

Abstract
The peroxisome proliferator-activated receptor γ coactivator 1-α (PGC-1α) interacts with various transcription factors involved in energy metabolism and in the regulation of mitochondrial biogenesis. PGC-1α mRNA levels are reduced in a number of neurodegenerative diseases and contribute to disease pathogenesis, since increased levels ameliorate behavioral defects and neuropathology of Huntington's disease, Parkinson's disease, and amyotrophic lateral sclerosis. PGC-1α and its downstream targets are reduced both in postmortem brain tissue of patients with Alzheimer's disease (AD) and in transgenic mouse models of AD. Therefore, we investigated whether increased expression of PGC-1α would exert beneficial effects in the Tg19959 transgenic mouse model of AD; Tg19959 mice express the human amyloid precursor gene (APP) with 2 familial AD mutations and develop increased β-amyloid levels, plaque deposition, and memory deficits by 2–3 mo of age. Rather than an improvement, the cross of the Tg19959 mice with mice overexpressing human PGC-1α exacerbated amyloid and tau accumulation. This was accompanied by an impairment of proteasome activity. PGC-1α overexpression induced mitochondrial abnormalities, neuronal cell death, and an exacerbation of behavioral hyperactivity in the Tg19959 mice. These findings show that PGC-1α overexpression exacerbates the neuropathological and behavioral deficits that occur in transgenic mice with mutations in APP that are associated with human AD.—Dumont, M., Stack, C., Elipenahli, C., Jainuddin, S., Launay, N., Gerges, M., Starkova, N., Starkov, A. A., Calingasan, N. Y., Tampellini, D., Pujol, A., Beal, M. F. PGC-1α overexpression exacerbates β-amyloid and tau deposition in a transgenic mouse model of Alzheimer's disease.
Keywords: Tg19959 mice, behavior, mitochondria, cell death
Alzheimer's disease (AD) is a neurodegenerative disorder characterized by the presence of amyloid deposition and neurofibrillary tangles. The disease is associated with synaptic deficits and neuronal degeneration leading to memory impairment (1). Early in disease pathogenesis, there is evidence of both increased oxidative stress and mitochondrial dysfunction in the brains of patients with AD and in transgenic mouse models of AD (2–5). These deleterious phenomena may originate from the disruption of several pivotal pathways, including mitochondrial bioenergetics, oxidative damage, and transcriptional regulation (6). Under adverse stimuli, specific regulatory proteins, also known as transcription factors, are recruited into the nucleus, where they bind to DNA and promote the expression of cytoprotective genes (7, 8).
The peroxisome proliferator-activated receptor γ coactivator 1-α (PGC-1α) acts as a transcriptional coactivator of peroxisome proliferator-activated receptor γ (PPARγ) (9). It regulates the expression of genes which play a role in energy metabolism, including mitochondrial biogenesis, adaptive thermogenesis, β-oxidation of fatty acids, and glucose metabolism (10). PGC-1α can interact with other transcription factors, such as the nuclear respiratory factors (11). The latter, in turn, promote the expression of both antioxidant enzymes, as well as several nuclear-encoded mitochondrial genes necessary for mitochondrial biogenesis, such as cytochrome c, and the mitochondrial transcription factor A (Tfam) (12, 13).
In the brains of human patients with AD, the expression of PGC-1α is reduced, and this correlates with elevations of β-amyloid (Aβ) levels (14). In neurons derived from Tg2576 mice, hyperglycemia produced a diminution of PGC-1α and an elevation of Aβ. Using adenoviral vectors, 2- to 3-fold overexpression of PGC-1α in these cells reduced the hyperglycemia-mediated increased Aβ concentrations, and restored levels of α-secretase cleaved amyloid precursor protein (APP) fragments known to be nonamyloidogenic (14). In addition, increased PGC-1α promoted the degradation of β-site APP cleaving enzyme 1 (BACE1) via mechanisms involving the Fbx2 gene (15). More recently, Sheng et al. (16) reported that in mouse AD brains and M17 cells transfected with the human APP gene harboring the Swedish mutation, there was decreased PGC-1α, which was accompanied by a reduction of mitochondrial biogenesis, as evidenced by diminished ATP content, cytochrome c, and the mitochondrial DNA/nuclear DNA ratio. Overexpressing PGC-1α in the M17 transfected cells rescued the impaired mitochondrial biogenesis and function (16).
Since PGC-1α plays an important role as a master regulator of cellular metabolic homeostasis and has been reported to produce beneficial effects on Aβ, we investigated the effects of its overexpression in a transgenic mouse model of AD, the Tg19959 mice. These mice, which express the human APP gene with 2 familial AD mutations (KM670/671NL and V717F), develop progressive Aβ deposition and synaptic defects starting at 2–3 mo of age (17, 18), which are associated with behavioral impairments (19, 20). We crossed the Tg19959 mice with a transgenic model constitutively overexpressing PGC-1α (21) and assessed Aβ deposition, tau levels, and behavioral deficits from 3 to 4 mo of age.
MATERIALS AND METHODS
Animals
Tg19959 mice were originally obtained from Dr. George Carlson (McLaughlin Research Institute, Great Falls, MT, USA). Tg19959 mice were constructed by injecting a cosmid insert containing human APP695 with 2 familial AD mutations (KM670/671NL and V717F), under the control of the hamster prion protein (PrP) promoter (17). Tg19959 mice were kept under the C57BL6/SJL background. PGC-1α mice were originally obtained from Dr. Walter F. Ward (University of Texas Health Science Center, San Antonio, TX, USA). The PGC-1α mice were constructed by injecting a purified human PGC-1α BAC DNA (21). PGC-1α mice were kept under the C57BL6 background.
For this study, male Tg19959 and female PGC-1α mice were bred to generate littermates of 4 different genotypes. Offspring were identified by PCR of tail DNA: wild-type, PGC-1α, Tg19959, and Tg19959xPGC-1α mice. All experiments were approved by the Weill Cornell Medical College Institutional Animal Care and Use Committee.
Behavior
At 3 mo of age, locomotor activity and exploration were assessed in the open field, as described previously (22). Briefly, mice were placed for a single trial of 5 min in the apparatus, and total distance traveled was recorded using a video tracking system (Ethovision 3.1, Noldus Technology, Attleborough, MA, USA).
At 3 mo of age, memory was assessed using the contextual fear-conditioning test. Experiments were performed using 4 standard conditioning chambers (Coulbourn Instruments, Allentown, PA, USA). On d 1, mice were placed in the conditioning chamber for 120 s and then exposed to foot shock (0.7 mA for 2 s with 1-min interval). After the last shock, mice were left in the chamber for 30 s. On d 2, mice were placed back into the same chambers, and contextual fear memory was measured by recording the freezing time for 180 s, using the FreezeFrame system (Coulbourn Instruments, Allentown, PA, USA).
