

Abstract
Neurokinin B (NKB) and its G-protein-coupled receptor, NK3R, have been implicated in the neuroendocrine control of GnRH release; however, little is known about the structure-function relationship of this ligand-receptor pair. Moreover, loss-of-function NK3R mutations cause GnRH deficiency in humans. Using missense mutations in NK3R we previously identified in patients with GnRH deficiency, we demonstrate that Y256H and Y315C NK3R mutations in the fifth and sixth transmembrane domains (TM5 and TM6), resulted in reduced whole-cell (79.3±7.2%) or plasma membrane (67.3±7.3%) levels, respectively, compared with wild-type (WT) NK3R, with near complete loss of inositol phosphate (IP) signaling, implicating these domains in receptor trafficking, processing, and/or stability. We further demonstrate in a FRET-based assay that R295S NK3R, in the third intracellular loop (IL3), bound NKB but impaired dissociation of Gq-protein subunits from the receptor compared with WT NK3R, which showed a 10.0 ± 1.3% reduction in FRET ratios following ligand binding, indicating activation of Gq-protein signaling. Interestingly, R295S NK3R, identified in the heterozygous state in a GnRH-deficient patient, also interfered with dissociation of G proteins and IP signaling from wild-type NK3R, indicative of dominant-negative effects. Collectively, our data illustrate roles for TM5 and TM6 in NK3R trafficking and ligand binding and for IL3 in NK3R signaling.—Noel, S. D., Abreu, A. P., Xu, S., Muyide, T., Gianetti, E., Tusset, C., Carroll, J., Latronico, A. C., Seminara, S. B., Carroll, R. S., Kaiser, U. B. TACR3 mutations disrupt NK3R function through distinct mechanisms in GnRH-deficient patients.
Keywords: GPCR, hypogonadotropic hypogonadism, neurokinin B, tachykinins
Neurokinin B (NKB), a neuropeptide belonging to the tachykinin family that also includes neurokinin A and substance P (1–4), has been implicated in various biological processes including menopause (5–7), cognitive function (8, 9), and smooth muscle contraction (8, 9). Perturbations in NKB activity or signaling have been implicated in preeclampsia (10), schizophrenia (11, 12), and more recently GnRH deficiency (13–17). The tachykinins bind to NK1R, NK2R, and NK3R (18–20), which are members of the rhodopsin superfamily of G-protein-coupled receptors (GPCRs), characterized by a common structure consisting of an amino terminus, 7 transmembrane (TM) domains connected by 3 extracellular loops (ELs) and intracellular loops (ILs), and a carboxyl terminus. NKB binds selectively to NK3R but can bind NK1R and NK2R with reduced affinity (21). NK3R, in turn, couples to Gq proteins and activates both inositol phosphate (IP) and protein kinase C signaling pathways following ligand stimulation (22). Although the crystal structure for NK3R has not yet been resolved, an increasing number of structural studies of GPCRs have provided additional insight into the roles of various domains of this family of GPCRs. In general, the amino terminus, ELs, and TM domains are typically involved in ligand recognition and binding (23, 24). The TM domains also play roles in receptor trafficking to the cell surface (25), while IL2 and IL3 are involved in GPCR coupling to G proteins and β-arrestins to activate signaling cascades (23) and to mediate receptor internalization, recycling, desensitization, and resensitization (26). Recently, a molecular model of an NKB/NK3R complex was generated, using the bovine rhodopsin crystal structure as a template for TM domains, NMR data of NK1R for EL2 and EL3, and computer-generated predictions for the conformations of EL1 and IL1–3 (27). Using this model, the authors were able to predict key amino acid residues involved in NKB binding to NK3R (27). Other bioinformatics programs, such as I-TASSER, have also been used to elucidate the 3-dimensional location and roles of amino acids within the NK3R structure (28).
Topaloglu et al. (13) first demonstrated the role of loss-of-function mutations in NKB and NK3R in the pathogenesis of GnRH deficiency, a condition characterized by a failure to undergo pubertal maturation associated with low gonadotropin and sex steroid hormone levels. We and others have subsequently reported additional loss-of-function mutations in these genes in cohorts of patients with GnRH deficiency, confirming that TAC3, encoding NKB, and TACR3, encoding NK3R, are critical regulators of normal reproductive development and function in humans (14–17). These findings, coupled with earlier reports that NKB and NK3R play roles in regulating LH secretion and mediating estrogen negative feedback mechanisms in postmenopausal women (5–7), highlight the importance of this ligand-receptor pair in reproduction.
The process through which NKB regulates GnRH release is not fully understood. NKB and NK3R are coexpressed with the potent GnRH secretagogue kisspeptin in neurons within the arcuate nucleus and are postulated to have an autocrine regulatory effect on kisspeptin release (29–32). These neurons project to KISS1R-expressing GnRH neurons to regulate GnRH release (33–35). In addition, early work demonstrated that GnRH axons express NK3R immunoreactivity (6, 36) and more recent studies have demonstrated direct NKB action on GnRH release from GFP-expressing GnRH neurons and from the GnRH neuronal GT1–7 cell line, providing evidence for multiple modes of NKB action on GnRH release (37, 38). A number of animal studies in monkeys, sheep, rats, and mice support a stimulatory effect of NKB on GnRH release (39–42), although other studies have demonstrated that NKB may inhibit or have no effect on LH secretion in rats and mice under certain physiological conditions (5, 43, 44). Interestingly, NK3R-deficient mice are fertile (45–47), although a detailed study recently demonstrated that these mice do manifest some reproductive defects, with knockout males having lower FSH levels and reduced testicular size and females having lower uterine weights, abnormal estrous cycles, reduced numbers of litters, and smaller litter sizes, compared with wild-type (WT) mice (48).
Mutations in genes encoding GPCRs can result in amino acid substitutions in the receptors that interfere with receptor expression, trafficking, ligand binding, or G-protein coupling and activation of signaling cascades, leading to disease. Although an increasing number of NK3R mutations have been reported in association with isolated hypogonadotropic hypogonadism (IHH; refs. 13–17), the mechanisms through which these mutations impair receptor function have not been fully elucidated. In our previous report (15), 2 homozygous missense mutations, Y256H and Y315C, and 3 heterozygous missense mutations, G18D, I249V, and R295S, were identified in NK3R in our cohort of patients with IHH. In vitro studies demonstrated that the Y256H, Y315C, and R295S mutant NK3Rs were loss-of-function mutations with reduced IP accumulation in response to 10−7 M NKB, compared with WT NK3R (15). In the current study, we hypothesized that mutations in the TM domains, such as Y256H and Y315C, may interfere with ligand binding or disrupt receptor trafficking to reduce cell surface receptor levels, whereas mutations in IL3, such as R295S, may affect receptor-mediated activation of signaling cascades. We have performed more extensive functional analyses to measure total cellular and plasma membrane receptor levels, ligand binding affinity and capacity, and dose-dependent ligand-stimulated activation of signal transduction pathways. We also examined whether deleterious mutants identified in patients with GnRH deficiency in the heterozygous state had dominant-negative effects on WT NK3R function to explain the manifestation of the reproductive phenotype. These studies serve to elucidate the molecular mechanisms by which these mutations cause GnRH deficiency and highlight structure-function relationships of the NKB receptor.
MATERIALS AND METHODS
Materials
Chemicals used were obtained from either Sigma Chemical (St. Louis, MO, USA) or Fisher Scientific (Pittsburgh, PA, USA). The following antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA): ERK, p-ERK, Gαq/11, β-actin, and Na+/K+-ATPase. Monoclonal anti-HA antibody was obtained from Covance (Berkeley, CA, USA), and anti-V5 antibody from Abcam (Cambridge, MA, USA). All secondary antibodies were purchased from Santa Cruz Biotechnology.
Site-directed mutagenesis
A hemagglutinin (HA) epitope-tagged human TACR3 cDNA was obtained from the Missouri University of Science and Technology (Missouri S&T) cDNA Resource Center (Rolla, MO, USA: http://www.cdna.org) and used as WT NK3R and as a template to generate the NK3R mutants using the QuikChange Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA, USA). cDNAs encoding WT and R295S NK3Rs were also subcloned into pCDNA3.1/V5HisD TOPO expression vector (Invitrogen). The primer sequences used for mutagenesis are as follows: G53A (G18D), 5′-GTGGAGGCGTGGATGCAGACGCCGT-3′ and 5′-ACGGCGTCTGCATCCACGCCTCCAC-3′; A745G (I249V), 5′-CCAAACAACATTTCACTTACCATGTTATCGTCATTATACTGGTGTAC-3′ and 5′-GTACACCAGTATAATGACGATAACATGGTAAGTGAAATGTTGTTTGG-3′; T766C (Y256H), 5′-TTATCGTATTATACTGGTGCACTGTTTCCCATTGCTCATC-3′ and 5′-GATGAGCAATGGGAAACAGT GCACCATATAAATGCGATAA-3′; A885C (R295S), 5′-GAGCAGCTAAAGGCCAAAAGCAAGGTTGTCAAAATGATGAT-3′ and 5′-ATCATCATTTTGACAACCTTGCTTTTGGCCTTTAGCTGCTC-3′; A944G (Y315C), 5′-CATTTGCTATGCTGGCTGCCCTGTCATATTTACTTCATTC-3′ and 5′-GAATGAAGTAAATATGACAGGGCAGCCAGCAGATAGCAAATG-3′. The sequences of the mutant cDNAs were confirmed by bidirectional sequencing.
Cell culture
Both HEK293 and COS-7 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) with 100 U/ml penicillin and 100 μg/ml streptomycin sulfate (Invitrogen, Carlsbad, CA, USA) and supplemented with 10% (v/v) fetal bovine serum (Omega, Tarzana, CA, USA) in 5% CO2 humidified air at 37°C.
IP assay
IP assays were performed as described previously with some modifications (49). COS-7 cells were plated into 6-well tissue culture dishes, and transient transfections were performed using GenePORTER (Gene Therapy Systems, San Diego, CA, USA) with 0.5 μg/well of WT NK3R, mutant NK3Rs, or control vector (pcDNA3). In all cases, total DNA transfected in each well was adjusted to 1 μg with control vector. After 48 h, medium was replaced with 1 ml of inositol-free DMEM for 2 h, which was then removed, and fresh inositol-free medium containing 2 μCi/well of myo-[2-3H]-inositol (Perkin-Elmer, Waltham, MA, USA) was added to cells for 15 min, followed by the addition of 10 mM LiCl. Cells were further incubated at 37°C for 16 h and then stimulated with increasing concentrations (10−10 to 10−6 M) of NKB (Genescript, Piscataway, NJ, USA) for 45 min. Cells were extracted twice on ice with 20 mM formic acid, and lysates were neutralized to pH 7.5 with 7.5 mM HEPES and 150 mM KOH and centrifuged at 14,000 g for 2 min. Supernatants were loaded onto 0.5 ml AG-×8 resin anion exchange columns (Bio-Rad, Hercules, CA, USA) previously equilibrated with 2 ml of 1 M NaOH, 2 ml of 1 M formic acid, and 5 × 5 ml of ddH2O. The columns were washed with 5 ml of ddH2O followed by 5 ml of 5 mM borax and 60 mM sodium formate, and IPs were eluted with 3 ml of 0.9 M ammonium formate and 0.1 M formic acid and counted using a scintillation counter. Data points were performed in triplicate, and each experiment was replicated ≥3 times. Dose-response curves were generated and EC50 values calculated using Prism 5.0 (GraphPad Software, San Diego, CA, USA).
