

Abstract
This study was performed to discover and characterize the first potent α3β2-subtype-selective nicotinic acetylcholine receptor (nAChR) ligand. A novel α4/7-conotoxin, α-CTxLvIA, was cloned from Conus lividus. Its pharmacological profile at Xenopus laevis oocyte-expressed rat nAChR subtypes was determined by 2-electrode voltage-clamp electrophysiology, and its 3-dimensional (3D) structure was determined by NMR spectroscopy. α-CTx LvIA is a 16-aa C-terminally-amidated peptide with 2-disulfide bridges. Using rat subunits expressed in Xenopus oocytes, we found the highest affinity of α-CTxLvIA was for α3β2 nAChRs (IC50 8.7 nM), where blockade was reversible within 2 min. IC50 values were >100 nM at α6/α3β2β3, α6/α3β4, and α3β4 nAChRs, and ≥3 μM at all other subtypes tested. α3β2 vs. α6β2 subtype selectivity was confirmed for human-subunit nAChRs with much greater preference (300-fold) for α3β2 over α6β2 nAChRs. This is the first α-CTx reported to show high selectivity for human α3β2 vs. α6β2 nAChRs. α-CTxLvIA adopts two similarly populated conformations water: one (assumed to be bioactive) is highly structured, whereas the other is mostly random coil in nature. Selectivity differences with the similarly potent, but less selective, α3β2 nAChR antagonist α-CTx PeIA probably reside within the three residues, which differ in loop 2, given their otherwise similar 3D structures. α4/7-CTx LvIA is a new, potent, selective α3β2 nAChR antagonist, which will enable detailed studies of α3β2 nAChR structure, function, and physiological roles.—Luo, S., Zhangsun, D., Schroeder, C. I., Zhu, X., Hu, Y., Wu, Y., Weltzin, M. M., Eberhard, S., Kaas, Q., Craik, D. J., McIntosh, J. M., Whiteaker, P. A novel α4/7-conotoxin LvIA from Conus lividus that selectively blocks α3β2 vs. α6/α3β2β3 nicotinic acetylcholine receptors.
Keywords: ligand-gated ion channel receptor, cholinergic, nuclear magnetic resonance spectroscopy, molecular modeling
Venoms of marine snails of the genus Conus are natural combinatorial peptide libraries. Different classes of conopeptides [conotoxins (CTxs)] have a high selectivity toward various ion channels and other targets (1). Among them, α-CTxs are short peptides of 12–20 aa residues and are effective antagonists of nicotinic acetylcholine receptors (nAChRs; ref. 2). The α-CTxs have 4 cysteine residues, resulting in 2 disulfide bonds that are arranged in a CC–Xm–C–Yn–C pattern with 2 loops connecting CI–CIII and CII–CIV. The first loop (Xm) contains 3 or 4 aa (m=3–4), and the second loop (Yn) consists of 3–7 aa (n=3–7). α-CTxs are divided into several structural subfamilies (3/5, 4/3, 4/4, 4/5, 4/6, and 4/7), according to the number of residues encompassed within the two loops (m/n). The pharmacological selectivity of different α-CTxs is roughly correlated with their loop sizes; α-CTxs with a 3/5 framework are generally active at fish and/or mammalian neuromuscular nAChRs, whereas the CTxs from the 4/3, 4/4, 4/5, 4/6, or 4/7 classes mainly block mammalian neuronal nAChRs (3).
nAChRs are pentameric membrane-bound proteins that are classified into 2 main groups; muscle and nonmuscle (neuronal) subtypes. Muscle-subtype nAChRs (α1β1δε or α1β1δγ) consist of 5 subunits: 2 α1, 1 β1, 1 ε (or γ), and 1 δ subunit. Neuronal nAChRs consist of combinations of α (α2–α10) and β (β2–β4) subunits and exist as a complex family of heteromeric and homooligomeric pentameric proteins (4). These subtypes are important in normal physiology and in a wide range of disease states, including pain, addiction, Alzheimer's disease, myasthenia gravis, schizophrenia, epilepsy, and breast and lung carcinoma (5, 6).
α-CTxs act at the nAChR acetylcholine (ACh) binding site as competitive antagonists and, in some cases, exhibit extraordinary subtype selectivity (7, 8). Accordingly, they have proved to be valuable tools for identifying and probing the composition and roles of nAChR subtypes (3, 9). In particular, α4/7-CTxs are very interesting because of their ability to discriminate between diverse neuronal α-β nAChR subunit combinations (see Fig. 1 for examples). In this study, we characterized a novel α4/7-CTx from Conus lividus. α-CTx LvIA is the first α-CTx isolated from C. lividus and is the first α-CTx that blocks human α3β2 with high selectivity over α6β2* nAChRs (where the asterisk denotes the possible presence of additional subunits; ref. 10).
Figure 1.
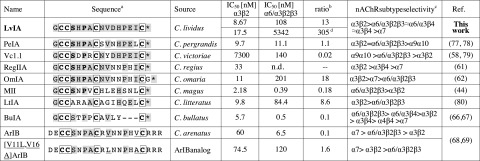
Sequence comparison of α-CTx LvIA with a variety of α-CTxs that have similar affinity for α3β2 and α6/α3β2β3nAChRs. aAmino acid conservations are denoted by light gray shade. Shaded amino acids are homologous within that locus of the set. The framework I scaffold formed by disulfide-bonded cysteines are in boldface and boxed. Disulfide connectivity of α-CTxs is between Cys1–Cys3 and Cys2–Cys4. Asterisks denote C-terminal carboxamides. The highly conserved Ser and Pro in loop 1 are in boldface. bRatio of α6/α3β2β3:α3β2 IC50 values. cNote that all nAChR subtypes were not tested in each instance. Only the subtypes for which the peptide has a value of IC50 < 10 μM are listed ranked by potency. Refer to cited literature for full details. n.d., not determined. dRatio is for human subunits.
MATERIALS AND METHODS
Materials
Specimens of C. lividus were collected from the South China Sea off Hainan Province. Unless otherwise stated, chemicals were analytical grade and were obtained from Sigma (St. Louis, MO, USA). Reverse-phase HPLC analytical Vydac C18 columns (5 μm, 4.6×250 mm, 300-Å pore size; cat. no. 218TP54) and preparative C18 Vydac columns (10 μm, 22×250 mm) were obtained from Grace Vydac (Hesperia, CA, USA). Absorbance monitor for analytical HPLC was a Waters 2996 photodiode array detector (Waters Corp., Milford, MA, USA). Reagents for peptide synthesis were obtained from GL Biochem (Shanghai, China). Acetonitrile was obtained from Fisher (Thermo Fisher Scientific, Waltham, MA, USA), and trifluoroacetic acid was from Tedia (Fairfield, OH, USA). Clones of rat α2–α7 and β2–β4, as well as mouse muscle α1β1δε cDNAs, were kindly provided by Stefan H. Heinemann (Salk Institute, San Diego, CA, USA). Clones of β2 and β3 subunits in the high-expressing pGEMHE vector were kindly provided by Charles W. Luetje (University of Miami, Miami, FL, USA). Human nAChR subunit clones were synthesized by GeneArt AG (Life Technologies, Grand Island, NY, USA).
Sequencing of α-CTx LvIA precursor gene
Venom gland bulbs and ducts were dissected from specimens of C. lividus, as described previously (11). Genomic DNA was isolated using a marine animal DNA isolation kit (Tiangen Biochemistry, Beijing, China). The procedure followed the kit manufacturer's suggested protocol for marine invertebrates, as described previously (12). The resulting genomic DNA was used as a template for PCR using oligonucleotide primers, corresponding to the 3′ end of the intron preceding the toxin region of α-CTx prepropeptides (primer 1) and the 3′-untranslated region (UTR) sequence of α-CTx prepropeptides (primer 2). The sequence of primer 1 was 5′-GTGGTTCTGGGTCCAGCA-3′. The sequence of primer 2 was 5′-GTCGTGGTTCAGAGGGTC-3′. PCR amplification was performed as described previously (13). Positive transformed colonies with conopeptide precursor DNA inserts were sequenced by Sangon (Shanghai, China). The mature conopeptide sequence was predicted using the online tool ProP 1.0 Server (14).
Peptide synthesis
The linear peptide was assembled by solid-phase methodology on an ABI 433A peptide synthesizer (Applied Biosystems Inc., Foster City, CA, USA) using FastMoc [N-(9-fluorenyl)methoxycarbonyl] chemistry and standard side-chain protection, except for cysteine residues. A 2-step oxidation protocol was used to fold the peptide selectively, as described previously (15). The folded peptide was purified by HPLC on a reversed-phase C18 Vydac column using a linear gradient of 0–40% buffer B (0.05% TFA and 90% acetonitrile in H2O) over 40 min. Buffer A was 0.075% TFA in H2O. Absorbance was monitored at 214 nm. Matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) mass spectrometry was utilized to confirm identity of the products.
