

Abstract
Deletion of copper-zinc superoxide dismutase (CuZnSOD) in Sod1−/− mice leads to accelerated loss of muscle mass and force during aging, but the losses do not occur with muscle-specific deletion of CuZnSOD. To determine the role of motor neurons in the muscle decline, we generated transgenic Sod1−/− mice in which CuZnSOD was expressed under control of the synapsin 1 promoter (SynTgSod1−/− mice). SynTgSod1−/− mice expressed CuZnSOD in brain, spinal cord, and peripheral nerve, but not in other tissues. Sciatic nerve CuZnSOD content in SynTgSod1−/− mice was ∼20% that of control mice, but no reduction in muscle mass or isometric force was observed in SynTgSod1−/− mice compared with control animals, whereas muscles of age-matched Sod1−/− mice displayed 30–40% reductions in mass and force. In addition, increased oxidative damage and adaptations in stress responses observed in muscles of Sod1−/− mice were absent in SynTgSod1−/− mice, and degeneration of neuromuscular junction (NMJ) structure and function occurred in Sod1−/− mice but not in SynTgSod1−/− mice. Our data demonstrate that specific CuZnSOD expression in neurons is sufficient to preserve NMJ and skeletal muscle structure and function in Sod1−/− mice and suggest that redox homeostasis in motor neurons plays a key role in initiating sarcopenia during aging.—Sakellariou, G. K., Davis, C. S., Shi, Y., Ivannikov, M. V., Zhang, Y., Vasilaki, A., Macleod, G. T., Richardson, A., Van Remmen, H., Jackson, M. J., McArdle, A., Brooks, S. V. Neuron-specific expression of CuZnSOD prevents the loss of muscle mass and function that occurs in homozygous CuZnSOD-knockout mice.
Keywords: SOD1, neuromuscular junction, heat-shock protein, oxidative stress
Aging of skeletal muscle is characterized by loss of mass and contractile force, which has a profound effect on the quality of life of older people (1). Loss of skeletal muscle begins in middle age and continues until the end of life (2). Studies of humans indicate that by the age of 70, there is a 25–30% reduction in the cross-sectional area (CSA) of skeletal muscle and a decline in muscle strength of 30–40% (3). The age-related reduction in the muscle fiber size and number (4) is a slow process with a complex and multifactorial etiology.
One factor in numerous tissues that is clearly associated with loss of function during aging is oxidative damage, and experimental evidence from humans and rodents has shown that skeletal muscles exhibit age-dependent increases in the products of oxidative damage to biomolecules including proteins, lipids, and nucleic acids (5–8). Recent reports have attributed the positive correlation between age and oxidative damage to age-related changes in production of reactive oxygen and nitrogen species (RONS), with skeletal muscles from old mice exhibiting a higher intracellular RONS generation in comparison to muscles from young mice (9, 10). The hypothesis that increased generation of oxidants in vivo plays a key role in age-related muscle dysfunction has been examined in transgenic and knockout mouse models, and work from our group has shown that deletion of the superoxide scavenging enzyme copper-zinc superoxide dismutase (CuZnSOD) in mice (Sod1−/− mice) induces an accelerated skeletal muscle aging phenotype (11–15). Many features of the muscles of Sod1−/− mice, including elevated oxidative damage, loss of contractile force, degeneration of neuromuscular junctions (NMJs), and loss of innervation resemble those observed in old wild-type (WT) mice (11–15). Hence, this knockout mouse represents a useful model for the study of the role of chronic oxidative stress in the loss of skeletal muscle that occurs with aging.
