

Abstract
Objectives:
The objective of this open-label study was primarily to assess the effect of taurine on adaptive behavior and secondarily to collect safety and tolerability data in patients with succinic semialdehyde dehydrogenase deficiency.
Methods:
In the current study, subjects were titrated weekly from a starting dose of 50 mg/kg/d to a target 200 mg/kg/d, and assessed for safety, tolerability, and adaptive functioning using age-normalized Adaptive Behavior Assessment Scales.
Results:
Eighteen patients (8 males/10 females, aged 0.5–28 years, mean 12 years) were recruited. Three subjects withdrew because of perceived lack of efficacy. One serious adverse event occurred (hospitalization for hypersomnia) on 16 g/d (200 mg/kg/d), leading to a dose-lowering paradigm with a maximum dose of 10 g/d. Results did not show clinically meaningful improvement in the adaptive domains after taurine therapy. Pre- and posttherapy adaptive scores also demonstrated no statistically significant difference (p > 0.18).
Conclusions:
Adaptive behavior did not improve significantly with taurine intervention. Further therapeutic clinical trials including an on-off paradigm using biomarkers are planned.
Classification of evidence:
This study provides Class IV evidence that for patients with succinic semialdehyde dehydrogenase deficiency, taurine does not significantly improve adaptive behavior. The study is rated Class IV because of the absence of a control group.
Succinic semialdehyde dehydrogenase (SSADH) deficiency (γ-hydroxybutyric aciduria) is a rare autosomal recessively inherited defect in γ-aminobutyric acid (GABA) catabolism (figure).1 This typically nonprogressive encephalopathy with late infantile onset presents with ataxia, hypotonia, expressive language deficits, intellectual disability, and in half of patients, seizures.2,3 Neuroimaging reveals dentato-pallido-luysian hyperintensity. Several hundred patients have been diagnosed worldwide.4
Figure. GABA catabolism pathway.

GABA (γ-aminobutyric acid) is normally converted via GABA-transaminase to succinate semialdehyde, which is then broken down to succinic acid by succinate semialdehyde dehydrogenase (SSADH). In the absence of SSADH, succinate semialdehyde is converted to γ-hydroxybutyric acid (GHB) rather than succinic acid, and this leads to a buildup of both GHB and GABA in the brain. Vigabatrin is an irreversible inhibitor of GABA-transaminase as shown.
No effective treatment exists. Improved survival has been demonstrated in the experimental homozygous mouse from the nonprotein amino acid taurine.5,6 This agent was originally selected because the suckling null mouse deteriorated with seizures upon weaning, and taurine represents the most concentrated constituent in murine breast milk.
Taurine has numerous neuromodulatory roles,5–8 including protection against free-radical damage, the latter prevalent in SSADH-deficient mice and patients.9–11 Taurine may act as a GABA transporter substrate,12,13 and GABA and taurine transporters may have reciprocal interrelationships. Vigabatrin (VGB)-induced retinotoxicity has been linked to corresponding taurine deficiency,14,15 of interest because VGB increases CNS GABA. Thus, our rationale for evaluating taurine centered on its successful use in the murine model, potential GABA transporter activity, and antioxidant effects.
A single case of taurine therapy (200 mg/kg/d) for 12 months in a 2-year-old boy with SSADH deficiency showed improved socialization, behavior, coordination, and activity.16 We undertook an open-label trial with collection of baseline and follow-up data, preparatory to a randomized controlled trial utilizing biomarkers. The aim of this study was to assess the effect of taurine on adaptive behavior in SSADH deficiency.
METHODS
This was an open-label study. Subjects with a diagnosis of SSADH deficiency, based on persistent 4-OH-butyric aciduria, typical phenotype, and when available genotype or enzymatic quantification, were eligible. Patients were identified during outpatient visits or by data from prior consented enrollment in a worldwide SSADH clinical database.
