

Abstract
Despite the global medical needs associated with Staphylococcus aureus infections, no licensed vaccines are currently available. We identified and characterized a protein annotated as an epidermin leader peptide processing serine protease (EpiP), as a novel S. aureus vaccine candidate. In addition, we determined the structure of the recombinant protein (rEpiP) by X-ray crystallography. The crystal structure revealed that rEpiP was cleaved somewhere between residues 95 and 100, and we found that the cleavage occurs through an autocatalytic intramolecular mechanism. The protein expressed by S. aureus cells also appeared to undergo a similar processing event. To determine whether the protein acts as a serine protease, we mutated the hypothesized catalytic serine 393 residue to alanine, generating rEpiP-S393A. The crystal structure of this mutant protein showed that the polypeptide chain was not cleaved and was not interacting stably with the active site. Indeed, rEpiP-S393A was shown to be impaired in its protease activity. Mice vaccinated with rEpiP were protected from S. aureus infection (34% survival, P=0.0054). Moreover, the protective efficacy generated by rEpiP and rEpiP-S393A was comparable, implying that the noncleaving mutant could be used for vaccination purposes.—Kuhn, M. L., Prachi, P., Minasov, G., Shuvalova, L., Ruan, J., Dubrovska, I., Winsor, J., Giraldi, M., Biagini, M., Liberatori, S., Savino, S., Bagnoli, F., Anderson, W. F., Grandi, G. Structure and protective efficacy of the Staphylococcus aureus autocleaving protease EpiP.
Keywords: vaccine, epidermin leader peptide processing serine protease, pathogenesis, lantibiotic, abscess, immunogenicity
Ribosomally synthesized and post-translationally modified peptides (RiPPs) are a class of natural products that exist in all forms of life (1). Many gram-positive bacteria produce RiPPs that have antimicrobial activity and are called lantibiotics. These lantibiotics are effective antibacterial agents against other gram-positive bacteria (2, 3) and have been investigated as possible alternatives for the treatment of bacterial infections (4, 5). A cascade of proteins is required to both post-translationally modify the lantibiotic into its mature form and to protect the producing organism from the effects of the lantibiotic via immunity proteins (6). Indeed, producing bacteria protect themselves from the lethal action of its own antimicrobial lantibiotics by expressing immunity proteins. Generally, the genes coding for these proteins are found in clusters on either plasmids or chromosomes (7, 8) and have been identified in several bacteria, such as Lactococcus, Bacillus, Staphylococcus, Streptococcus, and Enterococcus. Recently, in silico screenings have uncovered 49 unidentified clusters from bacteria not known to produce lantibiotics (9), which indicates that these clusters are more common than previously thought. Not all genes in the clusters are conserved, nor are they arranged in the same order among strains and species (10). Moreover, not all bacteria that have these clusters can produce active lantibiotic, and if a lantibiotic is produced, it does not always have antibacterial activity (5, 11).
Most lantibiotic gene clusters have a gene that codes for a lantibiotic leader peptide protease (10). Epidermin leader peptide processing serine protease (EpiP) is a subtilisin-like extracellular epidermin leader peptidase and is required for proteolytic processing of the mature lantibiotic epidermin in Staphylococcus epidermidis (6, 12); however, its function in Staphylococcus aureus remains unknown, since there has been debate regarding whether S. aureus produces epidermin (13–15) or just retains the lantibiotic immunity genes for self-protection against other lantibiotics or for increased virulence. It has been shown that bacteria that have these lantibiotic gene clusters, even if they do not produce the lantibiotic, have increased virulence and resistance (16, 17). Many lantibiotic peptidases are cytoplasmic, but some, like NisP, CylP, and EpiP, reside extracellularly (10). The regulation of expression of exoproteins, surface proteins, and virulence factors for S. epidermidis and S. aureus are controlled by the accessory gene regulator (agr) quorum-sensing system (18). When agr was deleted in S. epidermidis, a decrease in mature epidermin production was seen, which was due to the decreased ability of EpiP to process the propeptide of epidermin rather than a decrease in the transcription of genes in the lantibiotic cluster (18). Therefore, agr quorum sensing does not interfere with the transcription of epidermin biosynthetic genes but controls the extracellular processing of the N-terminal leader peptide by the EpiP protease.
S. aureus is one of the most common opportunistic pathogens of humans and causes a wide range of diseases, from mild skin infections to life-threatening diseases such as sepsis, pneumonia, endocarditis, and osteomyelitis (19). The burden of staphylococcal disease is increasing due to the ability of S. aureus to acquire resistance to various antibiotics, including methicillin and vancomycin (20–22). In fact, methicillin-resistant S. aureus (MRSA) has been recognized as a major cause of infections in healthcare settings and community environments (23, 24). For example, in 2005, it was estimated that invasive MRSA infections occurred at a rate of >30/100,000 U.S. citizens and caused >18,000 patient deaths in the United States alone (25). The alarming increase in multiantibiotic resistance of S. aureus together with the wide variety and severity of staphylococcal infections pose a threat to public health and challenge our ability to control the disease. In particular, this is due to the lack of medical treatment alternatives to antibiotics (26–28). Although several vaccine candidates have been proposed, and some of them have been tested in clinical trials using both active and passive immunization modalities (29), an effective vaccine is still missing.
Herein we characterized a S. aureus protein annotated as an EpiP. The epiP gene contains a peptidase_S8 domain that is present in subtilisin-like serine proteases and is also present in protective antigens of several other species (30–36). In addition, homologous proteins expressed by other bacterial species have been shown to play important roles during pathogenesis. In particular, the Streptococcus pyogenes homologue SpyCEP (also named ScpC) inactivates IL-8 by catalyzing its C-terminal cleavage (37). As a consequence, SpyCEP impairs the recruitment of neutrophils at the site of infection and subsequent bacterial clearance (38, 39). Given that neutrophils are probably one of the most important elements of the host immune system against S. aureus infections (40), it will be important to understand whether EpiP has similar pathogenic mechanisms to SpyCEP. Unfortunately, there are currently no reports describing structural and functional characterization of the S. aureus EpiP protein. Therefore, we decided to perform studies aimed at revealing the first fundamental biochemical and functional properties of this novel protein, as well as its potential as a vaccine antigen against S. aureus infection. Herein, we present the structure of EpiP, the first structure of a lantibiotic processing protease, and we show that mice vaccinated with it are protected against S. aureus infection.
MATERIALS AND METHODS
Bacterial strains and medium and growth conditions
S. aureus Newman (ST254), Los Angeles clone (LAC; USA300), Mu50 (USA100), and Δspa-Newman strains were used for the study of the epiP gene and protein expression. The strains Newman, LAC, and Mu50 were grown overnight in tryptic soy broth (TSB) under agitation at 250 rpm, 37°C, whereas the Δspa-Newman strain was grown overnight in TSB growth medium supplemented with 10 μg/ml erythromycin.
Cloning of EpiP wild-type (WT) and mutant genes
epiP was amplified by polymerase chain reaction (PCR) from the S. aureus NCTC8325 strain and was cloned in absence of its putative leader sequence (aa 1–27) as an N-terminal 6X-histidine-tag (His-tag) construct. The His-tagged construct was cloned using oligonucleotides rEpiP forward CTGTACTTCCAGGGCTCAGAAGAACTATATTACAGTGTTG and reverse AATTAAGTCGCGTTAACTTGCTTTTTGATTTGCTACATTTAATGCT. The His-tagged PCR product was subcloned into the pET-15b+ vector using the polymerase incomplete primer extension (PIPE) technique (41). The Stratagene QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA, USA) was used to construct the rEpiP-S393A and rEpiP-S393G mutants according to the procedure outlined in the manufacture's technical manual. The conditions for the PCR reaction were initial denaturation at 95°C for 5 min, followed by 18 cycles of denaturation at 95°C for 1 min, annealing at 55°C for 1 min, extension at 68°C for 7 min, and final extension at 68°C for 10 min, then cooling to 4°C. A silent mutation for L394 (TTA to CTG) was added to improve the AT-rich region of this gene for PCR. The following primers were used to construct the mutants: rEpiP-S393G, forward GAAGATATATTTATCAAGCTGGAACTGGCCTGGCCACACCTAAAGTTTCG and reverse CGAAACTTTAGGTGTGGCCAGGCCAGTTCCAGCTTGATAAATATATCTTC; rEpiP-S393A, forward GAAGATATATTTATCAAGCTGGAACTGCGCTGGCCACACCTAAAGTTTCG and reverse CGAAACTTTAGGTGTGGCCAGCGCAGTTCCAGCTTGATAAATATATCTTC.
Recombinant protein expression and purification
The ampicillin-resistant rEpiP or mutant His-tagged plasmids were transformed into kanamycin-resistant BL21 (DE3) Magic cells, grown and expressed in terrific broth (TB), and harvested according to previously described procedures (42, 43). Pelleted cells were resuspended in buffer and sonicated, and cleared lysates were loaded onto an immobilized metal ion affinity chromatography (IMAC) Ni2+-affinity sepharose column (GE His-trap HP; GE Healthcare, Pittsburgh, PA, USA) as described previously (42). Fractions of protein from IMAC were pooled and diluted with 10 mM Tris-HCl (pH 8.3) to reduce the salt concentration and then loaded onto a GE HiTrap SP 5-ml column for ion exchange (IEX) chromatography. The loading buffer was composed of 10 mM Tris-HCl (pH 8.3) and 5 mM β-mercaptoethanol, and the protein was eluted over 30 column volumes with a linear gradient from 0 to 1 M NaCl. rEpiP or mutant protein eluted at ∼0.27 M NaCl and was concentrated using Amicon protein concentrators (10,000 MWCO; Millipore, Billerica, MA, USA). Protein concentration was determined using the absorbance at 280 nm and the extinction coefficient 0.942 M−1 · cm−1. Protein purity was determined using SDS-PAGE and was purified to near homogeneity.