Elucidation of the primary mechanisms underlying sarcopenia has been challenging, in part because of the difficulty in unraveling the interplay between loss of muscle mass and loss of functional motor units, both of which occur with aging (16). Several experimental studies have demonstrated that manipulations that modify the integrity of NMJs lead to a phenotype that resembles the age-related loss of muscle mass and function (17–19). In addition, skeletal muscle atrophy in Sod1−/− mice is associated with NMJ alterations, including motor axon degeneration and postsynaptic end plate fragmentation (11, 20), indicating that NMJ degeneration may be a critical part of the muscle atrophy phenotype exhibited in the Sod1−/− mouse model; however, whether these degenerative changes are initiated by changes in redox homeostasis proximal and/or distal to the neuromuscular synapses is not known. To begin to tackle questions regarding the relative importance of pre- and postsynaptic changes in redox homeostasis, we recently examined whether specific Sod1 gene deletion targeted to skeletal muscle leads to muscle atrophy and showed that absence of CuZnSOD within the muscle tissue alone is not sufficient to induce muscle atrophy (21). These results therefore indicate that the muscle atrophy observed during aging of Sod1−/− mice is not primarily due to a deficiency in CuZnSOD in skeletal muscle; instead, failure of redox homeostasis in motor neurons may be the primary initiating factor.
To directly test whether the muscle atrophy phenotype exhibited by the Sod1−/− mice is related to redox changes in motor neurons, we generated a transgenic Sod1−/− mouse model in which CuZnSOD was expressed specifically in neurons under the control of the synapsin 1 promoter (SynTgSod1−/− mice). We hypothesized that if alterations in redox homeostasis within the motor neurons were the primary initiators of muscle atrophy, CuZnSOD-targeted expression in the nerve of Sod1−/− mice would prevent the overt phenotypic changes of accelerated skeletal muscle aging. In the present study we report the effects of nerve-specific rescue of CuZnSOD in Sod1−/− mice on muscle mass, morphology and function, oxidative damage, adaptive responses, and NMJ structure and function.
MATERIALS AND METHODS
Animals
All mice were generated at the University of Texas Health Science Center at San Antonio (UTHSCSA) as described below. In San Antonio, the mice were maintained under specific pathogen-free (SPF) conditions until they reached appropriate ages for the electrophysiological studies performed in San Antonio or were shipped to the University of Michigan (UM) for measurement of contractile properties and harvesting of tissues for histologic, biochemical, and molecular analyses. The mice were shipped at ∼12 mo of age and maintained under SPF conditions in the Unit for Laboratory Animal Medicine at UM until they were tested at an average age of 15.6 ± 0.4 mo. At both sites, the mice were fed standard NIH-31 chow ad libitum and were maintained under barrier conditions in microisolator cages on a 12-h dark/light cycle. For simple tissue collection, the mice were euthanized by CO2 inhalation followed by cervical dislocation, and the muscles and tissues were rapidly removed, snap-frozen, and stored at −80°C until analysis. For measurement of contractile properties, the mice were anesthetized with intraperitoneal injections of tribromoethanol (400 mg/kg), with supplemental injections provided to maintain an adequate level of anesthesia throughout the procedure. For in vivo electromyography studies, the mice were anesthetized with isoflurane. All procedures were approved by both the University Committee on the Use and Care of Animals at UM and the Institutional Animal Care and Use Committee at UTHSCSA and were in accordance with the U.S. National Institutes of Health (NIH) Guide for Care and Use of Laboratory Animals (Public Health Service 19965, NIH Publication 85-23).
Generation of a Sod1 nerve rescue Sod1−/− mouse model (SynTgSod1−/−)
Details of the generation and characterization of the Sod1−/− mouse model have been reported previously (13, 22, 23). For the present study, a transgenic mouse model with neuron-specific expression of human SOD1 (hSOD1) was generated on the C57BL/6 genetic background by using a transgene construct containing the hSOD1 cDNA driven by a neuron-specific synapsin promoter. The rescue mice were generated by crossing synapsin hSOD1 transgenic mice with Sod1−/− mice, resulting in mice with expression of hSOD1 restricted to neuronal tissue and completely lacking endogenous mouse Sod1 expression. To confirm the nerve-specific Sod1 expression, a variety of tissues were dissected and assayed for hSOD1 protein.