Participants were instructed to take taurine supplements (General Nutrition Center brand) in tablet form. Enrolled patients were titrated weekly from 50 mg/kg/d to the target 200 mg/kg/d, based on the previously published report.16 A maximum dosage of 10 g/d was established after a serious adverse event that prompted a dose-lowering paradigm. The titration schedule was implemented to allow monitoring for adverse effects before reaching the target dose. The trial and follow-up were conducted remotely, by phone and e-mail, because of the wide geographical distribution of patients. Patients' local physicians supervised therapy and monitored safety in conjunction with the principal investigator.
Patients were assessed for safety, tolerability, and adaptive functioning using the Adaptive Behavior Assessment Scale–II (ABAS-II),17 chosen because it is a comprehensive survey that provides general and subdomain scores, and has been standardized with normative data for pediatric, adolescent, and adult ages. Monthly progress report forms requesting data on side effects and observed changes were kept. The ABAS-II was administered at baseline and after 3, 6, and 12 months of treatment; additional follow-ups were performed every 6 months thereafter if families chose to continue therapy. The ABAS-II measured adaptive skills for conceptual, social, and practical domains, and combined these into an additional general score. The scale was normative, and separate questionnaires were administered for the age ranges 0–4, 5–15, and 16+ years. We used paired t tests to compare ABAS scores between the period of pre- and posttaurine use. This study provides Class IV evidence that for patients with SSADH deficiency, taurine does not significantly improve adaptive function.
