

Abstract
The purpose of this investigation was to determine the extent to which dystrophin insufficiency caused histomorphological changes in a novel pig model of Becker muscular dystrophy. In our procedures, we used a combination of biochemical approaches, including quantitative PCR and Western blots, along with a histological analysis using standard and immunohistological measures. We found that 8-wk-old male affected pigs had a 70% reduction in dystrophin protein abundance in the diaphragm, psoas major, and longissimus lumborum and a 5-fold increase in serum creatine kinase activity compared with healthy male littermates. Dystrophin insufficiency in the diaphragm and the longissimus resulted in muscle histopathology with disorganized fibrosis that often colocalized with fatty infiltration but not the psoas. Affected animals also had an 80–85% reduction in α-sarcoglycan localization in these muscles, indicating compromised assembly of the dystrophin glycoprotein complex. Controls used in this study were 4 healthy male littermates, as they are most closely related to the affected animals. We concluded that pigs with insufficient dystrophin protein expression have a phenotype consistent with human dystrophinopathy patients. Given that and their similarity in body size and physiology to humans, we further conclude that this pig line is an appropriate translational model for dystrophinopathies.—Hollinger, K., Yang, C. X., Montz, R. E., Nonneman, D., Ross, J. W., Selsby, J. T. Dystrophin insufficiency causes selective muscle histopathology and loss of dystrophin-glycoprotein complex assembly in pig skeletal muscle.
Keywords: Duchenne muscular dystrophy, DMD, Becker muscular dystrophy, BMD, animal model
Dystrophin mutations can result in substantial physical and locomotion deficits, leading to wheelchair confinement and early death due to respiratory and/or cardiac failure. The most severe form, Duchenne muscular dystrophy (DMD), is caused by a lack of dystrophin protein. The generally less severe form, Becker muscular dystrophy (BMD), is caused by insufficient dystrophin abundance and/or expression of a partially functional gene product.
Several existing dystrophin-deficient animal models are currently used in the study of dystrophinopathies. However, despite the many useful features of the different mouse and dog models a few significant drawbacks exist. For example, the disease phenotype exhibited by mdx mice is much milder than that of human patients with DMD (1, 2) in part due to increased abundance of utrophin, a dystrophin-like protein (3–5). To mitigate this potential confounding variable, mdx/utrophin−/− mice were developed (5). While the disease phenotype is more severe in these mice, the double-knockout approach allows for the possibility that muscle function and metabolism are affected independent of the dystrophin mutation. Additional dystrophin-deficient models with a secondary mutation have since been developed (5–7). Regardless of mouse model, there is a poor correlation between effectiveness of therapies in mouse models to that observed in human patients (8), as scaling from a mouse to a human is challenging for a variety of reasons, including expense, safety, and differences in body size.
In addition to mouse models, a golden retriever muscular dystrophy (GRMD) model has also been discovered and is the best characterized of identified dog models (9). GRMD dogs have a phenotype that is more similar in severity and in selective muscle injury compared with human patients than mdx mice (10, 11). However, GRMD dogs have a high degree of phenotypic variability (12, 13), and, as in mdx mice, limited adiposity is observed. Finally, dystrophic dogs treated with corticosteroids exhibited a greater frequency of calcified necrotic fibers and impairment of some measures of muscle function (14), which is contrary to the beneficial effects of steroid use in human patients (15). The objective of this project was to characterize the skeletal muscle phenotype of a recently discovered pig line with a spontaneously occurring substitution in exon 41 of the dystrophin gene causing an arginine to tryptophan amino acid change. This substitution leads to decreased dystrophin abundance in skeletal and cardiac muscle (16). To develop novel therapeutic strategies to treat dystrophinopathies, there must be animal models that accurately recapitulate the disease. Currently available models have been useful; however, their inherent limitations have hindered drug development and approval. The anatomy, physiology, and genetics of pigs are more similar to those of humans than are those of mice or dogs. These similarities increase the likelihood of a more accurate recapitulation of the human disease and also mitigate challenges in drug scale-up, as pigs are of human size. Pigs are currently being used widely for advancements in human health research (reviewed in ref. 17), so this suggestion is not unprecedented. In this report, we detail the early muscle response to dystrophin insufficiency in the diaphragm, psoas major, and longissimus lumborum. Our hope is that dystrophin-insufficient pigs will aid in the development of therapeutic strategies by supplementing currently available animal models.
MATERIALS AND METHODS
Animal use
All animal procedures were reviewed and approved by the U.S. Meat Animal Research Center Animal Care and Use Committee, and procedures for handling pigs complied with those specified in the Guide for the Care and Use of Agricultural Animals in Agricultural Research and Teaching. Animals were housed in 6.5- × 8-foot nursery pens by litter until 8 wk of age. Affected pigs were identified by genotyping with a Sequenome MassArray system (Sequenome, Inc., San Diego, CA, USA; ref. 16). At 8 wk of age, 4 male affected pigs and 4 male healthy littermates were euthanized by electrical stunning, followed by exsanguination, and ∼5-g portions of the medial diaphragm, psoas major, and longissimus lumborum at the last rib were collected within 15 min of stunning. Muscles were snap-frozen in liquid nitrogen for biochemical analyses or frozen in isopentane or fixed in 10% buffered formalin for histological analyses. Frozen samples were stored at −80°C until analyzed. The diaphragm was chosen because it is used in respiration and is a primary cause of mortality in patients with dystrophinopathy and therefore of importance for disease progression. The psoas and longissimus were chosen because of their differing uses and composition of different fiber types. Specifically, the psoas is generally lightly used and is comprised largely of type I fibers, while the longissimus is generally used more frequently and is comprised primarily of type II fibers.
Plasma creatine kinase activity
Plasma creatine phosphokinase was measured using a 2-part reagent system (Pointe Scientific, Canton, MI, USA), following the manufacturer's instructions, in a SpectraMax M5 microplate plate reader (Molecular Devices, Sunnyvale, CA, USA). Samples (25 μl) were measured in duplicate, and the rate of NADH formation was monitored at 340 nm at 37°C. Samples having >2500 U/L were diluted in PBS and assayed again.
mRNA quantification
Muscle samples were powdered with a dry-ice-chilled mortar and pestle. Total RNA was extracted from ∼50 mg of powdered muscle using Trizol (Invitrogen, Carlsbad, CA, USA), following the manufacturer's instructions with minor modifications. The RNA was then column purified (RNeasy Mini Kit, Qiagen, Valencia, CA, USA) to minimize organic carryover. On the column, and before RNA elution, DNase digestion (RNase free DNase set; Qiagen) was used to prevent DNA contamination of the sample. After quantification using a Nanodrop (Thermo Scientific, Waltham, MA, USA), 1 μg of RNA was reverse-transcribed (QuantiTect Reverse Transcription Kit; Qiagen) following the manufacturer's instructions, with the addition of reverse transcription primers for 18S ribosomal RNA. The 18S rRNA does not contain a poly-A tail, and the addition of the 18S reverse-transcription primers ensures that the 18S rRNA can be converted into cDNA. For quantitative PCR, equal amounts of cDNA, corresponding to 10 ng of reverse-transcribed mRNA, were loaded into each well. To measure gene expression, primer pairs (Table 1) were mixed together with a QuantiFast SYBR Green PCR kit (Qiagen) in a reaction volume of 12.5 μl, and gene expression was measured with a Mastercycler EP Realplex (Eppendorf, Hauppauge, NY, USA). At the end of the PCR program, melting curves for all amplicons were inspected to verify that a single product was amplified with each primer pair. The pig genome has been sequenced and is well annotated (18), allowing construction of appropriate primer pairs. All samples were measured in triplicate wells.