In IP assays used to assess dominant-negative activity, COS-7 cells were cotransfected with 100 ng (1×) of WT or mutant NK3R, 200 ng (2×) of WT NK3R, 100 ng of WT and mutant NK3R, 50 ng (0.5×) WT NK3R, or 50 ng of WT NK3R and 100 ng of mutant NK3R. Total cDNA transfected in each well was adjusted to 1 μg with control vector.
Receptor binding assays
Receptor binding was analyzed as described previously, with some modifications (49). COS-7 cells were plated into 6-well tissue culture dishes, and transient transfection was performed using GenePORTER with 0.5 μg/well of expression vectors encoding WT or mutant NK3Rs or control vector (pcDNA3). Total DNA transfected in each well was adjusted to 1 μg with control vector. After incubation for 24 h at 37°C, cells were rinsed with DMEM containing 0.1% BSA and incubated concomitantly with 105 cpm of 125I-NKB [specific activity 2200 Ci (81.4 TBq)/mmol; Perkin-Elmer] and increasing concentrations of unlabeled NKB (10−10 to 4×10−6 M) in DMEM and 0.1% BSA for 20 min at room temperature. Cells were rinsed twice with ice-cold PBS and lysed with 2 ml of 0.2 M NaOH, and radioactivity was measured in a γ counter. Affinity (50) and maximum binding (Bmax) were calculated based on nonlinear regression of homologous competition binding analysis using Prism 5.0. Data points were determined in triplicate, and each experiment was replicated ≥3 times.
In binding assays performed to assess dominant-negative activity, COS-7 cells were cotransfected with 300 ng (1×) of WT or R295S NK3R, 600 ng (2×) of WT NK3R, 300 ng of WT and R295S NK3R, or 300 ng WT NK3R and 900 ng of R295S NK3R. Total DNA transfected in each well was adjusted to 1.5 μg with control vector.
Western blot analyses
To analyze total cellular and plasma membrane NK3R protein levels, HEK293 cells plated in 6-well culture dishes were transiently transfected with expression vectors encoding WT or mutant NK3Rs, and 24 h later, total cellular or plasma membrane proteins were isolated using the Pierce Cell Surface Protein Isolation Kit (Thermo Scientific, Rockford, IL, USA). Proteins were subjected to SDS-PAGE with 20 μg protein loaded onto 10% polyacrylamide gels under reducing conditions at 100 V. Proteins were electrotransferred onto PVDF membranes (Millipore, Bedford, MA, USA). Nonspecific binding was blocked with 5% nonfat dry milk in Tris-buffered saline-Tween 20 (TBST) for 1 h at room temperature. Blots were then incubated overnight with anti-HA antibody (1:2000) at 4°C, rinsed 3× with 2% milk TBST, and incubated with an anti-mouse secondary antibody linked to horseradish peroxidase. After secondary antibody incubation, blots were rinsed 3×, and the immunoreactive bands were visualized using an enhanced chemiluminescence reagent (PerkinElmer) and then exposed to Kodak X-ray films (Kodak, Rochester, NY, USA). Molecular mass was approximated using molecular mass markers. Blots were stripped with Restore Western Blot Stripping Buffer (Thermo Scientific) and then total cellular protein blots were reprobed with β-actin antibody (1:10,000) and membrane protein blots reprobed with Na+/K+-ATPase antibody (1:1000) for normalization. All X-ray films were analyzed by densitometry using ImageJ software [Wayne Rasband, U.S. National Institutes of Health (NIH), Bethesda, MD, USA]. For analysis of whole-cell and membrane NK3R expression, both bands were quantified together.
Measurement of ERK activation
To analyze NKB-stimulated ERK phosphorylation, COS-7 cells were plated in 6-well culture dishes, and transient transfections were performed with expression vectors encoding WT or mutant NK3Rs. Cells were serum starved for 24 h, and then transfected cells were stimulated with 10−7 M NKB in a time course from 0 to 120 min. Cells were then lysed in RIPA buffer (PBS, 1% Nonidet P-40, 0.5% sodium deoxycholate, and 0.1% SDS) containing 0.1 mg/ml PMSF, 30 μl/ml aprotinin, and 1 mM sodium orthovanadate, and cell debris was pelleted by centrifugation at 4°C for 10 min. Protein lysates were separated on a 12% SDS-PAGE gel and electrotransferred onto PVDF membranes as described above. Membranes were incubated overnight at 4°C with mouse anti-phospho-ERK42/44 antibody (1:1000), washed 3× with TBS/0.05% Tween-20, and incubated for 1 h at room temperature with donkey anti-mouse antibody (1:2000) conjugated to horseradish peroxidase. After secondary antibody incubation, blots were rinsed 3×, and the immunoreactive bands were visualized using an enhanced chemiluminescence reagent (PerkinElmer) and then exposed to X-ray films (Kodak). Membranes were then stripped using Restore Western blot stripping buffer (Pierce) and incubated overnight at 4°C with mouse anti-ERK antibody (1:1000). Total immunoreactive ERK was detected as described above. X-ray films were analyzed by densitometry, and the intensity of pERK was normalized to total ERK.
Immunoprecipitation of Gαq and NK3R proteins
To determine whether NK3R dimerizes or whether WT and R295S NK3R can heterodimerize, cDNAs encoding WT and R295S NK3R were cloned into pCDNA3.1/V5HisD TOPO expression vector (Invitrogen). COS-7 cells were cotransfected with 500 ng of V5-tagged WT NK3R and 500 ng of either HA-tagged WT or R295S NK3R. Cells were lysed in buffer containing 0.5% Nonidet P-40, 50 mM Tris, and 5 mM EDTA. Lysates were immunoprecipitated with anti-V5 agarose beads (Bethyl Laboratories, Montgomery, TX, USA) according to manufacturer's instructions. Briefly, 500 μg of cell lysate was incubated with anti-V5 agarose beads for 3 h at room temperature. Beads were pelleted and washed 3× with lysis buffer, after which immunoprecipitated proteins were eluted using 4× sample buffer (Invitrogen), resolved on SDS-PAGE as described above, and immunoblotted with anti-HA antibody (1:1000), anti-V5 antibody (1:1000), and anti-β-actin (1:10,000). Fifteen percent of cellular lysates was used as input and subjected directly to Western blot analysis as a control.
To determine whether NK3R interacts with Gq protein, COS-7 cells transfected with HA-tagged WT and R295S NK3Rs were lysed with M-PER lysis buffer (Thermo Scientific), and lysates were incubated with Protein A/G-agarose beads (Invitrogen) for 1 h at 4°C on an orbital rocker, after which 1 μg/ml anti-Gαq/11 antibody was added and placed on the orbital rocker overnight at 4°C. Beads were pelleted and washed 3× with lysis buffer after which immunoprecipitated proteins were resolved on an SDS-PAGE gel as described above and immunoblotted with an anti-HA antibody.
Fluorescence resonance energy transfer (FRET)
FRET measurements for CFP and YFP were analyzed as described previously (51) from COS-7 cells transiently transfected with 250 ng Gαq-CFP, 250 ng Gβ-YFP [kindly provided by Dr. Bertil Hille (University of Washington School of Medicine, Seattle, WA, USA) with permission from Dr. Narasimhan Gautam (Washington University School of Medicine in St. Louis, St. Louis, MO, USA) and Dr. Stephen Ikeda (National Institute on Alcohol Abuse and Alcoholism, Division of Clinical and Biological Research, NIH)], 250 ng GRK2 (kindly provided by Dr. Moritz Buneman, Philipps-Universtitaet Marburg, Marburg, Germany) and 250 ng Gγ2 (obtained from the Missouri S&T cDNA Resource Center), and either 250 ng WT NK3R, 750 ng R295S NK3R, or both WT and R295S NK3R together at a ratio of 1:3. All wells were adjusted to a total of 2 μg of DNA using empty vector. GRK2 was added to transfections into cells, as this was previously reported to improve G-protein FRET amplitude ratios and decrease signal to noise ratios (51, 52). At 48 h after transfection, FRET measurements of 5–8 cells transfected with either WT NK3R, R295S NK3R, or both were imaged at the Harvard NeuroDiscovery Center optical imaging facility, and measurements were recorded using an Andor Revolution System with Yokogawa Spinning Disk with an Andor iXon3 EMCCD camera (Andor Technology, Belfast, UK). Recordings from YFP and CFP channels were done sequentially for each cell, with an exposure time of 200 ms for each channel and a time gap of 5 s between each recording. The images were taken with a 445-nm laser on CFP (468- to 499-nm) and YFP (529- to 555-nm) channels. Cells were imaged for 25 s before and 35 s after 10−7 M NKB application. After corrections for background fluorescence and bleedthrough, the YFP and CFP measurements were used to determine FRET ratios using the formula FRET ratio = FYFP/FCFP = 0.98(WL − 0.5WS)/WS, where WL is long wavelength channel and WS is short wavelength channel. Changes in FRET ratios are indicative of dissociation of CFP and YFP proteins.
Statistical analysis
IP and binding assays were subjected to nonlinear regression analysis using Prism 5.0 to calculate EC50 in the case of IP assays and Kd and Bmax in the case of binding assays. Western blot quantification was done using ImageJ. All statistical analyses were done using Prism 5.0 and were based on nonparametric t tests (P<0.05) or 1-way ANOVA with Tukey's multiple comparison test where applicable.
RESULTS
Computational modeling predicts that the Y256H, R295S, and Y315C mutations are deleterious to NK3R function
Three software programs, SIFT (53), PolyPhen (54), and Panther (55), were used to determine the degree of conservation at the positions of the amino acid substitutions and whether the mutations were predicted to have deleterious effects on NK3R function. The domains in which each mutation is located are indicated in Table 1. Panther analysis demonstrated that 4 of the 5 amino acid residues studied, I249, Y256, R295, and Y315, were highly conserved across several species, including humans, monkeys, mice, African clawed frogs (platannas), and zebrafish (Table 1). The tyrosine residue at position 256 was also highly conserved across the rhodopsin family of GPCRs (23) and is important for the active conformational state of the β-2 adrenergic receptor (56). The glycine at position 18 in the amino terminus was not conserved in the more primitive African clawed frogs and zebrafish, as these animals had more truncated amino termini. The prediction programs indicated the G18D and I249V substitutions would likely have no deleterious effect on NK3R function, while Y256H, R295S, and Y315C NK3R mutants were predicted to be pathological, in agreement with the results of our previous initial screen of these mutants (15). Further analysis using the I-TASSER bioinformatics program (28) predicted that Y256 and Y315, located in TM5 and TM6, respectively, are residues important for NKB binding to NK3R, a prediction replicated in an NKB/NK3R molecular modeling study of the NKB binding pocket of NK3R (27).