Using an SD of 15 percentile points on the ABAS, a pre- vs posttreatment study of 20 patients was considered sufficient to provide 80% power to detect a time-averaged pre-post difference of 6 percentile points based on 3 posttreatment assessments per participant assuming ABAS scores on the same participant were correlated at 60%. The sample size was inflated by 25% (+5 participants, n = 25) to account for dropouts. Under the same assumptions, this sample size had more than 90% power to detect a time-average pre vs post difference as small as 7.5.
Standard protocol approvals, registrations, and patient consents.
This study was approved by the Children's National Medical Center IRB (protocol 4650) and Washington State University IRB protocol 12678. The clinical trial identifier is NCT01608178. Written informed patient consent was received.
RESULTS
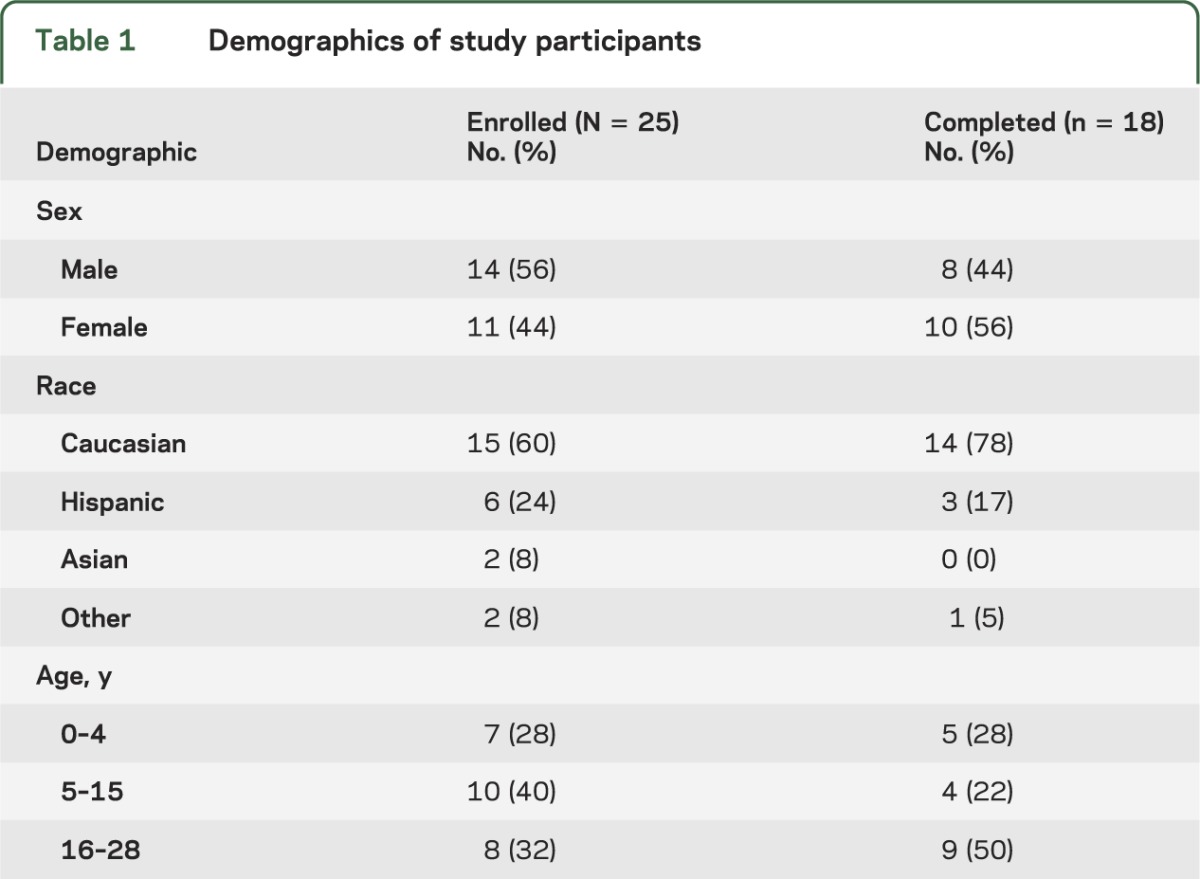
Twenty-five patients (14 males/11 females, age range 6 months to 28 years [mean age 12 years, median 9 years]) were recruited and provided baseline ABAS-II data. Eighteen patients provided follow-up data and were included in analysis (8 males/10 females, mean age 12 years, median 13 years). Demographic information is summarized in table 1. Sixteen unique subjects provided follow-up data for a range of 3 to 50 months of continuous treatment; length of treatment was median 8.5 months (mean 13.5 months). Two patients, females aged 17 and 19 years at initial enrollment, reenrolled in the trial after a 3-year hiatus.