Electrophysiology
Oocytes were harvested and injected with cRNA encoding nAChR subunits, as described previously (16). Capped cRNA for the various subunits was made using the mMessage mMachine in vitro transcription kit (Applied Biosystems/Ambion, Austin, TX, USA) following linearization of the plasmid. Purified rat mRNA (50 nl; or 81 nl of purified human mRNA) solution was injected into each Xenopus oocyte with a Drummond microdispenser (Drummond Scientific, Broomall, PA, USA) and incubated at 17°C. Voltage-clamp recordings were made 1–4 d postinjection. The membrane potential of the oocytes was clamped at −70 mV. All recordings were done at room temperature (∼22°C). Briefly, a 30-μl cylindrical oocyte recording chamber was gravity-perfused with ND96A (96.0 mM NaCl, 2.0 mM KCl, 1.8 mM CaCl2, 1.0 mM MgCl2, 1 μM atropine, 5 mM HEPES, 0.1 mg/ml BSA, pH 7.1–7.5) at a rate of ∼2 ml/min. In the case of the α9, α10, α7, and mouse muscle α1β1δε subtypes, the ND96 contained no atropine when recording. Oocytes were subjected 1×/min to a 1-s pulse of ACh (100 μM, with the following exceptions: α9α10 and muscle α1β1δε subtypes, 10 μM ACh; α7 subtype, 200 μM). For toxin concentrations of 10 μM and lower, once a stable baseline was achieved, either ND-96 alone or ND-96 containing CTx was manually preapplied for 5 min prior to the addition of the agonist.
Pharmacology data analysis
An average of 5 control responses just preceding a test response was used to normalize the test response to obtain a “100% control” response. Each data point of a dose-response curve represents the average ± sem of ≥3 oocytes. All statistical analyses were performed with Prism (Prism 4.0; GraphPad Software, La Jolla, CA, USA). IC50 values were determined by nonlinear regression analysis using Prism.
NMR spectroscopy
NMR measurements were made on peptide samples prepared in 90% H2O/10% D2O, 99.96% D2O, 30% trifluoroethanol (TFE)/70% H2O, or 30% acetonitrile (ACN)/70% H2O (Cambridge Isotope laboratories, Andover, MA, USA) at ∼1.5 mM and pH ∼4. Chemical shifts were referenced internally to sodium 2,2-dimethyl-2-silapentane-5-sulfonate (DSS). Spectra were recorded on a Bruker Avance-600 NMR spectrometer (Bruker Corp, Billerica, MA, USA) at 298 K or 280–315 K for variable-temperature experiments. Two-dimensional spectra were recorded in phase-sensitive mode using time-proportional phase incrementation for quadrature detection in the t1 dimension (17). Water suppression was achieved using excitation-sculpting gradients (18). Two-dimensional experiments included total correlation spectroscopy (TOCSY; ref. 19) using a MLEV-17 spin-lock sequence with an 80-ms mixing time; nuclear Overhauser effect spectroscopy (NOESY; ref. 20) with a 100, 200, or 300 ms mixing time; double-quantum-filtered correlated spectroscopy (DQF-COSY; ref. 21); exclusive correlation spectroscopy (ECOSY; ref. 22); and 13C heteronuclear single-quantum coherence (HSQC; ref. 23). Spectra were acquired with 4096 data points in the F2 dimension and 512 increments in the F1 dimension. The t1 dimension was zero-filled to 1024 real data points, and the F1 and F2 dimensions were multiplied by a sine-squared function prior to Fourier transformation. 3JHN–Hα coupling constants were measured from one-dimensional (1D) spectra or from antiphase cross-peak splitting in the DQF-COSY spectrum, and 3JHα–Hβ coupling constants were measured from the ECOSY spectrum and, together with intraresidue NOE intensities, were used for stereospecific assignments (24). Spectra were processed using TopSpin (Bruker) and assigned using Xeasy (25) and the sequential assignment protocol (26).
Amide protons involved in intramolecular hydrogen bonds were identified by their slow exchange after dissolution of the peptide in 99.96% D2O and the sensitivity of their chemical shift to temperature. Amide temperature coefficients at pH ∼4 were calculated from a series of TOCSY spectra acquired at 283–315 K, and amide protons with temperature coefficients more positive than −4.6 ppb/K were considered to be involved in hydrogen bonding interactions (27). Amide protons detected in the TOCSY spectrum >4 h after dissolution in D2O at 298 K were classified as slowly exchanging.
Structure calculations
Interproton distance restraints were derived from the intensity of cross peaks in NOESY spectra recorded with a mixing time of 300 ms at 298 K. A list of interproton distances was generated from the chemical shifts and NOE intensities using the CALIBA function in CYANA-3.0 (28). Pseudoatom corrections were applied to nonstereospecifically assigned protons. Structure calculations were then performed without automated assignment using the CALC function in CYANA-3.0. Restraints for the C-terminal amide, disulfide bonds, hydrogen bonds, and dihedral angles were added.
Constraints for the ϕ and ψ backbone dihedral angles were generated using TALOS+ (29) from the Hα, Cα, Cβ, and HN chemical shifts. The predicted angle ranges were checked for consistency with coupling constant information from the 1D spectra and NOE intensities in a 300-ms NOESY spectrum (24). Residues having a 3JHN–Hα ≤ 5 Hz were predicted to have a ϕ angle of −60 ± 30°, and residues with a 3JHN–Hα of ≥ 8 Hz were predicted to have a ϕ of −120 ± 30°. Side-chain χ1 angles and stereospecific assignments of methylene pairs that could be deduced from the 3JHα–Hβ coupling constants and Hα–HN sequential NOE peak intensities were included in the angle restraints. Disulfide bond restraints were included on the basis of sequence homology to previously reported α-CTxs, with disulfide connectivities between residues 2 and 8 and 3 and 16. Restraints for hydrogen bonding pairs were included for residues with amide protons that were slowly exchanging and had temperature coefficients more positive than −4.6 ppb/K (27).
The CALC function in CYANA (28) was used to perform 10,000 steps of torsion angle dynamics and generate an ensemble of 50 initial structures for analysis. Several rounds of structure calculations were performed to resolve restraint violations. Protocols from the RECOORD database (30) were then used to calculate an ensemble of 50 structures within CNS (31) using the force field distributed with Haddock 2.0 (32). The torsion angle simulated annealing protocol included a high-temperature phase of 5000 steps at 10000 K, followed by a cooling phase to 1000 K over 5000 steps, and a second cooling phase from 1000 K to 50 K over 5000 steps (all with 3-fs steps). The 50 structures generated were then further refined in a water shell (33). Each peptide was subjected to slow heating from 100 to 500 K in 100-K temperature steps, each comprising 200 steps of cartesian dynamics using a time step of 3 fs (during this phase, the harmonic position restraints for the heavy atoms are in place but slowly phased out); refinement at 500 K through 2000 cartesian dynamics steps (4-fs time steps); slow cooling from 500 to 25 K in 25-K steps, each comprising 2000 cartesian dynamics (4-fs time steps); and final minimization with 2000 steps of Powell minimization. A set of 27 structures with no violations >0.2 Å or >2° was selected for analysis using MolProbity (34). A final set of 20 structures was selected on the basis of MolProbity scores. The structures were visualized and figures were generated using MOLMOL (35).
Molecular modeling
The molecular model of the complex between α-CTx Vc1.1 and α9α10 nAChR was taken from Yu et al. (36). Briefly, this model was generated by homology using several templates: the NMR structure of Vc1.1 [Protein Data Bank (PDB) ID: 2H8S], the crystallographic structure of the isolated mouse α1 subunit (PDB ID: 2QC1), and the crystallographic structure (PDB ID: 2BR8) of a complex between α-CTx variant [A10L,D14K]PnIA and AChBP, which is a widely used structure surrogate of the nAChR ligand binding domain. This model was then refined using extensive molecular dynamics simulations.
The models of complexes involving LvIA and rat α9α10 nAChR, rat α3β2 nAChR, rat α3β4 nAChR, rat α6β2β3 nAChR, or rat α6β4 nAChR were generated by homology using Modeler 9v12 (37). For each complex, 100 models were initially computed, and the model displaying the lowest DOPE score was selected (38). More precisely, the model of Vc1.1/α9α10 nAChR complex (36) and the NMR solution structure of LvIA (present work) were used as templates for modeling the LvIA/α9α10 nAChR complex. The models of LvIA/α3β2 nAChR, LvIA/α3β4 nAChR, LvIA/α6β2β3 nAChR, and LvIA/α6β4 nAChR were created using several templates: the crystallographic structure of the [A10L,D14K]PnIA/AChBP complex (PDB ID: 2BR8), the X-ray structure of mouse α1 subunit (PDB ID: 2QC1) to refine the conformation of α subunits, the electron microscopy structure of Torpedo marmorata muscle type nAChR (PDB ID: 2BG9) to refine the conformation of β subunits, and the NMR solution structure of LvIA (present work). The pKa values of aspartate, glutamate, and histidine residues were predicted using PROPKA3.1 (39, 40).