Table 1.
Primer sequences used for quantitative PCR
| Primer | Sequence | Accession number | Amplicon length |
|---|---|---|---|
| DMD 5′ pig For (exon 9) | CCTCGGTTCAAGAGCTATGC | NM_001012408 | 128 |
| DMD 5′ pig Rev (exon 10) | TCCAACAATGAACTGCCAAA | ||
| DMD 5′ of SNP pig For (exon 37)a | AGCAAACTTGATGGCAAACC | NM_001012408 | 121 |
| DMD 5′ of SNP pig Rev (exon 38) | AATGGAGGCCTTTCCAGTCT | ||
| DMD across SNP pig For (exon 40) | TCAGTACAAGAGGCAGGCTG | NM_001012408 | 330 |
| DMD across SNP pig Rev (exon 42) | GGCATGTCTTCAGTCATCAC | ||
| DMD 3′ of SNP pig For (exon 41) | AATTTGCTCACTTTCGAAGA | NM_001012408 | 185 |
| DMD 3′ of SNP pig Rev (exon 42) | GAGGTCAGGAGCATTGAGAA | ||
| DMD 3′ pig For (exon 62/63) | CCACGAGACCCAAACAACTT | NM_001012408 | 153 |
| DMD 3′ pig Rev (exon 65) | AGGCTCAAGAGATCCAAGCA | ||
| TNF pig For | GCCCTTCCACCAACGTTTTC | NM_214022 | 158 |
| TNF pig Rev | TCCCAGGTAGATGGGTTCGT | ||
| IL1B pig For | AAGATAACACGCCCACCCTG | NM_214055 | 293 |
| IL1B pig Rev | TGTCAGCTTCGGGGTTCTTC | ||
| IL6 pig For | AGATGCCAAAGGTGATGCCA | NM_001252429 | 363 |
| IL6 pig Rev | CTCAGGGTCTGGATCAGTGC | ||
| MYOD1 pig For | CTACAGCGGTGACTCAGACG | NM_001002824 | 121 |
| MYOD1 pig Rev | GCTGTAATAGGTGCCGTCGT | ||
| DTNA pig For | ACTACCCACGGCAGTTTTTG | XM_003356399 | 110 |
| DTNA pig Rev | GCGTGTCCAAGAAACCATTT | ||
| SGCA pig For | AGGTCGAAAGGAAGGCGTAT | NM_001144122 | 131 |
| SGCA pig Rev | CATAGCAGGACAGCAGTGGA | ||
| NOS1 pig For | GGAAAACAGTCTCCCACCAA | XM_003132898 | 127 |
| NOS1 pig Rev | ATCCTGTTCCCAATGTGCTC | ||
| UTRN pig For | GAACGGATCATTGCTGACCT | XM_003121163 | 177 |
| UTRN pig Rev | CCTGAGGAGTTTGGCTTCTG | ||
| 18S RT primer | GAGCTGGAATTACCGCGGCT | ||
| 18S pig For | AAACGGCTACCACATCCAAG | NR_046261 | 141 |
| 18S pig Rev | TCGCGGAAGGATTTAAAGTG |
DTNA, α-dystrobrevin; For, forward; IL1B, interleukin 1β; IL6, interleukin 6; MYOD1, myogenic differentiation 1; NOS1, (neuronal) nitric oxide synthase 1; Rev, reverse; SGCA, α-sarcoglycan; TNF, tumor necrosis factor; UTRN, utrophin.
GeneBank dbSNP ss410758971.
Protein extraction
About 500 mg of powdered muscle was added into a glass Teflon Dounce homogenizer with 0.7 ml extraction buffer (2% SDS and 10 mM phosphate buffer, pH 7). Following ∼20 strokes of the homogenizer, the homogenizer was rinsed with 0.3 ml of extraction buffer, which was collected into the homogenized sample for a total dilution of 2:1. Samples were centrifuged at 1500 g for 15 min at room temperature to remove insoluble material. Protein concentration was measured with the BCA kit (Pierce, Rockford, IL, USA) at a 1:10 dilution in triplicate. All samples were diluted to a protein concentration of 3.5 μg/μl in loading buffer (62.5 mM Tris, pH 6.8; 2% SDS; 10% glycerol; 2.5% β-mercaptoethanol; and 0.002% bromophenole blue) and heated to 95°C for 5 min.
Western blot analysis
Protein (35 μg; 10 μl) was separated at 60 V for 15 min, followed by 1 h at 120 V in a 4–20% gradient polyacrylamide gel (Lonza, Rockland, ME, USA). Following separation, the protein was transferred to a nitrocellulose membrane (GE Water and Process Technologies, Feasterfille-Trevose, PA, USA) at 90 V for 90 min in the cold room. For blots intended to detect dystrophin or utrophin, protein was separated using 6% gels coupled with a 4% stacking gel. These separations were run at 30 V overnight and then continued as described previously, with the exception that PVDF membrane (Millipore, Billerica, MA, USA), rather than nitrocellulose membrane, was used. In our hands, PVDF performs better than nitrocellulose for detection of large proteins. All membranes were stained with Ponceau S to verify equal loading and transfer. Membranes were blocked for 30 min with 5% milk in Tris-buffered saline with 0.1% Tween 20 (TTBS). Membranes were subsequently incubated with primary antibody diluted in 1% milk in TTBS at 4°C overnight as follows: dystrophin [1:500; rabbit polyclonal (Abcam, Cambridge, MA, USA); ab15277; aa 3661–3677], α-sarcoglycan (1:1000; NCL-L-a-SARC; Novocastra, Newcastle, UK), and utrophin [1:250; Developmental Studies Hybridoma Bank (DSHB), University of Iowa, Iowa City, IA, USA; MANCHO3 (8A4) concentrate developed by G. E. Morris]. The next day, membranes were washed 3 times for 10 min in TTBS and incubated in secondary antibody: donkey anti-rabbit IgG horseradish peroxidase linked (1:200; GE Healthcare, Little Chalfont, UK) or sheep anti-mouse IgG horseradish peroxidase linked (1:2000; GE Healthcare) for 1 h at room temperature, as appropriate. Membranes were again washed 3 times for10 min with TTBS. After the last wash, ECL (Millipore) was added to the membranes, and emitted light was captured with film. To analyze the protein abundance, the film was scanned and digitized, and band density was measured using Carestream 5.0 molecular imaging software (Carestream Health, New Haven, CT, USA).