Table 1.
Demographics of study participants
The primary outcome of interest was efficacy, measured by change in adaptive behavior. At baseline, subjects' median ABAS percentile scores were as follows: general functioning 0.1 (range <0.1 to 47); conceptual domain 0.1 (range <0.1 to 34); social domain 3 (range <0.1 to 58); and practical domain 0.25 (range <0.1 to 45).
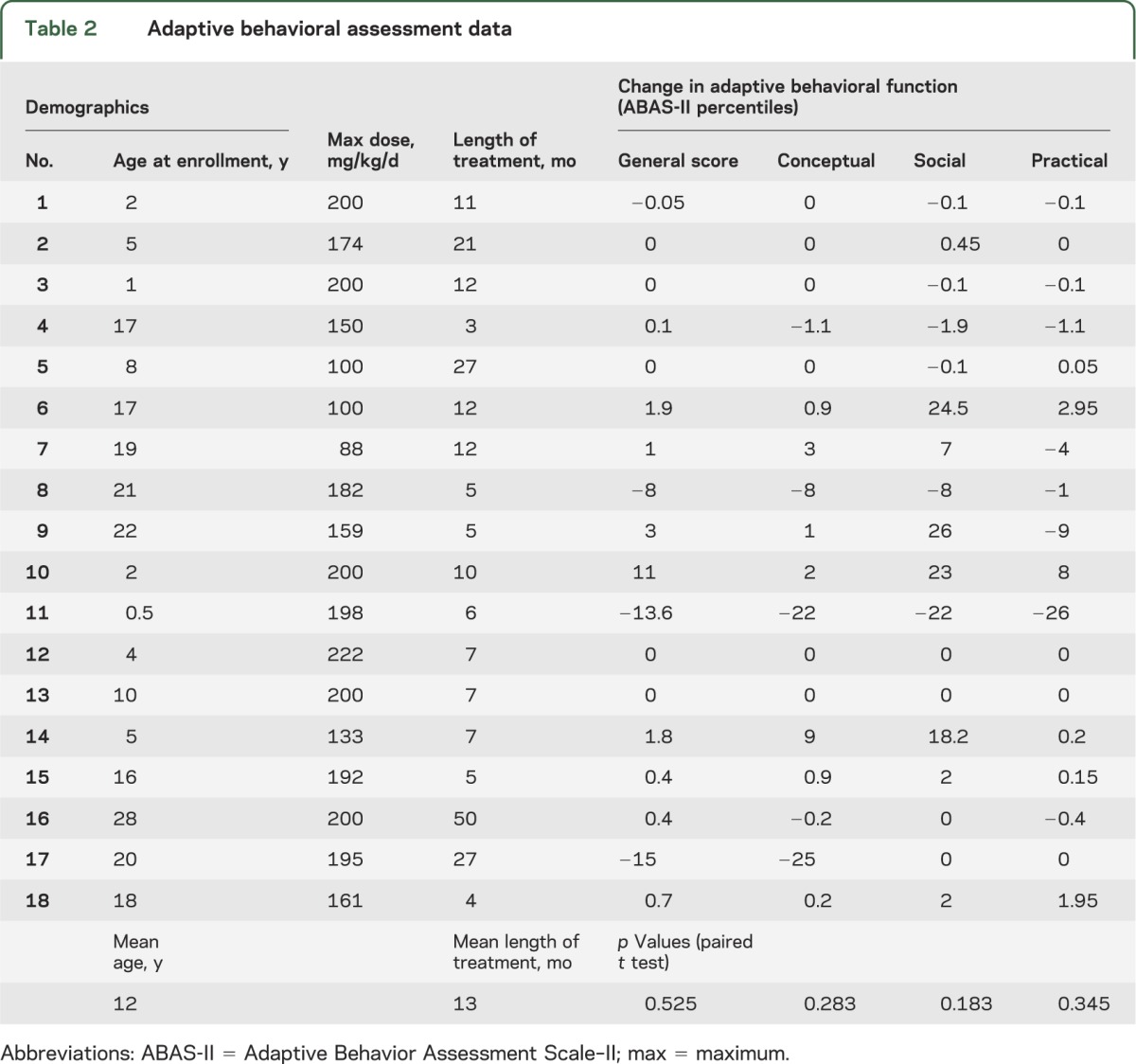
ABAS-II data did not show clinically meaningful or statistically significant improvement (p ≥ 0.18) in the general composite score or any of the 3 adaptive domains (table 2). In the majority of participants, adaptive behavior scores changed only a few percentile points in either a positive or negative direction, and often were only fractions of a percentile. Four individuals, however, did exhibit large improvements (≥18 percentile points) in the social domain. One patient showed a 22 percentile point decrease in conceptual function, and another a ≥22 percentile point decrease across all 3 domains. Specific changes reported in the social domain were improvements in eye contact and level of engagement.
Table 2.
Adaptive behavioral assessment data
Outside of ABAS-II measurements, 3 participants were reported by parents or close caretakers to have demonstrated significant qualitative improvement in their level of social engagement, communication, and eye contact within the first month of taurine therapy. These reports, however, did not have corresponding improvement in the individual ABAS-II scores. There were no reports of improvement in the cognitive or practical domains.