RESULTS
Identification of a novel α4/7-CTx LvIA by DNA cloning
A novel gene was isolated from C. lividus genomic DNA which, based on the conserved intron and 3′-UTR features of the α-CTx gene structure, was predicted to yield an α4/7-CTx. PCR amplification yielded a specific α-CTx gene product, which was cloned and sequenced. It represented the precursor gene (lv1a) of α-CTx LvIA, and is shown in Fig. 2A. The α-CTx LvIA precursor DNA sequence is composed of 168 bp and encodes 43 aa. It contains a 20-residue proregion sequence and a 16-residue mature toxin region sequence with a structure that is typical of α-CTx propeptides (41). The predicted mature peptide exhibited the common cysteine pattern of α-CTxs (CC–C–C). The EMBL accession number of lv1a is HF566436. The precursor of LvIA predicts the mature peptide to be C-terminally amidated, since Gly is present at the locus preceding the C-terminal cleavage site (42). The deduced mature toxin sequence of LvIA is GCCSHPACNVDHPEIC# (where # denotes C-terminal carboxamide). LvIA is a novel α4/7-CTx and a new member of the α-CTx family. Related α4/7 CTxs from different Conus species are shown for comparison in Fig. 1. There is highest sequence homology in the cysteine residues. In contrast, there is high divergence in the second loop of the peptide.
Figure 2.

DNA and prepropeptide sequences of α4/7-CTx LvIA from C. lividus and encoded toxin (A) and its mature peptide sequence with disulfide bond connectivity Cys 1–3, Cys 2–4 (B). A) EMBL accession number of α-CTx LvIA DNA is HF566436. Putative proteolytic processing site 1 (R) and amidated processing site 2 (#G) are indicated by arrows. The intro primer is shaded. The proregion is denoted by italic lettering. The mature toxin region is underscored. The first glycine following the C-terminal cysteine in the mature toxin is presumed to be processed to a C-terminal amide for LvIA. Cysteines are indicated in boldface. Stop codons are indicated by asterisks. B) Deduced mature toxin sequence of LvIA is GCCSHPACNVDHPEIC# (where # indicates C-terminal carboxamide).
Chemical synthesis and oxidative folding of α-CTx LvIA
Previously characterized α-CTxs purified from venom typically have a disulfide bond connectivity linking the first to the third Cys and the second to the fourth Cys (43, 44). Therefore, we synthesized α-CTx LvIA with Cys1-3 and Cys2-4 disulfide bonds using directed 2-step folding. The linear peptide corresponding to the deduced mature α-CTx LvIA sequence was successfully synthesized with Fmoc chemistry (Fig. 2B). We protected the cysteine side chains with two orthogonal protecting groups that can be removed selectively under different conditions, allowing the formation of one disulfide bridge at a time. For this purpose, Cys1 and Cys3 were introduced as S-trityl-protected amino acids, whereas S-(acetamidomethyl) cysteine was used for Cys2 and Cys 4 (Fig. 2B). The acid-labile protecting groups on Cys1 and Cys3 were removed first during cleavage from the resin. The linear peptide was then purified by HPLC. Ferricyanide was used to close the first disulfide bridge. Reversed-phase HPLC was used to purify the monocyclic peptide. Subsequently, the acid-stable acetamidomethyl groups were removed by iodine oxidation, which also closed the second disulfide bridge (Fig. 2B). The fully folded peptide was purified by HPLC. Laser desorption mass spectrometry (MALDI-TOF) of synthetic α-CTx LvIA was consistent with the amidated sequence. The monoisotopic mass of LvIA (observed 1678.8 Da; theoretical 1678.9 Da) is 4 Da less than the linear peptide mass (1682.9 Da), confirming the formation of 2 disulfide bonds. The synthetic peptide with this disulfide bond arrangement was used in all subsequent studies.
The quantity of peptide was calculated by injecting ∼1 × 10−9 mol of peptide dissolved in 10–20 μl of 0.1% TFA onto an analytical Vydac reversed-phase C-18 column that had a 5-μM particle, 300-Å pore size and was 4.6 mm in diameter × 250 mm in length. Peptide was eluted from the column using a linear gradient of 10% buffer B, 90% buffer A, 50% buffer B, and 50% buffer A over 40 min, where buffer A contained 0.1% TFA, and buffer B contained 0.092% TFA and 60% acetonitrile. Absorbance was monitored at 220 nm using a Waters 2996 photodiode array detector. Peptide peak area was integrated using Waters Empower 2 software with 1 × 10−9 mol defined as 1.5 × 106 U.
Effect of α-CTx LvIA on ACh-evoked nAChR-mediated currents
Synthetic α-CTx LvIA was tested on various subtypes of nAChRs. Figure 3 shows representative rat α3β2, α6/3β2β3, and α2β2 and mouse α1β1δε nAChR responses to ACh, in the presence and absence of α-CTx LvIA. α6/α3 denotes a chimeric nAChR subunit in which the N-terminal ligand binding domain of α6, at which α-CTxs interact, is fused to the remainder of an α3 subunit to enhance functional expression (45). Greater than 90% blockade of α3β2 nAChRACh-evoked currents was obtained with 100 nM α-CTx LvIA (Fig. 3A). As also shown, α-CTx LvIA blockade of α3β2 nAChRs was relatively quickly reversible, requiring only 2 min of toxin washout. α-CTx LvIA showed dose-dependent blockade of α3β2 receptors at low nanomolar concentrations with an IC50 of 8.7 (6.9–11.0) nM, and a Hill slope of 1.17 (0.88–1.46) (Table 1). In contrast, the same concentration (100 nM) of α-CTx LvIA blocked α6/3β2β3 nAChR by <50%, and even 10 μM α-CTx LvIA produced little or no blockade of α2β2 or adult muscle nAChRs (Fig. 3B–D).
Figure 3.

α-CTx LvIA selectively blocks rat α3β2 nAChRs. nAChR subtypes were expressed as described in Materials and Methods; C indicates control responses to ACh. Oocytes were then exposed to 100 nM or 10 μM peptide for 5 min, followed by application of ACh (arrows) and washing to allow recovery of blockade (if produced by α-CTx LvIA). Typical traces are shown. The peptide almost completely blocked α3β2 (A) but not α2β2 (B), α6/3β2β3 (C), or mouse α1βδε (D) nAChR subtypes.
Table 1.
IC50 and Hill slope values for blockade of rodent and human nAChR subtypes by α-CTx LvIA
| Subtype | IC50 (nM) | Ratio | Hill slope |
|---|---|---|---|
| α3β2 | 8.67 (6.9–11.0) | 1 | 1.17 (0.88–1.46) |
| α6/α3β2β3 | 108 (54–215) | 13 | 1.16 (0.34–1.97) |
| α6/α3β4 | 121 (86–170) | 14 | 0.94 (0.66–1.22) |
| α3β4 | 148 (103–213) | 17 | 1.14 (0.72–1.55) |
| α7 | 3000 (1797–4997) | 346 | 0.65 (0.43–0.87) |
| α2β4 | 15,520 (11,600–20,770) | 1790 | 1.13 (0.78–1.48) |
| α9α10 | >10,000 | ||
| α2β2 | >10,000 | ||
| α4β2 | >10,000 | ||
| α4β4 | >10,000 | ||
| Mα1β1δε | >10,000 | ||
| Hα3β2 | 17.5 (16.6–21.6) | 1a | 0.81 (0.44–1.18) |
| Hα6/α3β2β3 | 5342 (1763–8921) | 305c | 0.85 (0.55–1.15) |
Values in parentheses are 95% confidence intervals. Ratios are nAChR subtype:α3β2 IC50 values unless noted. M, mouse subunit; H, human subunit.
nAChR subtype:H3β2 IC50 values.
Concentration response relationships for α-CTx LvIA were subsequently assessed across a range of rodent nAChR subtypes (Fig. 4A, B). α-CTx LvIA has highest affinity for α3β2 nAChRs, with the next highest affinities being found at α6/α3β2β3 [IC50 108 (54–215) nM] and α6/α3β4 nAChRs [IC50 121 (86–170) nM]; α-CTx LvIA IC50 values at the remaining subtypes are summarized in Table 1. The ability of this peptide to potently and preferentially target α3β2 vs. α6β2* nAChR was further confirmed for human nAChR subtypes (Fig. 4C). The human α3β2 IC50 was similar to that seen at the same rat nAChR subtype at 17.5 (16.6–21.6) nM, while the human α6/α3β2β2 IC50 was considerably higher than at the rat equivalent: 5342 (1763–8921) nM. This resulted in an even higher (∼305-fold) affinity ratio. All IC50 values and affinity ratios relative to activity at α3β2 nAChRs are summarized in Table 1.
Figure 4.

α-CTx LvIA concentration-response data at multiple nAChR subtypes. Oocytes expressing 1 of 11 different rodent nAChR subtypes were voltage clamped and subjected to ACh pulses as described in Materials and Methods (A, B). Selectivity for rat α3β2 over α6/α3β2β3 nAChRs (A) was also confirmed for human subtypes (C). Values are expressed as means ± sem from 3–9 separate oocytes.