Histological staining
Fixed 5-μm muscle sections were deparaffinized and rehydrated by passing slides through 3 Citrisolv (Fisherbrand, King of Prussia, PA, USA) baths and 4 ethanol baths with decreasing percentages of ethanol (100, 100, 95, and 80%). Hematoxylin and eosin (H&E) staining was performed according to standard techniques. Briefly, sections were incubated in Mayer's hematoxylin, rinsed with tap water, counterstained with 1% eosin, and dehydrated, and coverslips were applied. To perform the trichrome stain, rehydrated sections were incubated in Bouin's solution overnight at room temperature. The next day, slides were thoroughly rinsed in tap water and stained with Weigert's hematoxylin. Slides were blued with tap water and stained with Biebrich scarlet. Excess stain was removed with distilled water before differentiating sections in a phosphotungstic/phoshomolybdic acid solution. After differentiation slides were stained with aniline blue. Extra stain was washed off with distilled water, and sections were differentiated in 1% acetic acid. Finally, acetic acid was rinsed off with distilled water, sections were dehydrated, and coverslips were applied. To assess muscle damage, slides were coded, and identifying information was removed. In a blinded fashion, the same trained technician subjectively sorted the slides according to increasing damage.
Immunohistochemistry (IHC)
Dystrophin expression and localization were measured using fixed sections. Muscle sections (5 μm) were deparaffinized, and antigen retrieval was performed by heating for 20 min at 95–100°C in Tris-EDTA buffer (10 mM Tris base, 1 mM EDTA solution, and 0.05% Tween 20, pH 9.0). After cooling to room temperature and 2 washes in PBS, slides were blocked with 5% BSA in PBS for 15 min at room temperature. Following blocking, slides were incubated overnight at 4°C with mouse monoclonal anti-dystrophin antibody (D8168; aa 1400–1505; Sigma, St. Louis, MO, USA) diluted 1:300 in 5% BSA. After 3 washes with 0.05% Tween-20 in PBS, sections were incubated with AlexaFluor 488-labeled goat anti-mouse IgG (1:100; Invitrogen) for 1 h at room temperature and then washed again. Slides were mounted with Slowfade Gold Antifade Reagent with DAPI (Invitrogen). Notably, for all IHC targets, some sections were incubated without a primary antibody or without secondary antibody as negative controls.
For detection of desmin, 10-μm frozen muscle sections were washed in PBS for 10 min before blocking with 5% BSA in PBS at room temperature for 15 min. Sections were incubated overnight at 4°C with rabbit anti-desmin serum (1:100; a gift from Dr. Ted Huiatt, Iowa State University, Ames, IA, USA). The next day, sections were washed 3 times with PBS and incubated with rhodamine-conjugated donkey anti-rabbit IgG (1:100; Millipore) for 1 h at room temperature in the dark. After the secondary incubation, slides were washed 3 times with PBS, and coverslips were mounted and sealed as above. To assess dystrophin-glycoprotein complex (DGC) stability in affected animals and healthy pigs, IHC for α-sarcoglycan (Novocastra, Newcastle, UK) expression and localization was measured in combination with laminin (NeoMarkers, Fremont, CA, USA). Sections were treated as described previously, and both primary antibodies were used at a concentration of 1:100. Secondary goat anti-mouse fluorescein-conjugated (Millipore) and goat anti-rabbit rhodamine-conjugated IgG (Millipore) were used at a dilution of 1:100 for 1 h at room temperature in the dark to detect α-sarcoglycan and laminin, respectively.
For analysis of protein abundance following IHC, 2 nonoverlapping ×200 pictures were randomly taken from each section with the appropriate filters. For each protein, images were collected on the same day for each muscle under identical exposure conditions in random order and in a blinded procedure by a technician. Digital images were transferred to Openlab (Perkin Elmer, Waltham, MA, USA) so that fluorescence intensity could be measured. The density slice function was used to transform the image into a binary image such that pixels were identified as either above or below threshold intensity. Threshold was determined by measuring pixel intensity of intracellular and extracellular areas, as well as at several locations on the sarcolemma in various random sections. All images were taken and processed under the same conditions. To measure minimal Feret diameter, the laminin images were converted into binary images as described above. Images were then exported to the ImagePro software (MediaCybernetics, Rockville, MD, USA), where the minimal Feret diameter tool was used to obtain the measurement. The measurements were exported to Excel (Microsoft, Redmond, WA, USA), where the data were binned and mean diameter and coefficient of variance were calculated for each muscle.
For determination of fiber type differences in affected animals compared with healthy littermates, IHC for slow myosin (A4.951) was performed in frozen sections in combination with laminin. The A4.651 serum (DSHB; developed by H. M. Blau, Stanford University, Stanford, CA, USA) was used undiluted. Type I myosin heavy chain (MHC) was detected using fluorescein conjugated goat anti-mouse IgG at 1:100 (Millipore), and laminin was detected as before. Slides were imaged at ×100, which resulted in ∼200–750 cells/image. For analyses, both the total number of cells per image and the number of positive staining cells per image were counted using ImageJ (19). The cell counts from 2 images/section were pooled, resulting in ∼500–1300 cells/section.
Statistics
All data are expressed as means ± se unless otherwise noted. Data were compared using t tests, and significance was set at P ≤ 0.05.
RESULTS
Dystrophin deficiency in pigs with an arginine to tryptophan (R1958W) substitution
Skeletal muscles from 8-wk-old male pigs with a missense mutation in the dystrophin gene were compared with healthy male littermates. Dystrophin protein expression was decreased by 70% in diaphragm, psoas, and longissimus collected from affected animals (tryptophan) compared with healthy littermates (arginine) (Fig. 1A). This decrease was confirmed by 70–90% reduction in expression by IHC in the diaphragm and the longissimus but was not significantly different in the psoas (Fig. 1B, C). Notably, full-length dystrophin protein was present in all muscles, and it was evenly localized to the sarcolemma, suggesting that these muscles are dystrophin insufficient as opposed to dystrophin deficient.
Figure 1.

Dystrophin protein abundance and localization. Dystrophin was assayed in the diaphragm, psoas, and longissimus in muscles taken from healthy pigs and pigs containing an arginine to tryptophan (R1958W) polymorphism in exon 41 of the dystrophin gene, DMD. A) By Western blot, dystrophin protein abundance was decreased by 70% in muscles from diseased animals compared with muscles from healthy animals. B) Representative ×400 images resulting from IHC targeted to dystrophin. C) Following fluorescence quantification, dystrophin abundance by IHC was significantly decreased in the diaphragm and longissimus (P<0.05), but failed to reach significance in the psoas, in part because variability in the control animals was high and in part because the magnitude of change in the psoas was not as substantial as in the diaphragm and longissimus. D) Dystrophin transcript abundance at 5 locations along DMD in the diaphragm. SNP, single-nucleotide polymorphism. E) Dystrophin transcript abundance at 5 locations along DMD in the psoas. F) Dystrophin transcript abundance at 5 locations along DMD in the longissimus. *P ≤ 0.05 vs. corresponding control.