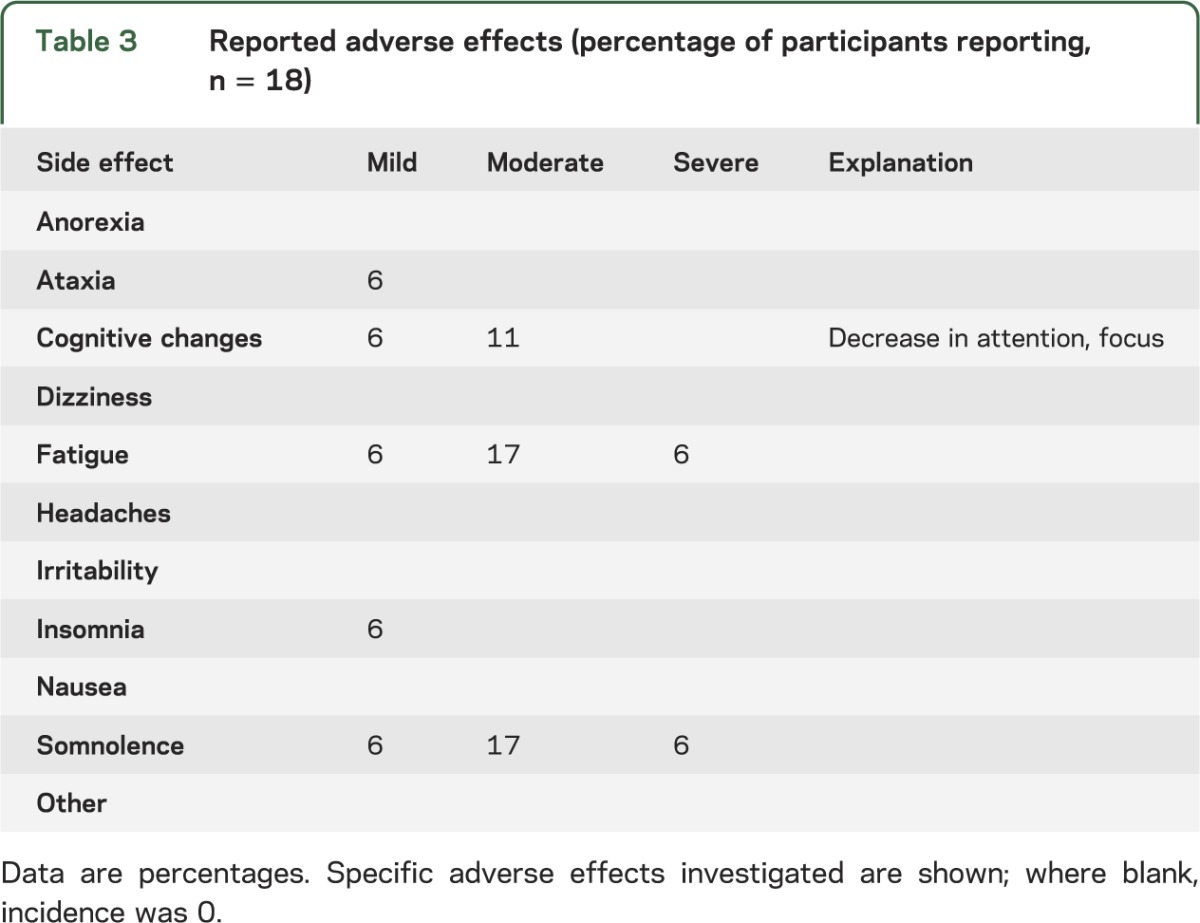
One severe adverse event, hospitalization for hypersomnia, occurred while a subject (aged 30 years) was on a dosage of 16 g/d (200 mg/kg/d). This prompted a dose-lowering paradigm establishing a maximum of 10 g/d for all patients. This new dose was well tolerated, with no further serious incidents reported. Other adverse events (table 3) reported with the most frequency included moderate fatigue, somnolence, and cognitive change (specified by the subjects' parents as diminished focus and attention). Mild insomnia, ataxia, somnolence, fatigue, and mildly diminished cognitive function were also noted for single individuals.
Table 3.
Reported adverse effects (percentage of participants reporting, n = 18)
Seven subjects who completed baseline ABAS-II forms did not return posttreatment data, and were therefore excluded from analyses. Three of these participants withdrew because of a perceived lack of efficacy. Data from one subject were invalidated because of an error in administering the ABAS-II (the wrong age-specific questionnaire was issued). The remaining 3 participants with incomplete data did not return their follow-up ABAS-II questionnaires.