Protein crystallization
The sitting-drop vapor-diffusion method was used for protein crystallization at 295 K in a 96-well plate format with 1 μl of precipitant and 1 μl of enzyme (7 mg/ml for rEpiP and 7.8 mg/ml for rEpiP-S393A). The Classics II, PACT, and JCSG+ commercial screens from Qiagen (Valencia, CA, USA) were used for crystallization trials. Several nicely diffracting crystals grew after 3–5 d for rEpiP in 0.2 M lithium sulfate, 0.1 M Bis-Tris (pH 5.5), and 25% (w/v) PEG 3350; however, only 1 crystal was obtained on a fiber after several weeks for the rEpiP-S393A in 0.2 M calcium chloride, 0.2 M Tris (pH 8.0), and 20% (w/v) PEG 6000. No protein crystals were observed even after 1 yr for the rEpiP-S393G. Single crystals were transferred to the corresponding mother liquor and flash-frozen in liquid nitrogen for data collection. The rEpiP had unit cell parameters of a = 85.5 Å, b = 94.7 Å, c = 123 Å, with α = 89.98°, β = 90.37°, and γ = 116.79°, and belonged to the P1 space group. The rEpiP-S393A had unit cell parameters of a = 71.23 Å, b = 68.47 Å, c = 97.72 Å, with α = 90°, β = 91.66° and γ = 90°, and belonged to the P21 space group.
Data collection and refinement
Data were collected at 100 K from single protein crystals at Argonne National Laboratory (Argonne, IL, USA) at the Advanced Photon Source (APS) on beamline 21-ID-F at a resolution of 2.05 Å and a wavelength of 0.97872 Å for rEpiP and on beamline 21-ID-G at a resolution of 1.95 Å and a wavelength of 0.97856 Å for rEpiP-S393A. The diffraction images for both data sets were indexed, integrated, and scaled with HKL-3000 (44). The structures of the rEpiP and rEpiP-S393A proteins were solved by molecular replacement using Protein Data Bank (PDB) codes 1THM and 3QFH, respectively, as the search model with Phaser (45). The initial model for rEpiP was rebuilt using ARP/wARP (46). Structures were refined using Refmac (47) and manually corrected using Coot (48). Both structures were checked and validated using Sfcheck (49) and MolProbity (50). The coordinates were deposited into PDB using accession codes 3QFH for rEpiP and 3T41 for rEpiP-S393A. Statistics for data collection are shown in Table 1. The PyMOL Molecular Graphics System 1.5.0.4 software (Schrodinger LLC, Portland, OR, USA) was used to create the figures.
Table 1.
Parameters for crystallography with statistics from data collection, refinement, and validation
| Parameter or statistic | WT | S393A |
|---|---|---|
| Data collection | ||
| Space group | P1 | P21 |
| Unit cell: a, b, c(Å) | 85.5, 94.7, 123.0 | 71.2, 68.5, 97.7 |
| α, β, γ (deg) | 90.0, 90.4, 116.8 | 90.0, 91.7, 90.0 |
| Wavelength (Å) | 0.97872 | 0.97856 |
| Resolution (Å) | 30-2.05 (2.09–2.05) | 30-1.95 (1.98–1.95) |
| Observed reflections (n) | 219,399 (10,837) | 63,885 (3419) |
| Rmerge (%) | 4.7 (35.4) | 4.5 (47.3) |
| Completeness (%) | 97.9 (97.0) | 98.0 (97.8) |
| I/σI | 15.0 (2.1) | 25.5 (3.0) |
| Phasing method | MR | MR |
| Refinement and validation | ||
| Rwork/Rfree (%) | 16.95/21.60 (24.8/27.2) | 16.60/19.48 (22.6/24.5) |
| AMP/GOL | 3 Na, EDO, 4 GOL, 38 SO4 | 6 Ca, 1 Cl |
| Solvent molecules | 1511 | 487 |
| Bond lengths (Å) | 0.011 | 0.011 |
| Bond angles (deg) | 1.35 | 1.38 |
| Ramachandran (%) | ||
| Most favored regions | 88.1 | 87.3 |
| Additionally allowed regions | 11.8 | 12.4 |
| Generously allowed regions | 0.1 | 0.3 |
| Disallowed regions | 0.0 | 0.0 |
| PDB ID | 3QFH | 3T41 |
High-resolution shell statistics are shown in parentheses. Total number of residues in the protein is 430 from residues 28–457. The 3QFH structure has 8 molecules in the asymmetric unit (chains A–H) and has 6 missing residues between the prodomain and protease domain for chain A, 5 for chain B, 9 for chains C and G, and 4 for chains D, E, F, and H. The 3T41 structure has 2 molecules in the asymmetric unit, where 7 residues are missing from chain A and 10 from chain B at the C-terminal end of the protein.
EpiP cleavage mechanism
For the evaluation of the cleavage mechanism of EpiP (whether it was intra- or intermolecular), rEpiP-S393A was incubated in 50 mM Tris buffer (pH 8) with rEpiP in an ∼0.25 M ratio of rEpiP to rEpiP-S393A in a final volume of 50 μl. Samples were incubated at 37°C for 1, 4, and 24 h and were analyzed by SDS-PAGE. The protein bands were visualized by Coomassie Brilliant Blue.
Mass spectrometry analyses
For intact mass measurement of rEpiP, protein was diluted in 0.1% formic acid. The acidified protein solutions were loaded onto a Protein MicroTrap cartridge (from 60 to 100 pmol; Michrom Bioresources, Inc., Auburn, CA, USA), desalted for 2 min with 0.1% formic acid at a flow rate of 200 ml/min, and eluted directly into the mass spectrometer using a step gradient of acetonitrile (55% acetonitrile and 0.1% formic acid). Spectra were acquired in positive mode on a SynaptG2 HDMS mass spectrometer (Waters Corp., Milford, MA, USA) equipped with a Z-spray electrospray ionization source. The quadrupole profile was optimized to ensure the best transmission of all ions generated during the ionization process.
For peptide mass fingerprinting (PMF) of rEpiP, spots of colloidal Coomassie-stained rEpiP bands were excised from the SDS-PAGE gel using a Pasteur pipette and destained overnight in 200 μl of 50% (v/v) acetonitrile and 50 mM ammonium bicarbonate. The spots were then washed with 200 μl of acetonitrile. The acetonitrile was discarded, and the spots were allowed to air dry. Modified trypsin (12 μg/ml) in 5 mM ammonium bicarbonate was added to each spot, and the enzymatic digestion was performed for 3 h at 37°C; 0.8 μl of the digestion was directly spotted on a prespotted anchor chip (PAC) target (PAC 96 set for proteomics; Bruker Daltonics, Bremen, Germany). The air-dried spots were washed with 0.6 μl of a solution of 70% (v/v) ethanol and 0.1% (v/v) trifluoroacetic acid (TFA). Peptide mass spectra were recorded with a matrix-assisted laser desorption ionization-time of flight (MALDI-TOF) mass spectrometer (UltraFlex; Bruker Daltonics). Ions generated by laser desorption at 337 nm (N2 laser) were recorded at an acceleration of 25 kV in the reflector mode. About 200 single spectra were accumulated for improving the signal/noise ratio and analyzed by Flex Analysis 2.4 (Bruker Daltonics). External calibration was performed using standard peptides prespotted on the target. Peptide identification was performed using BioTools and Sequence Editor 3.0 (Bruker Daltonics).
EpiP expression analysis in S. aureus
The expression of EpiP in S. aureus was evaluated in vitro, by Western blot and real-time quantitative reverse transcription PCR (qRT-PCR). For Western blot analysis, equal amounts of total cell proteins from the Δspa-Newman strain were separated on SDS-PAGE and transferred to nitrocellulose membranes. S. aureus Δspa-Newman strain was grown overnight in TSB (supplemented with 10 μg/ml erythromycin) at 250 rpm at 37°C using aerated Erlenmeyer flasks. The culture was centrifuged at 4000 rpm at 4°C for 15 min. Cell wall fractions were prepared by resuspending pellets from 5-ml overnight cultures in 500 μl TSM buffer (50 mM Tris HCl, pH 7.5; 10 mM MgCl2; and 0.5 M sucrose). Lysostaphin (50 μl, 5 μg/μl stock; Sigma-Aldrich, St. Louis, MO, USA) was added to samples and incubated for 1 h at 37°C at 400 rpm in a Thermomix (Vorwerk, Wuppertal, Germany). Samples were centrifuged at 4000 rpm for 15 min at 4°C, and supernatants containing the cell wall fraction were used for Western blot analysis using sera raised against EpiP. The supernatant protein (extracellular) was prepared by concentrating 10 ml of overnight culture supernatant up to 100× using a Vivapore 10/20 solvent absorption concentrator (7500-Da cutoff; Sigma-Aldrich).
To measure expression of the epiP gene, bacterial cultures at exponential and stationary phase were centrifuged at 4000 rpm for 15 min at 4°C. Bacterial pellets were resuspended in 200 μl TSM buffer and vigorously vortexed. A 10 μl aliquot of lysostaphin (5 μg/μl stock) was added to this suspension and incubated at 37°C for 15 min. After incubation, RNA extraction was performed with the RNeasy Mini kit (Qiagen) according to the manufacturer's instructions. RNA was quantified using a Bioanalyzer (Agilent, Santa Clara, CA, USA). To remove eukaryotic RNA contamination and to enrich the proportion of bacterial RNA, a MicrobeEnrich kit (Ambion, Austin, TX, USA) was used according to the manufacturer's instructions. Total RNA (1 μg) was reversed transcribed with 0.5 mM dNTP, 50 ng random hexamers, and 200 U of Superscript II reverse transcriptase (Invitrogen, Carlsbad, CA, USA), according to the manufacturer's recommendations. RNA was denatured and the cDNAs were purified with a QIAquick PCR purification kit (Qiagen). cDNA (1 μl)was amplified by real-time qRT-PCR on the Stratagene MX3000P (Stratagene) with the SYBR GreenER qPCR universal kit (Invitrogen) and 10 μM of the rEpiP forward CATAAAGCGCGCTATTATTAG and reverse CTTTATACACATCAAGCTCAC primers. Reaction mixtures were denatured for 10 min at 94°C, followed by 40 cycles of 30 s at 60°C and 1 min at 72°C, and finished with a dissociation ramp from 55 to 95°C. The level of expression of the epiP gene was calculated by using the cycle threshold (Ct) of the overnight-grown bacteria as the calibrator. epiP expression was normalized against 16s rRNA, for which expression was found to be constant throughout the S. aureus growth phase.