To better understand the mechanism leading to decreased dystrophin protein accumulation, we measured dystrophin gene expression (Fig. 1D–F and Table 1). Gene expression was similar at the 5′ end of the gene, immediately upstream of the substitution in exon 41, across the substitution, and immediately downstream of the substitution in the diaphragm and psoas. In the longissimus, there was a 50% reduction in gene expression at all 4 locations. Transcript abundance amplified by primers designed 3′ of the substitution was significantly lower in the diaphragm, psoas, and longissimus by 60, 45, and 70%, respectively, indicating decreased transcript stability.
Dystrophin insufficiency is associated with muscle histophatology
Dystrophin insufficiency was associated with a 5-fold increase in serum creatine kinase activity (Fig. 2A), which is consistent with other dystrophinopathy models and human patients with BMD or DMD patients. In addition, affected and healthy animals could be distinguished from one another during histological evaluation (evaluator blinded to the genotype of the animal) of the diaphragm and longissimus but not the psoas. Dystrophin insufficiency led to necrotic lesions in diaphragm and longissimus muscle, apparent in both H&E and trichrome images (Fig. 2B). Evident in these lesions was disorganized fibrosis and fatty infiltration that generally colocalized. Immune cell infiltration was only occasionally present along with immune cell attack of skeletal muscle cells. Gene expression indicating inflammation, including tumor necrosis factor (TNF), interleukin 1β (IL1B), and interleukin 6 (IL6) was similar between groups in all 3 muscles (Supplemental Table S1). Notably rare in these muscles was the appearance of centralized nuclei. Related, myogenic differentiation 1 (MYOD1) expression was similar between groups (Supplemental Table S1) for all muscles, suggesting that satellite cell activation is not widespread. While there were necrotic lesions evident in the longissimus and diaphragm from affected animals, there were also areas that appeared similar to healthy animals (Supplemental Fig. S1).
Figure 2.
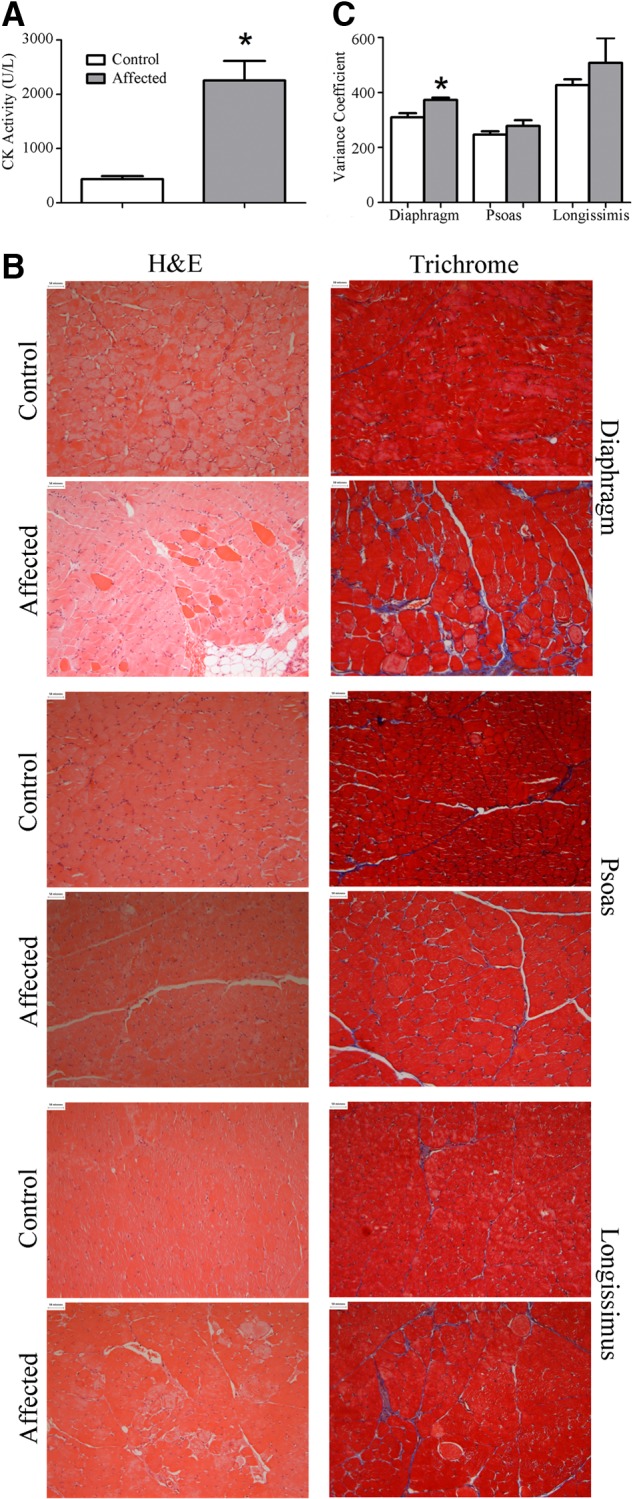
Dystrophin insufficiency leads to muscle injury. A) Serum creatine kinase activity was increased 4-fold in affected pigs compared with healthy littermates. B) Representative ×200 images of slides stained with H&E or trichrome. In a blinded fashion, diaphragm sections and longissimus sections were successfully identified as either from healthy or affected pigs by the appearance of necrotic lesions, while affected and healthy psoas muscles were indistinguishable. C) Minimum Feret diameter was determined on 500–1200 cells/muscle for the diaphragm, psoas, and longissimus taken from healthy and affected pigs, and the variance coefficient was calculated. Variance coefficient was increased in the diaphragm taken from affected animals compared with healthy animals but was similar in the longissimus and psoas. *P ≤ 0.05 vs. corresponding control.
Mean fiber diameter (minimum Feret diameter) was similar between healthy and affected pigs across the 3 muscles (Supplemental Fig. S2); however, the variance coefficient of fiber diameters was increased by ∼20% in the diaphragm providing an objective measure of muscle injury (Fig. 2C). Variance coefficient of fiber diameter in the psoas and longissimus was similar between groups.
Dystrophin insufficiency alters expression of DGC members but not other membrane proteins
In human patients and other animal models, a lack of dystrophin or insufficient dystrophin expression leads to a collapse of the DGC. Gene expression of α-sarcoglycan (SGCA), α-dystrobrevin (DTNA), and (neuronal) nitric oxide synthase 1 (NOS1; also known as nNOS) was similar in healthy and affected animals for all 3 muscles (Supplemental Table S1). Protein expression by Western blot, however, demonstrated a 50% reduction in SGCA abundance in all 3 muscles (Fig. 3A). Such discordant gene and protein expression for DGC components is consistent with other animal models as well as human patients with DMD (20). SGCA protein abundance was also measured by IHC and found to be decreased by 80–85% in the diaphragm and longissimus, while expression was similar in the psoas (Fig. 3B, C).
Figure 3.