DISCUSSION
Taurine is an aminosulfonic acid sold as a dietary supplement and is thought to have neuromodulatory and neuroprotective effects. This was a preliminary study to collect data on adaptive behavior, safety, and tolerability on taurine administration in a patient population that already had access to this amino acid. While taurine exhibited survival benefit in the SSADH-deficient mouse model, this open-label pilot trial of taurine in patients with SSADH deficiency did not lead to demonstrable clinical improvement in adaptive neurobehavioral function. Overall, changes in adaptive behavioral function as measured by ABAS-II were not statistically significant, although the small sample size (n = 18) would have rendered it difficult to conclude statistical significance for anything other than large effects (approximately ≥1 SD).
SSADH deficiency is not unique in the accumulation of GABA in CNS. Patients with heritable deficiency of GABA-transaminase accumulate supraphysiologic GABA levels, although few patients have been identified. As noted above, the use of VGB (a first-line intervention for infantile spasms and some forms of intractable epilepsy) leads to increased CNS GABA, and pharmacotherapeutics aimed to ameliorate VGB-associated retinal damage would be beneficial. Whereas most neurologic disorders, including major depressive disorder, feature decreased CNS GABA,18 human epileptic syndromes associated with mutations in GABAA and GABAB receptors19–21 could result in GABA increase within the synaptic cleft linked to reduced GABA binding and/or decreased GABA reuptake. Although these mutations invariably associate with altered GABAergic neurotransmission, quantitation of GABA in the CNS (either through CSF measures or magnetic resonance spectroscopy, or in vivo microdialysis evaluations in animal models) has not been reported. Accordingly, clinical trials in SSADH deficiency hold the potential to inform treatment concepts for other neurologic diseases in which increased CNS GABA may have a pathophysiologic role.
The patients in this trial did not all elect to continue the therapy, hence introducing variability in treatment duration, although only 2 were treated less than 5 months. The results of our pilot trial indicated that the majority of participants showed only small changes in social, conceptual, and practical adaptive behavior scores after taurine therapy. One-third of patients exhibited a change in at least 1 of the 3 ABAS domains, predominantly involving the social domain. Further therapeutic clinical trials focusing on SSADH deficiency are in progress, including a study of biomarkers (neuropsychological testing, transcranial magnetic stimulation, CSF metabolites) on controlled trials of taurine and an experimental GABAB receptor antagonist. Based on the present study, comprehensive neuropsychological testing measuring IQ, cognitive function, executive function, and hyperactivity are included in the future trials to enable heightened sensitivity in detection of change in clinical status.
GLOSSARY
- ABAS
Adaptive Behavior Assessment Scale
- GABA
γ-aminobutyric acid
- SSADH
succinic semialdehyde dehydrogenase
- VGB
vigabatrin
AUTHOR CONTRIBUTIONS
Phillip L. Pearl, MD: study design, data collection, and manuscript preparation. John Schreiber, MD, and William H. Theodore, MD: data collection and manuscript review. Robert McCarter, ScD: study design and statistical analysis. Emily S. Barrios, BA, Joe Yu, BA, and Edythe Wiggs, PhD: data collection and manuscript review. Jianping He, MS: statistical analysis. K. Michael Gibson, PhD: study design and manuscript preparation.
STUDY FUNDING
The trial was funded by NIH/NICHD R01 HD58553.