Western blot analysis
At the end of the behavioral testing, mice were euthanized by decapitation. Brains were collected, dissected, snap-frozen in liquid nitrogen, and stored at −80°C for biochemical studies. Cerebral cortices were homogenized in RIPA buffer or 6% sodium dodecyl sulfate (SDS) with protease and phosphatase inhibitors to extract soluble proteins (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Then, protein concentration was measured (DC Protein Measurement Kit; Bio-Rad, Hercules, CA, USA). Equal amounts of protein from the homogenates were electrophoresed through 4–12% Tri-Bis NuPage or 10–20% Tricine gels (Invitrogen, Carlsbad, CA, USA). After transfer to polyvinylidene fluoride (PVDF), membranes were blocked in 5% nonfat dry milk in phosphate-buffered saline (PBS) with 0.05% Tween 20 (PBST) and exposed overnight to the primary antibody at 4°C. Horseradish peroxidase (HRP)-conjugated secondary antibody binding was visualized with enhanced chemiluminescence (Pierce, Rockford, IL, USA). Films were scanned at 600 dpi, and densitometry was quantified with Scion Image 4.0.2 (Scion Corp., Frederick, MD, USA).
Primary antibodies and concentrations used for Western blot analysis were DA9 mouse monoclonal anti-total tau aa 102–140 (1:500), CP13 mouse monoclonal anti-human tau pSer202 (1:500), mouse monoclonal anti-human APP/Aβ 6E10 (1:500; Covance, Emeryville, CA, USA), rabbit polyclonal anti-insulin degrading enzyme (1:500; Abcam, Cambridge, MA, USA), rabbit polyclonal anti-human APP 369 (1:1000), rabbit polyclonal anti-presenilin 1 (PS1; 1:500; gift from Dr. Wenjie Luo, Weill Cornell Medical College, New York, NY, USA), rabbit polyclonal anti-nicastrin (1:1000, Cell Signaling, Danvers, MA, USA), and mouse monoclonal anti-β-actin (1:10,000, Sigma, St. Louis, MO, USA).
Dot blotting
RIPA homogenates from cortical tissues containing 20 μg of protein were spotted on nitrocellulose membrane (4 μl/sample). Once completely dry, membranes were blocked in 10% nonfat dry milk/Tris-buffered saline (TBS)-Tween 20 (TBST) for 1 h at room temperature. HRP-conjugated secondary antibody binding was visualized with enhanced chemiluminescence (Pierce, Rockford, IL, USA). Films were scanned at 600 dpi, and densitometry was quantified with Scion Image 4.0.2 (Scion Corp.).
Primary antibody and concentration was rabbit polyclonal anti-Aβ fibrils OC (1:1,000).
Aβ1–42 and Aβ1–40 enzyme-linked immunosorbent assay (ELISA)
Soluble Aβ1–42 and Aβ1–40 levels were analyzed on 6% SDS homogenates from cortical tissues using ELISA kits (Invitrogen) following the manufacturer's instructions.
Immunohistochemistry and histology
After decapitation, brains sections were dissected and postfixed in 4% paraformaldehyde followed by gradient sucrose (15 and 30%) and stored in cryoprotectant for immunohistochemical and histological studies.
NeuN staining
Sections were cut at 50-μm thickness and stained with the mouse anti-neuronal nuclear antigen antibody (NeuN, 1:1000; Millipore, Billerica, MA, USA). Immunolabeling was detected by the streptavidin-HRP method and visualized after 3,3′-diaminobenzidine (DAB) incubation for 30 s (Vector Laboratories, Burlingame, CA, USA). Sections were viewed with the ×10 and ×20 objectives on a Nikon Eclipse E600 microscope (Nikon, Tokyo, Japan). Digital images were captured using Stereo Investigator 9.12 (Microbrightfield, Burlington, VT, USA). Neuronal cell count was determined using five 50-μm serial nonadjacent sections per animal in the CA1 region of the hippocampus as well as the motor cortex (300 μm apart, from bregma −1.34 through bregma −2.84). The number of NeuN-positive neurons was measured using the optical fractionator of Stereo Investigator 9.12.
Congo red staining
Sections were mounted onto Superfrost Plus glass slides and air dried. After rinsing with distilled water, 1% Congo red solution in distilled water was applied to the sections for 30 min at 56°C. The sections were decolorized in 80% alcohol until clouds of stain no longer came out of the sections. Sections were then washed in distilled water, dehydrated, cleared, and coverslipped. Quantification was done using five 50-μm serial nonadjacent sections per animal (300 μm apart, from bregma −1.34 through bregma −2.84). The percentage area occupied by Congo red staining and the number of Congo red-positive plaques were quantified using Scion Image 4.0.2 (Scion Corp.).
Fluoro-Jade B
Adjacent sections containing the hippocampus and cerebral cortex were stained with Fluoro-Jade B (Millipore) following the manufacturer's instructions.
Mitochondria
Sample preparation
Dissected nonperfused cerebral cortex samples (∼30–55 mg) were stored frozen at −80°C until assaying. Before assays, tissue samples were thawed on ice and homogenized with a Dounce-type 2-ml homogenizer. Homogenates were centrifuged at 1000 g for 5 min to eliminate nuclear fraction and cell debris. Resulting supernatants were centrifuged at 14,000 g for 5 min. Pellets were collected and centrifuged again at 14,000 g for 5 min. Resulting pellets were resuspended in 20 mM HEPES (pH 7.8) and used for all assays.
Assays
All samples were assayed for the following: complex I activity (NADH:CoQ reductase, rotenone-sensitive; ref. 23), succinate dehydrogenase activity (succinate:CoQ:DCIP reductase, TTFA-sensitive; ref. 24), and citrate synthase activity (25). All activities were normalized by protein content in the sample (measured with BCA protein assay; Thermo Fisher Scientific, Waltham, MA, USA).
Proteasome activity assay
Assays
Tissue lysis and measurements of proteasome activity were carried out as described previously (26). Briefly, frozen brain tissues were homogenized in ice-cold buffer [0.25 M sucrose; 10 mM Tris–HCl, pH 7.8; 5 mM MgCl2; 0.5 mM EDTA; 1 mM dithiothreitol (DTT); and 2 mM ATP], using a Teflon-on-glass homogenizer, and cleared at 12,000 g for 10 min. Protein concentration was determined in duplicate on fresh lysate using the BCA protein assay kit (Thermo Fisher Scientific). Assays for proteasome activity were performed in duplicate on five frontal lobes of each genotype using the fluorogenic substrates for caspase-, chymotrypsin-, and trypsin-like activity. Equal amounts of extracts were incubated with the corresponding substrates (100 μM) in 100 μl proteasome activity assay buffer (10 mM Tris-HCl, pH 7.8; 5 mM MgCl2; 0.5 mM EDTA; 1 mM DTT; and 2 mM ATP) for 30 min at 37°C. Reactions were quenched by cold ethanol, and free 7-amino-4-methylcoumarin (AMC) fluorescence was quantified with a fluorescence multiplate reader FLUOstar Optima FL (BMG Labtech, Ortenberg, Germany) with excitation and emission wavelengths at 380 and 460 nm, respectively. Proteasome activity per milligram of protein per hour was calculated from fluorescence values, and all subsequent data were expressed as a ratio relative to wild-type value.