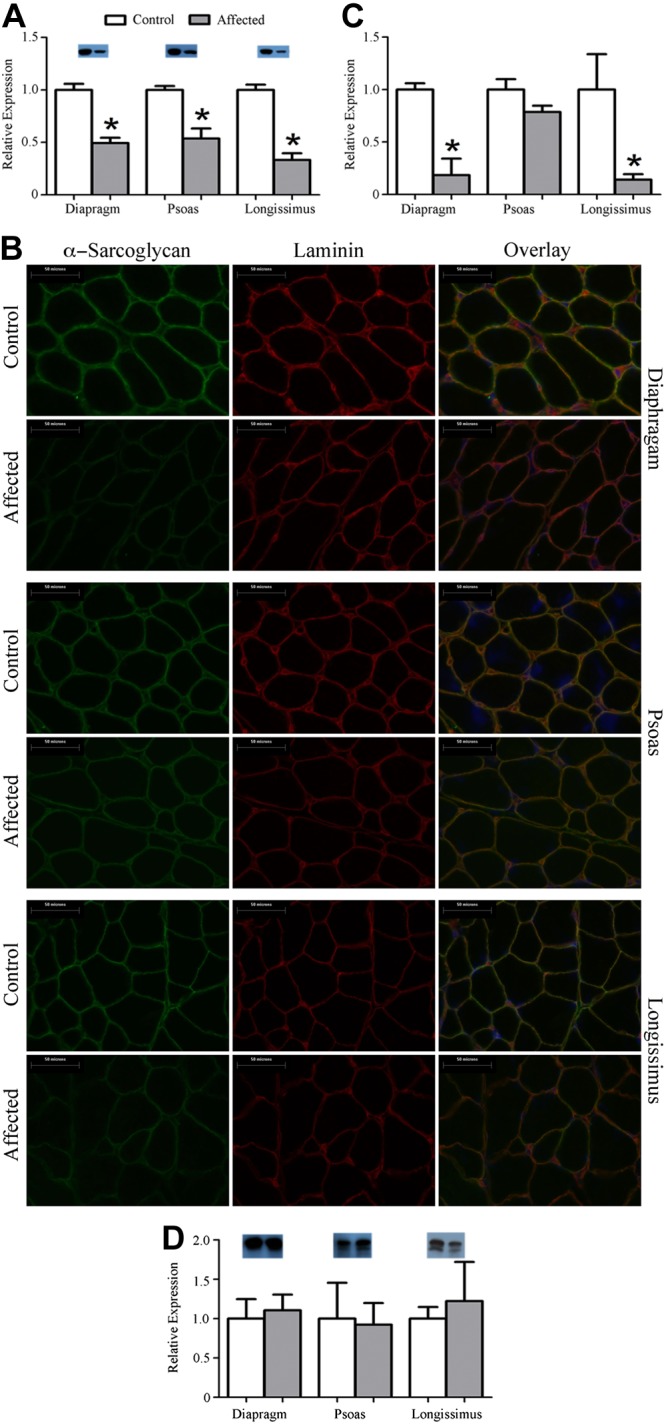
Dystrophin insufficiency leads to a reduction in α-sarcoglycan abundance but not utrophin abundance. A) Representative Western blot of α-sarcoglycan indicating an ∼50% reduction. B) Representative ×400 images for IHC of α-sarcoglycan. C) Fluorescent intensity was quantified and an 80–85% reduction was found in the diaphragm and the longissimus from affected animals while the psoas was similar between groups. D) Representative Western blot of utrophin and quantification. Utrophin was similar between groups. *P ≤ 0.05 vs. corresponding control.
In addition to SGCA, we also measured abundance and localization of several other membrane proteins. Abundance and localization of laminin (Fig. 3 and Supplemental Fig. S3) and desmin (Supplemental Fig. S4) were similar between healthy and affected animals for all 3 muscles. Notably, we found that utrophin gene expression (Supplemental Table S2), as well as protein expression, was similar between healthy and affected pigs for all 3 muscles (Fig. 3D). Hence, these muscles have a similar loss of expression and organization of the DGC, as is observed in human patients and other animal models, without observing compensatory utrophin expression, as in the mouse model.
Because in other animal models and human patients dystrophinopathy is associated with a progressive type I shift, we measured type I fiber distribution in these muscles. Surprisingly, at this early time point there was a 20% increase in type I fibers in the affected diaphragms compared with healthy muscle. Fiber type distribution in the longissimus and psoas was similar between groups (Fig. 4).
Figure 4.
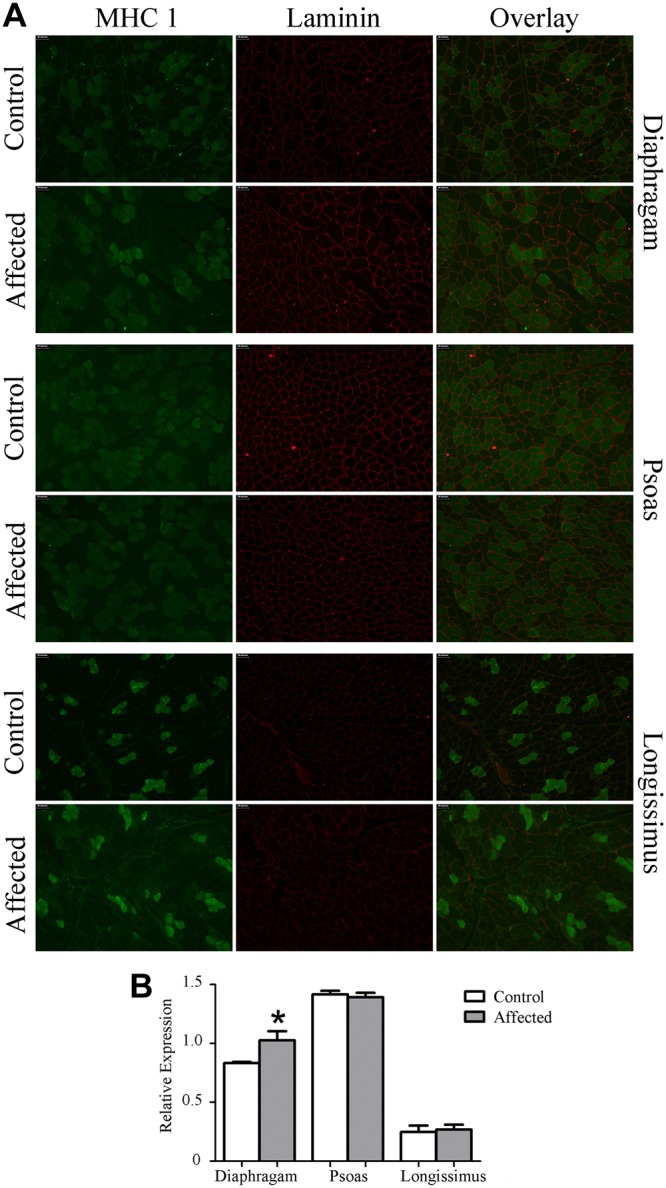
Dystrophin insufficiency alters fiber type distribution. A) Representative ×200 image of IHC for type I MHC. B) Type I MHC-positive cells were increased 20% in affected diaphragms compared with diaphragms from healthy animals, while the psoas and longissimus were similar between groups. *P ≤ 0.05 vs. corresponding control.