Muscle function analyses
Gastrocnemius (GTN) muscle contractile properties were measured in situ, as described by Larkin et al. (12). In anesthetized mice, the whole GTN muscle was isolated from surrounding muscle and connective tissue, and the distal tendon was severed and secured to the lever arm of a servomotor (model 305B; Aurora Scientific Inc., Aurora, ON, Canada). A continual drip of saline warmed to 37°C was applied to the GTN muscle to maintain its temperature. The muscle was activated by stimulation of the tibial nerve with a bipolar platinum wire electrode. The voltage of single 0.2 ms stimulation pulses was adjusted to give a maximum isometric twitch. Subsequently, muscle length was adjusted to the optimal length (Lo) at which twitch force was maximal. With the muscle held at Lo, 300-ms trains of pulses were applied at increasing stimulation frequencies until the maximum isometric tetanic force (Po) was achieved. The same procedure was then repeated, but, rather than activating the muscle via the tibial nerve, a cuff electrode was placed around the proximal and distal ends of the muscle for direct muscle stimulation. After force measurements, the GTN, anterior tibialis (AT), extensor digitorum longus (EDL), soleus (SOL), quadriceps (Quad), and plantaris (PLT) muscles were removed, and the deeply anesthetized mice were euthanized by pneumothorax. All muscles were trimmed off their tendons, blotted, and weighed. The muscles were either snap-frozen for protein or molecular analyses or coated in tissue-freezing medium (Triangle Biomedical Sciences, Durham, NC, USA) and rapidly frozen in isopentane cooled with liquid nitrogen for histologic analysis. GTN muscle fiber length (Lf) was calculated by multiplying Lo by 0.45. Total fiber CSA was calculated by dividing the muscle mass (in milligrams) by the product of Lf (in millimeters) and the density of mammalian skeletal muscle, 1.06 g/cm2. Specific Po (newtons per square centimeter) was calculated by dividing Po by total fiber CSA for each muscle.
In vivo electromyography (EMG)
Anesthetized animals were maintained at 33–34°C under a heat lamp. All experiments were performed by the Nicolet Viking Quest portable EMG apparatus (CareFusion, San Diego, CA, USA). The repetitive nerve stimulation (RNS) assay was performed according to the methods of Kaja et al. (24), with minor modifications. The evoked compound muscle action potentials (CMAPs) from the GTN were recorded with a subdermal electrode inserted at the midbelly region, and the stimulating electrode was inserted near the sciatic nerve in the thigh. The reference electrode was placed at the ankle. Trains of 10 stimuli were applied at various frequencies (0.2, 3, 5, and 10 Hz) with a 2 min recovery period. The CMAP decrement was reported as the percentage difference between the amplitude of the fifth evoked CMAP with the first CMAP.
Electrophysiological recordings from muscle
Intracellular recordings of miniature end plate potentials (mEPPs) and evoked end plate potentials (EPPs) in the EDL muscle were performed by standard techniques with a high-impedance amplifier (Axoclamp 900A; Molecular Devices, Sunnyvale CA, USA). For the EDL dissection, either the left or right hind limb was shaved and skinned. After removal of the biceps femoris muscle and sciatic nerve dissection, a complex of the EDL and AT muscles was dissected and pinned by the tendons to a dish. Recording microelectrodes were filled with a solution mix containing 1.5 M KCl and 1.5 M KAc, to give a final resistance of ∼40–50 MΩ. Data were digitized and recorded with Chart 5.5.6 software (AD Instruments, Colorado Springs CO, USA). To prevent muscle contractions, 1 μM μ-conotoxin was added to the recording solution (standard Kreb's solution continuously perfused with a 95% O2/5% CO2 gas mixture). Offline data analysis was performed with pCLAMP 10 (Molecular Devices).
CuZnSOD and manganese superoxide dismutase (MnSOD) enzymatic activity assays
The activities of CuZnSOD and MnSOD were measured in native gels, with negative staining, as described previously (25). The gel image was scanned and recorded with an Alpha Imager 3400 (Alpha Innotech, San Leandro, CA, USA) and analyzed with ImageQuant Software (GE Healthcare Life Sciences, Sunnyvale, CA, USA).
Lipid peroxidation assay
Lipid peroxidation was assessed in mouse skeletal muscle and brain by determining the levels of F2-isoprostanes by our published method (26). Mouse tissue (200 mg) was homogenized, and whole lipid was extracted with chloroform-heptane. The levels of F2-isoprostanes from brain and muscle tissue (esterified) were measured by gas chromatography-mass spectrometry (GC-MS), as previously described (26) and initially described by Morrow et al. (27). The levels of F2-isoprostanes are expressed as nanograms F2-isoprostanes (8-iso-PGF2α) per gram tissue.