Table 1.
Conservation of amino acids and prediction analysis of effects of mutations on NK3R function
| Nucleotide change | Amino acid change | Location | Amino acid conservation |
Prediction analysis |
|||
|---|---|---|---|---|---|---|---|
| Species | Amino acids | SIFT | PolyPhen | Panther | |||
| [c.53G>A]+[=] | [p.G18D] | NH2 terminus | Human | GGV G ADA | − | + | − |
| Monkey | GGV G ADA | ||||||
| Mouse | AGV G SHT | ||||||
| Rat | VEV G THT | ||||||
| Platanna | Absent in species | ||||||
| Zebrafish | Absent in species | ||||||
| [c.745A>G]+[=] | [p.I249V] | TM5 | Human | TYH I IVI | − | − | − |
| Monkey | TYH I IVI | ||||||
| Mouse | TYH I IVI | ||||||
| Rat | TYH I IVI | ||||||
| Platanna | MYH I IVT | ||||||
| Zebrafish | MYH I IVI | ||||||
| [c.766T>C]+[c.766T>C] | [p.Y256H] | TM5 | Human | ILV Y CFP | + | + | + |
| Monkey | ILV Y CFP | ||||||
| Mouse | ILV Y CFP | ||||||
| Rat | ILV Y CFP | ||||||
| Platanna | LLV Y VLP | ||||||
| Zebrafish | VLV Y MLP | ||||||
| [c.885A>C]+[=] | [p.R295S] | IL3 | Human | KAK R KVV | + | + | + |
| Monkey | KAK R KVV | ||||||
| Mouse | KAK R KVV | ||||||
| Rat | KAK R KVV | ||||||
| Platanna | RAK R KVV | ||||||
| Zebrafish | RAK R KVV | ||||||
| [c.944A>G]+[c.944A>G] | [p.Y315C] | TM6 | Human | WLP Y HIY | + | + | + |
| Monkey | WLP Y HVY | ||||||
| Mouse | WLP Y HIY | ||||||
| Rat | WLP Y HIY | ||||||
| Platanna | WLP Y HIF | ||||||
| Zebrafish | WLP Y HIY | ||||||
NKB-mediated activation of IP and ERK signaling is impaired for Y256H, R295S, and Y315C NK3Rs
To assess activation of NK3R signaling by NKB and to generate full dose-response curves for EC50 calculations, expression vectors encoding WT or mutant NK3Rs were transiently transfected into COS-7 cells, and NKB-stimulated IP accumulation was measured using increasing concentrations (10−10 to 10−6 M) of NKB (Fig. 1). G18D and I249V NK3R had dose-response curves similar to WT NK3R (P>0.05; EC50: WT 1.31×10−8 M, G18D 1.72×10−8 M, and I249V 4.76×10−9 M). However, stimulation of Y256H, R295S, and Y315C NK3Rs by NKB resulted in significantly reduced maximal IP responses compared with WT NK3R (P<0.001), with EC50 values of 1.36 × 10−7, 8.94 × 10−8, and 1.44 × 10−7 M, respectively (Fig. 1). These findings were consistent with our original report indicating that Y256H, R295S, and Y315C NK3R mutants had impaired NK3R activation (15).
Figure 1.
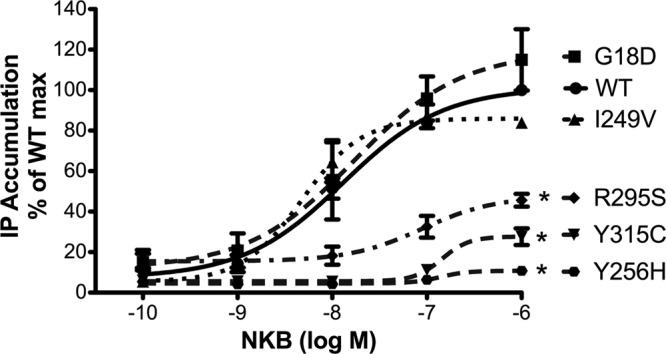
IP accumulation following NKB stimulation is impaired for Y256H, R295S, and Y315C NK3Rs. COS-7 cells were transfected with vectors encoding WT or mutant NK3Rs as indicated and stimulated with increasing concentrations of NKB (10−10 to 10−6 M) for 45 min, after which IP accumulation was measured to generate IP dose-response curves and calculate EC50 values and maximal responses. Data are pooled from ≥3 independent experiments, each performed in triplicate. Results are expressed as percentage of maximal IP accumulation for WT NK3R. Data at each point are shown as means ± se. Significant differences between maximal IP responses were measured using Student's t test. *P < 0.001 vs. WT.
The effect of NK3R mutations on ERK phosphorylation (P-ERK) following NKB stimulation was also assessed. COS-7 cells expressing either WT or mutant NK3Rs were treated with 10−7 M NKB in a time course from 0 to 120 min. In COS-7 cells expressing WT, G18D, or I249V NK3R, NKB rapidly stimulated P-ERK within 5 min, with maximal levels obtained 10 min after treatment, and P-ERK levels remained elevated above basal levels for as long as 120 min post-treatment, the longest time point studied (Fig. 2A). In contrast, the increase in P-ERK following NKB stimulation was more modest and less sustained for R295S and Y315C NK3Rs, and there was no P-ERK activation observed for Y256H NK3R, indicating that these mutations blunted P-ERK activation by NKB (Fig. 2A). The data were corrected for total ERK and normalized to P-ERK levels 10 min after NKB stimulation in cells expressing WT NK3R to compare the extent of NKB-stimulated ERK phosphorylation for each receptor. Statistical analyses showed that NKB treatment elicited a significant increase in P-ERK after 10 min over baseline for WT, G18D, and I249V NK3R (P<0.05), whereas the increases in P-ERK for Y256H, R295S, and Y315C NK3R were not significant compared with baseline, and were significantly reduced compared with WT NK3R (Fig. 2B).
Figure 2.

ERK phosphorylation following NKB stimulation is impaired for Y256H, R295S, and Y315C NK3Rs. COS-7 cells expressing WT or mutant NK3Rs as indicated were serum starved for 24 h and then treated with 10−7 M NKB. MAPK activation was assessed by measurement of ERK1/2 phosphorylation by Western blot analysis. A) Time course of NKB-stimulated ERK phosphorylation, from 0 to 120 min. Figures are representative of 3 independent experiments. B) Graph of levels of ERK phosphorylation 10 min after NKB treatment from 3 independent experiments, each normalized to P-ERK levels in cells transfected with WT NK3R. Significant differences, indicated by different letters, were measured using 1-way ANOVA with a post hoc Tukey's multiple comparison test.
Two mutant NK3Rs with signaling defects, Y256H and Y315C, fail to bind NKB
The Y256H, R295S, and Y315C mutants failed to stimulate IP accumulation or P-ERK activation at a level comparable to WT NK3R. We therefore sought to examine whether these signaling defects resulted from a loss of NKB binding to these mutant receptors. In silico structural analysis implicated Y256 and Y315 as important residues for NKB binding to NK3R (27, 28). Radioligand binding assays were performed by incubating COS-7 cells expressing WT or mutant NK3Rs concomitantly with 125I-radiolabeled NKB (125I-NKB) and increasing concentrations (10−10 to 5×10−6 M) of unlabeled NKB to produce competitive binding curves (Fig. 3). The WT, G18D, and I249V NK3Rs all bound 125I-NKB, producing binding curves with similar Kd (Kd: WT 1.0×10−7 M, G18D 1.1×10−7 M, and I249V 7.8×10−8 M) and Bmax values (Bmax: WT 1040±26 cpm, G18D 1107±113 cpm, and I249V 1164±165 cpm). Interestingly, R295S NK3R, which had markedly reduced IP accumulation and ERK phosphorylation, bound 125I-NKB with a Kd (3.9×10−7 M) not significantly different from WT NK3R, although Bmax (782±105 cpm) was modestly but significantly reduced (P<0.05). There was no detectable specific binding of 125I-NKB to Y256H and Y315C NK3R, consistent with their lack of signaling activity.
Figure 3.
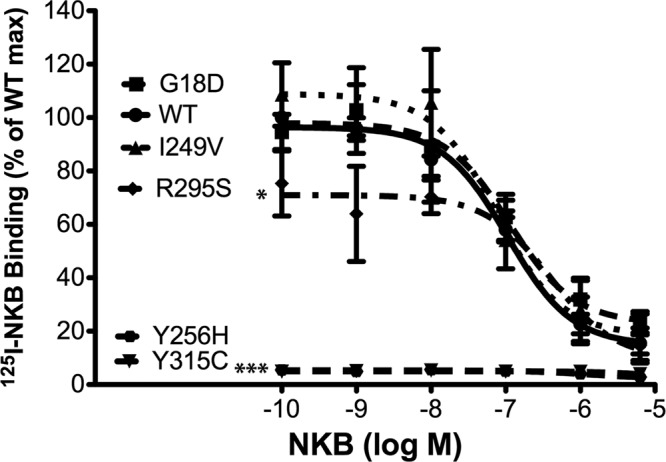
Y256H and Y315C NK3R, but not R295S NK3R, fail to bind 125I-NKB. Competitive displacement curves were generated from radiolabeled binding assays. COS-7 cells expressing WT or mutant NK3R were incubated concomitantly with 125I-NKB and increasing concentrations of unlabeled NKB (10−10 to 4×10−6 M). Displacement curves were used to calculate Kd and Bmax values. Data were pooled from ≥3 independent experiments, each performed in triplicate. Results are expressed as the percentage of maximal binding for WT receptor. Data at each point represent means ± se. Significant differences between Kd and Bmax values for mutant receptors compared with WT NK3R were calculated using Student's t test. *P < 0.05, ***P < 0.001 vs. WT.