Fluorogenic peptides
Z-Leu-Leu-Glu-AMC (Z-LLE-AMC), Suc-Leu-Leu-Val-Tyr-AMC (Suc-LLVT-AMC), Z-Ala-Arg-Arg-AMC (Z-AAA-AMC), and AMC were obtained from Calbiochem (Millipore).
Gene expression by real-time PCR
Frozen cortices stored at −80°C were processed for RNA extraction (Qiagen kit). Quantitative real-time PCR was performed at the Weill Cornell Medical College Microarray Core Facility using SYBR Green assays with the ABI Prism 7900HT sequence detection system (Applied Biosystems, Foster City, CA, USA) for the following genes: BACE1, Neprilysin, and GAPDH as control. Quantitative real-time PCR was also performed using TaqMan assays with the ABI Prism 7900HT sequence detection system for the following genes: human PGC1α and GAPDH as a control.
Statistical analysis
All data are expressed as means ± se. ANOVA followed by post hoc Fisher's protected least significant difference (PLSD) test was used to compare all 4 groups: wild-type, PGC-1α, Tg19959, and Tg19959xPGC-1α mice. When only 2 groups were involved in the study (Tg19959 mice and Tg19959xPGC-1α littermates), 2-tailed unpaired t tests were used (StatView 5.0.1; SAS Institute, Cary, NC, USA).
RESULTS
PGC-1α overexpression promoted amyloid and tau pathology in Tg19959 mice
To determine the effects of a chronic, mild, whole-body overexpression of PGC-1α on disease pathogenesis in Tg19959 mice, we crossed the Tg19959 mice with BAC transgenic mice constitutively overexpressing the human PGC-1α gene under its own endogenous promoter (21). Thus, we generated 4 different genotypes: wild-type, PGC-1α, Tg19959, and Tg19959xPGC-1α. As described previously (21), PGC-1α mice showed about a 1.5-fold increase in human PGC-1α gene expression in the cerebral cortex as compared to their wild-type littermates. A similar increase was also seen in the Tg19959xPGC-1α mice (n=5–10; Supplemental Fig. S1).
To assess amyloid pathology at ∼4 mo of age, we examined Congo red staining in the cerebral cortex and hippocampus of Tg19959 mice and their Tg19959xPGC-1α littermates (Fig. 1A–C). Both plaque burden (Fig. 1B) and plaque number (Fig. 1C) in the cerebral cortex and hippocampus were significantly increased in Tg19959xPGC-1α mice as compared to their Tg19959 littermates (unpaired t tests, P<0.05, n=6). No Aβ accumulation was found in the PGC-1α mice alone, as these mice do not express Aβ. These data were confirmed by dot blots using the anti-human Aβ fibril OC antibody (Fig. 1D). PGC-1α overexpression increased levels of Aβ fibrils in Tg19959 mice (unpaired t tests, P<0.05; n=6).
Figure 1.

PGC-1α overexpression promoted amyloid plaque deposition in Tg19959 mice. A) Congo red staining of Tg19959 and Tg19959xPGC-1α mouse brains. B, C) Amyloid plaque burden (B) and plaque number (C) in the cerebral cortex and hippocampus of Tg19959 mice (n=6) and Tg19959xPGC-1α littermates (n=6). D) Dot blots of Aβ fibrils using OC in the cerebral cortex of Tg19959 mice (n=6) and Tg19959xPGC-1α littermates (n=6). Quantifications are expressed as a percentage of control (control Tg19959 mice). PGC-1α overexpression increased amyloid plaque burden and number as well as Aβ fibrils in Tg19959 mice. *P < 0.05; unpaired t test.
Cortical levels of both soluble Aβ1–40 and Aβ1–42 were measured by ELISA. We observed a trend toward an increase of soluble Aβ1–42 and a decrease of Aβ1–40 with PGC-1α overexpression in Tg19959 mice (unpaired t tests, P=0.06; n=5; Fig. 2A, B). Since we found increased Aβ levels in Tg19959-PGC-1α mice, we investigated whether the amyloidogenic processing of APP was affected. We did not observe changes in mRNA levels of BACE1 (unpaired t tests, P>0.05; n=5; Supplemental Fig. S2A) and protein levels of full-length and C-terminal fragment of APP (data not shown). On the other hand, we measured protein levels of the γ-secretase catalytic subunit PS1 using a noncommercially available antibody (27) and of nicastrin. While full-length PS1 levels were unchanged (data not shown), we found increased levels of PS1 N-terminal fragment, the most active form of PS1, in Tg19959-PGC-1α mice as compared to Tg19959 mice (unpaired t tests, P=0.05; n=6; Fig. 2C;). There was a trend toward an increase in nicastrin levels in Tg19959-PGC-1α mice as compared to Tg19959 mice (unpaired t tests, P>0.05; n=6; Supplemental Fig. S3A). In addition, there was no difference in PS1 and nicastrin levels in PGC-1α mice as compared to wild-type littermates (unpaired t tests, P>0.05; n=8; Supplemental Fig. S3B, C).
Figure 2.
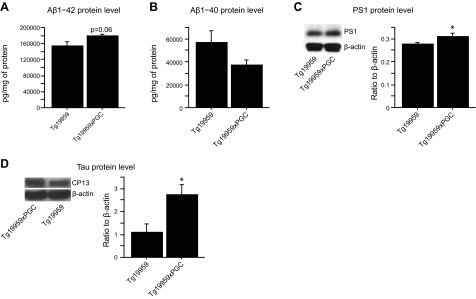
PGC-1α overexpression increased soluble Aβ and tau levels in Tg19959 mice. A, B) Aβ1–42 (A) and Aβ1–40 (B) levels by ELISA in Tg19959 mice (n=6) and Tg19959xPGC-1α littermates (n=6). C) Western blots of PS1 in Tg19959 mice (n=5) and Tg19959xPGC-1α littermates (n=7). D) Western blots of tau using CP13 antibody in Tg19959 mice (n=6) and Tg19959xPGC-1α littermates (n=6). Quantifications are expressed as ratios to β-actin. PGC-1α overexpression resulted in increased Aβ1–42 and CP13 levels in Tg19959 mice. *P < 0.05; unpaired t test.
We also explored the effects of PGC-1α on Aβ degradation. PGC-1α overexpression did not alter levels of either neprilysin- or insulin-degrading enzyme (IDE) in Tg19959 mice (unpaired t tests, P>0.05; n=6; Supplemental Fig. 2C, D).
Cortical levels of soluble tau were evaluated by Western blots using the following antibodies: DA9 (anti-total tau aa 102–140) and CP13 (anti-human tau pSer202). PGC-1α overexpression promoted tau accumulation, as evidenced by the increased levels of CP13, a marker of early to mature tau pathology (unpaired t tests, P<0.05, n=6; Fig. 2D), without affecting levels of total tau (data not shown).
PGC-1α overexpression induced cell death and mitochondrial defects in Tg19959 mice
We used stereological procedures to count the number of NeuN-stained neurons in the cerebral cortex and hippocampus of Tg19959 mice (Fig. 3A). The numbers of neurons in the cerebral cortex (Fig. 3B) and hippocampus (Fig. 3C) were significantly reduced by ∼20% in Tg19959xPGC-1α mice, as compared to their Tg19959 littermates (Fisher's PLSD test, P<0.05; n=6). No significant differences were observed between wild-type mice and their PGC-1α littermates (Fig. 3). However, there was a trend toward an additive effect of both genes on hippocampal neuronal loss (Fisher's PLSD test, P=0.081; n=6). To assess whether cell death was related to increased neurodegeneration, we stained adjacent sections with Fluoro-Jade B; we found no positive staining in any of the 4 groups (data not shown).