NMR studies
Assignment of LvIA was carried out by identification of sequential NH-NHi+1, Hα-NHi+1, and Hβ-NHi+1 NOEs, combined with identification of individual spin systems in the TOCSY spectrum. Two distinct conformations with approximately equal populations were observed in the NMR spectra (Supplemental Fig. S1). The relative populations of the two conformers did not detectably change over the temperature range 280–315 K (data not shown), but the addition of 30% TFE to the solution significantly reduced the relative population of conformer 2, most notably at lower temperatures (Supplemental Fig. S2). A similar result was observed on addition of 30% ACN (Supplemental Fig. S3). For conformer 1, amide spin systems could be identified in the TOCSY spectra for all residues, with the exception of Pro6 and Pro13 (which lack amide protons) and for Asp11 in H2O at 298 K and Cys2 in 30% TFE at 305 K. In conformer 2, all amide spin systems were identified (Supplemental Fig. S1) only in spectra recorded in H2O. A sequential walk identifying neighboring residues via Hα-NHi+1 NOE connectivities in the NOESY spectra recorded in H2O at 298 K for conformers 1 and 2 identified all residues with the exception of Pro6 and Pro13 (Supplemental Fig. S1). In conformer 1, the amide spin system of Asp11 could not be identified, but the Hα-NHi+1NOE between Asp11 and His12 was observed in the amide region of the NOESY spectrum, and Hα-Hβ NOEs of Asp11 were identified in the aliphatic region (Supplemental Fig. S4). In conformer 1, both proline residues were determined to be in a trans conformation based on the observation of strong Hα-Nδi+1 NOEs (Supplemental Fig. S4). However, in conformer 2, only Pro13 could be unambiguously identified and was determined to be in a trans conformation based on strong Hα-Nδi+1 NOEs. Unambiguous assignment of Pro6 was not possible, but it was assumed to be in the cis form, as the differentiating feature between conformers 1 and 2. NMR chemical shift analysis indicated negative secondary chemical shifts for conformer 1 in H2O, TFE, and ACN for Cys2-Ser-4 and Pro6-Val10, suggesting helical characteristics across these two segments (Fig. 5). This suggestion was further supported with observed small 3Jα N coupling constants in these regions. The suggested helical stretch across residues 6–10 is consistent with previously reported α-CTx structures (46, 47) and a secondary shift comparison with Vc1.1 further supports this helicity (Fig. 5). In contrast, secondary chemical shift analysis of conformer 2 did not display the stretches of negative secondary shifts traditionally observed in α4/7-CTxs (Fig. 5 and ref. 48), and this conformer had poor dispersion of amide signals, consistent with a more random coil structure. Structure determination was, therefore, only carried out on conformer 1.
Figure 5.

Secondary shift analysis of α-CTx LvIA conformer 1 (trace with squares), conformer 2 (trace with circles; present study), and Vc1.1 (trace with diamonds; PDB ID: 2H8S) (46). Graph highlights the similarities between the Hα secondary chemical shifts when comparing LvIA to Vc1.1 and showcases the differences between the two conformations of LvIA, with the Hα secondary shift values for the second conformation being close to random coil.
Structure determination of LvIA
The solution structure of LvIA (conformer 1) in 90% H2O/10% D2O was determined by simulated annealing using experimental NOESY cross peaks and dihedral restraints derived from coupling constants. A total of 27 dihedral restraints and 117 distance restraints, including 50 sequential and 26 medium- and long-range NOEs, were used for structural calculations. NOESY spectra with various mixing times were run to confirm that medium- and long-range NOEs were not due to spin diffusion. Fifty structures were calculated, and the 20 with the lowest energy combined with the best MolProbity scores (34) were chosen as a representative structure for LvIA (Fig. 6A). NMR refinement and structural statistics are reported in Table 2. The RMSD was 0.50 ± 0.14 Å across the backbone atoms and 0.85 ± 0.19 Å across all heavy atoms. The percentage of residues in the Ramachandran-favored regions was 99.6% ± 1.6, with no Ramachandran outliers identified, and an overall MolProbity score of 1.50 ± 0.13. The α-CTxLvIA structure has been submitted to the PDB and Biological Magnetic Resonance Bank (BMRB; University of Wisconsin, Madison, WI, USA) databanks (PDB ID:2MQD, BMRB ID: 19501).
Figure 6.
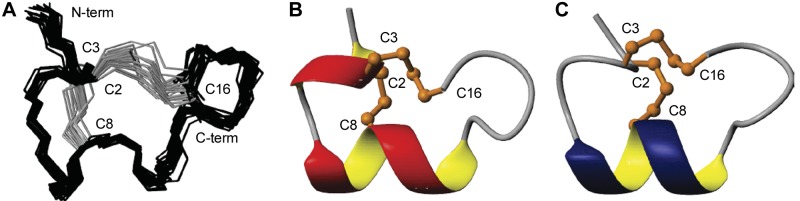
A) Overlay of the 20-lowest energy structures of LvIA (PDB ID: 2MDQ). B, C) Ribbon representations of LvIA (present study; PDB ID: 2MDQ; B) and Vc1.1 (PDB ID: 2H8S; C), highlighting the similarities between the structures, as well as pointing out the different orientation of the C termini. Disulfide bonds are shown in stick representation; and α-helical turn segments are shown as thickened ribbons.
Table 2.
NMR and refinement and structural statistics
| Parameter | Value |
|---|---|
| Distance and dihedral constraints (n) | |
| Total NOEs | 117 |
| Intraresidue | 41 |
| Interresidue | 76 |
| Sequential: |i − j| = 1 | 50 |
| Medium- and long-range: |i − j| ≥ 2 | 26 |
| Hydrogen bond restraintsa | 4 |
| Disulfide bond restraints | 4 |
| Total dihedral angle restraints | 27 |
| ϕ | 11 |
| ψ | 11 |
| χ1 | 5 |
| Mean NOE violations >0.2 | 0 |
| Mean dihedral violations >2.0 | 2 |
| Structural statistic | |
| RMSD from mean structure (Å) | |
| Backbone atoms | 0.50 ± 0.14 |
| Heavy atoms | 0.85 ± 0.19 |
| Stereochemical quality | |
| Ramachandran favored (%) | 99.6 ± 1.60 |
| Ramachandran outliers (%) | 0 |
| Clash score, all atomsb | 11.3 ± 3.87 |
| Overall Mol Probity score | 1.5 ± 0.13 |
| Percentile | 92.8 ± 3.35 |
Statistics are given as means ± sd. Stereochemical quality is according to MolProbity (http://molprobity.biochem.duke.edu).
Two restraints per hydrogen bond were used.
Defined as the number of steric overlaps of >0.4 Å/103 atoms.
DISCUSSION
In this study, we used a conserved intron sequence primer 1 (Fig. 2A) found within the genes that encode α-CTx precursors and a PCR-based technique to identify a novel peptide from the worm-hunting species C. lividus. Previously discovered CTxs and precursors from C. lividus are O- and M-gene superfamily members (49). Here, we describe a novel α4/7-CTx, LvIA. To our knowledge, this is the first member of the A-gene superfamily, and, in particular, of the α-CTx family, to be functionally characterized from C. lividus. We note that native LvIA has not yet been isolated from venom. Instead, the LvIA peptide was chemically synthesized using the Cys 1–3, Cys 2–4 disulfide connectivity configuration of previously characterized α4/7-CTxs (8). As detailed in Results section, LvIA is selective for α3β2 vs. α6β2* nAChRs. Multiple α-CTxs have been discovered that target both subtypes with high affinity. Several α6β2* subtype-preferring α-CTxs have been characterized (15, 50, 51). However, this is the first report of an α-CTx with a strong α3β2 nAChR preference (which is maintained between α3β2 vs. α6/α3β2β3 nAChRs expressed using either rat or human subunits).
Interestingly, synthetic LvIA shows two distinct conformations by NMR in aqueous solution, despite showing a single peak on RP-HPLC and only a single mass observed by mass spectrometry. The conformations exist in a near 1:1 ratio, and can both be readily identified using the sequential walk in the fingerprint region of the NOESY spectrum (Supplemental Fig. S1). The presence of additional conformations of CTxs appears to be quite common, but tends to go unreported if the major conformation is readily assignable and similar to a previously published structure and the minor form is less well defined. However, there are now several reports of CTxs showing 2 (or more) conformations in solution (52–54), where it has been possible to modulate the relative populations of the conformers by the addition of cosolvents to help define them. For example, Nielsen et al. (54) noted that for CTx PIIIA, the addition of ACN (up to 50%) suppressed a minor conformation. The solution structure of BuIA showed 3 distinct conformations associated with cis-trans isomerization of Pro residues (52). The addition of ACN and recording NMR spectra at various temperatures and pHs assisted in assigning the 3 conformations (52). For LvIA, we similarly observed suppression of a second conformation on addition of ACN or TFE. TFE is known to stabilize α helices (55) and because a large proportion of the globular fold of α4/7-CTxs is helical, the relative stabilization of conformer 1 on organic cosolvent addition is consistent with it being the native fold. Interestingly, additional conformations, including cis/trans isomers, were not reported for α-CTxs Vc1.1 or PeIA, the two peptides most similar in sequence to LvIA (56, 57).