Plasma membrane receptor levels of Y256H, R295S, and Y315C NK3R mutants are reduced compared with WT NK3R
Amino acid residues, particularly those in TM domains, also play roles in receptor folding and trafficking, which can result in reduced receptor levels at the cell surface and may contribute to the observed decrease in ligand binding for some of the mutant receptors. We therefore examined the effects of the mutations on total cellular and membrane receptor levels. Western blot analysis was performed to analyze whole-cell and membrane protein fractions from HEK293 cells transiently transfected with expression vectors encoding N-terminally HA-tagged WT or mutant NK3Rs. Whole-cell receptor levels were similar for WT, G18D, I249V, R295S, and Y315C NK3Rs (P>0.05), producing 2 bands (expected size 52 kDa), which may represent both mature and immature or less modified forms of NK3R. However, the Y256H NK3R had a 79.3 ± 7.2% decrease in receptor levels (based on the combined intensity of both bands) compared with WT NK3R (Fig. 4A, C). Membrane receptor levels for G18D NK3R were also similar to those for WT NK3R. However, membrane levels of I249V and R295S NK3R were modestly but significantly decreased (P<0.05), by 24.5 ± 4.2 and 35.3 ± 3.4%, respectively, compared with WT NK3R. Interestingly, the Y315C mutant NK3R had a more marked 67.3 ± 7.3% decrease in membrane levels despite normal total cellular levels, indicating that this mutant had impaired trafficking to plasma membranes (Figs. 4B, D). The Y256H NK3R mutant had an 87.6 ± 5.2% decrease in membrane levels, consistent with its reduced total cellular expression, suggesting that the primary defect for this mutant may be in protein processing and/or stability. The reductions in R295S, Y256H, and Y315C NK3R membrane protein levels are consistent with the reduced Bmax levels determined in the binding assays for these mutants.
Figure 4.

Mutant NK3Rs have reduced total whole-cell and membrane levels. Western blot analysis of total cellular or membrane fraction lysates extracted from cells expressing WT or mutant NK3R. A) Representative Western blot of total cellular levels for WT and mutant NK3Rs, probed with an anti-HA antibody and an anti-β-actin antibody (used as a loading control). B) Representative Western blot of membrane fraction proteins for WT and mutant NK3Rs, probed with an anti-HA antibody and an anti-Na+/K+-ATPase antibody (a membrane protein marker, used as a loading control). C, D) Bar graphs were generated from pooled data from 3 independent experiments for total cellular (C) and membrane (D) receptor levels, normalized to WT NK3R protein levels. Bars represent means ± se. Significant differences between WT and mutant receptor levels were calculated using Student's t test. *P < 0.05 vs. WT.
R295S NK3R exerts dominant-negative effects on WT NK3R signaling but not by interfering with NKB binding to WT NK3R
NKB-stimulated IP accumulation was impaired for the R295S, Y256H, and Y315C mutants compared with WT NK3R. The patients expressing the Y256H and Y315C mutants were homozygous for these changes, so the loss of NKB signaling can explain the reproductive phenotype in these patients (15). However, the patient harboring the R295S NK3R mutation was heterozygous for this variant (15); therefore, the WT NK3R encoded by the unaffected allele might be expected to compensate for the loss of function of the mutant R295S receptor to prevent manifestations of the GnRH-deficient phenotype. We hypothesized that the R295S NK3R mutant may have dominant-negative effects on WT receptor function to result in the reproductive phenotype in this heterozygous patient. To test this hypothesis, IP assays were performed after ligand stimulation of COS-7 cells cotransfected with expression vectors encoding both WT and R295S NK3R in varying ratios. Cells transfected with equivalent amounts of both receptors showed a 42.7 ± 3.5% reduction (P<0.05) in NKB-stimulated IP accumulation compared with cells expressing WT NK3R alone (Fig. 5A). These effects were even more marked, with a 79.7 ± 1.0% reduction (P<0.001) in IP accumulation, when the R295S:WT ratio was increased to 2:1 (Fig. 5A). These results are consistent with a dominant-negative effect of R295S NK3R on signal transduction by the WT NK3R. In contrast, both G18D and I249V NK3R, also identified in the heterozygous state in patients with GnRH deficiency, failed to exert any detectable dominant-negative effects on WT NK3R IP signaling (data not shown).
Figure 5.
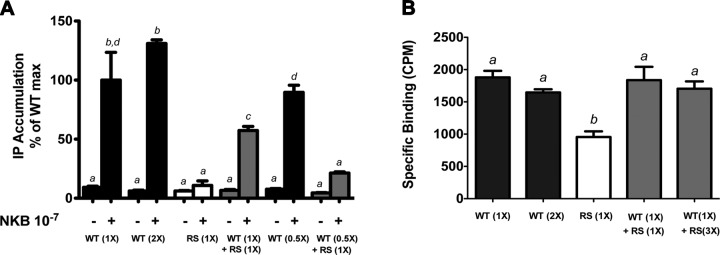
R295S NK3R disrupts NKB-stimulated IP accumulation by the WT NK3R but does not interfere with ligand binding to WT NK3R. Cells expressing WT NK3R (solid bars), R295S NK3R (open bars), or both (shaded bars) were used to assess whether the R295S mutant exerted dominant-negative effects on WT NK3R signal transduction or ligand binding. A) IP accumulation in COS-7 cells expressing WT NK3R, R295S NK3R (RS), or both in varying ratios, after treatment with 10−7 M NKB. Results are expressed as percentage of maximal IP accumulation for 1X WT NK3R (X=100 ng). B) Specific binding of 125I-NKB to COS-7 cells expressing WT NK3R, R295S NK3R (RS), or both in varying ratios (X=300ng). Bars represent means ± se of 3 independent experiments, each in triplicate. Significant differences, indicated by different letters, were calculated using 1-way ANOVA with a post hoc Tukey's multiple comparison test.
To determine whether the R295S mutant interfered with WT NK3R trafficking to the plasma membrane and/or ligand binding, we further examined whether R295S NK3R altered NKB binding to WT NK3R. Binding assays were performed using COS-7 cells cotransfected with WT and R295S NK3R in varying ratios. COS-7 cells transfected with equivalent amounts of both receptors bound 125I-NKB similarly to cells expressing WT NK3R alone; even increasing the ratio of R295S:WT NK3R to 3:1 failed to disrupt ligand binding (Fig. 5B). These results indicate that the R295S NK3R mutant did not interfere with WT NK3R expression, plasma membrane trafficking, or ligand binding, and suggest that the mutant receptor exerts its dominant-negative effects by interfering with NK3R-mediated activation of signaling pathways following ligand stimulation.
R295S NK3R heterodimerizes with WT NK3R but does not impair interaction with Gq protein
Because the R295S mutant had markedly impaired activation of signal transduction cascades despite relatively preserved membrane receptor levels and ligand binding, we hypothesized that the mutation might disrupt interactions with Gq proteins to result in impaired signaling. Furthermore, we hypothesized that the R295S mutant receptor might heterodimerize with WT NK3R and interfere with WT NK3R function to result in the observed dominant-negative effects on WT NK3R signaling. To test for the ability of NK3R to dimerize, lysates from COS-7 cells coexpressing V5-tagged WT NK3R and HA-tagged WT or R295S NK3R were immunoprecipitated with an anti-V5 antibody and immunoblotted with an anti-HA antibody. Immunoreactive bands were present in lysates from cells coexpressing V5- and HA-tagged WT NK3Rs but not from control untransfected COS-7 cells, demonstrating that NK3R can form dimers (Fig. 6A). Furthermore, immunoreactive bands were also observed in lysates from cells coexpressing V5-tagged WT NK3R and HA-tagged R295S NK3R, indicating that the R295S mutant receptor can form heterodimers with the WT NK3R (Fig. 6A).
Figure 6.
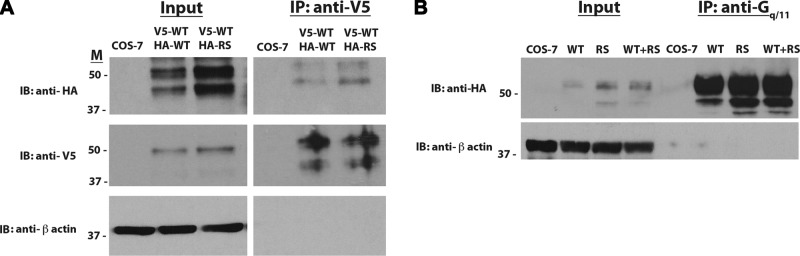
WT and R295S NK3Rs dimerize and both receptors interact with Gαq protein. A) COS-7 cells coexpressing V5-tagged WT NK3R and HA-tagged WT or R295S (RS) NK3R were lysed and lysates were used in a coimmunoprecipitation (IP) assay. Proteins were immunoprecipitated with an anti-V5 antibody (right panel) and subjected to Western blot analysis and immunoblotting (IB) with an anti-HA (top panels), anti-V5 (middle panels), or anti-β-actin (bottom panels) antibody. Fifteen percent of the lysates were used as input and subjected directly to immunoblotting (left panels). B) Cell lysates from COS-7 cells expressing WT or R295S NK3R, or both, were prepared, and immunoprecipitation was performed with an anti-Gαq/11 antibody coupled to agarose G beads (right lanes). Immunoprecipitated proteins were subjected to Western blot analysis and immunoblotted with an anti-HA antibody (top panel) or an anti-β-actin antibody (bottom panel). One percent of the lysates was used as input and subjected directly to immunoblotting (left lanes).
To determine whether the R295S NK3R mutation disrupted its ability to interact with Gq proteins, lysates from COS-7 cells expressing either HA-tagged WT NK3R, HA-tagged R295S NK3R, or both were immunoprecipitated using an anti-Gq antibody and then immunoblotted with an anti-HA antibody. Both WT and R295S NK3Rs interacted with Gq proteins, as indicated by the immunoreactive bands detected in lanes containing lysates derived from cells transfected with either HA-tagged WT or R295S NK3R (Fig. 6B). In addition, cotransfection of both WT and R295S NK3Rs had no effect on the interaction with Gq proteins (Fig. 6B). Taken together, these results indicate that the signaling defect observed for R295S NK3R was not a consequence of impaired interaction with Gq proteins and that the R295S NK3R mutant did not disrupt WT NK3R interactions with Gq proteins to cause dominant-negative effects on WT NK3R signaling.
R295S mutation impairs ligand-induced dissociation of Gq proteins from NK3R
To further investigate the loss of receptor signaling for R295S NK3R, we used a FRET-based assay to monitor receptor–Gq-protein interaction kinetics following ligand binding (51). With the use of confocal microscopy, this method allows visualization of dissociation of the Gαq subunit from the Gβγ subunits that form a heterotrimeric complex associated with GPCRs. COS-7 cells expressing Gαq-CFP, Gβ-YFP, GRK2, Gγ2, and WT NK3R, R295S NK3R, or both were used to measure FRET ratios every 5 s for 30 s before and 30 s after application of 10−7 M NKB. Gαq-CFP and Gβ-YFP were confirmed by fluorescence microscopy to be coexpressed in the same cells (data not shown) and only cells with strong CFP and YFP signals were used for FRET analyses. FRET ratios for all cells were similar before NKB application. However, within 5 s after NKB treatment, cells expressing WT NK3R showed a 10% reduction in the FRET ratio, indicating dissociation of the Gαq-CFP and Gβ-YFP subunits following NKB application (Fig. 7). In contrast, cells expressing R295S NK3R showed no change in the FRET ratio, indicating that this mutation impairs dissociation of Gαq-CFP and Gβ-YFP subunits, thereby inhibiting receptor-mediated activation of intracellular signaling (Fig. 7). Moreover, cells expressing both WT and R295S NK3R also showed no significant changes in FRET ratios after NKB stimulation (Fig. 7). These results suggest that the R295S NK3R impairs Gαq-Gβγ dissociation from WT NK3R after ligand binding, thus disrupting WT NK3R-G-protein kinetics when coexpressed within the same cell.