Enzymatic assays
Samples for testing activity of EpiP toward casein or collagen were prepared by addition of 1 mg of rEpiP protein to 200 μl of agarose-collagen or agarose-casein suspension (Sigma-Aldrich) that was washed with buffer (50 mM Tris-HCl, pH 8.0, and 2 mM CaCl2) in a final volume of 1 ml. Samples were incubated at 37°C with shaking at 125 rpm overnight and then centrifuged for 3 min at 13,000 rpm on a tabletop centrifuge to pellet the unreacted immobilized substrate. The pellet was washed with 1 ml of buffer and centrifuged again to obtain any additional proteolytic fragments, and 15-μl aliquots of the supernatant of each sample were used for SDS-PAGE.
Proteolytic activity of the rEpiP protein was tested against azocoll (dye-impregnated collagen; Sigma-Aldrich) using a modified procedure (51). To begin, 0.25 g of azocoll was washed twice in 50 ml buffer (50 mM Tris HCl, pH 7.8, and 1 mM CaCl2) in a 100-ml beaker and stirred rapidly for 2 h. The azocoll was filtered using Whatman no. 3 filter paper (Whatman, Maidstone, UK) and was resuspended in fresh buffer immediately before using in the assay. The suspended azocoll was dispensed as 1-ml aliquots into glass test tubes using a 5-ml syringe; 0.04 mg of enzyme was added to each reaction, and the tubes were incubated at 37°C with shaking for 24 h. Approximately 4 mg of azocoll was in each 1-ml reaction. After the reaction, samples were centrifuged, the supernatant was removed, and the absorbance at A550nm was measured using a Bio-Tek ELx808 microplate reader equipped with filters (Bio-Tek Instruments, Inc., Winooski, VT, USA). The negative control for the reaction included the azocoll suspension without EpiP enzyme, and the positive control was subtilisin from Bacillus licheniformis (85968; Sigma-Aldrich). The reactions were performed in triplicate. A unit is defined as the change in A550nm per hour per milliliter of reaction.
Ethics statement
Mice were monitored daily and euthanized at the appearance of humane endpoints in accordance with the Novartis Animal Welfare Policies and Italian law. Experimental protocols were reviewed and approved by the Italian National Institute of Health, Istituto Superiore di Sanità (ISS), protocol 136/2010-B, for mouse studies.
Immunogenicity assay
EpiP antibody titers present in sera of immunized mice were measured by Luminex technology (Luminex 200; Luminex Corp., Austin, TX, USA). The protein was covalently conjugated to the free carboxyl groups of microspheres using an N-hydroxysulfosuccinimide-enhanced carbodiimide-mediated conjugation chemistry. Antigen-specific antibodies were revealed by phycoerythrin-labeled secondary antibodies. The assay readout is a measure of fluorescence intensity expressed as arbitrary relative luminex units (RLU) per milliliter.
Peritonitis model
Immunized animals were challenged on d 24 by intraperitoneal injection of a lethal dose of S. aureus. Mice were monitored daily for 7 d. TSB cultures of S. aureus were centrifuged, washed twice, and diluted in PBS before challenge. Further dilutions were needed for the desired inoculum, which was experimentally verified by agar plating and colony counting. Mice were infected with ∼5 × 108 colony-forming units of the S. aureus Newman strain.
Statistical analysis
Two independent experiments, run under the same conditions, were performed to assess protective efficacy of EpiP in the peritonitis model. Experiments were analyzed using Fisher's exact test.
RESULTS
In silico analysis predicted EpiP to be surface located and identified a signature of protection
The reverse vaccinology approach was applied to the S. aureus NCTC8325 genome, and several bioinformatic algorithms were used to select surface-associated and secreted proteins as described elsewhere (52–54). Among the selected antigens, we focused on the sequence SAOUHSC_01949, annotated as EpiP. EpiP was predicted to have an extracellular localization due to the presence of a leader peptide and the lack of other known signals for membrane or cell wall anchoring. EpiP also showed a peptidase_S8 domain organized with the catalytic triad Asp, His, and Ser, which is typical for the widespread subtilisin-like family of serine proteases (ref. 55 and Fig. 1). The analysis of the 177 completely annotated S. aureus genomes available through the UniProt Knowledgebase (release 2013_02) revealed that epiP is a dispensable gene, and it is either well conserved (present in 49 strains with >90% identity) or absent (Supplemental Tables S1 and S2). The analysis of the genomic organization showed that epiP is a part of an 8-gene operon (epiABCDPFEG), similar to the one previously reported for the lantibiotic genes in S. epidermidis (56). Figure 2A reports the epi operon region view in a representative subset of S. aureus strains, showing that when epiP is present, the entire operon organization is also conserved. Among strains where epiP was absent, we wanted to investigate whether the epi operon was also absent. To this aim, we selected strains for which a complete genome set is available, i.e., with high-quality reviewed annotation. We found that in most strains (9 of 13) the epi operon was completely absent. On the other hand, the remaining 4 strains showed a truncated epiG gene in the corresponding epi region (Fig. 2B).
Figure 1.
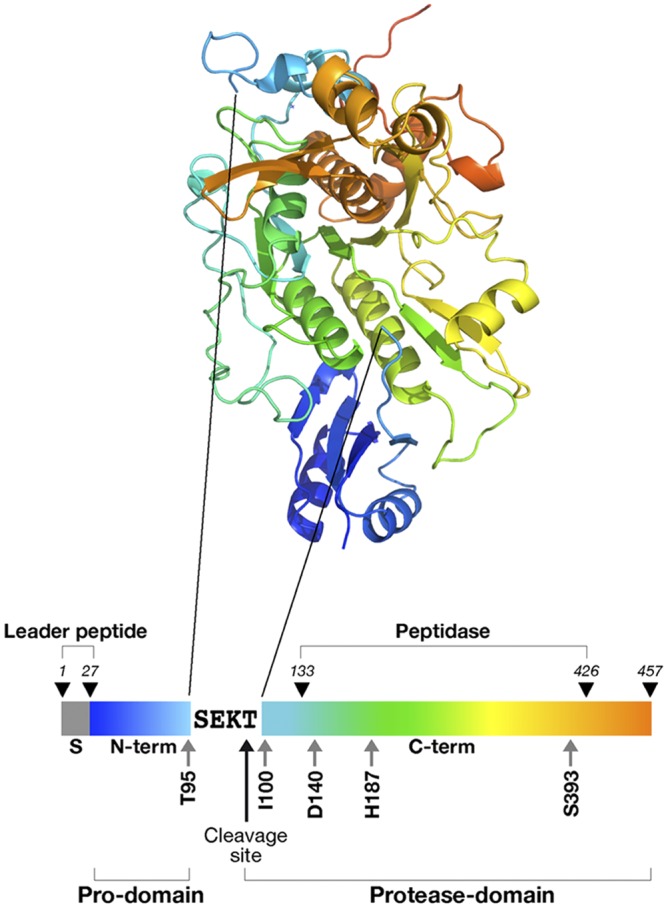
EpiP organization and its corresponding 3-dimensional structure. Layout of the EpiP protein, including the leader peptide, prodomain, cleavage site, and protease domain, is shown. The catalytic triad includes residues D140, H187, and S393, and the peptidase_S8_lantibiotic-specific protease domain includes residues 133–426. To obtain soluble protein, the leader peptide (residues 1–27) was removed and replaced by a polyhistidine tag. The cleavage site between the prodomain and protease domain is depicted on the bar beneath the structure, and is also indicated on the structure with arrows. Residues SEKT are not always included in the electron density of each chain of the rEpiP crystal structure. The coloration of the bar representing the primary sequence is equivalent to the coloration seen in the structure. There is no cell wall anchoring domain predicted in the sequence.
Figure 2.

Schematic representation of the genomic organization of the epi region in S. aureus strains. A) S. aureus strains where epiP is present. B) Corresponding region in strains carrying a truncated epiG gene. Open reading frames are represented as arrows showing the direction of transcription, with the size reflecting the relative length of the genes and the different colors reflecting the gene family.
Bacterial peptidase_S8 proteins have been shown to play important roles during pathogenesis by cleaving neutrophil chemoattractant factors such as IL-8 and C5a, thus impairing the recruitment of neutrophils at the site of infection (37, 38, 57). In addition, members of this family have been identified as protective antigens and proposed as vaccine candidates. These include the Streptococcus agalactiae C5a peptidases, the S. pyogenes SpyCEP, the Streptococcus pneumoniae PrtA, and the Neisseria meningitidis NMB1969 (AspA) (30, 38, 57–59). Therefore, we hypothesized that the peptidase_S8 domain may represent a signature of protection and that EpiP could be a protective antigen against S. aureus infection. Hence, we performed a more thorough investigation into structural, functional, and immunogenic properties of this protein.
Structure of EpiP
Subtilisin-like serine proteases typically cleave themselves into 2 domains: an N-terminal prodomain and a C-terminal protease domain, which contains catalytic residues that are used to cleave specific substrates. We have determined the 3-dimensional structure of the rEpiP from S. aureus in its cleaved form with the prodomain noncovalently interacting with the protease domain (Figs. 1 and 3). The prodomain is composed of 4 β strands flanked by 2 α helices, and the protease domain has a fold that is characteristic of a subtilisin-like protease. The electrostatic surface interactions between these 2 domains are tight and complementary (Fig. 4), and attempts to clone these domains individually or to chromatographically separate them when they were produced from the full-length construct were unsuccessful. There are 8 molecules in the asymmetric unit for this structure, and all of the molecules are cleaved somewhere between residues 95 and 100 (TSEKTI; Fig. 1). The electron density surrounding this cleavage site is only present for residues CSTCxxxxxTI in chain A, CSTCIxxxxTI in chain B, CxxxxxxxxxI in chains C and G, and CSTCITxxxxI in chains D–F and H, where x indicates lack of density for that particular residue. Due to the lack of and inconsistent electron density for residues in this region, it is not obvious what the cleavage specificity is from the structure alone.