DISCUSSION
Because of inherent limitations in existing animal models of dystrophinopathy, there has been great interest in establishing a novel large animal model of the disease. A genetic line of pigs was recently discovered that was more susceptible to stress-mediated death. A genome-wide association identified a defect in the dystrophin gene, which was associated with a reduction in dystrophin protein accumulation in skeletal muscle (16). In this investigation, we evaluated the expression and abundance of dystrophin and DGC components in diaphragm, psoas major, and longissimus lumborum muscle from healthy and affected 8-wk-old male pigs. For pigs, the use of the muscles examined in this study vary greatly in their function, such that the diaphragm is used during respiration; the longissimus is a heavily used antigravity muscle and is involved in transitioning from laying to standing, during standing, in shifting weight from one leg to another, and in lying down; the psoas, a hip flexor, is only lightly used. We found that in these muscles, dystrophin protein accumulation was decreased in affected pigs, as was expression of SGCA, a DGC component, compared with healthy male littermates. This was associated with muscle histopathology consisting of necrotic lesions, fatty infiltration, fibrosis, and increased fiber-size variability in a muscle specific fashion. Notably, expression of utrophin, a protein that can substitute for the missing dystrophin protein, was similar between groups.
At 8 wk of age, affected pigs had a 70% reduction of DMD in the diaphragm, psoas, and longissimus compared with healthy littermates that resulted from a uniform reduction in dystrophin localization to the sarcolemma. Because each muscle contained residual full-length dystrophin, these pigs are representative of a Becker phenotype, not Duchenne. While the Leiden database does not yet have record of a BMD-causing missense mutation at this precise location, there are numerous documented instances of missense mutations leading to BMD (21–24). To better understand the molecular cause of decreased dystrophin protein accumulation, dystrophin gene DMD expression was measured using primers against multiple sites along the DMD gene. Gene expression in the longissimus was significantly reduced along the entire gene, while the diaphragm and the psoas only show a reduction at the 3′ end of the gene, suggesting that the point mutation in the dystrophin gene makes the transcript less stable. This 5′–3′ transcript imbalance also has been observed in patients with BMD (25). How a substitution in the amino acid sequence alters susceptibility to proteolysis is unknown. To gain additional information regarding the probability of calpain degradation, using CaMPDB (26), we performed in silico digests of aa 1867–2097, which is inclusive of the amino acid substitution. We found that the affected sequence was predicted to be more susceptible to calpain degradation than sequence from control animals. Also, the vastly different amino acid properties of arginine and tryptophan likely contribute to aberrant protein folding or a resultant conformational change causing reduced protein stability (22). Alternatively, altered electrostatic interactions in the rod domain may also destabilize the dystrophin protein (27). These data suggest that dystrophin insufficiency results from decreased transcript stability and, speculatively, increased protein degradation.
Like in other animal models (11, 28, 29) and human patients (30), failure to accumulate sufficient dystrophin protein leads to a systemic collapse of muscle stability, resulting in a 4-fold increase in serum creatine phosphokinase. H&E and trichrome staining revealed injury in both the diaphragm and longissimus of affected animals, including fatty infiltration, fibrosis, and necrotic fibers. A progressive increase in adiposity is a hallmark of dystrophinopathy in human patients (31), although it is not present in the mdx mouse or the GRMD dog (1). At this early time point, and similar to muscle from patients, we did not see accumulation of adipocytes and fibrosis throughout the entire muscle but rather colocalized within foci of necrosis. Within these foci, fibrosis and infiltrating adiposity appeared disorganized, in contrast to the high degree of organization found in healthy regions of muscles from affected animals and muscles from healthy animals.
Fiber-size variation is another indicator of muscle injury, and degeneration/regeneration cycles where larger variance coefficients are indicative of disease in other animal models and patients with dystrophinopathy (32). Consistent with our subjective histological evaluation, the objectively measured variance coefficients in fiber-size distribution were significantly increased in diaphragm from affected pigs compared with healthy littermates. Finally, dystrophic muscle undergoes a progressive type I shift as this fiber type is generally less susceptible to disease-related injury (33, 34). While unexpected at this early time point, the frequency of type I fibers was increased in diaphragms from affected animals compared with healthy animals.
Despite being dystrophin insufficient like the diaphragm and longissimus, the psoas did not show signs of increased muscle histopathology, as assessed through subjective histological evaluation or, more objectively, by fiber diameter variance. The mechanism leading to preservation of the psoas is of potential therapeutic interest. Like in the other muscles investigated, DGC abundance was decreased in psoas from affected animals; however, localization of DGC components was similar. Hence, these data suggest that in the psoas a greater proportion of these proteins are resident at the sarcolemma than in the diaphragm or longissimus. It is reasonable to suggest then that the similar DGC localization between the psoas from affected and health pigs protects the psoas from injury. Alternatively, the pattern of use of the psoas in pigs relative to the diaphragm and longissimus may differ resulting in less injury and subsequently allowing dystrophin accumulation at the sarcolemma. If this were the case, it would suggest that increased use, such as during exercise, could hasten disease progression (35, 36). Finally, the psoas is predominantly composed of type I fibers, which are generally more disease resistant than type II fibers, and may, therefore, offer protection from disease.
The absence or reduction of dystrophin protein accumulation inhibits assembly of the DGC in dystrophic muscle (28–30). In the affected diaphragm, longissimus, and psoas, gene expression of SGCA, NOS1, and DTNA was similar to healthy littermates, which is consistent with other animal models and human patients (20). Dysregulation becomes apparent when evaluating protein accumulation and localization of these products. Protein expression and localization of SGCA was decreased in the diaphragm and longissimus. Surprisingly, the reduction in SGCA accumulation by Western blot measured in the psoas was not well matched with expression measured by IHC. This was consistent, however, with dystrophin measurement by IHC in this muscle. Collectively, these observations indicate that in the diaphragm and longissimus, DGC component proteins are being translated (albeit less than in healthy muscle) but are failing to integrate into the membrane. Conversely, in the psoas, translated DGC components appear to integrate at an abundance that is similar to healthy muscle. Apparent DGC fidelity is consistent with muscle injury, such that, at 8 wk of age, muscle injury in the diaphragm and longissimus was apparent; however, the psoas was not distinguishable between diseased and healthy animals.
In addition, expression and localization of laminin and desmin were similar between groups, indicating that there is not a wholesale collapse of membrane structure. Also, utrophin expression was similar between groups for all muscles, eliminating one of the compensatory mechanisms and, hence, limitations noted in other animal models.
In total, these data indicate that these pigs have muscle histopathology consistent with a dystrophinopathy. That full-length dystrophin is present at the membrane further implicates these pigs as a novel large animal model of BMD. Given the genetic, anatomical, and physiological similarities between pigs and humans and the fact that these pigs recapitulate many hallmarks of disease, there is the promise that the BMD pig will result in a translational disease model capable of filling voids left by currently available dystrophinopathy models.
Supplementary Material
Acknowledgments
The authors are grateful for insights provided by Dr. Ted Huiatt.
This project was partially supported by the U.S. National Institutes of Health, grants RR-030232 (to J.W.R.) and NS-079603 (to J.T.S.).
Mention of trade names or commercial products in this publication is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the U.S. Department of Agriculture (USDA).