Figure 7.
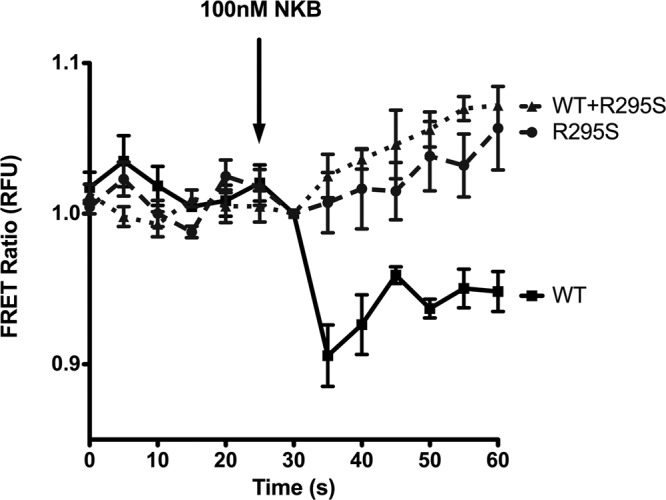
R295S NK3R disrupts NKB-induced Gαq activation. FRET ratios derived from COS-7 cells expressing Gαq-CFP, Gβ-YFP, GRK2, Gγ2, and either WT NK3R, R295S NK3R, or both. Cells were excited with a 445-nm laser, and images were recorded for 25 s before NKB application and for 35 s thereafter on CFP (468- to 499-nm) and YFP (529- to 555-nm) channels. Data at each point are shown as means ± se from 2 individual experiments, each using 5–8 cells from each experimental group.
DISCUSSION
The GPCR NK3R mediates the effects of NKB in biological processes, including reproductive development. However, little is known about the structural domains of NK3R and their roles in receptor function. Extensive studies have determined the crystal structure of >16 GPCRs and have demonstrated key amino acid residues and helices that are important for proper receptor conformation, ligand binding, and G-protein docking (57). From these studies, it is clear is that there is a high degree of diversity in the functional roles for amino acids within the various domains of GPCR family members, making it difficult to predict the importance of specific amino acids within each domain of a GPCR. However, there are some highly conserved amino acids across class 1 GPCRs (23). Inactivating mutations in GPCRs associated with human disease have focused attention on amino acids involved in receptor processing, stability, trafficking, ligand binding, or signaling (58). For NK3R, the roles of specific amino acids are largely unknown; however, a recent study in patients with GnRH deficiency proposed that a tyrosine at position 267 in TM5 may be important for receptor folding (17). In the current study, using mutations in NK3R identified in patients with GnRH deficiency, we elucidated roles for Y256, R295, and Y315 in NK3R structure and function and identified the mechanisms through which mutations at these positions contribute to the reproductive phenotype observed in these patients.
Functional analysis of the 5 missense NK3R mutations previously identified in patients with GnRH deficiency (15) confirmed the deleterious computational predictions for Y256H, R295S, and Y315C (Table 1), as these mutants had significantly impaired activation of intracellular signaling cascades compared with WT NK3R (Figs. 1 and 2). Our results demonstrated that the R295S mutant, harboring a mutation in IL3, bound NKB but failed to induce IP accumulation in response to ligand stimulation. Over the past 2 decades, several investigators have shown the importance of IL3 of GPCRs in G-protein coupling and activation. In 1988, using flash photolysis, Franke et al. (59) demonstrated that a mutation of K248 in IL3 of rhodopsin did not affect G-protein (transducin) binding but impaired G-protein activation. Since this early report, other studies have shown the importance of IL3 in G-protein binding and activation, including a recent report on the corticotropin-releasing hormone receptor that showed the importance of several arginine and lysine residues in this domain in the activation of G-protein-mediated signaling cascades (60). Although an increasing number of studies have implicated arginine residues in ILs as important for GPCR signaling, no consensus sequence for G-protein coupling has been identified, likely due to the high sequence diversity within these loops among GPCRs. The third intracellular loop is also important for tachykinin receptor interactions with β-arrestins and activation of intracellular signaling cascades (61–63). Schmidlin et al. (62) demonstrated that the third IL and carboxyl terminus of NK1R and NK3R were important for β-arrestin specificity and determined the rate of resensitization for each receptor. NK1R interacts with β-arrestin 1 and 2 and is resensitized at a slower rate compared with NK3R, which only transiently associates with β-arrestin 1 after ligand activation (62). Our finding for R295S NK3R is consistent with these earlier reports establishing IL3 as important for NK3R signaling but highlights the significance of the R295 residue in G-protein binding and activation.
The R295S mutation was identified in the heterozygous state in a patient with GnRH deficiency. The WT NK3R encoded by the unaffected allele in this individual would be expected to be capable of mediating effects of NKB, thereby preventing any deleterious effects of the mutant receptor. The presence of the mutation in the heterozygous state in this affected patient suggests that R295S may have dominant-negative effects on WT NK3R. IP assays performed in COS-7 cells expressing both WT and R295S NK3R demonstrated that the R295S mutant receptor interferes with NKB stimulation of IP accumulation by the WT NK3R, supporting the dominant-negative model. Interestingly, the R295S NK3R did not interfere significantly with 125I-NKB binding by WT NK3R, suggesting that the R295S mutant does not interfere with WT NK3R expression, trafficking to the plasma membrane, or ligand binding, but rather interferes specifically with NKB-mediated NK3R signaling. To explore this signaling defect further, we utilized a FRET-based approach (51), with NK3R coupled to fluorescent protein-tagged Gαq and Gβ subunits, to investigate Gq-protein activation following NKB treatment. WT NK3R coupled to fluorescent protein-tagged Gαq and Gβ resulted in a FRET ratio that decreased following NKB treatment, reflecting the uncoupling and dissociation of Gαq and Gβ from the receptor and from each other following receptor activation, leading to activation of intracellular signal transduction cascades. In contrast, although R295S NK3R produced a resting FRET ratio similar to that of WT NK3R, there was no decrease in this ratio following NKB treatment, suggesting that this mutation prevented ligand-dependent activation of Gq and the resultant dissociation of Gαq and Gβ subunits from the NK3R and from each other, leading to failure of this mutant receptor to activate signal transduction pathways. Moreover, there was no decrease in FRET ratios following NKB treatment of cells cotransfected with WT and R295S NK3R, demonstrating that R295S NK3R has dominant-negative effects on WT NK3R-mediated G-protein activation and signaling. We further showed by immunoprecipitation that WT and R295S NK3Rs interact, providing a mechanism through which R295S NK3R exerts its dominant effects on WT NK3R, contributing to the abnormal reproductive phenotype.
Dominant-negative mutations have been reported for other GPCR family members including the α1-adrenergic receptor, although these α1-adrenergic receptor mutants were not derived from clinical observations (64) and the calcium-sensing receptor (CaR; refs. 65, 66). To date, extensive studies of heterozygous CaR mutants demonstrated that these mutants disrupt WT receptor expression and function by reducing cell surface expression, impairing receptor binding, altering G-protein coupling, or increasing rate of degradation (67). The CaR forms homodimers and these dimers are important for agonist-dependent activation of the receptor (68, 69). Therefore, mutant CaRs could cause a conformational change in dimer structure in patients with heterozygous mutations, resulting in impaired expression, binding or signaling. Our study demonstrates that Gq proteins fail to dissociate from R295S NK3R and that both WT and R295S NK3R form dimers, although it is unclear whether dimers are critical for NK3R function. We hypothesize that heterodimerization of WT and R295S NK3R results not only in failure of the G protein to dissociate from R295S NK3R after ligand binding but that it similarly constrains the conformation of the WT NK3R heterodimer partner to impair G-protein dissociation. Further studies will be necessary to confirm this model.
The G18D and I249V mutations were not predicted to be pathological, and our functional studies confirmed that these 2 mutant NK3Rs bound NKB and stimulated IP accumulation similar to WT NK3R in response to NKB stimulation and therefore may not be involved in causing GnRH deficiency. However, it is possible that the G18D and I249V mutations could affect other functions of the receptor or could disrupt other yet unidentified signaling pathways activated by NK3R. In contrast, the Y256H and Y315C NK3R mutations were predicted to be pathological mutations. Molecular modeling indicated that the residues Y256 (28) and Y315 (27, 28) in TM5 and TM6, respectively, were involved in forming the binding pocket for NKB. Studies conducted by Gether et al. (21) demonstrated that TMs 5 and 7 are important for the recognition of NK3R ligands. Therefore, we hypothesized that these mutations would affect ligand binding to the mutated receptors. Indeed, binding was impaired, but in addition, Western blot analysis demonstrated that Y256H NK3R levels were markedly impaired in both total cellular and cell surface membrane fractions (Fig. 4), indicating that this mutation may interfere with receptor processing, stability, or folding, resulting in targeting of the receptor for degradation. Although Y256 is highly conserved across the rhodopsin family of GPCRs (23) and has been shown to be important in the formation of the active conformational state of the β-2 adrenergic receptor (56), there are no previous reports of this residue playing a role in receptor processing or stability. The quality control mechanism of the endoplasmic reticulum plays an integral role in identifying and routing misfolded proteins to the ER-associated degradation pathway (70–72). Mutations in the vasopressin receptor, delta opioid receptor, and the GnRHR have resulted in misfolded receptors that were targeted and removed by this quality control mechanism (73–75).