The Hα NMR secondary shifts of conformer 1 are similar to previously assigned α4/7-CTxs, including Vc1.1. Conformer 2 is less structured, with Hα chemical shifts close to random coil values, and low spectral dispersion in the amide region compared to conformer 1. It was not possible to definitively determine whether the second conformation of LvIA reflects cis-trans isomerization of a proline residue, but this seems highly likely. Proline-13 was identified in a trans conformation, leaving open the possibility that proline 6 could be cis (both Pro residues are trans in conformer 1).
Full structural characterization was only carried out on the structured conformer (conformer 1). Like previously reported α4/7-CTxs, conformer 1 of LvIA has a compact structure (Fig. 6B, C) with an α-helical motif across the middle of the molecule (Pro6-Val10), although the helical region is slightly shorter than in α-CTxVc1.1 (46). α-CTxLvIA also includes an α-helical turn across residues Cys2–Ser4. As in the case of other α-CTxs, the compact structure is enhanced by the globular disulfide bond connectivity. The Cys2–Cys8 disulfide bond pulls the α helix toward the N terminus, as confirmed by a series of NOEs between Cys3 and Asn9, whereas the Cys3–Cys16 disulfide bond pulls the N and C termini into proximity. A series of medium-range NOEs between His12-Ile15 and Pro13-Cys16 shows that the C terminus is pulled toward the α helix, further contributing to the compactness of the α-CTxLvIA structure. The highly conserved nature of the overall α-CTxLvIA backbone structure reinforces the central role of side-chain residue interactions with the nAChR binding pocket in determining the subtype selectivity of α-CTxs.
On the basis of the molecular models shown in Figs. 7 and 8, we propose molecular explanations of the difference of specificity for the nAChR subtypes between LvIA and other α-CTxs with similar sequences. As noted previously, α-CTx Vc1.1 and PeIA share high sequence identity (Fig. 1), as well as a very similar structure with LvIA. Comparison between these toxins is interesting because Vc1.1 has a similar loop 2 sequence compared with LvIA but a different sequence in loop 1, and, conversely, loop 1 of PeIA is identical to that of LvIA, but their loop 2 sequences are more divergent.
Figure 7.
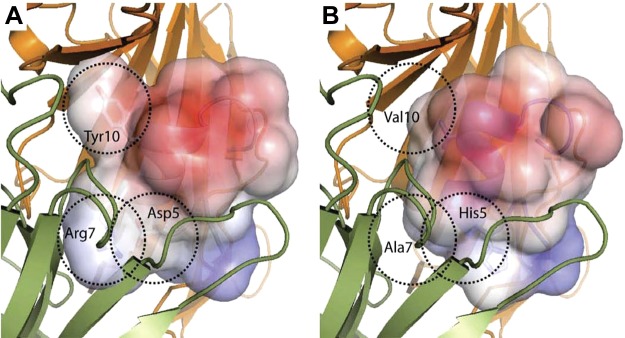
Electrostatic surface rendering and docking of Vc1.1 (PDB ID: 2H8S; A) and LvIA (present study; PDB ID: 2MDQ; B) with a molecular model of the α9/α10 nAChR rat subunit taken from Yu et al. (36). Principal subunit α10 containing the C loop is shown in green; complementary subunit α9 is in orange. Dashed circles highlight the differences in sequence between Vc1.1 and LvIA.
Figure 8.
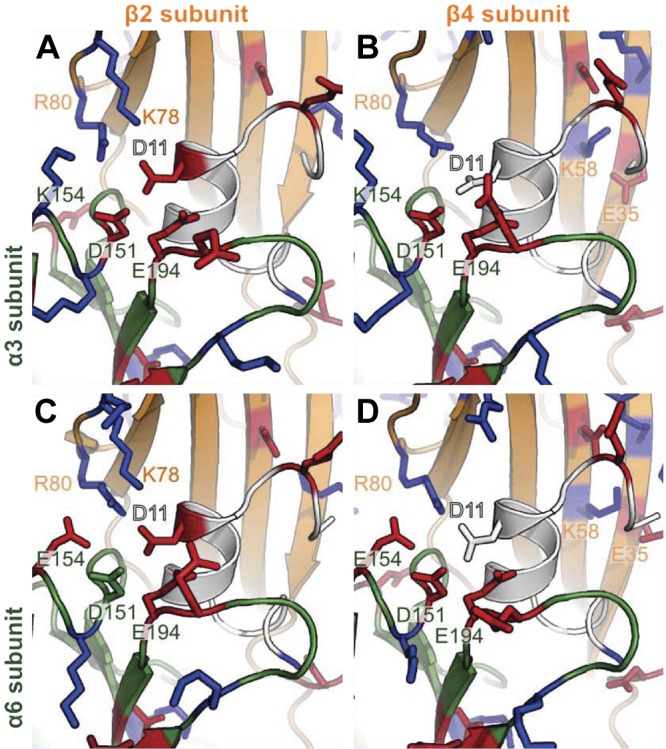
Homology models of the interactions involving α-CTx LvIA and α3β2 nAChR (A), α3β4 nAChR (B), α6β2β3 nAChR (C), and α6β4 nAChR (D). LvIA is in white, α3 and α6 subunits are in dark green, and β2 and β4 subunits are in orange. Positively and negatively charged residues, as predicted by PROPKA3.1 at pH 7.0, are shown as blue and red sticks, respectively. Important residues that were not predicted to be charged in some complexes, including D11 and D151, are shown as sticks. Numbering of positions is according to the α3 and β2 ligand-binding domains.
CTx Vc1.1 is well documented for its high affinity for the rat α9α10 nAChR (58–60), and, here, we suggest that the lack of activity of LvIA at this subtype comes from a different shape complementarity, as highlighted in Fig. 7. A recent docking study shows that Arg7 and Tyr10 of Vc1.1 are deeply buried at the interface with α9α10 nAChR (36), and both of these positions are occupied by smaller hydrophobic residues, i.e., Ala7 and Val10, in LvIA (Fig. 7). Arg7 was shown experimentally to be essential for Vc1.1 activity at the α9α10 nAChR because its substitutions by Ala or Lys lead to a dramatic loss of activity (59). In the molecular model of Yu et al. (36), the guanidinium group of Arg7 is involved in a network of hydrogen bond interactions at the interface, which cannot be established by Ala7. Interestingly, PeIA has similar residues to LvIA at position 7 and 10 but has been shown to still have some activity at α9α10 nAChR. We propose that this discrepancy with LvIA activity could be explained by PeIA binding at a different binding site. The heteropentamer α9α10 nAChR displays two potential binding sites located at the α9α10 and at the α10α9 interfaces. The later binding site displays more charged residues than the former, and it was found to be the most probable binding site of Vc1.1, which has 4 charged side chains (36). By contrast, PeIA has only one charged side chain, Glu14, and could potentially bind to the mostly hydrophobic α9α10 pocket. LvIA retains an additional charge at Asp11 that could reduce affinity at the α9α10 pocket, whereas binding to the α10α9 pocket is unlikely due to poor shape complementarity.
Molecular models of the interactions of LvIA with other nAChR subtypes than α9α10 suggest that the specificity of LvIA for α3β2 nAChR, at least in part, may arise from electrostatic interactions between LvIA Asp11 and the receptor. The LvIA/α3β2 nAChR model illustrated in Fig. 8A shows that the negatively charged Asp11 is buried in a cluster of charged residues, including Asp151, Lys154, and Glu194 of the α3 subunit, and Lys78 and Arg80 of the β2 subunit. This cluster of residues forms a globally electropositive environment, which is favorable for an interaction with a negatively charged Asp11, as suggested by the predicted pKa value of 3.4 of this residue in the context of the complex. Figure 8B–D shows that the three other nAChR subtypes, i.e., α3β4, α6β2β3, and α6β4, display an equivalent cluster with more negative charges, possibly decreasing the affinity for LvIA. Indeed, position 154 of the α6 subunit is occupied by a negatively charged Glu residue, whereas the α3 subunit has a positively charged Lys residue at this position, and the β4 subunit has a neutral Ile residue at position 78, whereas the β2 subunit has a positively charged Lys residue. The predicted pKa values of Asp11 when in complex with α3β4, α6β2β3, and α6β4 are 9.4, 5.5 and 9.8, respectively, indicating a substantial perturbation of the electrostatic environment compared to the α3β2 interface, where the predicted pKa value of Asp11 is 3.4. A salt bridge between Lys58 and Glu35 of the β4 subunit becomes buried when the CTx binds, and the corresponding cost in desolvation energy might also account for the decreased binding of β4-containing subtypes compared to the α3β2 nAChR.