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
- BMD
- Becker muscular dystrophy
- DGC
- dystrophin glycoprotein complex
- DMD
- Duchenne muscular dystrophy
- DTNA
- α-dystrobrevin
- GRMD
- golden retriever muscular dystrophy
- H&E
- hematoxylin and eosin
- IHC
- immunohistochemistry
- MHC
- myosin heavy chain
- NOS1
- (neuronal) nitric oxide synthase 1
- SGCA
- α-sarcoglycan
REFERENCES
- 1. Banks G. B., Chamberlain J. S. (2008) The value of mammalian models for duchenne muscular dystrophy in developing therapeutic strategies. Curr. Top Dev. Biol. 84, 431–453 [DOI] [PubMed] [Google Scholar]
- 2. Chamberlain J. S., Metzger J., Reyes M., Townsend D., Faulkner J. A. (2007) Dystrophin-deficient mdx mice display a reduced life span and are susceptible to spontaneous rhabdomyosarcoma. FASEB J. 21, 2195–2204 [DOI] [PubMed] [Google Scholar]
- 3. Law D. J., Allen D. L., Tidball J. G. (1994) Talin, vinculin and DRP (utrophin) concentrations are increased at mdx myotendinous junctions following onset of necrosis. J. Cell Sci. 107, 1477–1483 [DOI] [PubMed] [Google Scholar]
- 4. Rafael J. A., Townsend E. R., Squire S. E., Potter A. C., Chamberlain J. S., Davies K. E. (2000) Dystrophin and utrophin influence fiber type composition and post-synaptic membrane structure. Hum. Mol. Genet. 9, 1357–1367 [DOI] [PubMed] [Google Scholar]
- 5. Deconinck A. E., Rafael J. A., Skinner J. A., Brown S. C., Potter A. C., Metzinger L., Watt D. J., Dickson J. G., Tinsley J. M., Davies K. E. (1997) Utrophin-dystrophin-deficient mice as a model for Duchenne muscular dystrophy. Cell 90, 717–727 [DOI] [PubMed] [Google Scholar]
- 6. Sacco A., Mourkioti F., Tran R., Choi J., Llewellyn M., Kraft P., Shkreli M., Delp S., Pomerantz J. H., Artandi S. E., Blau H. M. (2010) Short telomeres and stem cell exhaustion model duchenne muscular dystrophy in mdx/mTR mice. Cell 143, 1059–1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mangner N., Adams V., Sandri M., Hoellriegel R., Hambrecht R., Schuler G., Gielen S. (2012) Muscle function and running activity in mouse models of hereditary muscle dystrophy: impact of double knockout for dystrophin and the transcription factor MyoD. Muscle Nerve 45, 544–551 [DOI] [PubMed] [Google Scholar]
- 8. Wells D. J., Wells K. E. (2005) What do animal models have to tell us regarding Duchenne muscular dystrophy? Acta Myol. 24, 172–180 [PubMed] [Google Scholar]
- 9. Kornegay J. N., Bogan J. R., Bogan D. J., Childers M. K., Li J., Nghiem P., Detwiler D. A., Larsen C. A., Grange R. W., Bhavaraju-Sanka R. K., Tou S., Keene B. P., Howard J. F., Jr., Wang J., Fan Z., Schatzberg S. J., Styner M. A., Flanigan K. M., Xiao X., Hoffman E. P. (2012) Canine models of Duchenne muscular dystrophy and their use in therapeutic strategies. Mamm. Genome 23, 85–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kornegay J. N., Cundiff D. D., Bogan D. J., Bogan J. R., Okamura C. S. (2003) The cranial sartorius muscle undergoes true hypertrophy in dogs with golden retriever muscular dystrophy. Neuromuscul. Disord. 13, 493–500 [DOI] [PubMed] [Google Scholar]
- 11. Cooper B. J., Winand N. J., Stedman H., Valentine B. A., Hoffman E. P., Kunkel L. M., Scott M. O., Fischbeck K. H., Kornegay J. N., Avery R. J., Williams J. R., Schmickel R. D., Sylvester J. E. (1988) The homologue of the Duchenne locus is defective in X-linked muscular dystrophy of dogs. Nature 334, 154–156 [DOI] [PubMed] [Google Scholar]
- 12. Ambrosio C. E., Valadares M. C., Zucconi E., Cabral R., Pearson P. L., Gaiad T. P., Canovas M., Vainzof M., Miglino M. A., Zatz M. (2008) Ringo, a golden retriever muscular dystrophy (GRMD) dog with absent dystrophin but normal strength. Neuromuscul. Disord. 18, 892–893 [DOI] [PubMed] [Google Scholar]
- 13. Zucconi E., Valadares M. C., Vieira N. M., Bueno C. R., Jr., Secco M., Jazedje T., da Silva H. C., Vainzof M., Zatz M. (2010) Ringo: discordance between the molecular and clinical manifestation in a golden retriever muscular dystrophy dog. Neuromuscul. Disord. 20, 64–70 [DOI] [PubMed] [Google Scholar]
- 14. Liu J. M., Okamura C. S., Bogan D. J., Bogan J. R., Childers M. K., Kornegay J. N. (2004) Effects of prednisone in canine muscular dystrophy. Muscle Nerve 30, 767–773 [DOI] [PubMed] [Google Scholar]
- 15. Drachman D. B., Toyka K. V., Myer E. (1974) Prednisone in Duchenne muscular dystrophy. Lancet 2, 1409–1412 [DOI] [PubMed] [Google Scholar]
- 16. Nonneman D. N., Brown-Brandl T., Jones S. A., Wiedmann R. T., Rohrer G. A. (2012) A defect in dystrophin causes a novel porcine stress syndrome. BMC Genomics 13, 233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Prather R. S., Lorson M., Ross J. W., Whyte J. J., Walters E. (2013) Genetically engineered pig models for human diseases. Ann. Rev. Anim. Biosci. 1, 203–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Groenen M. A., Archibald A. L., Uenishi H., Tuggle C. K., Takeuchi Y., Rothschild M. F., Rogel-Gaillard C., Park C., Milan D., Megens H. J., Li S., Larkin D. M., Kim H., Frantz L. A., Caccamo M., Ahn H., Aken B. L., Anselmo A., Anthon C., Auvil L., Badaoui B., Beattie C. W., Bendixen C., Berman D., Blecha F., Blomberg J., Bolund L., Bosse M., Botti S., Bujie Z., Bystrom M., Capitanu B., Carvalho-Silva D., Chardon P., Chen C., Cheng R., Choi S. H., Chow W., Clark R. C., Clee C., Crooijmans R. P., Dawson H. D., Dehais P., De Sapio F., Dibbits B., Drou N., Du Z. Q., Eversole K., Fadista J., Fairley S., Faraut T., Faulkner G. J., Fowler K. E., Fredholm M., Fritz E., Gilbert J. G., Giuffra E., Gorodkin J., Griffin D. K., Harrow J. L., Hayward A., Howe K., Hu Z. L., Humphray S. J., Hunt T., Hornshøj H., Jeon J. T., Jern P., Jones M., Jurka J., Kanamori H., Kapetanovic R., Kim J., Kim J. H., Kim K. W., Kim T. H., Larson G., Lee K., Lee K. T., Leggett R., Lewin H. A., Li Y., Liu W., Loveland J. E., Lu Y., Lunney J. K., Ma J., Madsen O., Mann K., Matthews L., McLaren S., Morozumi T., Murtaugh M. P., Narayan J., Nguyen D. T., Ni P., Oh S. J., Onteru S., Panitz F., Park E. W., Park H. S., Pascal G., Paudel Y., Perez-Enciso M., Ramirez-Gonzalez R., Reecy J. M., Rodriguez-Zas S., Rohrer G. A., Rund L., Sang Y., Schachtschneider K., Schraiber J. G., Schwartz J., Scobie L., Scott C., Searle S., Servin B., Southey B. R., Sperber G., Stadler P., Sweedler J. V., Tafer H., Thomsen B., Wali R., Wang J., White S., Xu X., Yerle M., Zhang G., Zhang J., Zhao S., Rogers J., Churcher C., Schook L. B. (2012) Analyses of pig genomes provide insight into porcine demography and evolution. Nature 491, 393–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rasband W. S. (1997–2012) ImageJ. U.S. National Institutes of Health, Bethesda, MD, USA [Google Scholar]