The mechanism through which the Y315C mutation impairs NK3R function is somewhat different from the Y256H mutation. Total cellular receptor levels for the Y315C mutant were similar to WT NK3R levels, yet cell surface levels were significantly reduced (Fig. 3) and mutant receptors present at the cell surface failed to bind NKB (Fig. 2), suggesting this mutated NK3R is also targeted by the ER quality control system but leads to intracellular retention of Y315C NK3R, resulting in impaired trafficking of the receptor to the cell surface. Interestingly, the upper immunoreactive bands for both Y256H and Y315C are reduced to a greater extent than the lower bands in membrane fractions, which suggests that this upper band may represent mature receptor that is trafficked to the cell surface membrane while the lower band may represent immature receptors associated with intracellular membranes. In addition, the Y315C NK3R present at the cell surface does not show appreciable binding of NKB. This absence of measurable binding activity may reflect the predicted role Y315 plays in ligand binding. Alternatively, the reduced number of receptors present on the cell surface may be insufficient to result in detectable binding levels above background. While the specific accessory proteins or chaperones involved in the trafficking of NK3R to the cell surface are unknown, several proteins, including BiP/GRP78, may facilitate translocation of GPCRs through the ER, as both the thyrotropin-releasing hormone receptor and the luteinizing hormone receptor have been demonstrated to interact with BiP (76–79). During ER translocation, GPCRs also undergo post-translational modifications including glycosylation, which enable interactions with carbohydrate-binding chaperones such as calnexin and calreticulin (80, 81). It should be noted that prolonged association with chaperones may also enhance degradation of GPCRs (80). Other chaperones important for transportation of GPCRs to the cell surface include receptor-activity modifying proteins (RAMPs), which regulate trafficking of several GPCRS to the cell surface including the calcitonin receptor-like receptor (82), the glucagon receptors (83), and the calcium sensing receptor (84). Members of the receptor transporting protein (RTP) family are also critical for expression of odorant receptors when expressed in heterologous cell systems (85). Pharmacological chaperones have rescued cell surface expression and function of several mutant GPCRs that had impaired receptor trafficking and signaling, including the vasopressin receptor (73) and the GnRHR (86, 87). These data suggest that although mutations may interfere with receptor interactions with endogenous chaperones, pharmacoperones can either restore folding of the mutated receptors to restore receptor trafficking and function, or these receptors maintain functional capabilities and once restored to the cell surface by pharmacoperones, are able to function normally. Another possibility for the reduced trafficking of Y315C is that this mutation prevents masking of ER retention motifs, thereby trapping the mutant receptor in the ER. However, such motifs have not yet been identified for NK3R. Ongoing studies should determine the precise intracellular localization of Y315C NK3R.
Collectively, our data show that mutations in different domains of NK3R impair receptor function through distinct mechanisms, indicating a role for R295 in IL3 in receptor signaling and G-protein activation, for Y256 in TM5 in receptor folding and/or stability, and for Y315 in TM6 in folding or trafficking to the plasma membrane. In addition, R295S, Y256H, and Y315C are loss-of-function mutations in NK3R that could contribute to GnRH deficiency and the abnormal reproductive phenotype.
Supplementary Material
Acknowledgments
The authors thank Dr. John Gill and Dr. Ann-Marie Zavacki for helpful discussions. The authors also thank Drs. Bertil Hille (University of Washington School of Medicine, Seattle, WA, USA), Narasimhan Gautam (Washington University School of Medicine in St. Louis, St. Louis, MO, USA), Stephen Ikeda [National Institute on Alcohol Abuse and Alcoholism, Division of Clinical and Biological Research, U. S. National Institutes of Health (NIH), Bethesda, MD, USA] and Moritz Buneman (Philipps-Universtitaet Marburg, Marburg, Germany) for providing DNA constructs.
This research was supported by the Eunice Kennedy Shriver National Institute of Child Health and Human Development, NIH, through cooperative agreement U54 HD028138 as part of the Specialized Cooperative Centers Program in Reproduction and Infertility Research (to U.B.K. and S.B.S.), by R01 HD019938 (to U.B.K.), by a T32 DK007529-25 Ruth L. Kirschstein National Research Service Postdoctoral Award (to S.D.N.), by a F05 HD072773-01 NIH Fellowship (to A.P.A.), and by R01 HD019938-21S1 (to J.C.).
Author contributions: S.D.N., R.S.C., and U.B.K. designed the study; S.D.N, A.P.A, S.X, T.M., E.G., and J.C. performed experiments; S.D.N. collected and analyzed data; E.G., C.T., A.C.L., S.B.S., R.S.C., and U.B.K. provided materials and reagents; S.D.N. and U.B.K. wrote the manuscript; A.C.L., S.B.S., R.S.C., and U.B.K. gave technical support and conceptual advice. The authors declare no conflicts of interest.
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
- DMEM
- Dulbecco's modified Eagle's medium
- EL
- extracellular loop
- FRET
- fluorescence resonance energy transfer
- GPCR
- G-protein-coupled receptor
- HA
- hemagglutinin
- IHH
- isolated hypogonadotropic hypogonadism
- IL
- intracellular loop
- IP
- inositol phosphate
- NKB
- neurokinin B
- TM
- transmembrane
- WT
- wild-type
REFERENCES
- 1. Chang M. M., Leeman S. E., Niall H. D. (1971) Amino-acid sequence of substance P. Nat. New Biol. 232, 86–87 [DOI] [PubMed] [Google Scholar]
- 2. Nawa H., Doteuchi M., Igano K., Inouye K., Nakanishi S. (1984) Substance K: a novel mammalian tachykinin that differs from substance P in its pharmacological profile. Life Sci. 34, 1153–1160 [DOI] [PubMed] [Google Scholar]
- 3. Kangawa K., Minamino N., Fukuda A., Matsuo H. (1983) Neuromedin K: a novel mammalian tachykinin identified in porcine spinal cord. Biochem. Biophys. Res. Commun. 114, 533–540 [DOI] [PubMed] [Google Scholar]
- 4. Severini C., Improta G., Falconieri-Erspamer G., Salvadori S., Erspamer V. (2002) The tachykinin peptide family. Pharmacol. Rev. 54, 285–322 [DOI] [PubMed] [Google Scholar]
- 5. Sandoval-Guzman T., Rance N. E. (2004) Central injection of senktide, an NK3 receptor agonist, or neuropeptide Y inhibits LH secretion and induces different patterns of Fos expression in the rat hypothalamus. Brain Res. 1026, 307–312 [DOI] [PubMed] [Google Scholar]
- 6. Krajewski S. J., Anderson M. J., Iles-Shih L., Chen K. J., Urbanski H. F., Rance N. E. (2005) Morphologic evidence that neurokinin B modulates gonadotropin-releasing hormone secretion via neurokinin 3 receptors in the rat median eminence. J. Comp. Neurol. 489, 372–386 [DOI] [PubMed] [Google Scholar]
- 7. Rance N. E. (2009) Menopause and the human hypothalamus: evidence for the role of kisspeptin/neurokinin B neurons in the regulation of estrogen negative feedback. Peptides 30, 111–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Osakada F., Kubo K., Goto K., Kanazawa I., Munekata E. (1986) The contractile activities of neurokinin A, B and related peptides on smooth muscles. Eur. J. Pharmacol. 120, 201–208 [DOI] [PubMed] [Google Scholar]
- 9. D'Orleans-Juste P., Dion S., Drapeau G., Regoli D. (1986) Different receptors are involved in the endothelium-mediated relaxation and the smooth muscle contraction of the rabbit pulmonary artery in response to substance P and related neurokinins. Eur. J. Pharmacol. 125, 37–44 [DOI] [PubMed] [Google Scholar]
- 10. Page N. M., Woods R. J., Gardiner S. M., Lomthaisong K., Gladwell R. T., Butlin D. J., Manyonda I. T., Lowry P. J. (2000) Excessive placental secretion of neurokinin B during the third trimester causes pre-eclampsia. Nature 405, 797–800 [DOI] [PubMed] [Google Scholar]
- 11. Simonsen K. B., Juhl K., Steiniger-Brach B., Nielsen S. M. (2010) Novel NK(3) receptor antagonists for the treatment of schizophrenia and other CNS indications. Curr. Opin. Drug Discov. Devel. 13, 379–388 [PubMed] [Google Scholar]
- 12. Dawson L. A., Smith P. W. (2010) Therapeutic utility of NK3 receptor antagonists for the treatment of schizophrenia. Curr. Pharm. Des. 16, 344–357 [DOI] [PubMed] [Google Scholar]
- 13. Topaloglu A. K., Reimann F., Guclu M., Yalin A. S., Kotan L. D., Porter K. M., Serin A., Mungan N. O., Cook J. R., Ozbek M. N., Imamoglu S., Akalin N. S., Yuksel B., O'Rahilly S., Semple R. K. (2009) TAC3 and TACR3 mutations in familial hypogonadotropic hypogonadism reveal a key role for neurokinin B in the central control of reproduction. Nat. Genet. 41, 354–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Guran T., Tolhurst G., Bereket A., Rocha N., Porter K., Turan S., Gribble F. M., Kotan L. D., Akcay T., Atay Z., Canan H., Serin A., O'Rahilly S., Reimann F., Semple R. K., Topaloglu A. K. (2009) Hypogonadotropic hypogonadism due to a novel missense mutation in the first extracellular loop of the neurokinin B receptor. J. Clin. Endocrinol. Metab. 94, 3633–3639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gianetti E., Tusset C., Noel S. D., Au M. G., Dwyer A. A., Hughes V. A., Abreu A. P., Carroll J., Trarbach E., Silveira L. F., Costa E. M., de Mendonca B. B., de Castro M., Lofrano A., Hall J. E., Bolu E., Ozata M., Quinton R., Amory J. K., Stewart S. E., Arlt W., Cole T. R., Crowley W. F., Kaiser U. B., Latronico A. C., Seminara S. B. (2010) TAC3/TACR3 mutations reveal preferential activation of gonadotropin-releasing hormone release by neurokinin B in neonatal life followed by reversal in adulthood. J. Clin. Endocrinol. Metab. 95, 2857–2867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Young J., Bouligand J., Francou B., Raffin-Sanson M. L., Gaillez S., Jeanpierre M., Grynberg M., Kamenicky P., Chanson P., Brailly-Tabard S., Guiochon-Mantel A. (2010) TAC3 and TACR3 defects cause hypothalamic congenital hypogonadotropic hypogonadism in humans. J. Clin. Endocrinol. Metab. 95, 2287–2295 [DOI] [PubMed] [Google Scholar]