The absence of selectivity between α3β2 and α6/α3β2β3 nAChR subtypes of CTx PeIA supports our hypothesis that Asp11 plays a crucial role for the selectivity of LvIA. Indeed, the sequence of CTx PeIA only differs from that of LvIA at 2 positions, i.e., positions 9 and 11, with the Asp11 of LvIA being replaced by a neutral Asn11 in PeIA. The binding of the negatively charged Asp11 should distinguish the different electrostatic properties of nAChR subtypes, whereas the neutral Asn11 would not, in agreement with the measured affinities. Similarly to PeIA, CTxs RegIIA (61) and OmIA (62) have identical loop 1 sequences to LvIA and display a neutral Asn11 but still have moderate selectivity for α3β2 (Fig. 1). These two peptides have additional differences at the end of loop 2, i.e., Asn12 and His14 instead of His12 and Glu14 found in LvIA, and as a result, RegIIA and OmIA do not bear any charge on their side chains. The specificity of these peptides cannot be explained by charge-charge interactions, as is possible for LvIA. Instead, differences of affinity between receptor subtypes are likely to result from distinct van der Waals and hydrogen bonds interactions. These interactions are more subtle and harder to predict when using models established by remote homology, and the structural models have typically to be refined by extensive conformational search, for example using molecular dynamic simulations (63). Carrying out these simulations is time consuming and was not attempted here, but in their absence we could easily foresee that RegIIA and OmIA could adopt slightly different orientations than LvIA in the binding pocket (because the differences in loop 2 sequence probably cause a different packing with the β subunit). The delicate complementary shapes of the ligand and receptor can easily be upset by the presence of an additional amino acid or the lack of appropriate interactions between ligand and receptor (64). Because of this, it is well known that as little as one residue difference in sequence can shift the selectivity profile of α-CTxs (65–68).
The extraordinary nAChR subtype selectivity of α-CTxs has made them indispensable tools for investigating the distribution and roles of nAChR subtypes (3, 9). The unique α3β2 vs. α6β2* nAChR subtype selectivity of α-CTx LvIA is likely to prove very valuable in this regard. In dopaminergic regions, α6β2* nAChRs predominate. Much of our understanding of the composition, properties, and physiological roles of this receptor population has been dependent on the use of α-CTx MII (which is poorly selective between α3β2 vs. α6β2* nAChRs), or an assortment of more α6β2*-selective α-CTxs (15, 50, 69–72). However, α3β2* nAChR expression overlaps with that of α6β2* nAChRs in optic and habenulopeduncular tract nuclei and likely predominates in the latter (72–74). In addition, α3β2* nAChRs in the spine have been implicated in the transmission of nociceptive stimuli (75). The availability of a truly α3β2* nAChR-selective probe should permit, for the first time, a much more comprehensive understanding of the significance of this subtype in normal and disease physiology.
Supplementary Material
Acknowledgments
This work was supported, in part, by the National Natural Science Foundation of China, grants 81160503 and 41366002; State High-Tech Research and Development Project (863) of the Ministry of Science and Technology of China, grant 2012AA021706; Program for International Science and Technology Cooperation Program of China, grant 2011DFR31210; and Changjiang Scholars and Innovative Research Team in University, grants PCSIRT and IRT1123. This work was also supported by U.S. National Institutes of Health grants DA012242, GM103801, and GM48677, and Australian Research Council grant 1093115. D.J.C. is an Australian National Health and Medical Research Council Professorial Fellow (APP1026501). A preliminary account of some of this work was presented in the patent literature (76).
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
- 1D
- 1-dimensional
- Ach
- acetylcholine
- ACN
- acetonitrile
- CTx
- conotoxin
- DQF-COSY
- double-quantum-filtered correlated spectroscopy
- ECOSY
- exclusive correlation spectroscopy
- nAChR
- nicotinic acetylcholine receptor
- MALDI-TOF
- matrix-assisted laser desorption ionization time-of-flight
- NOE
- nuclear Overhauser effect
- NOESY
- nuclear Overhauser effect spectroscopy
- PDB
- Protein Data Bank
- TFE
- trifluoroethanol
- TOCSY
- total correlation spectroscopy
- UTR
- untranslated region
REFERENCES
- 1. Norton R. S., Olivera B. M. (2006) Conotoxins down under. Toxicon 48, 780–798 [DOI] [PubMed] [Google Scholar]
- 2. McIntosh J. M., Santos A. D., Olivera B. M. (1999) Conus peptides targeted to specific nicotinic acetylcholine receptor subtypes. Annu. Rev. Biochem. 68, 59–88 [DOI] [PubMed] [Google Scholar]
- 3. Kasheverov I. E., Utkin Y. N., Tsetlin V. I. (2009) Naturally occurring and synthetic peptides acting on nicotinic acetylcholine receptors. Curr. Pharm. Des. 15, 2430–2452 [DOI] [PubMed] [Google Scholar]
- 4. Gotti C., Clementi F., Fornari A., Gaimarri A., Guiducci S., Manfredi I., Moretti M., Pedrazzi P., Pucci L., Zoli M. (2009) Structural and functional diversity of native brain neuronal nicotinic receptors. Biochem. Pharm. 78, 703–711 [DOI] [PubMed] [Google Scholar]
- 5. Gotti C., Moretti M., Bohr I., Ziabreva I., Vailati S., Longhi R., Riganti L., Gaimarri A., McKeith I. G., Perry R. H., Aarsland D., Larsen J. P., Sher E., Beattie R., Clementi F., Court J. A. (2006) Selective nicotinic acetylcholine receptor subunit deficits identified in Alzheimer's disease, Parkinson's disease and dementia with Lewy bodies by immunoprecipitation. Neurobiol. Dis. 23, 481–489 [DOI] [PubMed] [Google Scholar]
- 6. Gotti C., Clementi F. (2004) Neuronal nicotinic receptors: from structure to pathology. Prog. Neurobiol. 74, 363–396 [DOI] [PubMed] [Google Scholar]
- 7. Terlau H., Olivera B. M. (2004) Conus venoms: A rich source of novel ion channel-targeted peptides. Physiol. Rev. 84, 41–68 [DOI] [PubMed] [Google Scholar]
- 8. Muttenthaler M., Akondi K. B., Alewood P. F. (2011) Structure-activity studies on alpha-conotoxins. Curr. Pharm. Des. 17, 4226–4241 [DOI] [PubMed] [Google Scholar]
- 9. Nicke A., Wonnacott S., Lewis R. J. (2004) alpha-Conotoxins as tools for the elucidation of structure and function of neuronal nicotinic acetylcholine receptor subtypes. Eur. J. Biochem. 271, 2305–2319 [DOI] [PubMed] [Google Scholar]
- 10. Lukas R. J., Changeux J. P., Le Novere N., Albuquerque E. X., Balfour D. J. K., Berg D. K., Bertrand D., Chiappinelli V. A., Clarke P. B. S., Collins A. C., Dani J. A., Grady S. R., Kellar K. J., Lindstrom J. M., Marks M. J., Quik M., Taylor P. W., Wonnacott S. (1999) International Union of Pharmacology. XX. Current status of the nomenclature for nicotinic acetylcholine receptors and their subunits. Pharm. Rev. 51, 397–401 [PubMed] [Google Scholar]
- 11. Luo S., Zhang B., Zhangsun D. (2004) Optimization of conus genomic DNA isolation methods. Chin. J. Mar. Drugs 23, 21–25 [Google Scholar]
- 12. Zheng X., Gao B., Li B., Peng C., Wu A., Zhu X., Chen X. N., Zhangsun D., Luo S. (2011) Primer screening for new α-conotoxin gene cloning. Bio/Technology 21, 40–44 [Google Scholar]
- 13. McIntosh J. M., Dowell C., Watkins M., Garrett J. E., Yoshikami D., Olivera B. M. (2002) alpha-Conotoxin GIC from Conus geographus, a novel peptide antagonist of nicotinic acetylcholine receptors. J. Biol. Chem. 277, 33610–33615 [DOI] [PubMed] [Google Scholar]
- 14. Duckert P., Brunak S., Blom N. (2004) Prediction of proprotein convertase cleavage sites. Protein Eng. Des. Sel. 17, 107–112 [DOI] [PubMed] [Google Scholar]
- 15. Dowell C., Olivera B. M., Garrett J. E., Staheli S. T., Watkins M., Kuryatov A., Yoshikami D., Lindstrom J. M., McIntosh J. M. (2003) α-Conotoxin PIA is selective for α6 subunit-containing nicotinic acetylcholine receptors. J. Neurosci. 23, 8445–8452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cartier G. E., Yoshikami D. J., Gray W. R., Luo S. Q., Olivera B. M., McIntosh J. M. (1996) A new alpha-conotoxin which targets α3β2 nicotinic acetylcholine receptors. J. Biol. Chem. 271, 7522–7528 [DOI] [PubMed] [Google Scholar]