Figure 3.
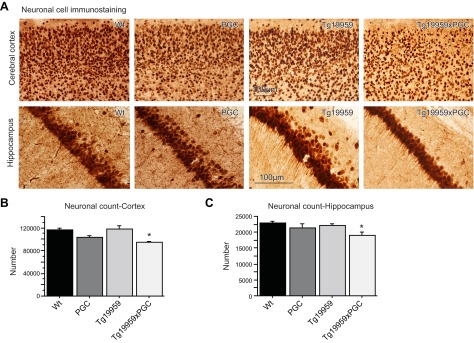
PGC-1α overexpression induced cell death in Tg19959 mice. A) NeuN staining of wild-type, PGC-1α, Tg19959, and Tg19959xPGC-1α mouse brains. B, C) Neuronal cell count in the cerebral cortex (B) and hippocampus (C) of wild-type (n=4), PGC-1α (n=4), Tg19959 mice (n=7), and Tg19959xPGC-1α littermates (n=6). There is a significant reduction of neuronal cells in Tg19959xPGC-1α mice relative to Tg19959 littermates. *P < 0.05; Fisher's PLSD test.
We also investigated the role of PGC-1α on the activities of key mitochondrial enzymes, such as citrate synthase (Fig. 4A), complex I (Fig. 4B), and succinate dehydrogenase (Fig. 4C). There were significant reductions of citrate synthase, complex I, and succinate dehydrogenase activities by PGC-1α overexpression in Tg19959 mice (Fisher's PLSD test, P<0.05; n=5–14).
Figure 4.
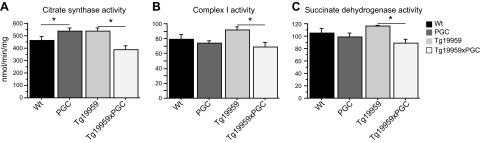
PGC-1α overexpression reduced mitochondrial enzyme activities in Tg19959 mice. Enzymatic activity of citrate synthase (A), complex I (B), and succinate dehydrogenase (C) in wild-type (n=14), PGC-1α (n=10), Tg19959 (n=6), and Tg19959xPGC-1α (n=5) mice. Enzymatic activities of citrate synthase, complex I, and succinate dehydrogenase were decreased in Tg19959xPGC-1α as compared to Tg19959 mice. *P < 0.05; Fisher's PLSD test.
PGC-1α overexpression inhibited proteasome activity in Tg19959 mice
To determine whether the abnormal protein accumulation was due to decreased proteasomal activity mediated by PGC-1α overexpression, we studied the caspase-, chymotrypsin-, and trypsin-like activities of the proteasome (28), using fluorogenic peptides as described previously (26, 29). In PGC-1α mice, chymotrypsin and trypsin-like activities in the cortex were significantly reduced as compared to their wild-type littermates (Fisher's PLSD test, P<0.05; n=5; Fig. 5). In Tg19959xPGC-1α mice, all of the proteasomal activities were significantly diminished as compared to Tg19959 mice (Fisher's PLSD test, P<0.05; n=5; Fig. 5). Also, in Tg19959 mice, we observed an increase in trypsin-like activity as compared to wild-type littermates (Fisher's PLSD test, P<0.05; n=5).
Figure 5.
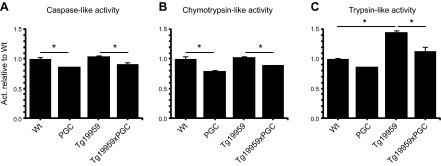
PGC-1α overexpression inhibited proteasomal activity in Tg19959 mice. Caspase-like (A), chymotrypsin-like (B), and trypsin-like (C) activities of the proteasome system in wild-type (n=5), PGC-1α (n=5), Tg19959 (n=5), and Tg19959xPGC-1α (n=5) mice. Proteasome activity was reduced by PGC-1α overexpression. *P < 0.05; Fisher's PLSD test.
PGC-1α overexpression exacerbated behavioral deficits in Tg19959 mice
To determine whether PGC-1α overexpression had any effects on behavioral impairments, we conducted both the open field and contextual fear-conditioning tests at 3 mo of age, to study locomotor activity and memory, respectively (Fig. 6). In both tasks, Tg19959 mice were impaired as compared to wild-type littermates, as evidenced by increased distance traveled in the open field (Fig. 6A) and decreased time spent freezing in the fear-conditioning chamber (Fig. 6B). PGC-1α overexpression in the Tg19959 mice exacerbated the hyperactivity seen in Tg19959 mice during open field testing (Fisher's PLSD test, P<0.05; n=6–14).
Figure 6.

PGC-1α overexpression exacerbated behavioral deficit in Tg19959 mice. A) Distance traveled during the open field test in wild-type (n=14), PGC-1α (n=12), Tg19959 (n=9), and Tg19959xPGC-1α (n=6) mice. B) Percentage of time spent freezing during the contextual fear-conditioning test in wild-type (n=14), PGC-1α (n=12), Tg19959 (n=8) and Tg19959xPGC-1α (n=6) mice. PGC-1α overexpression increased hyperactivity in Tg19959 mice in the open field. In addition, PGC-1α overexpression did not improve memory deficits in Tg19959 mice. *P < 0.05; Fisher's PLSD test.
DISCUSSION
Transcriptional regulation plays a key role in protective responses to cellular damage (6). PGC-1α is a transcriptional coactivator that regulates the expression of transcriptional factors involved in lipid and energy metabolism, inflammation, mitochondrial enzymes, and redox homeostasis (9, 10, 30). It plays an important role in maintaining metabolic homeostasis, as well as in both cell growth and survival.
Reduced transcription and/or protein expression of PGC-1α plays a role in the pathogenesis of neurodegenerative diseases, including Huntington's disease (HD), Parkinson's disease (PD), and amyotrophic lateral sclerosis (ALS) (31, 32). Mice deficient in PGC-1α exhibit reduced numbers of mitochondria, increased locomotor activity, and striatal pathology (33). Subsequent studies in patients with HD and in transgenic mouse models showed reduced levels of PGC-1α (34, 35) and its downstream targets, such as nuclear respiratory factor 1 and 2 (NRF1 and NRF2), and Tfam (36, 37). We also observed reductions in both mitochondrial DNA (mtDNA) copy number and numbers of mitochondria in HD transgenic mice (36, 37), and in striatal spiny neurons from HD postmortem brain tissue (38). Increasing levels of PGC1α using bezafibrate, a pan- PPAR agonist, restored the numbers of mitochondria in R6/2 HD transgenic mice, while ameliorating the behavioral and neuropathological deficits and improving survival (39).