Figure 3.
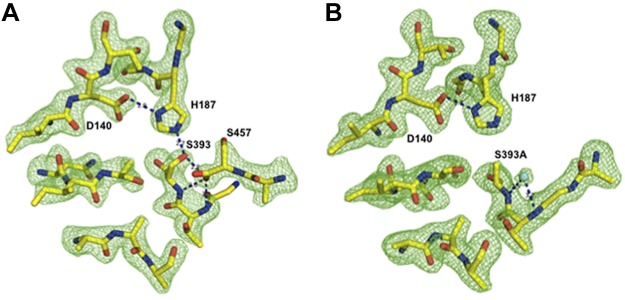
Active site comparison of WT and S393A mutant EpiP. Ball-and-stick models of the catalytic triad (D140, H187, and S393) and selected residues from the active site are presented. To indicate the high quality of the refinement and the final models, Fo-Fc omit maps (green) contoured at the 3σ level for the WT (A) and mutant (B) proteins are shown. In the active site of the WT protein, S457 of the C terminus of a neighboring WT protein molecule is bound in the active site. In the mutant protein, a water molecule resides in the same location as one of the oxygen atoms of the C-terminal carboxyl group of S457. Hydrogen bonds are shown by black dashes, nitrogen atoms are shown in blue, oxygen atoms in red, carbon atoms in yellow, and water in cyan.
Figure 4.
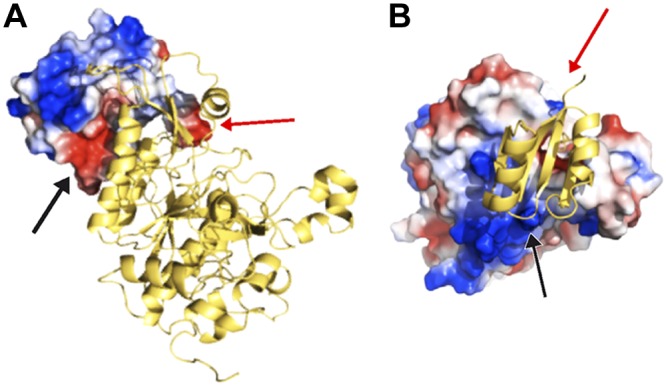
Complementary electrostatic charge interactions of the surfaces of the prodomain and protease domain in rEpiP protein. A) Prodomain as an electrostatic surface and the protease domain as a ribbon diagram. B) Prodomain as ribbon diagram and protease domain is an electrostatic surface. Arrows indicate corresponding interactions of the 2 domains in each panel. Electrostatic surfaces are shown in red for negative charge, blue for positive charge, and white for neutral charge.
We were curious whether EpiP cleaved itself into the 2 separate domains or whether there was another protein involved in processing the prodomain, so we mutated the conserved serine residue at position 393 to alanine (S393A) and determined the structure of the mutant protein. rEpiP-S393A crystallized in a different space group than rEpiP, and it contained 2 molecules in the asymmetric unit compared with 8 (Table 1). The structure of rEpiP-S393A showed that the protein remained fully intact, which indicates that this serine residue is indeed involved in the self-cleavage mechanism for EpiP. In the rEpiP structure, the polypeptide chain that links the prodomain and protease domain acts like a rubber band and snaps once it is cleaved to expose the active site of the protease domain (Fig. 5). In the rEpiP-S393A mutant structure, however, this polypeptide chain hovers above the active site and does not interact with it directly (Figs. 5 and 6). Both molecules in the asymmetric unit of rEpiP-S393A have disordered regions of this polypeptide chain, which include 102–106 for one molecule and 100–106 for another molecule (Table 1).
Figure 5.
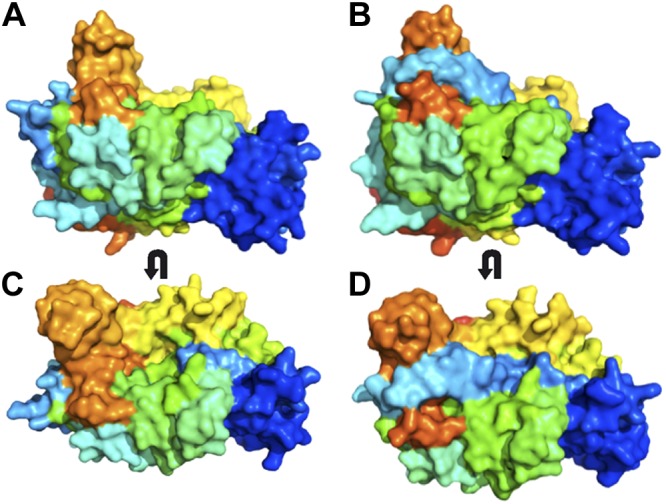
Surface representations of the rEpiP and rEpiP-S393A proteins. A, B) WT protein. C, D) S393A mutant. A, C) Side view of the protein. B, D) Top view of the protein. Coloration of the surface diagrams is the same as in Fig. 1. Polypeptide chain that links the prodomain and protease domains is shown in light blue.
Figure 6.
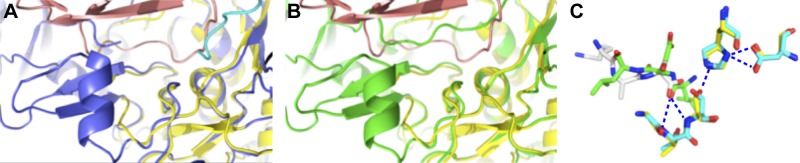
Comparison of active sites of rEpiP and rEpiP-S393A proteins with a thermitase-eglin-c complex. A) Overlay of rEpiP and a thermitase-eglin-c complex (PDB ID: 2TEC). B) Overlay of the rEpiP-S393A and thermitase-eglin-c complex. rEpiP is shown in blue, rEpiP-S393A in green, and the thermitase-eglin-c complex in pink. The C terminus of a neighboring molecule in the rEpiP active site is shown in cyan. C) Ball-and-stick representation of the superposition of the active sites of the thermitase-eglin-c complex and rEpiP. Nitrogen atoms are shown in blue, oxygen atoms in red, carbon atoms for the thermitase in yellow and for rEpiP (chain A) in cyan, the thermitase-eglin-c complex in green, and the C-terminal portion of chain H of rEpiP in gray. Despite the fact that peptide chains for thermitase-eglin-c complex and rEpiP chain H run in the opposite direction, carbonyl oxygen atoms are pointed in the same direction. The position of one of the C-terminal oxygens of rEpiP chain H matches the position of the carbonyl oxygen of the thermitase-eglin-c complex, which is bound in the oxyanion hole. There are almost no differences in the active sites of rEpiP and rEpiP-S393A, so the mutant is not shown here. In the crystal structure of rEpiP, the active site of the protein contains the C terminus of a neighboring molecule as follows: in the active site of A–H is the C terminus of chains H, D, F, G, B, A, E, and C, respectively.
In rEpiP, the active site contains the C terminus of a neighboring molecule of the asymmetric unit (Fig. 6). The placement of this polypeptide chain in the active site is in the reverse direction of what would be expected for an actual substrate. This is because the C-terminal carboxyl group of S457 on the chain of the neighboring molecule does not mimic the proper conformation of this group (see structural comparison of rEpiP with 2TEC in Fig. 6). However, the active site of EpiP has a high affinity for a carboxyl group because it pulls the C-terminal tail of a neighboring molecule into its active site. The S393 residue of the catalytic triad of rEpiP interacts with the Oγ side chain and an oxygen atom of the terminal carboxyl group of residue S457 from a neighboring molecule. Other active site interactions with the neighboring molecule include N290 with an oxygen atom of the terminal carboxyl group of S457 in a different manner than the S393 interactions and the carbonyl group of A390 with the nitrogen atom of S457. The catalytic H187 residue of rEpiP has one nitrogen atom that interacts with Oγ of S393 and another nitrogen that interacts with the oxygen atoms of the catalytic D140 residue (Fig. 6). rEpiP-S393A does not generate this type of interaction between neighboring molecules of the asymmetric unit. In fact, the C-terminal end of the molecule is disordered, and the last residue with electron density is either A452 or Q454.
The prodomain of rEpiP contains 2 cysteine residues (C90 and C93) that reside directly upstream of the polypeptide cleavage site between the 2 domains of the protein. In the rEpiP structure, the presence of a disulfide bond between C90 and C93 of the prodomain varies among chains in the asymmetric unit. In chains E and F a disulfide bond exists between these 2 residues but is absent in chains A, B, D, G, and H. Although chain H does not have a disulfide bond, these 2 cysteine residues face each other and their electron density is connected. There is no electron density for residue C90 in chain C, and thus lacks a disulfide bond. In the rEpiP-S393A, however, these 2 cysteine residues form disulfide bonds and are present in both chains of the asymmetric unit. It is not clear what the role of these cysteine residues is in EpiP, but they are only conserved in the lantibiotic protease from S. epidermidis and S. aureus.
Cleavage mechanism of the polypeptide extension between the prodomain and protease domain of EpiP
EpiP deprived of its putative leader sequence (aa 1–27) was expressed in, and purified from, Escherichia coli-soluble extracts. rEpiP appeared as 3 bands migrating with apparent molecular weights of ∼ 50, 39, and 8 kDa by SDS-PAGE (Fig. 7A). PMF of the 3 bands separated by SDS-PAGE identified peptides specific for the 3 fragments (Fig. 7B). More specifically, peptides covering both the N and C terminus of the protein were identified in the upper 50-kDa band, while coverage of the 39-kDa fragment spanned amino acids starting from threonine 88 to the end of the C terminus, and in the 8-kDa fragment, identified peptides mapping in the first 74 aa residues of rEpiP.
Figure 7.