- 17. Francou B., Bouligand J., Voican A., Amazit L., Trabado S., Fagart J., Meduri G., Brailly-Tabard S., Chanson P., Lecomte P., Guiochon-Mantel A., Young J. (2011) Normosmic congenital hypogonadotropic hypogonadism due to TAC3/TACR3 mutations: characterization of neuroendocrine phenotypes and novel mutations. PLoS One 6, e25614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gerard N. P., Eddy R. L., Jr., Shows T. B., Gerard C. (1990) The human neurokinin A (substance K) receptor. Molecular cloning of the gene, chromosome localization, and isolation of cDNA from tracheal and gastric tissues. J. Biol. Chem. 265, 20455–20462 [PubMed] [Google Scholar]
- 19. Takeda Y., Chou K. B., Takeda J., Sachais B. S., Krause J. E. (1991) Molecular cloning, structural characterization and functional expression of the human substance P receptor. Biochem. Biophys. Res. Commun. 179, 1232–1240 [DOI] [PubMed] [Google Scholar]
- 20. Takahashi K., Tanaka A., Hara M., Nakanishi S. (1992) The primary structure and gene organization of human substance P and neuromedin K receptors. Eur. J. Biochem. 204, 1025–1033 [DOI] [PubMed] [Google Scholar]
- 21. Gether U., Johansen T. E., Schwartz T. W. (1993) Chimeric NK1 (substance P)/NK3 (neurokinin B) receptors. Identification of domains determining the binding specificity of tachykinin agonists. J. Biol. Chem. 268, 7893–7898 [PubMed] [Google Scholar]
- 22. Khawaja A. M., Rogers D. F. (1996) Tachykinins: receptor to effector. Int. J. Biochem. Cell Biol. 28, 721–738 [DOI] [PubMed] [Google Scholar]
- 23. Wess J. (1998) Molecular basis of receptor/G-protein-coupling selectivity. Pharmacol. Ther. 80, 231–264 [DOI] [PubMed] [Google Scholar]
- 24. Peeters M. C., van Westen G. J., Guo D., Wisse L. E., Muller C. E., Beukers M. W., Ijzerman A. P. (2011) GPCR structure and activation: an essential role for the first extracellular loop in activating the adenosine A2B receptor. FASEB J. 25, 632–643 [DOI] [PubMed] [Google Scholar]
- 25. Tan C. M., Brady A. E., Nickols H. H., Wang Q., Limbird L. E. (2004) Membrane trafficking of G protein-coupled receptors. Annu. Rev. Pharmacol. Toxicol. 44, 559–609 [DOI] [PubMed] [Google Scholar]
- 26. Ferguson S. S., Zhang J., Barak L. S., Caron M. G. (1998) Molecular mechanisms of G protein-coupled receptor desensitization and resensitization. Life Sci. 62, 1561–1565 [DOI] [PubMed] [Google Scholar]
- 27. Ganjiwale A. D., Rao G. S., Cowsik S. M. (2011) Molecular modeling of neurokinin B and tachykinin NK(3) receptor complex. J. Chem. Inf. Model. 51, 2932–2938 [DOI] [PubMed] [Google Scholar]
- 28. Zhang Y. (2009) I-TASSER: fully automated protein structure prediction in CASP8. Proteins 77(Suppl. 9), 100–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Goodman R. L., Lehman M. N., Smith J. T., Coolen L. M., de Oliveira C. V., Jafarzadehshirazi M. R., Pereira A., Iqbal J., Caraty A., Ciofi P., Clarke I. J. (2007) Kisspeptin neurons in the arcuate nucleus of the ewe express both dynorphin A and neurokinin B. Endocrinology 148, 5752–5760 [DOI] [PubMed] [Google Scholar]
- 30. Navarro V. M., Gottsch M. L., Chavkin C., Okamura H., Clifton D. K., Steiner R. A. (2009) Regulation of gonadotropin-releasing hormone secretion by kisspeptin/dynorphin/neurokinin B neurons in the arcuate nucleus of the mouse. J. Neurosci. 29, 11859–11866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wakabayashi Y., Nakada T., Murata K., Ohkura S., Mogi K., Navarro V. M., Clifton D. K., Mori Y., Tsukamura H., Maeda K., Steiner R. A., Okamura H. (2010) Neurokinin B and dynorphin A in kisspeptin neurons of the arcuate nucleus participate in generation of periodic oscillation of neural activity driving pulsatile gonadotropin-releasing hormone secretion in the goat. J. Neurosci. 30, 3124–3132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Eghlidi D. H., Haley G. E., Noriega N. C., Kohama S. G., Urbanski H. F. (2010) Influence of age and 17beta-estradiol on kisspeptin, neurokinin B, and prodynorphin gene expression in the arcuate-median eminence of female rhesus macaques. Endocrinology 151, 3783–3794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rance N. E., Krajewski S. J., Smith M. A., Cholanian M., Dacks P. A. (2010) Neurokinin B and the hypothalamic regulation of reproduction. Brain Res. 1364, 116–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Amstalden M., Coolen L. M., Hemmerle A. M., Billings H. J., Connors J. M., Goodman R. L., Lehman M. N. (2010) Neurokinin 3 receptor immunoreactivity in the septal region, preoptic area and hypothalamus of the female sheep: colocalisation in neurokinin B cells of the arcuate nucleus but not in gonadotrophin-releasing hormone neurones. J. Neuroendocrinol. 22, 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kallo I., Vida B., Deli L., Molnar C. S., Hrabovszky E., Caraty A., Ciofi P., Coen C. W., Liposits Z. (2012) Co-localisation of kisspeptin with galanin or neurokinin B in afferents to mouse GnRH neurones. J. Neuroendocrinol. 24, 464–476 [DOI] [PubMed] [Google Scholar]
- 36. Todman M. G., Han S. K., Herbison A. E. (2005) Profiling neurotransmitter receptor expression in mouse gonadotropin-releasing hormone neurons using green fluorescent protein-promoter transgenics and microarrays. Neuroscience 132, 703–712 [DOI] [PubMed] [Google Scholar]
- 37. Glidewell-Kenney C. A., Shao P. P., Iyer A. K., Grove A. M., Meadows J. D., Mellon P. L. (2013) Neurokinin B causes acute GnRH secretion and repression of GnRH transcription in GT1-7 GnRH neurons. Mol. Endocrinol. 27, 437–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gaskins G. T., Glanowska K. M., Moenter S. M. (2013) Activation of neurokinin 3 receptors stimulates GnRH release in a location-dependent but kisspeptin-independent manner in adult mice. Endocrinology 154, 3984–3989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Billings H. J., Connors J. M., Altman S. N., Hileman S. M., Holaskova I., Lehman M. N., McManus C. J., Nestor C. C., Jacobs B. H., Goodman R. L. (2010) Neurokinin B acts via the neurokinin-3 receptor in the retrochiasmatic area to stimulate luteinizing hormone secretion in sheep. Endocrinology 151, 3836–3846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ramaswamy S., Seminara S. B., Ali B., Ciofi P., Amin N. A., Plant T. M. (2010) Neurokinin B stimulates GnRH release in the male monkey (Macaca mulatta) and is colocalized with kisspeptin in the arcuate nucleus. Endocrinology 151, 4494–4503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Navarro V. M., Castellano J. M., McConkey S. M., Pineda R., Ruiz-Pino F., Pinilla L., Clifton D. K., Tena-Sempere M., Steiner R. A. (2011) Interactions between kisspeptin and neurokinin B in the control of GnRH secretion in the female rat. Am. J. Physiol. Endocrinol. Metab. 300, E202–E210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Navarro V. M., Gottsch M. L., Wu M., Garcia-Galiano D., Hobbs S. J., Bosch M. A., Pinilla L., Clifton D. K., Dearth A., Ronnekleiv O. K., Braun R. E., Palmiter R. D., Tena-Sempere M., Alreja M., Steiner R. A. (2011) Regulation of NKB pathways and their roles in the control of Kiss1 neurons in the arcuate nucleus of the male mouse. Endocrinology 152, 4265–4275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kinsey-Jones J. S., Grachev P., Li X. F., Lin Y. S., Milligan S. R., Lightman S. L., O'Byrne K. T. (2012) The inhibitory effects of neurokinin B on GnRH pulse generator frequency in the female rat. Endocrinology 153, 307–315 [DOI] [PubMed] [Google Scholar]
- 44. Corander M. P., Challis B. G., Thompson E. L., Jovanovic Z., Loraine Tung Y. C., Rimmington D., Huhtaniemi I. T., Murphy K. G., Topaloglu A. K., Yeo G. S., O'Rahilly S., Dhillo W. S., Semple R. K., Coll A. P. (2010) The effects of neurokinin B upon gonadotrophin release in male rodents. J. Neuroendocrinol. 22, 181–187 [DOI] [PubMed] [Google Scholar]
- 45. Kung T. T., Crawley Y., Jones H., Luo B., Gilchrest H., Greenfeder S., Anthes J. C., Lira S., Wiekowski M., Cook D. N., Hey J. A., Egan R. W., Chapman R. W. (2004) Tachykinin NK3-receptor deficiency does not inhibit pulmonary eosinophilia in allergic mice. Pharmacol. Res. 50, 611–615 [DOI] [PubMed] [Google Scholar]
- 46. Siuciak J. A., McCarthy S. A., Martin A. N., Chapin D. S., Stock J., Nadeau D. M., Kantesaria S., Bryce-Pritt D., McLean S. (2007) Disruption of the neurokinin-3 receptor (NK3) in mice leads to cognitive deficits. Psychopharmacology (Berl.) 194, 185–195 [DOI] [PubMed] [Google Scholar]
- 47. Nordquist R. E., Delenclos M., Ballard T. M., Savignac H., Pauly-Evers M., Ozmen L., Spooren W. (2008) Cognitive performance in neurokinin 3 receptor knockout mice. Psychopharmacology (Berl.) 198, 211–220 [DOI] [PubMed] [Google Scholar]
- 48. Yang J. J., Caligioni C. S., Chan Y. M., Seminara S. B. (2012) Uncovering novel reproductive defects in neurokinin B receptor null mice: closing the gap between mice and men. Endocrinology 153, 1498–1508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bedecarrats G. Y., Linher K. D., Janovick J. A., Beranova M., Kada F., Seminara S. B., Michael Conn P., Kaiser U. B. (2003) Four naturally occurring mutations in the human GnRH receptor affect ligand binding and receptor function. Mol. Cell. Endocrinol. 205, 51–64 [DOI] [PubMed] [Google Scholar]