In PD, a metaanalysis of microarray data from brain tissue, white blood cells and laser-dissected dopaminergic neurons showed reduced expression of PGC-1α (32). Furthermore mutations in ubiquitin E3 ligase parkin are the most common genetic cause of early onset PD, and they result in accumulation of PARIS, which represses expression of PGC-1α (40). Conditional inactivation of parkin causes an increase in PARIS, a depletion of PGC-1α, and selective loss of dopaminergic neurons in the substantia nigra in adult mice (40). In transgenic mouse models of ALS, motor impairment, survival, and loss of motor neurons are improved by increased PGC-1α (41).
The expression levels of PGC-1α were reduced in the brains of both human patients with AD and transgenic mouse models (14). The reductions of PGC-1α in patients with AD were inversely correlated with the premortem clinical dementia rating score, as well as with the numbers of neurofibrillary tangles (14). Increasing mitochondrial biogenesis and expression of antioxidant enzymes has been shown to be beneficial in mouse models of AD (20, 42). Reconstitution of PGC-1α in primary neurons from Tg2576 embryos attenuated hyperglycemic-mediated increases in Aβ, decreasing expression of the forkhead-like transcription factor 1 (FoxO3a), and stimulating α-secretase processing of the APP (16). In addition, PGC-1α downstream targets, such as NRF1, NRF2, and Tfam, are markedly reduced in the brains of human patients with AD and in M17 cells transfected with a human APP gene mutation linked to familial AD (16). The defects of mitochondrial biogenesis and function were rescued by PGC-1α overexpression (16). In another study of mouse N2a neuroblastoma cells transfected with the human APP gene with the Swedish mutation linked to AD, both Aβ levels and BACE1 activity were inhibited by PGC-1α overexpression (43). Therefore, increasing PGC-1α appears to be a promising therapeutic target for neurodegenerative diseases associated with mitochondrial dysfunction and oxidative damage (32, 44–46). Therefore, we examined whether a mild, sustained increased expression of PGC-1α would produce beneficial effects in the Tg19959 transgenic mouse model of AD.
We crossed Tg19959 mice with PGC-1α-overexpressing mice and conducted behavioral and pathological assays from 3 to 4 mo of age, which is when Tg19959 mice develop Aβ deposition and exhibit behavioral deficits (19, 20). Both the PGC-1α and Tg19959xPGC-1α mice showed about a 1.5-fold increase in human PGC-1α gene expression in the brain, as previously reported (21). Although we expected to detect beneficial effects, we observed that overexpression of PGC-1α exacerbated both Aβ deposition and fibrillarization in Tg19959xPGC-1α mice. This increased amyloid pathology was not due to changes in α- or β-secretase cleavage on APP, or to the reduction of Aβ-degrading enzymes, neprilysin and IDE. We found that it was caused by increased N-terminal fragment of PS1, the catalytic subunit of the γ-secretase complex, enhancing the production of Aβ.
We examined the effects of PGC-1α on tau pathology by measuring levels of phosphorylated tau using the CP13 antibody, a marker of early to mature stage neurofibrillary tangles. A significant increase of CP13 immunoreactivity was observed in the brains of Tg19959xPGC-1α mice, which may be a secondary consequence of increased Aβ levels. Deleterious effects of PGC-1α overexpression were also present at the behavioral and cellular level. We observed a significant reduction in the numbers of neurons in the cerebral cortex and hippocampus of the Tg19959-PGC-1α mice. Neuronal cell death was not associated with neurodegeneration, since we did not find positive Fluoro-Jade B cells with PGC-1α overexpression. As a consequence, we cannot exclude that Tg19959xPGC-1α mice may already have fewer neurons at birth. PGC-1α overexpression did not worsen memory deficits, but it exacerbated hyperactivity. The lack of worsening in the fear-conditioning task may be explained by the already very profound deficit in the Tg19959 mice. We believe that we have reached a “floor effect” by which we could not discriminate further between Tg19959 mice and Tg19959xPGC-1α littermates. Sustained high levels of overexpression of PGC-1α were previously shown to produce toxic effects in muscle and heart, which are accompanied by extensive mitochondrial proliferation and myopathy (47–49). Extremely high levels of PGC-1α (∼400-fold increases) using viral vectors are toxic to dopaminergic neurons; however in the striatum, 4-fold increases were not toxic (42). There is, therefore, little precedent for the toxic effects that we encountered.
PGC-1α overexpression reduces protein degradation by inhibiting autophagy and proteasome activation in muscle after exercise, which prevents muscle wasting (50, 51). We hypothesized that inhibition of protein degradation may have occurred in the brains of the Tg19959 mice, which overexpress PGC-1α, leading to an exacerbation of protein aggregation and amyloid deposition. Consistent with this, we observed that PGC-1α overexpression markedly decreased proteasomal activity in both the PGC-1α and Tg19959xPGC-1α mice. Previous studies of human patients with AD and transgenic AD mice showed impaired proteasomal activity, which may contribute to the accumulation of tau aggregates in AD brains (52, 53). We found that at early disease stages (4 mo of age), the trypsin-like activity of the proteasome was increased as compared to wild-type mice, indicating a possible compensatory mechanism, which is in accordance with another study showing that moderate phosphorylation of tau can activate trypsin-like activity of the proteasome (54). However, at this early disease stage, Aβ deposition was also increased, indicating that the stimulation of proteasomal activity was not sufficient to clear Aβ.
Our observations suggest that PGC-1α overexpression induces accumulation of proteins prone to aggregate, such as Aβ, by disrupting their clearance by the proteasome. Our results are in line with those reported by Brault et al. (50), who showed that PGC-1α overexpression inhibited both the proteasomal and the autophagic/lysosomal protein degradation systems in muscle, as a mechanism to impede muscle wasting. Although the precise molecular mechanisms remain elusive at present, we are tempted to speculate that redox signaling may be involved. Indeed, increased reactive oxygen species (ROS) production stimulates proteasome-dependent protein degradation by activating expression of the rate-limiting enzymes of the ubiquitination and proteasomal degradation process such as E3 ligases (55). Moreover, PGC-1α directly inactivates FOXO3a signaling (56), a transcription factor that up-regulates the E3 ligase ZNF216, a major player in the recognition and delivery of ubiquitinated proteins to the proteasome (57), thus directly limiting the rate of the proteasomal degradation process.
Conversely, a recent report using HD transgenic mice showed that overexpression of PGC-1α was neuroprotective by inducing autophagy and reducing huntingtin aggregates and oxidative stress (58). This discrepancy with our results may be due to the different proteins and pathophysiology involved in these two disease models, in particular, regarding mitochondria dysfunction, as well as different expression levels of PGC-1α.
A number of studies showed that Aβ can be localized to mitochondria, where it is reported to bind to the Aβ peptide alcohol dehydrogenase (59, 60), increasing the production of ROS and contributing to synaptic deficits (61). We found that levels of mtDNA and cytochrome c were increased in Tg19959 mice at baseline and that PGC-1α overexpression produced no further increase. Levels of NRF1 and Tfam were similar in all experimental groups (data not shown). In Tg19959xPGC-1α, the activities of mitochondrial enzymes such as complex I, citrate synthase, and succinate dehydrogenase were diminished compared to Tg19959 mice alone. Because we did not observe an additive effect of PGC-1α and APP genes in two of these enzymes, our data suggest the hypothesis of a primary effect of PGC-1α on mitochondrial function. Studies at multiple ages would be of interest to determine whether there is evidence of progressive changes with age, in the Tg19959-PGC-1α mice; unfortunately, a high rate of mortality in the double transgenic mice did not make this possible in the current study.