Recombinant EpiP is composed of 3 polypeptide fragments. Analysis of recombinant EpiP purified from E. coli extracts through SDS-PAGE, and mass spectrometry. A) In SDS-PAGE stained with Coomassie, the proteins migrated with 3 species (bands 1–3). Molecular mass of the bands from top to bottom is comparable with the size of the mature protein (≈50 kDa), the protease domain (≈39 kDa), and the prodomain (≈8 kDa), respectively. B) Peptides identified by PMF in bands i–iii in panel A are indicated on the sequence of rEpiP: red indicates peptides identified in band i, green indicates peptides from band ii, and blue indicates peptides from band iii. C) Experimental mass observed by intact mass measurement of the rEpiP fits with band ii in panel A. Sequence starts with threonine 88 of rEpiP, indicating that the cleavage site of the protein occurred between lysine 87 and threonine 88, corresponding to residues 98 and 99, respectively, of EpiP. Asterisk indicates loss of H2O.
The theoretical mass of rEpiP is 49744.27 Da (aa 1–446), but by intact mass spectrometry we observed an experimental mass of 39694.48 Da (aa 88–446; 30 ppm tolerance; Fig. 7C) for the most abundant species in the sample starting with amino acid threonine at position 88. This observation fits well with the pattern obtained by SDS-PAGE in which the first peptide identified by PMF in the band migrating at 39 kDa begins with threonine 88. Altogether, these data indicate that rEpiP is composed of 3 polypeptides resulting from the cleavage of the protein between amino acids lysine 87 and threonine 88, corresponding to residues 98 and 99 of the WT protein.
We next investigated whether cleavage of EpiP could be due to autoproteolysis, as reported for other extracellular bacterial proteases (58). To address this point, we generated a rEpiP protein in which the serine at position 393 of the predicted catalytic triad was substituted with alanine (rEpiP-S393A). The purified mutant protein was then subjected to SDS-PAGE analysis to investigate whether the mutation impaired the cleavage pattern observed with rEpiP. As shown in Fig. 8, the rEpiP-S393A appeared as a single band with an electrophoretic mobility comparable to that of the noncleaved protein band of rEpiP.
Figure 8.

EpiP cleavage is dependent on serine residue 393 and occurs through an autocatalytic intramolecular mechanism. Analysis of the autoproteolytic activity of the recombinant purified EpiP and its mutant derivative by a time course coincubation of the 2 proteins in a 0.25 M ratio. The 2 recombinant proteins (rEpiP and rEpiP-S393A) were used as molecular mass controls (lanes 1 and 2). No decrease in the intensity of the noncleaved EpiP-S393A was observed throughout the 24 h incubation time, which indicates that EpiP uses an intramolecular autocleavage to remove the prodomain.
The combination of SDS-PAGE analysis and the 3D structure of the rEpiP-S393A mutant shows that the full-length protein remains intact in the absence of this serine residue. To understand whether the cleavage of EpiP occurs through an inter- or an intramolecular mechanism, we tested whether rEpiP could cleave the prodomain of rEpiP-S393A. rEpiP and rEpiP-S393A were mixed in a 0.25 M ratio, incubated for varying lengths of time, and then analyzed using SDS-PAGE. During this experiment, we observed no increase in intensity of the protease domain of the protein by SDS-PAGE (Fig. 8), indicating that rEpiP does not cleave rEpiP-S393A. We also tested the effect of varying concentrations of rEpiP in the presence of constant concentrations of rEpiP-S393A for up to 24 h and did not observe a decrease in the intensity of the full-length (mutant) band (data not shown). Given that rEpiP did not cleave rEpiP-S393A during extended periods of time or increased concentrations of rEpiP, these data suggest that the cleavage event of EpiP occurs through an intramolecular autocatalytic mechanism.
Enzymatic activity of the recombinant EpiP
Although EpiP is annotated as being responsible for proteolytically processing mature epidermin in S. epidermidis, this lantibiotic is not produced in S. aureus (14). Thus, it is not clear what the function of this protease is in S. aureus. To investigate a potential functional role of this protein that is alternative to epidermin cleavage, we subjected rEpiP to in vitro proteolytic assays. The S1 pocket of EpiP is relatively shallow and hydrophobic and resembles that of an elastase-like serine protease. Based on this information, we tailored the enzymatic assays to investigate whether the enzyme had activity toward an elastase-like protease substrate in addition to its own intramolecular cleavage.
We determined that the rEpiP protein was able to cleave both casein and collagen, indicating that the enzyme is capable of proteolytically processing substrates other than itself. First, we tested EpiP activity toward casein or collagen that was immobilized onto agarose beads and visualized the cleavage products via SDS-PAGE. Some of the proteolytic fragments of casein were large enough to be seen on the gel, but collagen was not (data not shown). At this point, it was not clear whether collagen was being proteolyzed into smaller fragments than what we could visualize on SDS-PAGE, or whether it was not a substrate. To address this question, we used a colorimetric assay to determine whether EpiP could cleave azocoll, a dye-impregnated collagen, with subtilisin from B. licheniformis as a positive control. Compared with subtilisin, the activity of the rEpiP enzyme toward azocoll was 2.4-fold lower (0.31 compared with 0.73 U/mg); however, it was of the same order of magnitude as subtilisin. The subtilisin enzyme began cleaving the collagen more quickly than EpiP, so the activity reported for subtilisin after 24 h may be artificially lower than expected. It is not clear whether the decrease in activity of rEpiP compared with subtilisin is due to the presence of the prodomain of rEpiP or whether the enzyme is just less active than subtilisin. Based on the structure of the rEpiP, it is likely that this prodomain does not need to be removed for catalysis to occur. One reason is that once the polypeptide chain that links the prodomain and protease domains is cleaved, the structure becomes more open and exposes the active site. Another reason is that we were not able to separate the 2 domains of the protein; therefore EpiP is constantly in the presence of its prodomain. Since the enzyme is active, the presence of its prodomain does not seem to be inhibitory for EpiP compared with other types of subtilisin-like proteases that must have their prodomains removed before they become active.
A comparison of the sequences of EpiP from S. epidermidis and S. aureus within 4 Å of the conserved catalytic triad residues indicates that 72% are identical and the remaining residues have similar characteristics, with the exception of K185 and G186 of S. aureus (L192 and N193 for S. epidermidis, respectively). Based on the high sequence identity and similarity of the residues in the active sites of EpiP from S. aureus and S. epidermidis, we cannot rule out the possibility that EpiP from S. aureus may indeed cleave epidermin produced by S. epidermidis. However, further experiments will be necessary to confirm this hypothesis.
EpiP is expressed and processed in S. aureus cells
To assess the expression of the epiP gene, real-time qRT-PCR was performed on RNA isolated from bacteria grown in TSB, which is the same medium used to prepare challenge inocula for infection experiments in the animal model. Under these conditions, the gene was found to be expressed and increased gene expression was observed in exponential compared with stationary growth phase (Supplemental Fig. S1). Moreover, we also looked for the epiP gene expression in different S. aureus strains (i.e., Newman, LAC, and Mu50) by qRT-PCR with RNA isolated from bacteria grown to exponential phase in TSB. Under the condition tested, the epiP gene was expressed in Newman and LAC strains but not in Mu50. Furthermore, LAC showed ∼2-fold increased gene expression as compared with Newman (Fig. 9A).
Figure 9.

EpiP expression and processing in S. aureus. A) epiP gene was expressed in the Newman and LAC strains, whereas no expression was detected in the Mu50 strain, as expected. Furthermore, LAC showed ∼2 fold increased gene expression as compared with the Newman strain. B) Analysis of culture supernatant, cell wall fraction and protoplast of S. aureus strain Newman by Western blot using an EpiP mouse antiserum. The 3 polypeptides (noncleaved, protease domain, and prodomain) of the purified recombinant protein were recognized by the serum; however, the predominant bands that were recognized by the serum had a molecular mass compatible with the full-length protein and that of the prodomain of EpiP. In the supernatant preparation, a faint band with a molecular mass congruous with the noncleaved protein was visible as well an immunoreactive band with a size similar to the prodomain. An additional band was visible which does not correspond to any of the 3 polypeptides of EpiP. No immunoreactivity was detected in the lane where the cell wall preparation was loaded. A major immunoreactive band was present in the protoplast preparation comparable to the molecular mass of the noncleaved protein. Other minor protein species were also detected that appeared similar to the ones observed in the lower molecular mass range of the culture supernatant preparation.
The S. aureus EpiP lacks a known localization signal (Fig. 1), and it may either be released in the extracellular milieu or be anchored to the cell wall through an unknown mechanism, as reported for EpiP of S. epidermidis (6, 8, 60, 61). To experimentally verify its localization, we isolated the cell wall fraction and the culture supernatant, as well as protoplasts of S. aureus cells, and analyzed them by immunoblot using an EpiP mouse antiserum. For this experiment, we decided to use the S. aureus strain Newman deficient for SpA (SEJ2; ref. 62). This strain was selected to reduce the nonspecific staining in Western blot analyses due to the binding of IgGs mediated by SpA (62, 63). The anti-EpiP serum identified several immunoreactive bands, including one at ∼50 kDa and another at 8 kDa. These bands appear to have molecular mass comparable to the noncleaved EpiP and the prodomain, respectively. Furthermore, this indicates that the serum mainly detects the prodomain of EpiP (Fig. 9B). Lack of detection of the protease domain (the 39-kDa band in Fig. 9B) may be due to poor recognition by the serum that we have used in the immunoblot. Indeed, with the purified recombinant protein, the protease domain also appears as a faint band as compared with the noncleaved protein and the prodomain. At this point, the identity of the 17-kDa immunoreactive band is not clear. No immunoreactive bands were detected in the cell wall fraction, suggesting that the protein was mainly released into the extracellular milieu. Interestingly, different patterns were observed in the supernatant and protoplast preparations. In particular, a band migrating at a molecular mass comparable to that of the noncleaved form was more abundant in the cytoplasm, while the prodomain appeared more prevalent in the supernatant. This suggests that the protein was produced in its noncleaved form and then was processed during or after its release into the extracellular milieu.