DISCLOSURE
The authors report no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Pearl PL, Novotny EJ, Acosta MT, et al. Succinic semialdehyde dehydrogenase deficiency in children and adults. Ann Neurol 2003;54(suppl 6):S73–S80 [DOI] [PubMed] [Google Scholar]
- 2.Pearl PL, Gibson KM, Acosta MT, et al. Clinical spectrum of succinic semialdehyde dehydrogenase deficiency. Neurology 2003;60:1413–1417 [DOI] [PubMed] [Google Scholar]
- 3.Pearl PL, Shukla L, Theodore WH, et al. Epilepsy in succinic semialdehyde dehydrogenase deficiency, a disorder of GABA metabolism. Brain Dev 2011;33:796–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pearl PL, Dorsey AM, Barrios ES, Gibson KM. Succinic semialdehyde dehydrogenase deficiency. In: Pagon RA, Bird TD, Dolan CR, et al., editors. GeneReviews [Internet]. Seattle: University of Washington; 1993. Available at: http://www.ncbi.nlm.nih.gov/books/NBK1195/. Accessed February 3, 2014 [Google Scholar]
- 5.Gupta M, Greven R, Jansen EE, et al. Therapeutic intervention in mice deficient for succinate semialdehyde dehydrogenase (gamma-hydroxybutyric aciduria). J Pharmacol Exp Ther 2002;302:180–187 [DOI] [PubMed] [Google Scholar]
- 6.Vogel KR, Pearl PL, Theodore WH, et al. Thirty years beyond discovery: clinical trials in succinic semialdehyde dehydrogenase deficiency, a disorder of GABA metabolism. J Inherit Metab Dis 2013;36:401–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bidri M, Choay P. Taurine: a particular amino acid with multiple functions [in French]. Ann Pharm Fr 2003;61:385–391 [PubMed] [Google Scholar]
- 8.Olive MF. Interactions between taurine and ethanol in the central nervous system. Amino Acids 2002;23:345–357 [DOI] [PubMed] [Google Scholar]
- 9.Latini A, Scussiato K, Leipnitz G, Gibson KM, Wajner M. Evidence for oxidative stress in tissues derived from succinate semialdehyde dehydrogenase-deficient mice. J Inherit Metab Dis 2007;30:800–810 [DOI] [PubMed] [Google Scholar]
- 10.Sauer SW, Kölker S, Hoffmann GF, et al. Enzymatic and metabolic evidence for a region specific mitochondrial dysfunction in brains of murine succinic semialdehyde dehydrogenase deficiency (Aldh5a1−/− mice). Neurochem Int 2007;50:653–659 [DOI] [PubMed] [Google Scholar]
- 11.Niemi AK, Brown C, Moore T, Enns GM, Cowan TM. Low glutathione levels in a patient with succinic semialdehyde dehydrogenase (SSADH) deficiency. Mol Genet Metab 2012;105:345 Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tomi M, Tajima A, Tachikawa M, et al. Function of taurine transporter (Slc6a6/TauT) as a GABA transporting protein and its relevance to GABA transport in rat retinal capillary endothelial cells. Biochim Biophys Acta 2008;1778:2138–2142 [DOI] [PubMed] [Google Scholar]
- 13.Oermann E, Warskulat U, Heller-Stilb B, et al. Taurine-transporter gene knockout-induced changes in GABA(A), kainate and AMPA but not NMDA receptor binding in mouse brain. Anat Embryol 2005;210:363–372 [DOI] [PubMed] [Google Scholar]
- 14.Jammoul F, Degardin J, Gondouin P, et al. Taurine deficiency damages photoreceptors and retinal ganglion cells in vigabatrin-treated neonatal rats. Mol Cell Neurosci 2010;43:414–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jammoul F, Wang Q, Nabbout R, et al. Taurine deficiency is a cause of vigabatrin-induced retinal phototoxicity. Ann Neurol 2009;65:98–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Saronwala A, Tournay A, Gargus JJ. Taurine treatment of succinate semialdehyde dehydrogenase (SSADH) deficiency reverses MRI-documented globus lesions and clinical syndrome. Proc Am Coll Med Genet 2008:103 Abstract [Google Scholar]
- 17.Harrison PL, Oakland T. Adaptive Behavior Assessment System Manual. San Antonio: Harcourt; 2000 [Google Scholar]
- 18.Luscher B, Shen Q, Sahir N. The GABAergic deficit hypothesis of major depressive disorder. Mol Psychiatry 2011;16:383–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tian M, Mei D, Feri E, et al. Impaired surface αβγ GABAA receptor expression in familial epilepsy due to a GABARG2 frameshift mutation. Neurobiol Dis 2013;50:135–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Galanopoulou AS. Mutations affecting GABAergic signaling in seizures and epilepsy. Pflugers Arch 2010;460:505–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Enna SJ, Bowery NG. GABAB receptor alterations as indicators of physiological and pharmacological function. Biochem Pharmacol 2004;68:1541–1548 [DOI] [PubMed] [Google Scholar]