Our results underscore that preserving the exquisite balance of PGC-1α expression and function may be critical to achieve beneficial effects, which may be of relevance when designing therapeutic strategies.
Supplementary Material
Acknowledgments
This work was supported by the U.S. National Institutes of Health (NIH), grant 5P01 (AG14930). The work performed at the Neurometabolic Diseases Laboratory was carried out thanks to grants from the Spanish National Institute of Health Carlos III (FIS PI11/01043) and the Catalan Government Group of Excellence (2009SGR85) to A.P. N.L. holds a postdoctoral fellowship from Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER), an initiative of Instituto de Salud Carlos III (ISCIII).
The authors thank Peter Davies (Albert Einstein College of Medicine, New York, NY, USA), Rakez Kayed (University of Texas Medical Branch, Galveston, TX, USA), and Wenjie Luo (Weill Cornell Medical College, New York, NY, USA) for providing the tau (CP13 and DA9), amyloid (OC), and PS1 antibodies, respectively. The authors declare no conflicts of interest.
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
- Aβ
- β-amyloid
- AD
- Alzheimer disease
- ALS
- amyotrophic lateral sclerosis
- AMC
- 7-amino-4-methylcoumarin
- APP
- amyloid precursor protein
- BACE1
- β-site amyloid precursor protein cleaving enzyme 1
- DTT
- dithiothreitol
- ELISA
- enzyme-linked immunosorbent assay
- FOXO3a
- forkhead-like transcription factor 1
- HD
- Huntington disease
- HRP
- horseradish peroxidase
- IDE
- insulin-degrading enzyme
- mtDNA
- mitochondrial DNA
- NRF1/2
- nuclear respiratory factor 1/2
- PBS
- phosphate-buffered saline
- PD
- Parkinson disease
- PGC-1α
- peroxisome proliferator-activated receptor γ coactivator 1-α
- PLSD
- protected least significant difference
- PPARγ
- peroxisome proliferator-activated receptor γ
- PS1
- presenilin 1
- ROS
- reactive oxygen species
- SDS
- sodium dodecyl sulfate
- TBS
- Tris-buffered saline
- Tfam
- mitochondrial transcription factor A
REFERENCES
- 1. Terry R. D., Masliah E., Salmon D. P., Butters N., DeTeresa R., Hill R., Hansen L. A., Katzman R. (1991) Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann. Neurol. 30, 572–580 [DOI] [PubMed] [Google Scholar]
- 2. Higgins G. C., Beart P. M., Shin Y. S., Chen M. J., Cheung N. S., Nagley P. (2010) Oxidative stress: emerging mitochondrial and cellular themes and variations in neuronal injury. J. Alzheimers. Dis. 20(Suppl. 2), S453–S473 [DOI] [PubMed] [Google Scholar]
- 3. Lin M. T., Beal M. F. (2006) Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443, 787–795 [DOI] [PubMed] [Google Scholar]
- 4. Morais V. A., De Strooper B. (2010) Mitochondria dysfunction and neurodegenerative disorders: cause or consequence. J. Alzheimers Dis. 20(Suppl. 2), S255–S263 [DOI] [PubMed] [Google Scholar]
- 5. Muller W. E., Eckert A., Kurz C., Eckert G. P., Leuner K. (2010) Mitochondrial dysfunction: common final pathway in brain aging and Alzheimer's disease–therapeutic aspects. Mol. Neurobiol. 41, 159–171 [DOI] [PubMed] [Google Scholar]
- 6. Shi Q., Gibson G. E. (2007) Oxidative stress and transcriptional regulation in Alzheimer disease. Alzheimer Dis. Assoc. Disord. 21, 276–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Go Y. M., Jones D. P. (2010) Redox control systems in the nucleus: mechanisms and functions. Antioxid. Redox Signal. 13, 489–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ma Q. (2010) Transcriptional responses to oxidative stress: pathological and toxicological implications. Pharmacol. Ther. 125, 376–393 [DOI] [PubMed] [Google Scholar]
- 9. Puigserver P., Wu Z., Park C. W., Graves R., Wright M., Spiegelman B. M. (1998) A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell 92, 829–839 [DOI] [PubMed] [Google Scholar]
- 10. Puigserver P., Spiegelman B. M. (2003) Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1 alpha): transcriptional coactivator and metabolic regulator. Endocr. Rev. 24, 78–90 [DOI] [PubMed] [Google Scholar]
- 11. Lin J., Handschin C., Spiegelman B. M. (2005) Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 1, 361–370 [DOI] [PubMed] [Google Scholar]
- 12. Scarpulla R. C. (2008) Nuclear control of respiratory chain expression by nuclear respiratory factors and PGC-1-related coactivator. Ann. N. Y. Acad. Sci. 1147, 321–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wu Z., Puigserver P., Andersson U., Zhang C., Adelmant G., Mootha V., Troy A., Cinti S., Lowell B., Scarpulla R. C., Spiegelman B. M. (1999) Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 98, 115–124 [DOI] [PubMed] [Google Scholar]
- 14. Qin W., Haroutunian V., Katsel P., Cardozo C. P., Ho L., Buxbaum J. D., Pasinetti G. M. (2009) PGC-1alpha expression decreases in the Alzheimer disease brain as a function of dementia. Arch. Neurol. 66, 352–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gong B., Chen F., Pan Y., Arrieta-Cruz I., Yoshida Y., Haroutunian V., Pasinetti G. M. (2010) SCFFbx2-E3-ligase-mediated degradation of BACE1 attenuates Alzheimer's disease amyloidosis and improves synaptic function. Aging Cell. 9, 1018–1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sheng B., Wang X., Su B., Lee H. G., Casadesus G., Perry G., Zhu X. (2012) Impaired mitochondrial biogenesis contributes to mitochondrial dysfunction in Alzheimer's disease. J. Neurochem. 120, 419–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chishti M. A., Yang D. S., Janus C., Phinney A. L., Horne P., Pearson J., Strome R., Zuker N., Loukides J., French J., Turner S., Lozza G., Grilli M., Kunicki S., Morissette C., Paquette J., Gervais F., Bergeron C., Fraser P. E., Carlson G. A., George-Hyslop P. S., Westaway D. (2001) Early-onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein 695. J. Biol. Chem. 276, 21562–21570 [DOI] [PubMed] [Google Scholar]
- 18. Li F., Calingasan N. Y., Yu F., Mauck W. M., Toidze M., Almeida C. G., Takahashi R. H., Carlson G. A., Flint Beal M., Lin M. T., Gouras G. K. (2004) Increased plaque burden in brains of APP mutant MnSOD heterozygous knockout mice. J. Neurochem. 89, 1308–1312 [DOI] [PubMed] [Google Scholar]