EpiP vaccination protects mice against lethal S. aureus infection
Given that EpiP appears to be secreted, it might be exposed to the immune system during infection. Therefore, we asked whether EpiP immunization could confer protection against staphylococcal infection in a murine peritonitis model (64). Five-week-old CD1 mice were immunized intraperitoneally with a prime booster injection of 20 μg purified recombinant EpiP adsorbed to aluminum hydroxide adjuvant (alum; 2 mg/ml) with a 14 d interval. Control mice received equal amounts of PBS and alum adjuvant. Animals were bled immediately before the first immunization and 23 d thereafter, and sera were examined for IgG antibodies directed against rEpiP. Vaccination generated significant IgG titers against the protein (geometric mean ± se[scap]m: 1319±644). Mice were challenged intraperitoneally on d 24 with a lethal injection of a bacterial suspension of S. aureus Newman. After the challenge, survival was monitored for 7 d. EpiP vaccination significantly protected mice from the lethal challenge (2 independent experiments for a total of 32 mice/group), with greater survival rate at d 7 after challenge as compared with the control group (Table 2).
Table 2.
Protective efficacy of EpiP vaccination in the peritonitis model
| Treatment | Survived/total animals | % | PE | P |
|---|---|---|---|---|
| Alum | 2/32 | 6 | – | – |
| EpiP | 11/32 | 34 | 30 | 0.0054 |
| EpiP-S393A | 10/31 | 32 | 28 | 0.0093 |
A lethal dose of S. aureus Newman strain was used to challenge CD1 mice (n=32/group, 2 separate experiments), and survival rates were followed for 1 wk. Mice were vaccinated intraperitoneally and challenged with the S. aureus strain Newman. Statistical analysis was performed by 1-tailed Fisher's exact test; comparisons are vs. alum treatment. Protective efficacy (PE) = 1 − (DV/DC), where DV is dead vaccinated (%), and DC is dead control (%).
At this point, we asked whether the noncleaving rEpiP-S393A was as efficacious as the rEpiP in eliciting protection against S. aureus infection. Protective efficacy of the 2 proteins was compared in the peritonitis model against lethal challenge with strain Newman. No significant difference was observed between the groups of mice immunized with the 2 proteins (Table 2).
DISCUSSION
Based on bioinformatics antigen prediction (reverse vaccinology) and sequence homology, we identified the S. aureus EpiP as a novel vaccine candidate.
Like other extracellular bacterial proteases, EpiP has an N-terminal leader peptide involved in translocation across the cell membrane; however, it does not contain a known cell wall anchoring motif. Indeed, our data indicate that the protein is released into the extracellular milieu. Furthermore, we found that the recombinant purified protein undergoes an autocatalytic intramolecular cleavage, which separates an N-terminal propeptide from the rest of the molecule. The protein expressed by staphylococcal cells appears to be similarly processed. Most likely the autocatalytic intramolecular cleavage occurs after protein translocation beyond the cell membrane. Indeed, while most of the EpiP in the protoplast preparation is present with a molecular weight compatible with the full-length protein in the extracellular fraction the most abundant reactive band appears to be the propeptide. The biological meaning of these observations requires further investigation.
It is puzzling as to why S. aureus retains EpiP, since it does not produce epidermin and lacks several of the genes for lantibiotic production. Daly et al. (13) determined that retention of lantibiotic immunity genes in S. aureus was not sufficient to protect it from the bactericidal effects of other lantibiotics, as previously hypothesized. Additional studies have shown that S. aureus also does not produce a bacteriocin (bsa) but rather a phenol-soluble modulin (PSM) with the same molecular weight as the previously identified bsa (13, 14). In addition to its intramolecular cleavage between residues Lys98 and Thr99 to remove its prodomain, we found that rEpiP is capable of cleaving both collagen and casein and may have an alternative functional role. The site of autoproteolysis does not necessarily represent the preferred cleavage specificity or the sites of cleavage in collagen because it is potentially a local concentration effect. Thus, a more thorough analysis of its proteolytic activity will be necessary to elucidate its in vivo function in S. aureus.
EpiP from S. aureus is structurally similar to other subtilisin-like serine proteases, including several that are involved in a variety of bacterial infections. Some of the structures in PDB that are similar include one from Bacillus lentus (PDB ID: 1GCI; ref. 65), Tk-subtilisin from Thermosococcus kodakaensis (PDB ID: 3AFG, 2Z2Z; ref. 66, 67), the A subunit of AB5 toxin in E. coli (PDB ID: 2IY9; ref. 68), the cytobactin from Cyanothece PCC 7425 (PDB ID: 3ZXY; ref. 69), AprV2 (PDB ID: 3LPC; ref. 70), and PCSK9 (PDB ID: 2W2N, 2QTW, 2P4E; refs. 71–73). The inability to separate the domains is not unique, and the structures of other prodomains are quite similar to the prodomain of EpiP. Other examples of proteases that retain interactions with their prodomain have been described in the literature (66, 67, 72, 74, 75), although the way in which the 2 domains interact can vary (e.g., be inhibitory or have complementary charge interactions). The structure of EpiP is unique because it is the first structure of a lantibiotic peptide processing protease.
As predicted, mice vaccinated with EpiP were protected from staphylococcal lethal infection. However, our analysis shows that epiP is not fully conserved; therefore, in order to develop a broadly protective vaccine, EpiP must be combined with other antigens. Given that the protein was released extracellularly, protection might be associated with EpiP antibodies blocking the function of the protein. This advocates in favor of a role of EpiP in invasive infection, as has been shown for some of its homologs. The function of the S. pyogenes homologue SpyCEP in impairing the recruitment of neutrophils at the site of infection (38, 39) is particularly intriguing in this regard. Given the prominent role of neutrophils against S. aureus infections, we are now investigating whether EpiP has a similar function.
Protective efficacy of rEpiP and rEpiP-S393A was found to be comparable. In addition, a comparison of the crystal structure of rEpiP and rEpiP-S393A shows that the mutation does not alter the protein conformation, thus providing an explanation for the protective equivalence of the 2 proteins. The mutant rEpiP-S393A may be better suited for vaccine development, because it consists of a single polypeptide, while the WT may present some issues, particularly in terms of product characterization. Therefore, these observations highlight the importance of structural biology in antigen design.
Supplementary Material
Acknowledgments
The authors thank Dr. Elisabetta Sabini for her tireless efforts and organization to enable a productive collaboration between our groups.
Use of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory (Argonne, IL, USA), was supported by the U.S. DOE under contract DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and Michigan Technology Tri-Corridor (grant 085P100817). This project has been funded in whole or in part with federal funds from the U.S. National Institute of Allergy and Infectious Diseases, U.S. National Institutes of Health, U.S. Department of Health and Human Services, under contracts HHSN272200700058C and HHSN272201200026C.
The authors are grateful to Dr. Ravi P. N Mishra, Dr. Luigi Fiaschi, and Dr. Fabiana Falugi for their contribution in antigen identification and cloning and RNA extraction and real-time PCR experiments.
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
- Agr
- accessory gene regulator
- EpiP
- epidermin leader peptide processing serine protease
- LAC
- Los Angeles clone
- PCR
- polymerase chain reaction
- PIPE
- polymerase incomplete primer extension
- PDB
- Protein Data Bank
- PMF
- peptide mass fingerprinting
- qRT-PCR
- quantitative reverse transcription polymerase chain reaction
- RiPP
- ribosomally synthesized and post-translationally modified peptide
- TSB
- tryptic soy broth
- WT
- wild type
REFERENCES
- 1. Arnison P. G., Bibb M. J., Bierbaum G., Bowers A. A., Bugni T. S., Bulaj G., Camarero J. A., Campopiano D. J., Challis G. L., Clardy J., Cotter P. D., Craik D. J., Dawson M., Dittmann E., Donadio S., Dorrestein P. C., Entian K. D., Fischbach M. A., Garavelli J. S., Goransson U., Gruber C. W., Haft D. H., Hemscheidt T. K., Hertweck C., Hill C., Horswill A. R., Jaspars M., Kelly W. L., Klinman J. P., Kuipers O. P., Link A. J., Liu W., Marahiel M. A., Mitchell D. A., Moll G. N., Moore B. S., Muller R., Nair S. K., Nes I. F., Norris G. E., Olivera B. M., Onaka H., Patchett M. L., Piel J., Reaney M. J., Rebuffat S., Ross R. P., Sahl H. G., Schmidt E. W., Selsted M. E., Severinov K., Shen B., Sivonen K., Smith L., Stein T., Sussmuth R. D., Tagg J. R., Tang G. L., Truman A. W., Vederas J. C., Walsh C. T., Walton J. D., Wenzel S. C., Willey J. M., van der Donk W. A. (2013) Ribosomally synthesized and post-translationally modified peptide natural products: overview and recommendations for a universal nomenclature. Nat. Prod. Rep. 30, 108–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Schnell N., Entian K. D., Schneider U., Gotz F., Zahner H., Kellner R., Jung G. (1988) Prepeptide sequence of epidermin, a ribosomally synthesized antibiotic with four sulphide-rings. Nature 333, 276–278 [DOI] [PubMed] [Google Scholar]
