parkinson's and Huntington's diseases, known to show a differential spread of pathology within the BG.7 In particular, the BG appear to play a key role in sequence and category learning, planning and set shifting.8 Significantly, motor symptoms were not noted to emerge during the period of follow up.
Our case is notable also for the hippocampal involvement identified on MRI. The hippocampal formation is noted in the literature to be relatively rarely involved in acute cyanide poisoning despite being vulnerable in anoxic brain damage. The important role of the medial temporal lobe, including the hippocampus, in human memory has been repeatedly and convincingly demonstrated since the days of the famous amnestic patient HM, who had bilateral temporal lobe resection for control of seizures.9 The prominence of memory deficits in this case is well explained by evident hippocampal involvement.
The pathomechanisms of cyanide toxicity are varied, resulting in some variability in the resulting clinical phenotype. The primary effect of cyanide is exerted though the blockade of the cellular respiratory chain at the level of cytochrome oxidase, a terminal enzyme in the respiratory electron transport chain.10 This disruption of cellular aerobic respiration leads rapidly to respiratory arrest. Cyanide, though, also has a particular predilection for affecting the central nervous system. In cultured neurons, cyanide induced N-methyl d-aspartate (NMDA) receptor activation induces apoptotic cell death.11 In addition, cyanide increases the generation of reactive oxygen species, contributing to necrotic cell death through membrane lipid peroxidation implicating oxidative stress as a key driver of cyanide-induced neurotoxicity. In this animal study, the differential vulnerability of brain regions in response to oxidative stress was also highlighted.12
This report highlights the relative unpredictability of the effects of acute cyanide poisoning on brain structure and function consistent with the multiple intracellular mechanisms involved in cyanide-induced neurotoxicity. In a case series of two patients with acute cyanide poisoning, the authors of that paper make note of a distinct clinical and neuropsychological presentation, explained by characteristic lesional patterns on structural and functional neuroimaging.2 These predictions appear not to have been borne out in our case, arguing for individual variance in brain susceptibility to insult in this context. We have also presented a detailed and longitudinal neuropsychological investigation of such a case, which to the best of our knowledge, has not been reported in the literature thus far.
Learning points.
Cyanide-induced neurotoxicity is mediated by a complex series of intracellular mechanisms and biochemical cascades with resulting neuropathological variance.
Detailed neuropsychological investigation and follow-up are key, given the involvement of key cognitive areas within the brain.
Movement disorders, though more common, are not a necessary sequel of cyanide poisoning in survivors.
Acknowledgments
The authors would like to thank Sophia Dean who assisted with the preparation of the manuscript.
Footnotes
Contributors: AM, TL and PS were involved in managing the case and subsequent follow-up, and all three authors contributed equally to the planning and execution of the project.
Competing interests: None.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Rachinger J, Fellner FA, Stieglbauer K, et al. MR changes after acute cyanide intoxication. Am J Neuroradiol 2002;23:1398–401 [PMC free article] [PubMed] [Google Scholar]
- 2.Rosenow F, Herholz K, Lanfermann H, et al. Neurological sequelae of cyanide intoxication—the patterns of clinical, magnetic resonance imaging, and positron emission tomography findings. Ann Neurol 1995;38:825–8 [DOI] [PubMed] [Google Scholar]
- 3.Borgohain R, Singh AK, Radhakrishna H, et al. Delayed onset generalised dystonia after cyanide poisoning. Clin Neurol Neurosurg 1995;97:213–15 [DOI] [PubMed] [Google Scholar]
- 4.Kasamo K, Okuhata Y, Satoh R, et al. Chronological changes of MRI findings on striatal damage after acute cyanide intoxication: pathogenesis of the damage and its selectivity, and prevention for neurological sequelae: a case report. Eur Arch Psychiatry Clin Neurosci 1993;243:71–4 [DOI] [PubMed] [Google Scholar]
- 5.Carella F, Grassi MP, Savoiardo M, et al. Dystonic-parkinsonian syndrome after cyanide poisoning: clinical and MRI findings. J Neurol Neurosurg Psychiatry 1988;51:1345–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Seger CA. The basal ganglia in human learning. Neuroscientist 2006;12:285–90 [DOI] [PubMed] [Google Scholar]
- 7.Dauer W, Przedborski S. Parkinson's disease: mechanisms and models. Neuron 2003;39:889–909 [DOI] [PubMed] [Google Scholar]
- 8.McNab F, Klingberg T. Prefrontal cortex and basal ganglia control access to working memory. Nature Neurosci 2008;11:103–7 [DOI] [PubMed] [Google Scholar]
- 9.Squire LR, Wixted JT. The cognitive neuroscience of human memory since H.M. Annu Rev Neurosci 2011;34:259–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nelson L. Acute cyanide toxicity: mechanisms and manifestations. J Emerg Nurse 2006;32(4 Suppl):S8–11 [DOI] [PubMed] [Google Scholar]
- 11.Gunasekar PG, Sun PW, Kanthasamy AG, et al. Cyanide-induced neurotoxicity involves nitric oxide and reactive oxygen species generation after N-methyl-D-aspartate receptor activation. J Pharmacol Exp Ther 1996;277:150–5 [PubMed] [Google Scholar]
- 12.Prabhakaran K, Li L, Borowitz JL, et al. Cyanide induces different modes of death in cortical and mesencephalon cells. J Pharmacol Exp Ther 2002;303:510–19 [DOI] [PubMed] [Google Scholar]