- 19. Dumont M., Wille E., Calingasan N. Y., Tampellini D., Williams C., Gouras G. K., Liby K., Sporn M., Nathan C., Flint Beal M., Lin M. T. (2009) Triterpenoid CDDO-methylamide improves memory and decreases amyloid plaques in a transgenic mouse model of Alzheimer's disease. J. Neurochem. 109, 502–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dumont M., Wille E., Stack C., Calingasan N. Y., Beal M. F., Lin M. T. (2009) Reduction of oxidative stress, amyloid deposition, and memory deficit by manganese superoxide dismutase overexpression in a transgenic mouse model of Alzheimer's disease. FASEB J. 23, 2459–2466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Liang H., Balas B., Tantiwong P., Dube J., Goodpaster B. H., O'Doherty R. M., DeFronzo R. A., Richardson A., Musi N., Ward W. F. (2009) Whole body overexpression of PGC-1α has opposite effects on hepatic and muscle insulin sensitivity. Am. J. Physiol. Endocrinol. Metab. 296, E945–E954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dumont M., Wille E., Calingasan N. Y., Nathan C., Flint Beal M., Lin M. T. N-iminoethyl-L-lysine improves memory and reduces amyloid pathology in a transgenic mouse model of amyloid deposition. Neurochem. Int. 56, 345–351 [DOI] [PubMed] [Google Scholar]
- 23. Degli Esposti M., Ghelli A., Crimi M., Estornell E., Fato R., Lenaz G. (1993) Complex I and complex III of mitochondria have common inhibitors acting as ubiquinone antagonists. Biochem. Biophys. Res. Commun. 190, 1090–1096 [DOI] [PubMed] [Google Scholar]
- 24. Arrigoni O., Singer T. P. (1962) Limitations of the phenazine methosulphate assay for succinic and related dehydrogenases. Nature 193, 1256–1258 [DOI] [PubMed] [Google Scholar]
- 25. Srere P. A. (1969) Citrate synthase. Methods Enzymol. 13, 3–11 [Google Scholar]
- 26. Cheroni C., Peviani M., Cascio P., Debiasi S., Monti C., Bendotti C. (2005) Accumulation of human SOD1 and ubiquitinated deposits in the spinal cord of SOD1G93A mice during motor neuron disease progression correlates with a decrease of proteasome. Neurobiol. Dis. 18, 509–522 [DOI] [PubMed] [Google Scholar]
- 27. Luo W. J., Wang H., Li H., Kim B. S., Shah S., Lee H. J., Thinakaran G., Kim T. W., Yu G., Xu H. (2003) PEN-2 and APH-1 coordinately regulate proteolytic processing of presenilin 1. J. Biol. Chem. 278, 7850–7854 [DOI] [PubMed] [Google Scholar]
- 28. Ciechanover A. (1994) The ubiquitin-proteasome proteolytic pathway. Cell 79, 13–21 [DOI] [PubMed] [Google Scholar]
- 29. Launay N., Ruiz M., Fourcade S., Schlüter A., Guilera C., Ferrer I., Knecht E., Pujol A. (2013) Oxidative stress regulates the ubiquitin–proteasome system and immunoproteasome functioning in a mouse model of X-adrenoleukodystrophy. Brain 136, 891–904 [DOI] [PubMed] [Google Scholar]
- 30. Handschin C., Spiegelman B. M. (2006) Peroxisome proliferator-activated receptor gamma coactivator 1 coactivators, energy homeostasis, and metabolism. Endocr. Rev. 27, 728–735 [DOI] [PubMed] [Google Scholar]
- 31. Rona-Voros K., Weydt P. (2010) The role of PGC-1alpha in the pathogenesis of neurodegenerative disorders. Curr. Drug Targets 11, 1262–1269 [DOI] [PubMed] [Google Scholar]
- 32. Zheng B., Liao Z., Locascio J. J., Lesniak K. A., Roderick S. S., Watt M. L., Eklund A. C., Zhang-James Y., Kim P. D., Hauser M. A., Grunblatt E., Moran L. B., Mandel S. A., Riederer P., Miller R. M., Federoff H. J., Wullner U., Papapetropoulos S., Youdim M. B., Cantuti-Castelvetri I., Young A. B., Vance J. M., Davis R. L., Hedreen J. C., Adler C. H., Beach T. G., Graeber M. B., Middleton F. A., Rochet J. C., Scherzer C. R. (2010) PGC-1α, a potential therapeutic target for early intervention in Parkinson's disease. Sci Transl. Med. 2, 52ra73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lin J., Wu P. H., Tarr P. T., Lindenberg K. S., St-Pierre J., Zhang C. Y., Mootha V. K., Jager S., Vianna C. R., Reznick R. M., Cui L., Manieri M., Donovan M. X., Wu Z., Cooper M. P., Fan M. C., Rohas L. M., Zavacki A. M., Cinti S., Shulman G. I., Lowell B. B., Krainc D., Spiegelman B. M. (2004) Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1α-null mice. Cell 119, 121–135 [DOI] [PubMed] [Google Scholar]
- 34. Weydt P., Pineda V. V., Torrence A. E., Libby R. T., Satterfield T. F., Lazarowski E. R., Gilbert M. L., Morton G. J., Bammler T. K., Strand A. D., Cui L., Beyer R. P., Easley C. N., Smith A. C., Krainc D., Luquet S., Sweet I. R., Schwartz M. W., La Spada A. R. (2006) Thermoregulatory and metabolic defects in Huntington's disease transgenic mice implicate PGC-1α in Huntington's disease neurodegeneration. Cell Metab. 4, 349–362 [DOI] [PubMed] [Google Scholar]
- 35. Zhai W., Jeong H., Cui L., Krainc D., Tjian R. (2005) In vitro analysis of huntingtin-mediated transcriptional repression reveals multiple transcription factor targets. Cell 123, 1241–1253 [DOI] [PubMed] [Google Scholar]
- 36. Chaturvedi R. K., Adhihetty P., Shukla S., Hennessy T., Calingasan N., Yang L., Starkov A., Kiaei M., Cannella M., Sassone J., Ciammola A., Squitieri F., Beal M. F. (2009) Impaired PGC-1α function in muscle in Huntington's disease. Hum. Mol. Genet. 18, 3048–3065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chaturvedi R. K., Calingasan N. Y., Yang L., Hennessey T., Johri A., Beal M. F. (2010) Impairment of PGC-1α expression, neuropathology and hepatic steatosis in a transgenic mouse model of Huntington's disease following chronic energy deprivation. Hum. Mol. Genet. 19, 3190–3205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kim J., Moody J. P., Edgerly C. K., Bordiuk O. L., Cormier K., Smith K., Beal M. F., Ferrante R. J. (2010) Mitochondrial loss, dysfunction and altered dynamics in Huntington's disease. Hum. Mol. Genet. 19, 3919–3935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Johri A., Calingasan N. Y., Hennessey T. M., Sharma A., Yang L., Wille E., Chandra A., Beal M. F. (2012) Pharmacologic activation of mitochondrial biogenesis exerts widespread beneficial effects in a transgenic mouse model of Huntington's disease. Hum. Mol. Genet. 