- 50. Gardner T. A., Ferreira J., Barlow J., Lees A. C., Parry L., Vieira I. C., Berenguer E., Abramovay R., Aleixo A., Andretti C., Aragao L. E., Araujo I., de Avila W. S., Bardgett R. D., Batistella M., Begotti R. A., Beldini T., de Blas D. E., Braga R. F., Braga Dde L., de Brito J. G., de Camargo P. B., Campos dos Santos F., de Oliveira V. C., Cordeiro A. C., Cardoso T. M., de Carvalho D. R., Castelani S. A., Chaul J. C., Cerri C. E., Costa Fde A., da Costa C. D., Coudel E., Coutinho A. C., Cunha D., D'Antona A., Dezincourt J., Dias-Silva K., Durigan M., Esquerdo J. C., Feres J., Ferraz S. F., Ferreira A. E., Fiorini A. C., da Silva L. V., Frazao F. S., Garrett R., Gomes Ados S., Goncalves Kda S., Guerrero J. B., Hamada N., Hughes R. M., Igliori D. C., Jesus Eda C., Juen L., Junior M., de Oliveira Junior J. M., de Oliveira Junior R. C., Souza Junior C., Kaufmann P., Korasaki V., Leal C. G., Leitao R., Lima N., Almeida Mde F., Lourival R., Louzada J., Mac Nally R., Marchand S., Maues M. M., Moreira F. M., Morsello C., Moura N., Nessimian J., Nunes S., Oliveira V. H., Pardini R., Pereira H. C., Pompeu P. S., Ribas C. R., Rossetti F., Schmidt F. A., da Silva R., da Silva R. C., da Silva T. F., Silveira J., Siqueira J. V., de Carvalho T. S., Solar R. R., Tancredi N. S., Thomson J. R., Torres P. C., Vaz-de-Mello F. Z., Veiga R. C., Venturieri A., Viana C., Weinhold D., Zanetti R., Zuanon J. (2013) A social and ecological assessment of tropical land uses at multiple scales: the Sustainable Amazon Network. Philos. Trans. R. Soc. Lond. B Biol. Sci. 368, 20120166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wacker J. L., Feller D. B., Tang X. B., Defino M. C., Namkung Y., Lyssand J. S., Mhyre A. J., Tan X., Jensen J. B., Hague C. (2008) Disease-causing mutation in GPR54 reveals the importance of the second intracellular loop for class A G-protein-coupled receptor function. J. Biol. Chem. 283, 31068–31078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Schliefenbaum J., Kreile A. K., Lohse M. J., Bunemann M. (2008) G Protein coupled receptor kinase 2/3 separate Galphaq and Gbetagamma subunits during G protein activation. Biophys. J. 94, 665–667 [Google Scholar]
- 53. Ng P. C., Henikoff S. (2001) Predicting deleterious amino acid substitutions. Genome Res. 11, 863–874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sunyaev S., Ramensky V., Koch I., Lathe W., 3rd, Kondrashov A. S., Bork P. (2001) Prediction of deleterious human alleles. Hum. Mol. Genet. 10, 591–597 [DOI] [PubMed] [Google Scholar]
- 55. Thomas P. D., Kejariwal A., Campbell M. J., Mi H., Diemer K., Guo N., Ladunga I., Ulitsky-Lazareva B., Muruganujan A., Rabkin S., Vandergriff J. A., Doremieux O. (2003) PANTHER: a browsable database of gene products organized by biological function, using curated protein family and subfamily classification. Nucleic Acids Res. 31, 334–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Rasmussen S. G., DeVree B. T., Zou Y., Kruse A. C., Chung K. Y., Kobilka T. S., Thian F. S., Chae P. S., Pardon E., Calinski D., Mathiesen J. M., Shah S. T., Lyons J. A., Caffrey M., Gellman S. H., Steyaert J., Skiniotis G., Weis W. I., Sunahara R. K., Kobilka B. K. (2011) Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature 477, 549–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Katritch V., Cherezov V., Stevens R. C. (2013) Structure-function of the G protein-coupled receptor superfamily. Annu. Rev. Pharmacol. Toxicol. 53, 531–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Tao Y. X. (2006) Inactivating mutations of G protein-coupled receptors and diseases: structure-function insights and therapeutic implications. Pharmacol. Therap. 111, 949–973 [DOI] [PubMed] [Google Scholar]
- 59. Franke R. R., Sakmar T. P., Oprian D. D., Khorana H. G. (1988) A single amino acid substitution in rhodopsin (lysine 248–leucine) prevents activation of transducin. J. Biol. Chem. 263, 2119–2122 [PubMed] [Google Scholar]
- 60. Punn A., Chen J., Delidaki M., Tang J., Liapakis G., Lehnert H., Levine M. A., Grammatopoulos D. K. (2012) Mapping structural determinants within third intracellular loop that direct signaling specificity of type 1 corticotropin-releasing hormone receptor. J. Biol. Chem. 287, 8974–8985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. McConalogue K., Dery O., Lovett M., Wong H., Walsh J. H., Grady E. F., Bunnett N. W. (1999) Substance P-induced trafficking of beta-arrestins. The role of beta-arrestins in endocytosis of the neurokinin-1 receptor. J. Biol. Chem. 274, 16257–16268 [DOI] [PubMed] [Google Scholar]
- 62. Schmidlin F., Roosterman D., Bunnett N. W. (2003) The third intracellular loop and carboxyl tail of neurokinin 1 and 3 receptors determine interactions with beta-arrestins. Am. J. Physiol. Cell Physiol. 285, C945–C958 [DOI] [PubMed] [Google Scholar]
- 63. Cezanne L., Lecat S., Lagane B., Millot C., Vollmer J. Y., Matthes H., Galzi J. L., Lopez A. (2004) Dynamic confinement of NK2 receptors in the plasma membrane. Improved FRAP analysis and biological relevance. J. Biol. Chem. 279, 45057–45067 [DOI] [PubMed] [Google Scholar]
- 64. Chen S., Lin F., Xu M., Hwa J., Graham R. M. (2000) Dominant-negative activity of an alpha(1B)-adrenergic receptor signal-inactivating point mutation. EMBO J. 19, 4265–4271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ward B. K., Magno A. L., Blitvich B. J., Rea A. J., Stuckey B. G., Walsh J. P., Ratajczak T. (2006) Novel mutations in the calcium-sensing receptor gene associated with biochemical and functional differences in familial hypocalciuric hypercalcaemia. Clin. Endocrinol. (Oxf.) 64, 580–587 [DOI] [PubMed] [Google Scholar]
- 66. Al-Salameh A., Cetani F., Pardi E., Vulpoi C., Pierre P., de Calan L., Guyetant S., Jeunemaitre X., Lecomte P. (2011) A novel mutation in the calcium-sensing receptor in a French family with familial hypocalciuric hypercalcaemia. Eur. J. Endocrinol. 165, 359–363 [DOI] [PubMed] [Google Scholar]
- 67. Pidasheva S., D'Souza-Li L., Canaff L., Cole D. E., Hendy G. N. (2004) CASRdb: calcium-sensing receptor locus-specific database for mutations causing familial (benign) hypocalciuric hypercalcemia, neonatal severe hyperparathyroidism, and autosomal dominant hypocalcaemia. Hum. Mut. 24, 107–111 [DOI] [PubMed] [Google Scholar]
- 68. Bai M., Trivedi S., Kifor O., Quinn S. J., Brown E. M. (1999) Intermolecular interactions between dimeric calcium-sensing receptor monomers are important for its normal function. Proc. Natl. Acad. Sci. U. S. A. 96, 2834–2839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Bai M., Trivedi S., Brown E. M. (1998) Dimerization of the extracellular calcium-sensing receptor (CaR) on the cell surface of CaR-transfected HEK293 cells. J. Biol. Chem. 273, 23605–23610 [DOI] [PubMed] [Google Scholar]
- 70. Tsai B., Ye Y., Rapoport T. A. (2002) Retro-translocation of proteins from the endoplasmic reticulum into the cytosol. Nat. Rev. Mol. Cell Biol. 3, 246–255 [DOI] [PubMed] [Google Scholar]
- 71. Gow A., Friedrich V. L., Jr., Lazzarini R. A. (1994) Many naturally occurring mutations of myelin proteolipid protein impair its intracellular transport. J. Neurosci. Res. 37, 574–583 [DOI] [PubMed] [Google Scholar]
- 72. Cheng S. H., Gregory R. J., Marshall J., Paul S., Souza D. W., White G. A., O'Riordan C. R., Smith A. E. (1990) Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell 63, 827–834 [DOI] [PubMed] [Google Scholar]
- 73. Morello J. P., Salahpour A., Laperriere A., Bernier V., Arthus M. F., Lonergan M., Petaja-Repo U., Angers S., Morin D., Bichet D. G., Bouvier M. (2000) Pharmacological chaperones rescue cell-surface expression and function of misfolded V2 vasopressin receptor mutants. J. Clin. Invest. 105, 887–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Conn P. M., Janovick J. A. (2009) Trafficking and quality control of the gonadotropin releasing hormone receptor in health and disease. Mol. Cell. Endocrinol. 299, 137–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Petaja-Repo U. E., Hogue M., Bhalla S., Laperriere A., Morello J. P., Bouvier M. (2002) Ligands act as pharmacological chaperones and increase the efficiency of delta opioid receptor maturation. EMBO J. 21, 1628–1637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Hebert D. N., Molinari M. (2007) In and out of the ER: protein folding, quality control, degradation, and related human diseases. Physiol. Rev. 87, 1377–1408 [DOI] [PubMed] [Google Scholar]
- 77. Ellgaard L., Molinari M., Helenius A. (1999) Setting the standards: quality control in the secretory pathway. Science 286, 1882–1888 [DOI] [PubMed] [Google Scholar]
- 78. Horwich A. (2002) Protein aggregation in disease: a role for folding intermediates forming specific multimeric interactions. J. Clin. Invest. 110, 1221–1232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Siffroi-Fernandez S., Giraud A., Lanet J., Franc J. L. (2002) Association of the thyrotropin receptor with calnexin, calreticulin and BiP. Efects on the maturation of the receptor. Eur. J. Biochem. 269, 4930–4937 [DOI] [PubMed] [Google Scholar]
- 80. Achour L., Labbe-Jullie C., Scott M. G., Marullo S. (2008) An escort for GPCRs: implications for regulation of receptor density at the cell surface. Trends Pharmacol. Sci. 29, 528–535 [DOI] [PubMed] [Google Scholar]
- 81. Mizrachi D., Segaloff D. L. (2004) Intracellularly located misfolded glycoprotein hormone receptors associate with different chaperone proteins than their cognate wild-type receptors. Mol. Endocrinol. 18, 1768–1777 [DOI] [PubMed] [Google Scholar]
- 82. McLatchie L. M., Fraser N. J., Main M. J., Wise A., Brown J., Thompson N., Solari R., Lee M. G., Foord S. M. (1998) RAMPs regulate the transport and ligand specificity of the calcitonin-receptor-like receptor. Nature 393, 333–339 [DOI] [PubMed] [Google Scholar]
- 83. Christopoulos A., Christopoulos G., Morfis M., Udawela M., Laburthe M., Couvineau A., Kuwasako K., Tilakaratne N., Sexton P. M. (2003) Novel receptor partners and function of receptor activity-modifying proteins. J. Biol. Chem. 278, 3293–3297 [DOI] [PubMed] [Google Scholar]
- 84. Bouschet T., Martin S., Henley J. M. (2005) Receptor-activity-modifying proteins are required for forward trafficking of the calcium-sensing receptor to the plasma membrane. J. Cell Sci. 118, 4709–4720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Saito H., Kubota M., Roberts R. W., Chi Q., Matsunami H. (2004) RTP family members induce functional expression of mammalian odorant receptors. Cell 119, 679–691 [DOI] [PubMed] [Google Scholar]
- 86. Janovick J. A., Patny A., Mosley R., Goulet M. T., Altman M. D., Rush T. S., 3rd, Cornea A., Conn P. M. (2009) Molecular mechanism of action of pharmacoperone rescue of misrouted GPCR mutants: the GnRH receptor. Mol. Endocrinol. 23, 157–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Maya-Nunez G., Ulloa-Aguirre A., Janovick J. A., Conn P. M. (2012) Pharmacological chaperones correct misfolded gpcrs and rescue function: protein trafficking as a therapeutic target. Subcell. Biochem. 63, 263–289 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.