- 17. Marion D., K. W. (1983) Application of phase sensitive two-dimensional correlated spectroscopy (COSY) for measurement of 1H-1H spin-spin coupling constants in proteins. Biochem. Biophys. Res. Commun. 113, 967–974 [DOI] [PubMed] [Google Scholar]
- 18. Hwang T. L., Shaka A. J. (1995) Water suppression that works: Excitation sculpting using arbitraty wave-forms and pulse-field gradients. J. Magn. Reson. 112, 275–279 [Google Scholar]
- 19. Braunschweiler L., Ernst R. R. (1983) Coherence transfer by isotropic mixing: application to proton correlation spectroscopy. J. Magn. Reson. 53, 521–528 [Google Scholar]
- 20. Jeener J., Meier B. H., Bachman P., Ernst R. R. (1979) Investigation of exchange processs by two-dimensional NMR spectrscopy. J. Chem. Phys. 71, 4546–4553 [Google Scholar]
- 21. Rance M., Sørensen O. W., Bodenhausen G., Wagner G., Ernst R. R., Wüthrich K. (1983) Improved spectral resolution in cosy 1H NMR spectra of proteins via double quantum filtering. Biochem. Biophys. Res. Commun. 117, 479–485 [DOI] [PubMed] [Google Scholar]
- 22. Griesinger C., Sørensen O. W., Ernst R. R. (1987) Practical aspects of the E.COSY technique, measurement of scalar spin-spin coupling constants in peptides. J. Magn. Reson. 75, 474–492 [Google Scholar]
- 23. Palmer A. G., Cavanagh J., Wright P. E., Rance M. (1991) Sensitivity improvement in proton-detected 2-dimensional heteronuclear correlation NMR-spectroscopy. J. Magn. Reson. 93, 151–170 [Google Scholar]
- 24. Wagner G. (1990) NMR investigations of protein structure. Prog. NMR Spectros. 22, 101–139 [Google Scholar]
- 25. Bartels C., Xia T.-H., Billeter M., Güntert P., Wüthrich K. (1995) The program XEASY for computer-supported NMR spectral analysis of biological macromolecules. J. Biomol. NMR 6, 1–10 [DOI] [PubMed] [Google Scholar]
- 26. Wüthrich K. (1986) NMR of Proteins and Nucleic Acids, Wiley-Interscience, New York [Google Scholar]
- 27. Cierpicki T., Otlewski J. (2001) Amide proton temperature coefficients as hydrogen bond indicators in proteins. J. Biomol. NMR 21, 249–261 [DOI] [PubMed] [Google Scholar]
- 28. Güntert P., Mumenthaler C., Wüthrich K. (1997) Torsion angle dynamics for NMR structure calculation with the new program DYANA. J. Mol. Biol. 273, 283–298 [DOI] [PubMed] [Google Scholar]
- 29. Shen Y., Delaglio F., Cornilescu G., Bax A. (2009) TALOS plus: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J. Biomol. NMR 44, 213–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nederveen A. J., Doreleijers J. F., Vranken W., Miller Z., Spronk C., Nabuurs S. B., Guntert P., Livny M., Markley J. L., Nilges M., Ulrich E. L., Kaptein R., Bonvin A. (2005) RECOORD: A recalculated coordinate database of 500+ proteins from the PDB using restraints from the BioMagResBank. Proteins Struct. Funct. Bioinform. 59, 662–672 [DOI] [PubMed] [Google Scholar]
- 31. Brunger A. T., Adams P. D., Clore G. M., DeLano W. L., Gros P., Grosse-Kunstleve R. W., Jiang J. S., Kuszewski J., Nilges M., Pannu N. S., Read R. J., Rice L. M., Simonson T., Warren G. L. (1998) Crystallography and NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54, 905–921 [DOI] [PubMed] [Google Scholar]
- 32. Dominguez C., Boelens R., Bonvin A. (2003) HADDOCK: A protein-protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 125, 1731–1737 [DOI] [PubMed] [Google Scholar]
- 33. Linge J. P., Nilges M. (1999) Influence of non-bonded parameters on the quality of NMR structures: a new force field for NMR structure calculation. J. Biomol. NMR 13, 51–59 [DOI] [PubMed] [Google Scholar]
- 34. Chen V. B., Arendall W. B., Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Koradi R., Billeter M., Wüthrich K. (1996) MOLMOL: a program for display and analysis of macromolecular structures. J. Mol. Graph. 14, 29–32 [DOI] [PubMed] [Google Scholar]
- 36. Yu R., Kompella S. N., Adams D. J., Craik D. J., Kaas Q. (2013) Determination of the α-conotoxin Vc1.1 binding site on the α9α10 nicotinic acetylcholine receptor. J. Med. Chem. 56, 3557–3567 [DOI] [PubMed] [Google Scholar]
- 37. Sali A., Blundell T. L. (1993) Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815 [DOI] [PubMed] [Google Scholar]
- 38. Shen M.-Y., Sali A. (2006) Statistical potential for assessment and prediction of protein structures. Prot. Sci. 15, 2507–2524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Olsson M. H. M., Søndergaard C. R., Rostkowski M., Jensen J. H. (2011) PROPKA3: Consistent treatment of internal and surface residues in empirical pKa predictions. J. Chem. Theor. Comp. 7, 525–537 [DOI] [PubMed] [Google Scholar]
- 40. Søndergaard C. R., Olsson M. H. M., Rostkowski M., Jensen J. H. (2011) Improved treatment of ligands and coupling effects in empirical calculation and rationalization of pKa values. J. Chem. Theor. Comp. 7, 2284–2295 [DOI] [PubMed] [Google Scholar]
- 41. Santos A. D., McIntosh J. M., Hillyard D. R., Cruz L. J., Olivera B. M. (2004) The A-superfamily of conotoxins—structural and functional divergence. J. Biol. Chem. 279, 17596–17606 [DOI] [PubMed] [Google Scholar]
- 42. Corpuz G. P., Jacobsen R. B., Jimenez E. C., Watkins M., Walker C., Colledge C., Garrett J. E., McDougal O., Li W. Q., Gray W. R., Hillyard D. R., Rivier J., McIntosh J. M., Cruz L. J., Olivera B. M. (2005) Definition of the M-conotoxin superfamily: characterization of novel peptides from molluscivorous Conus venoms. Biochemistry 44, 8176–8186 [DOI] [PubMed] [Google Scholar]
- 43. Armishaw C. J., Dutton J. L., Craik D. J., Alewood P. F. (2010) Establishing regiocontrol of disulfide bond isomers of alpha-conotoxin ImI via the synthesis of N-to-C cyclic analogs. Biopolymers 94, 307–313 [DOI] [PubMed] [Google Scholar]
- 44. Gehrmann J., Alewood P. F., Craik D. J. (1998) Structure determination of the three disulfide bond isomers of alpha-conotoxin GI: amodel for the role of disulfide bonds in structural stability. J. Mol. Biol. 278, 401–415 [DOI] [PubMed] [Google Scholar]
- 45. Kuryatov A., Olale F., Cooper J., Choi C., Lindstrom J. (2000) Human alpha 6 AChR subtypes: subunit composition, assembly, and pharmacological responses. Neuropharmacology 39, 2570–2590 [DOI] [PubMed] [Google Scholar]
- 46. Clark R. J., Fischer H., Nevin S. T., Adams D. J., Craik D. J. (2006) The synthesis, structural characterization, and receptor specificity of the α-conotoxin Vc1.1. J. Biol. Chem. 281, 23254–23263 [DOI] [PubMed] [Google Scholar]
- 47. Dutton J. L., Craik D. J. (2001) α-Conotoxins: nicotinic acetylcholine receptor antagonists as pharmacological tools and potential drug leads. Curr. Med. Chem. 8, 327–344 [DOI] [PubMed] [Google Scholar]
- 48. Kaas Q., Westermann J.-C., Halai R., Wang C. K. L., Craik D. J. (2008) ConoServer, a database for conopeptide sequences and structures. Bioinformatics 24, 445–446 [DOI] [PubMed] [Google Scholar]
- 49. Kaas Q., Yu R. L., Jin A. H., Dutertre S., Craik D. J. (2012) ConoServer: updated content, knowledge, and discovery tools in the conopeptide database. Nucleic Acids Res. 40, D325–D330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. McIntosh J. M., Azam L., Staheli S., Dowell C., Lindstrom J. M., Kuryatov A., Garrett J. E., Marks M. J., Whiteaker P. (2004) Analogs of α-conotoxin MII are selective for α6-containing nicotinic acetylcholine receptors. Mol. Pharmacol. 65, 944–952 [DOI] [PubMed] [Google Scholar]