21, 1124–1137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shin J. H., Ko H. S., Kang H., Lee Y., Lee Y. I., Pletinkova O., Troconso J. C., Dawson V. L., Dawson T. M. (2011) PARIS (ZNF746) repression of PGC-1alpha contributes to neurodegeneration in Parkinson's disease. Cell 144, 689–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhao W., Varghese M., Yemul S., Pan Y., Cheng A., Marano P., Hassan S., Vempati P., Chen F., Qian X., Pasinetti G. M. (2011) Peroxisome proliferator activator receptor gamma coactivator-1α (PGC-1α) improves motor performance and survival in a mouse model of amyotrophic lateral sclerosis. Mol. Neurodegener. 6, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Melov S., Adlard P. A., Morten K., Johnson F., Golden T. R., Hinerfeld D., Schilling B., Mavros C., Masters C. L., Volitakis I., Li Q. X., Laughton K., Hubbard A., Cherny R. A., Gibson B., Bush A. I. (2007) Mitochondrial oxidative stress causes hyperphosphorylation of tau. PLoS ONE 2, e536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Katsouri L., Parr C., Bogdanovic N., Willem M., Sastre M. (2011) PPARγ coactivator-1α (PGC-1α) reduces amyloid-beta generation through a PPARγ-dependent mechanism. J. Alzheimers Dis. 25, 151–162 [DOI] [PubMed] [Google Scholar]
- 44. Beal M. F. (2009) Therapeutic approaches to mitochondrial dysfunction in Parkinson's disease. Parkinsonism Relat. Disord. 15(Suppl. 3), S189–S194 [DOI] [PubMed] [Google Scholar]
- 45. Keeney P. M., Quigley C. K., Dunham L. D., Papageorge C. M., Iyer S., Thomas R. R., Schwarz K. M., Trimmer P. A., Khan S. M., Portell F. R., Bergquist K. E., Bennett J. P., Jr. (2009) Mitochondrial gene therapy augments mitochondrial physiology in a Parkinson's disease cell model. Hum. Gene Ther. 20, 897–907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. McGill J. K., Beal M. F. (2006) PGC-1α, a new therapeutic target in Huntington's disease? Cell 127, 465–468 [DOI] [PubMed] [Google Scholar]
- 47. Ciron C., Lengacher S., Dusonchet J., Aebischer P., Schneider B. L. (2012) Sustained expression of PGC-1α in the rat nigrostriatal system selectively impairs dopaminergic function. Hum. Mol. Genet. 21, 1861–1876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Miura S., Tomitsuka E., Kamei Y., Yamazaki T., Kai Y., Tamura M., Kita K., Nishino I., Ezaki O. (2006) Overexpression of peroxisome proliferator-activated receptor gamma co-activator-1α leads to muscle atrophy with depletion of ATP. Am. J. Pathol. 169, 1129–1139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Russell L. K., Mansfield C. M., Lehman J. J., Kovacs A., Courtois M., Saffitz J. E., Medeiros D. M., Valencik M. L., McDonald J. A., Kelly D. P. (2004) Cardiac-specific induction of the transcriptional coactivator peroxisome proliferator-activated receptor gamma coactivator-1α promotes mitochondrial biogenesis and reversible cardiomyopathy in a developmental stage-dependent manner. Circ. Res. 94, 525–533 [DOI] [PubMed] [Google Scholar]
- 50. Brault J. J., Jespersen J. G., Goldberg A. L. (2010) Peroxisome proliferator-activated receptor gamma coactivator 1α or 1β overexpression inhibits muscle protein degradation, induction of ubiquitin ligases, and disuse atrophy. J. Biol. Chem. 285, 19460–19471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wenz T., Rossi S. G., Rotundo R. L., Spiegelman B. M., Moraes C. T. (2009) Increased muscle PGC-1α expression protects from sarcopenia and metabolic disease during aging. Proc. Natl. Acad. Sci. U. S. A. 106, 20405–20410 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 52. Cripps D., Thomas S. N., Jeng Y., Yang F., Davies P., Yang A. J. (2006) Alzheimer disease-specific conformation of hyperphosphorylated paired helical filament-Tau is polyubiquitinated through Lys-48, Lys-11, and Lys-6 ubiquitin conjugation. J. Biol. Chem. 281, 10825–10838 [DOI] [PubMed] [Google Scholar]
- 53. Ii K., Ito H., Tanaka K., Hirano A. (1997) Immunocytochemical co-localization of the proteasome in ubiquitinated structures in neurodegenerative diseases and the elderly. J. Neuropathol. Exp. Neurol. 56, 125–131 [DOI] [PubMed] [Google Scholar]
- 54. Ren Q. G., Liao X. M., Chen X. Q., Liu G. P., Wang J. Z. (2007) Effects of tau phosphorylation on proteasome activity. FEBS Lett. 581, 1521–1528 [DOI] [PubMed] [Google Scholar]
- 55. Li Y. P., Chen Y., Li A. S., Reid M. B. (2003) Hydrogen peroxide stimulates ubiquitin-conjugating activity and expression of genes for specific E2 and E3 proteins in skeletal muscle myotubes. Am. J. Physiol. Cell Physiol. 285, C806–C812 [DOI] [PubMed] [Google Scholar]
- 56. Geng T., Li P., Yin X., Yan Z. (2011) PGC-1α promotes nitric oxide antioxidant defenses and inhibits FOXO signaling against cardiac cachexia in mice. Am. J. Pathol. 178, 1738–1748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hishiya A., Iemura S., Natsume T., Takayama S., Ikeda K., Watanabe K. (2006) A novel ubiquitin-binding protein ZNF216 functioning in muscle atrophy. EMBO J. 25, 554–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Tsunemi T., Ashe T. D., Morrison B. E., Soriano K. R., Au J., Roque R. A., Lazarowski E. R., Damian V. A., Masliah E., La Spada A. R. PGC-1α rescues Huntington's disease proteotoxicity by preventing oxidative stress and promoting TFEB function. Sci. Transl. Med. 4, 142ra197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lustbader J. W., Cirilli M., Lin C., Xu H. W., Takuma K., Wang N., Caspersen C., Chen X., Pollak S., Chaney M., Trinchese F., Liu S., Gunn-Moore F., Lue L. F., Walker D. G., Kuppusamy P., Zewier Z. L., Arancio O., Stern D., Yan S. S., Wu H. (2004) ABAD directly links Abeta to mitochondrial toxicity in Alzheimer's disease. Science 304, 448–452 [DOI] [PubMed] [Google Scholar]
- 60. Yao J., Du H., Yan S., Fang F., Wang C., Lue L. F., Guo L., Chen D., Stern D. M., Gunn Moore F. J., Xi Chen J., Arancio O., Yan S. S. (2011) Inhibition of amyloid-β (Aβ) peptide-binding alcohol dehydrogenase-Aβ interaction reduces Aβ accumulation and improves mitochondrial function in a mouse model of Alzheimer's disease. J. Neurosci. 31, 2313–2320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Du H., Guo L., Yan S. S. (2012) Synaptic mitochondrial pathology in Alzheimer's disease. Antioxid. Redox Signal. 16, 1467–1475 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.