- 20. Porter J. D., Khanna S., Kaminski H. J., Rao J. S. N., Merriam A. P., Richmonds C. R., Leahy P., Li J. J., Andrade F. H. (2002) A chronic inflammatory response dominates the skeletal muscle molecular signature in dystrophin-deficient mdx mice. Hum. Mol. Genet. 11, 263–272 [DOI] [PubMed] [Google Scholar]
- 21. Aartsma-Rus A., Van Deutekom J. C., Fokkema I. F., Van Ommen G. J., Den Dunnen J. T. (2006) Entries in the Leiden Duchenne muscular dystrophy mutation database: an overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle Nerve 34, 135–144 [DOI] [PubMed] [Google Scholar]
- 22. Acsadi G., Moore S. A., Cheron A., Delalande O., Bennett L., Kupsky W., El-Baba M., Le Rumeur E., Hubert J. F. (2012) Novel mutation in spectrin-like repeat 1 of dystrophin central domain causes protein misfolding and mild Becker muscular dystrophy. J. Biol. Chem. 287, 18153–18162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tuffery-Giraud S., Beroud C., Leturcq F., Yaou R. B., Hamroun D., Michel-Calemard L., Moizard M. P., Bernard R., Cossee M., Boisseau P., Blayau M., Creveaux I., Guiochon-Mantel A., de Martinville B., Philippe C., Monnier N., Bieth E., Khau Van Kien P., Desmet F. O., Humbertclaude V., Kaplan J. C., Chelly J., Claustres M. (2009) Genotype-phenotype analysis in 2,405 patients with a dystrophinopathy using the UMD-DMD database: a model of nationwide knowledgebase. Hum. Mutat. 30, 934–945 [DOI] [PubMed] [Google Scholar]
- 24. Hamed S., Sutherland-Smith A., Gorospe J., Kendrick-Jones J., Hoffman E. (2005) DNA sequence analysis for structure/function and mutation studies in Becker muscular dystrophy. Clin. Genet. 68, 69–79 [DOI] [PubMed] [Google Scholar]
- 25. Spitali P., van den Bergen J. C., Verhaart I. E., Wokke B., Janson A. A., van den Eijnde R., den Dunnen J. T., Laros J. F., Verschuuren J. J., 't Hoen P. A., Aartsma-Rus A. (2013) DMD transcript imbalance determines dystrophin levels. FASEB J. 27, 4909–4916 [DOI] [PubMed] [Google Scholar]
- 26. DuVerle D., Takigawa I., Ono Y., Sorimachi H., Mamitsuka H. (2010) CaMPDB: a resource for calpain and modulatory proteolysis. Genome Inform. 22, 202–213 [PubMed] [Google Scholar]
- 27. Legardinier S., Legrand B., Raguenes-Nicol C., Bondon A., Hardy S., Tascon C., Le Rumeur E., Hubert J. F. (2009) A two-amino acid mutation encountered in Duchenne muscular dystrophy decreases stability of the rod domain 23 (R23) spectrin-like repeat of dystrophin. J. Biol. Chem. 284, 8822–8832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Petrof B. J., Shrager J. B., Stedman H. H., Kelly A. M., Sweeney H. L. (1993) Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc. Natl. Acad. Sci. U. S. A. 90, 3710–3714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ervasti J. M., Roberds S. L., Anderson R. D., Sharp N. J., Kornegay J. N., Campbell K. P. (1994) Alpha-dystroglycan deficiency correlates with elevated serum creatine kinase and decreased muscle contraction tension in golden retriever muscular dystrophy. FEBS Lett. 350, 173–176 [DOI] [PubMed] [Google Scholar]
- 30. Welch E. M., Barton E. R., Zhuo J., Tomizawa Y., Friesen W. J., Trifillis P., Paushkin S., Patel M., Trotta C. R., Hwang S., Wilde R. G., Karp G., Takasugi J., Chen G., Jones S., Ren H., Moon Y.-C., Corson D., Turpoff A. A., Campbell J. A., Conn M. M., Khan A., Almstead N. G., Hedrick J., Mollin A., Risher N., Weetall M., Yeh S., Branstrom A. A., Colacino J. M., Babiak J., Ju W. D., Hirawat S., Northcutt V. J., Miller L. L., Spatrick P., He F., Kawana M., Feng H., Jacobson A., Peltz S. W., Sweeney H. L. (2007) PTC124 targets genetic disorders caused by nonsense mutations. Nature 447, 87–U86 [DOI] [PubMed] [Google Scholar]
- 31. Pena S. D., Karpati G., Carpenter S., Fraser F. C. (1987) The clinical consequences of X-chromosome inactivation: Duchenne muscular dystrophy in one of monozygotic twins. J. Neurol. Sci 79, 337–344 [DOI] [PubMed] [Google Scholar]
- 32. Briguet A., Courdier-Fruh I., Foster M., Meier T., Magyar J. P. (2004) Histological parameters for the quantitative assessment of muscular dystrophy in the mdx-mouse. Neuromuscul. Disord. 14, 675–682 [DOI] [PubMed] [Google Scholar]
- 33. Webster C., Silberstein L., Hays A. P., Blau H. M. (1988) Fast muscle fibers are preferentially affected in Duchenne muscular dystrophy. Cell 52, 503–513 [DOI] [PubMed] [Google Scholar]
- 34. Moens P., Baatsen P. H., Marechal G. (1993) Increased susceptibility of EDL muscles from mdx mice to damage induced by contractions with stretch. J. Muscle Res. Cell Motil. 14, 446–451 [DOI] [PubMed] [Google Scholar]
- 35. Brereton D., Plochocki J., An D., Costas J., Simons E. (2012) The effects of glucocorticoid and voluntary exercise treatment on the development of thoracolumbar kyphosis in dystrophin-deficient mice. PLoS Currents 4, e4ffdff160de168b [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Selsby J. T., Acosta P., Sleeper M. M., Barton E. R., Sweeney H. L. (2013) Long-term wheel running compromises diaphragmatic function but improves cardiac and plantarflexor function in the mdx mouse. J. Appl. Physiol. 115, 660–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.