- 3. Willey J. M., van der Donk W. A. (2007) Lantibiotics: peptides of diverse structure and function. Annu. Rev. Microbiol. 61, 477–501 [DOI] [PubMed] [Google Scholar]
- 4. Cotter P. D., Hill C., Ross R. P. (2005) Bacteriocins: developing innate immunity for food. Nat. Rev. Microbiol. 3, 777–788 [DOI] [PubMed] [Google Scholar]
- 5. Smith L., Hillman J. (2008) Therapeutic potential of type A (I) lantibiotics, a group of cationic peptide antibiotics. Curr. Opin. Microbiol. 11, 401–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schnell N., Engelke G., Augustin J., Rosenstein R., Ungermann V., Gotz F., Entian K. D. (1992) Analysis of genes involved in the biosynthesis of lantibiotic epidermin. Eur. J. Biochem. 204, 57–68 [DOI] [PubMed] [Google Scholar]
- 7. Kuipers O. P., Rollema H. S., de Vos W. M., Siezen R. J. (1993) Biosynthesis and secretion of a precursor of nisin Z by Lactococcus lactis, directed by the leader peptide of the homologous lantibiotic subtilin from Bacillus subtilis. FEBS Lett. 330, 23–27 [DOI] [PubMed] [Google Scholar]
- 8. Augustin J., Rosenstein R., Wieland B., Schneider U., Schnell N., Engelke G., Entian K. D., Gotz F. (1992) Genetic analysis of epidermin biosynthetic genes and epidermin-negative mutants of Staphylococcus epidermidis. Eur. J. Biochem. 204, 1149–1154 [DOI] [PubMed] [Google Scholar]
- 9. Marsh A. J., O'Sullivan O., Ross R. P., Cotter P. D., Hill C. (2010) In silico analysis highlights the frequency and diversity of type 1 lantibiotic gene clusters in genome sequenced bacteria. BMC Genomics 11, 679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Siezen R. J., Kuipers O. P., de Vos W. M. (1996) Comparison of lantibiotic gene clusters and encoded proteins. Antonie van Leeuwenhoek 69, 171–184 [DOI] [PubMed] [Google Scholar]
- 11. Bierbaum G., Sahl H. G. (2009) Lantibiotics: mode of action, biosynthesis and bioengineering. Curr. Pharm. Biotechnol. 10, 2–18 [DOI] [PubMed] [Google Scholar]
- 12. Geissler S., Gotz F., Kupke T. (1996) Serine protease EpiP from Staphylococcus epidermidis catalyzes the processing of the epidermin precursor peptide. J. Bacteriol. 178, 284–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Daly K. M., Upton M., Sandiford S. K., Draper L. A., Wescombe P. A., Jack R. W., O'Connor P. M., Rossney A., Gotz F., Hill C., Cotter P. D., Ross R. P., Tagg J. R. (2010) Production of the Bsa lantibiotic by community-acquired Staphylococcus aureus strains. J. Bacteriol. 192, 1131–1142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Joo H. S., Cheung G. Y., Otto M. (2011) Antimicrobial activity of community-associated methicillin-resistant Staphylococcus aureus is caused by phenol-soluble modulin derivatives. J. Biol. Chem. 286, 8933–8940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Otto M., Gotz F. (2001) ABC transporters of staphylococci. Res. Microbiol. 152, 351–356 [DOI] [PubMed] [Google Scholar]
- 16. Moore P. C., Lindsay J. A. (2001) Genetic variation among hospital isolates of methicillin-sensitive Staphylococcus aureus: evidence for horizontal transfer of virulence genes. J. Clin. Microbiol. 39, 2760–2767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Highlander S. K., Hulten K. G., Qin X., Jiang H., Yerrapragada S., Mason E. O., Jr., Shang Y., Williams T. M., Fortunov R. M., Liu Y., Igboeli O., Petrosino J., Tirumalai M., Uzman A., Fox G. E., Cardenas A. M., Muzny D. M., Hemphill L., Ding Y., Dugan S., Blyth P. R., Buhay C. J., Dinh H. H., Hawes A. C., Holder M., Kovar C. L., Lee S. L., Liu W., Nazareth L. V., Wang Q., Zhou J., Kaplan S. L., Weinstock G. M. (2007) Subtle genetic changes enhance virulence of methicillin resistant and sensitive Staphylococcus aureus. BMC Microbiol. 7, 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kies S., Vuong C., Hille M., Peschel A., Meyer C., Gotz F., Otto M. (2003) Control of antimicrobial peptide synthesis by the agr quorum sensing system in Staphylococcus epidermidis: activity of the lantibiotic epidermin is regulated at the level of precursor peptide processing. Peptides 24, 329–338 [DOI] [PubMed] [Google Scholar]
- 19. Lowy F. D. (1998) Staphylococcus aureus infections. N. Engl. J. Med. 339, 520–532 [DOI] [PubMed] [Google Scholar]
- 20. Smith T. L., Jarvis W. R. (1999) Antimicrobial resistance in Staphylococcus aureus. Microbes Infect. 1, 795–805 [DOI] [PubMed] [Google Scholar]
- 21. Rybak M. J., Akins R. L. (2001) Emergence of methicillin-resistant Staphylococcus aureus with intermediate glycopeptide resistance: clinical significance and treatment options. Drugs 61, 1–7 [DOI] [PubMed] [Google Scholar]
- 22. Webb G. F., Horn M. A., D'Agata E. M., Moellering R. C., Jr., Ruan S. (2010) Competition of hospital-acquired and community-acquired methicillin-resistant Staphylococcus aureus strains in hospitals. J. Biol. Dynamics 4, 115–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Anonymous. (1989) Methicillin-resistant Staphylococcus aureus. N. Engl. J. Med. 321, 1342–1345 [DOI] [PubMed] [Google Scholar]
- 24. Yamamoto T., Nishiyama A., Takano T., Yabe S., Higuchi W., Razvina O., Shi D. (2010) Community-acquired methicillin-resistant Staphylococcus aureus: community transmission, pathogenesis, and drug resistance. J. Infect. Chemo. 16, 225–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Klevens R. M., Morrison M. A., Nadle J., Petit S., Gershman K., Ray S., Harrison L. H., Lynfield R., Dumyati G., Townes J. M., Craig A. S., Zell E. R., Fosheim G. E., McDougal L. K., Carey R. B., Fridkin S. K. (2007) Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA 298, 1763–1771 [DOI] [PubMed] [Google Scholar]
- 26. Maskalyk J. (2002) Antimicrobial resistance takes another step forward. CMAJ 167, 375. [PMC free article] [PubMed] [Google Scholar]
- 27. Shorr A. F. (2007) Epidemiology and economic impact of meticillin-resistant Staphylococcus aureus: review and analysis of the literature. Pharmacoeconomics 25, 751–768 [DOI] [PubMed] [Google Scholar]
- 28. Kuklin N. A., Clark D. J., Secore S., Cook J., Cope L. D., McNeely T., Noble L., Brown M. J., Zorman J. K., Wang X. M., Pancari G., Fan H., Isett K., Burgess B., Bryan J., Brownlow M., George H., Meinz M., Liddell M. E., Kelly R., Schultz L., Montgomery D., Onishi J., Losada M., Martin M., Ebert T., Tan C. Y., Schofield T. L., Nagy E., Meineke A., Joyce J. G., Kurtz M. B., Caulfield M. J., Jansen K. U., McClements W., Anderson A. S. (2006) A novel Staphylococcus aureus vaccine: iron surface determinant B induces rapid antibody responses in rhesus macaques and specific increased survival in a murine S. aureus sepsis model. Infect. Immun. 74, 2215–2223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Patti J. M. (2011) Will we ever see the approval of a Staphylococcus aureus vaccine? Exp. Rev. Anti-infective Therap. 9, 845–846 [DOI] [PubMed] [Google Scholar]
- 30. Rodriguez-Ortega M. J., Norais N., Bensi G., Liberatori S., Capo S., Mora M., Scarselli M., Doro F., Ferrari G., Garaguso I., Maggi T., Neumann A., Covre A., Telford J. L., Grandi G. (2006) Characterization and identification of vaccine candidate proteins through analysis of the group A Streptococcus surface proteome. Nat. Biotechnol. 24, 191–197 [DOI] [PubMed] [Google Scholar]
- 31. Turner C. E., Kurupati P., Wiles S., Edwards R. J., Sriskandan S. (2009) Impact of immunization against SpyCEP during invasive disease with two streptococcal species: Streptococcus pyogenes and Streptococcus equi. Vaccine 27, 4923–4929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cleary P. P., Matsuka Y. V., Huynh T., Lam H., Olmsted S. B. (2004) Immunization with C5a peptidase from either group A or B streptococci enhances clearance of group A streptococci from intranasally infected mice. Vaccine 22, 4332–4341 [DOI] [PubMed] [Google Scholar]
- 33. Cheng Q., Debol S., Lam H., Eby R., Edwards L., Matsuka Y., Olmsted S. B., Cleary P. P. (2002) Immunization with C5a peptidase or peptidase-type III polysaccharide conjugate vaccines enhances clearance of group B streptococci from lungs of infected mice. Infect. Immun. 70, 6409–6415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ji Y., Carlson B., Kondagunta A., Cleary P. P. (1997) Intranasal immunization with C5a peptidase prevents nasopharyngeal colonization of mice by the group A Streptococcus. Infect. Immun. 65, 2080–2087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Santillan D. A., Andracki M. E., Hunter S. K. (2008) Protective immunization in mice against group B streptococci using encapsulated C5a peptidase. Am. J. Obstetrics Gynecol. 198, 114.e1–114.e6 [DOI] [PubMed] [Google Scholar]
- 36. Bethe G., Nau R., Wellmer A., Hakenbeck R., Reinert R. R., Heinz H. P., Zysk G. (2001) The cell wall-associated serine protease PrtA: a highly conserved virulence factor of Streptococcus pneumoniae. FEMS Microbiol. Lett. 205, 99–104 [DOI] [PubMed] [Google Scholar]
- 37. Edwards R. J., Taylor G. W., Ferguson M., Murray S., Rendell N., Wrigley A., Bai Z., Boyle J., Finney S. J., Jones A., Russell H. H., Turner C., Cohen J., Faulkner L., Sriskandan S. (2005) Specific C-terminal cleavage and inactivation of interleukin-8 by invasive disease isolates of Streptococcus pyogenes. J. Infect. Dis. 192, 783–790 [DOI] [PubMed] [Google Scholar]