- 51. Luo S., Zhangsun D., Wu Y., Zhu X., Hu Y., McIntyre M., Christensen S., Akcan M., Craik D. J., McIntosh J. M. (2013) Characterization of a novel α-conotoxin from Conus textile that selectively targets α6/α3β2β3 nicotinic acetylcholine receptors. J. Biol. Chem. 288, 894–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Jin A. H., Brandstaetter H., Nevin S. T., Tan C. C., Clark R. J., Adams D. J., Alewood P. F., Craik D. J., Daly N. L. (2007) Structure of α-conotoxin BuIA: influences of disulfide connectivity on structural dynamics. BMC Struct. Biol. 7, 28–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Peng C., Chen W. H., Han Y. H., Sanders T., Chew G., Liu J., Hawrot E., Chi C. W., Wang C. G. (2009) Characterization of a novel alpha 4/4-conotoxin, Qc1.2, from vermivorous Conus quercinus. Acta Bioch. Biophys. Sinica 41, 858–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Nielsen K. J., Watson M., Adams D. J., Hammarstrom A. K., Gage P. W., Hill J. M., Craik D. J., Thomas L., Adams D., Alewood P. F., Lewis R. J. (2002) Solution structure of mu-conotoxin PIIIA, a preferential inhibitor of persistent tetrodotoxin-sensitive sodium channels. J. Biol. Chem. 277, 27247–27255 [DOI] [PubMed] [Google Scholar]
- 55. Shiraki K., Nishikawa K., Goto Y. (1995) Trifluoroethanol-induced stabilization of the alpha-helical structure of β-lactoglobulin—implication for non-heirarchical protein-folding. J. Mol. Biol. 245, 180–194 [DOI] [PubMed] [Google Scholar]
- 56. Clark R. J., Jensen J., Nevin S. T., Callaghan B. P., Adams D. J., Craik D. J. (2010) The engineering of an orally active conotoxin for the treatment of neuropathic pain. Angew. Chem. Int. Ed. Engl. 49, 6545–6548 [DOI] [PubMed] [Google Scholar]
- 57. Daly N. L., Callaghan B., Clark R. J., Nevin S. T., Adams D. J., Craik D. J. (2011) Structure and activity of α-conotoxin PeIA at nicotinic acetylcholine receptor subtypes and GABAB receptor-coupled N-type calcium channels. J. Biol. Chem. 286, 10233–10237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Vincler M., Wittenauer S., Parker R., Ellison M., Olivera B. M., McIntosh J. M. (2006) Molecular mechanism for analgesia involving specific antagonism of alpha 9 alpha 10 nicotinic acetylcholine receptors. Proc. Natl. Acad. Sci. U. S. A. 103, 17880–17884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Halai R., Clark R. J., Nevin S. T., Jensen J. E., Adams D. J., Craik D. J. (2009) Scanning mutagenesis of α-conotoxin Vc1.1 reveals residues crucial for activity at the α9α10 nicotinic acetylcholine receptor. J. Biol. Chem. 284, 20275–20284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Nevin S. T., Clark R. J., Klimis H., Christie M. J., Craik D. J., Adams D. J. (2007) Are α9α10 nicotinic acetylcholine receptors a pain target for α-conotoxins? Mol. Pharmacol. 72, 1406–1410 [DOI] [PubMed] [Google Scholar]
- 61. Franco A., Kompella S. N., Akondi K. B., Melaun C., Daly N. L., Luetje C. W., Alewood P. F., Craik D. J., Adams D. J., Mari F. (2012) RegIIA: An alpha 4/7-conotoxin from the venom of Conus regius that potently blocks α3β4 nAChRs. Biochem. Pharmacol. 83, 419–426 [DOI] [PubMed] [Google Scholar]
- 62. Talley T. T., Olivera B. M., Han K. H., Christensen S. B., Dowell C., Tsigelny I., Ho K. Y., Taylor P., McIntosh J. M. (2006) α-Conotoxin OmIA is a potent ligand for the acetylcholine-binding protein as well as alpha 3 beta 2, and alpha 7 nicotinic acetylcholine receptors. J. Biol. Chem. 281, 24678–24686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Yu R., Craik D. J., Kaas Q. (2011) Blockade of neuronal α7-nAChR by α-conotoxin ImI explained by computational scanning and energy calculations. PLoS Comput. Biol. l7, e1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Celie P. H. N., Kasheverov I. E., Mordvintsev D. Y., Hogg R. C., van Nierop P., van Elk R., van Rossum-Fikkert S. E., Zhmak M. N., Bertrand D., Tsetlin V., Sixma T. K., Smit A. B. (2005) Crystal structure of nicotinic acetylcholine receptor homolog AChBP in complex with an alpha-conotoxin PnIA variant. Nat. Struct. Mol. Biol. 12, 582–588 [DOI] [PubMed] [Google Scholar]
- 65. Hogg R. C., Miranda L. P., Craik D. J., Lewis R. J., Alewood P. F., Adams D. J. (1999) Single amino acid substitutions in alpha-conotoxin PnIA shift selectivity for subtypes of the mammalian neuronal nicotinic acetylcholine receptor. J. Biol. Chem. 274, 36559–36564 [DOI] [PubMed] [Google Scholar]
- 66. Hogg R. C., Hopping G., Alewood P. F., Adams D. J., Bertrand D. (2003) α-conotoxins PnIA and A10L PnIA stabilize different states of the α7-L247T nicotinic acetylcholine receptor. J. Biol. Chem. 278, 26908–26914 [DOI] [PubMed] [Google Scholar]
- 67. Jacobsen R. B., De la Cruz R. G., Grose J. H., McIntosh J. M., Yoshikami D., Olivera B. M. (1999) Critical residues influence the affinity and selectivity of α-conotoxin MI for nicotinic acetylcholine receptors. Biochemistry 38, 13310–13315 [DOI] [PubMed] [Google Scholar]
- 68. Teichert R. W., Lopez-Vera E., Gulyas J., Watkins M., Rivier J., Olivera B. M. (2006) Definition and characterization of the short alpha A-conotoxins: a single residue determines dissociation kinetics from the fetal muscle nicotinic acetylcholine receptor. Biochemistry 45, 1304–1312 [DOI] [PubMed] [Google Scholar]
- 69. Quik M., Perez X. A., Grady S. R. (2011) Role of alpha 6 nicotinic receptors in CNS dopaminergic function: relevance to addiction and neurological disorders. Biochem. Pharm. 82, 873–882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Letchworth S. R., Whiteaker P. (2011) Progress and challenges in the study of alpha 6-containing nicotinic acetylcholine receptors. Biochem. Pharm. 82, 862–872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Champtiaux N., Gotti C., Cordero-Erausquin M., David D. J., Przybylski C., Lena C., Clementi F., Moretti M., Rossi F. M., Le Novere N., McIntosh J. M., Gardier A. M., Changeux J. P. (2003) Subunit composition of functional nicotinic receptors in dopaminergic neurons investigated with knock-out mice. J. Neurosci. 23, 7820–7829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Whiteaker P., McIntosh J. M., Luo S. Q., Collins A. C., Marks M. J. (2000) I-125-alpha-conotoxin MII identifies a novel nicotinic acetylcholine receptor population in mouse brain. Mol. Pharm. 57, 913–925 [PubMed] [Google Scholar]
- 73. Whiteaker P., Peterson C. G., Xu W., McIntosh J. M., Paylor R., Beaudet A. L., Collins A. C., Marks M. J. (2002) Involvement of the alpha 3 subunit in central nicotinic binding populations. J. Neurosci. 22, 2522–2529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. McClure-Begley T. D., Wageman C. R., Grady S. R., Marks M. J., McIntosh J. M., Collins A. C., Whiteaker P. (2012) A novel alpha-conotoxin MII-sensitive nicotinic acetylcholine receptor modulates H-3-GABA release in the superficial layers of the mouse superior colliculus. J. Neurochem. 122, 48–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Young T., Wittenauer S., McIntosh J. M., Vincler M. (2008) Spinal α3β2* nicotinic acetylcholine receptors tonically inhibit the transmission of nociceptive mechanical stimuli. Brain Res. 1229, 118–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Luo S., Zhangsun D., Hu Y., Zhu X., Wu Y., McIntosh J. M. (2012) α-Conotoxin LvIA/LVD21, its drug combination and application. Chinese Pat. Lit. CN(201210347966.3)-A [Google Scholar]
- 77. McIntosh J. M., Plazas P. V., Watkins M., Gomez-Casati M. E., Olivera B. M., Elgoyhen A. B. (2005) A novel α-conotoxin, PeIA, cloned from Conus pergrandis, discriminates between rat alpha 9 alpha 10 and alpha 7 nicotinic cholinergic receptors. J. Biol. Chem. 280, 30107–30112 [DOI] [PubMed] [Google Scholar]
- 78. Hone A. J., Scadden M., Gajewiak J., Christensen S., Lindstrom J., McIntosh J. M. (2012) α-Conotoxin PeIA [S9H,V10A,E14N] potently and selectively blocks alpha 6 beta 2 beta 3 versus alpha 6 beta 4 nicotinic acetylcholine receptors. Mol. Pharm. 82, 972–982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Clarke P. B. S., Schwartz R. D., Paul S. M., Pert C. B., Pert A. (1985) Nicotinic binding in rat-brain - autoradiographic comparison of [H3] acetylcholine, [H3] nicotine, and [I125] alpha-bungarotoxin. J. Neurosci. 5, 1307–1315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Luo S. L., Akondi K. B., Zhangsun D. T., Wu Y., Zhu X. P., Hu Y. Y., Christensen S., Dowell C., Daly N. L., Craik D. J., Wang C. I. A., Lewis R. J., Alewood P. F., McIntosh J. M. (2010) Atypical alpha-conotoxin LtIA from Conus litteratus targets a novel microsite of the alpha3beta 2 nicotinic receptor. J. Biol. Chem. 285, 12355–12366 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.