- 38. Zinkernagel A. S., Timmer A. M., Pence M. A., Locke J. B., Buchanan J. T., Turner C. E., Mishalian I., Sriskandan S., Hanski E., Nizet V. (2008) The IL-8 protease SpyCEP/ScpC of group A streptococcus promotes resistance to neutrophil killing. Cell Host Microbe 4, 170–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hidalgo-Grass C., Dan-Goor M., Maly A., Eran Y., Kwinn L. A., Nizet V., Ravins M., Jaffe J., Peyser A., Moses A. E., Hanski E. (2004) Effect of a bacterial pheromone peptide on host chemokine degradation in group A streptococcal necrotising soft-tissue infections. Lancet 363, 696–703 [DOI] [PubMed] [Google Scholar]
- 40. Andrews T., Sullivan K. E. (2003) Infections in patients with inherited defects in phagocytic function. Clin. Microbiol. Rev. 16, 597–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Klock H. E., Lesley S. A. (2009) The Polymerase Incomplete Primer Extension (PIPE) method applied to high-throughput cloning and site-directed mutagenesis. Methods Mol. Biol. 498, 91–103 [DOI] [PubMed] [Google Scholar]
- 42. Millard C. S., Stols L., Quartey P., Kim Y., Dementieva I., Donnelly M. I. (2003) A less laborious approach to the high-throughput production of recombinant proteins in Escherichia coli using 2-liter plastic bottles. Protein Expr. Purif. 29, 311–320 [DOI] [PubMed] [Google Scholar]
- 43. Kuhn M. L., Majorek K. A., Minor W., Anderson W. F. (2013) Broad-substrate screen as a tool to identify substrates for bacterial Gcn5-related N-acetyltransferases with unknown substrate specificity. Protein Sci. 22, 222–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Minor W., Cymborowski M., Otwinowski Z., Chruszcz M. (2006) HKL-3000: the integration of data reduction and structure solution–from diffraction images to an initial model in minutes. Acta Crystallogr. D Biol. Crystallogr. 62, 859–866 [DOI] [PubMed] [Google Scholar]
- 45. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Perrakis A., Morris R., Lamzin V. S. (1999) Automated protein model building combined with iterative structure refinement. Nat. Struct. Biol. 6, 458–463 [DOI] [PubMed] [Google Scholar]
- 47. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 48. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Vaguine A. A., Richelle J., Wodak S. J. (1999) SFCHECK: a unified set of procedures for evaluating the quality of macromolecular structure-factor data and their agreement with the atomic model. Acta Crystallogr. D Biol. Crystallogr. 55, 191–205 [DOI] [PubMed] [Google Scholar]
- 50. Chen V. B., Arendall W. B., 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chavira R., Jr., Burnett T. J., Hageman J. H. (1984) Assaying proteinases with azocoll. Anal. Biochem. 136, 446–450 [DOI] [PubMed] [Google Scholar]
- 52. Bagnoli F., Baudner B., Mishra R. P., Bartolini E., Fiaschi L., Mariotti P., Nardi-Dei V., Boucher P., Rappuoli R. (2011) Designing the next generation of vaccines for global public health. OMICS 15, 545–566 [DOI] [PubMed] [Google Scholar]
- 53. Rappuoli R. (2001) Reverse vaccinology, a genome-based approach to vaccine development. Vaccine 19, 2688–2691 [DOI] [PubMed] [Google Scholar]
- 54. Palumbo E., Fiaschi L., Brunelli B., Marchi S., Savino S., Pizza M. (2012) Antigen identification starting from the genome: a “reverse vaccinology” approach applied to MenB. Methods Mol. Biol. 799, 361–403 [DOI] [PubMed] [Google Scholar]
- 55. Rawlings N. D., Barrett A. J. (1994) Families of serine peptidases. Methods Enzymol. 244, 19–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ten Broeke-Smits N. J., Pronk T. E., Jongerius I., Bruning O., Wittink F. R., Breit T. M., van Strijp J. A., Fluit A. C., Boel C. H. (2010) Operon structure of Staphylococcus aureus. Nucleic Acids Res. 38, 3263–3274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hidalgo-Grass C., Mishalian I., Dan-Goor M., Belotserkovsky I., Eran Y., Nizet V., Peled A., Hanski E. (2006) A streptococcal protease that degrades CXC chemokines and impairs bacterial clearance from infected tissues. EMBO J. 25, 4628–4637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zingaretti C., Falugi F., Nardi-Dei V., Pietrocola G., Mariani M., Liberatori S., Gallotta M., Tontini M., Tani C., Speziale P., Grandi G., Margarit I. (2010) Streptococcus pyogenes SpyCEP: a chemokine-inactivating protease with unique structural and biochemical features. FASEB J. 24, 2839–2848 [DOI] [PubMed] [Google Scholar]
- 59. Turner D. P., Wooldridge K. G., Ala'Aldeen D. A. (2002) Autotransported serine protease A of Neisseria meningitidis: an immunogenic, surface-exposed outer membrane, and secreted protein. Infect. Immun. 70, 4447–4461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Van der Meer J. R., Polman J., Beerthuyzen M. M., Siezen R. J., Kuipers O. P., De Vos W. M. (1993) Characterization of the Lactococcus lactis nisin A operon genes nisP, encoding a subtilisin-like serine protease involved in precursor processing, and nisR, encoding a regulatory protein involved in nisin biosynthesis. J. Bacteriol. 175, 2578–2588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Engelke G., Gutowski-Eckel Z., Kiesau P., Siegers K., Hammelmann M., Entian K. D. (1994) Regulation of nisin biosynthesis and immunity in Lactococcus lactis 6F3. Appl. Environ. Microbiol. 60, 814–825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Stranger-Jones Y. K., Bae T., Schneewind O. (2006) Vaccine assembly from surface proteins of Staphylococcus aureus. Proc. Natl. Acad. Sci. U. S. A. 103, 16942–16947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Atkins K. L., Burman J. D., Chamberlain E. S., Cooper J. E., Poutrel B., Bagby S., Jenkins A. T., Feil E. J., van den Elsen J. M. (2008) S. aureus IgG-binding proteins SpA and Sbi: host specificity and mechanisms of immune complex formation. Mol. Immunol. 45, 1600–1611 [DOI] [PubMed] [Google Scholar]
- 64. Mishra R. P., Mariotti P., Fiaschi L., Nosari S., Maccari S., Liberatori S., Fontana M. R., Pezzicoli A., De Falco M. G., Falugi F., Altindis E., Serruto D., Grandi G., Bagnoli F. (2012) Staphylococcus aureus FhuD2 is involved in the early phase of staphylococcal dissemination and generates protective immunity in mice. J. Infect. Dis. 206, 1041–1049 [DOI] [PubMed] [Google Scholar]
- 65. Kuhn P., Knapp M., Soltis S. M., Ganshaw G., Thoene M., Bott R. (1998) The 0.78 A structure of a serine protease: Bacillus lentus subtilisin. Biochemistry 37, 13446–13452 [DOI] [PubMed] [Google Scholar]
- 66. Tanaka S., Matsumura H., Koga Y., Takano K., Kanaya S. (2007) Four new crystal structures of Tk-subtilisin in unautoprocessed, autoprocessed and mature forms: insight into structural changes during maturation. J. Mol. Biol. 372, 1055–1069 [DOI] [PubMed] [Google Scholar]
- 67. Foophow T., Tanaka S., Angkawidjaja C., Koga Y., Takano K., Kanaya S. (2010) Crystal structure of a subtilisin homologue, Tk-SP, from Thermococcus kodakaraensis: requirement of a C-terminal beta-jelly roll domain for hyperstability. J. Mol. Biol. 400, 865–877 [DOI] [PubMed] [Google Scholar]
- 68. Paton A. W., Beddoe T., Thorpe C. M., Whisstock J. C., Wilce M. C., Rossjohn J., Talbot U. M., Paton J. C. (2006) AB5 subtilase cytotoxin inactivates the endoplasmic reticulum chaperone BiP. Nature 443, 548–552 [DOI] [PubMed] [Google Scholar]
- 69. Houssen W. E., Koehnke J., Zollman D., Vendome J., Raab A., Smith M. C., Naismith J. H., Jaspars M. (2012) The discovery of new cyanobactins from Cyanothece PCC 7425 defines a new signature for processing of patellamides. Chembiochem 13, 2683–2689 [DOI] [PubMed] [Google Scholar]
- 70. Kennan R. M., Wong W., Dhungyel O. P., Han X., Wong D., Parker D., Rosado C. J., Law R. H., McGowan S., Reeve S. B., Levina V., Powers G. A., Pike R. N., Bottomley S. P., Smith A. I., Marsh I., Whittington R. J., Whisstock J. C., Porter C. J., Rood J. I. (2010) The subtilisin-like protease AprV2 is required for virulence and uses a novel disulphide-tethered exosite to bind substrates. PLoS Pathogens 6, e1001210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Bottomley M. J., Cirillo A., Orsatti L., Ruggeri L., Fisher T. S., Santoro J. C., Cummings R. T., Cubbon R. M., Lo Surdo P., Calzetta A., Noto A., Baysarowich J., Mattu M., Talamo F., De Francesco R., Sparrow C. P., Sitlani A., Carfi A. (2009) Structural and biochemical characterization of the wild type PCSK9-EGF(AB) complex and natural familial hypercholesterolemia mutants. J. Biol. Chem. 284, 1313–1323 [DOI] [PubMed] [Google Scholar]
- 72. Hampton E. N., Knuth M. W., Li J., Harris J. L., Lesley S. A., Spraggon G. (2007) The self-inhibited structure of full-length PCSK9 at 1.9 A reveals structural homology with resistin within the C-terminal domain. Proc. Natl. Acad. Sci. U. S. A. 104, 14604–14609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Cunningham D., Danley D. E., Geoghegan K. F., Griffor M. C., Hawkins J. L., Subashi T. A., Varghese A. H., Ammirati M. J., Culp J. S., Hoth L. R., Mansour M. N., McGrath K. M., Seddon A. P., Shenolikar S., Stutzman-Engwall K. J., Warren L. C., Xia D., Qiu X. (2007) Structural and biophysical studies of PCSK9 and its mutants linked to familial hypercholesterolemia. Nat. Struct. Mol. Biol. 14, 413–419 [DOI] [PubMed] [Google Scholar]
- 74. Comellas-Bigler M., Maskos K., Huber R., Oyama H., Oda K., Bode W. (2004) 1.2 A crystal structure of the serine carboxyl proteinase pro-kumamolisin; structure of an intact pro-subtilase. Structure 12, 1313–1323 [DOI] [PubMed] [Google Scholar]
- 75. Jain S. C., Shinde U., Li Y., Inouye M., Berman H. M. (1998) The crystal structure of an autoprocessed Ser221Cys-subtilisin E-propeptide complex at 2.0 A resolution. J. Mol. Biol. 284, 137–144 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.