Ultrastructural analysis by electron microscopy (EM)
GTN muscles were dissected, carefully cut into ∼1- × 2-mm fragments, and incubated overnight with 2.5% glutaraldehyde in 0.1 M Sorensen's buffer (pH 7.4). The samples were then rinsed 3 times for 15 min each in 0.1 M Sorensen's buffer and stored at 4°C. The tissue was postfixed for 1 h in 1% osmium tetroxide in the same buffer, rinsed in Sorensen's buffer, and dehydrated in ascending concentrations of ethanol, transitioned through propylene oxide, and infiltrated into and embedded in Epon epoxy resin. The tissue was ultrathin sectioned (70 nm), mounted on copper grids, and poststained with uranyl acetate and lead citrate. The sections were examined with a CM-100 transmission electron microscope at 60 kV (Philips, Eindhoven, The Netherlands). The images were recorded digitally with an ORCA-HR digital camera system (Hamamatsu, Hamamatsu City, Japan), operated by AMT software (Advanced Microscopy Techniques Corp., Danvers, MA, USA).
Immunohistochemistry (IHC) of the NMJs
To visualize the NMJs, AT muscles from Sod1−/−, WT, and SynTgSod1−/− mice were fixed in 4% paraformaldehyde (PFA) for 30 min and sectioned on a cryostat (Bright Instrument Co., Huntingdon, UK). The longitudinal sections (35 μm) were blocked and permeabilized in PBS, 4% BSA, and 1% Triton X-100 for 3 h at 25°C and then incubated with the primary antibodies anti-pan-axonal neurofilament marker (Covance, Princeton NJ, USA) and synaptotagmin 2 (Abcam, Cambridge, UK) for 36 h in 4% BSA and 1% Triton X-100 in PBS at 4°C. The sections were then washed for 2 h in PBS and incubated for 2 h at 25°C with secondary antibody (488 anti-mouse IgG; Invitrogen, Carlsbad CA, USA) and AlexaFluor 594-conjugated bungarotoxin (Molecular Probes, Eugene, OR, USA) diluted in PBS, 4% BSA, and 1% Triton X-100. After the sections were washed for 1 h in PBS, they were mounted in Vectashield (Vector Laboratories, Peterborough, UK), and immunofluorescence images of the NMJs were obtained on an LSM 510 Meta confocal microscope (Zeiss GmbH, Oberkochen, Germany) and analyzed with ZEN acquisition software (Zeiss).
IHC of spinal cords
Mice were perfused with 4% PFA, and the spinal cords were dissected and placed in 4% PFA overnight. The tissues were then transferred to glucose for 48 h. After cryosectioning, transverse lumbar spinal cord sections of 20 μm thickness were permeabilized in 0.1% Tween-20 in PBS for 5 min and blocked in PBS, 5% BSA for 1 h. The sections were probed with primary antibodies and incubated overnight at 4°C. The following antibodies were used: anti-glial fibrillary acidic protein (GFAP; Cell Signaling, Danvers, Mass, USA) and anti-CuZnSOD (Enzo Life Sciences, Ltd., Exeter, UK). The sections were washed 3 times for 5 min each in PBS and counterstained for 1 h at 25°C with AlexaFluor-conjugated secondary antibodies (Molecular Probes) diluted in PBS and 5% BSA. They were then washed again (3×5 min in PBS) and mounted on slides with Prolong Gold Antifade reagent containing DAPI (Molecular Probes). Spinal cord immunofluorescence images were visualized with an Eclipse TE2000-U fluorescence microscope (Nikon Inc., Tokyo, Japan).
Western blot analysis of muscle proteins
For assessment of specific proteins in muscle, 30 μg of total protein was applied to an 8–15% polyacrylamide gel with a 4% stacking gel (National Diagnostics Ltd., Atlanta, GA, USA). The separated proteins were transferred onto nitrocellulose membranes by Western blot analysis. The membranes were probed with antibodies against CuZnSOD (SOD1) and MnSOD (SOD2) (Stressgen Inc., Victoria, BC, Canada); heat-shock protein (HSP)25, HSP60, and heat shock cognate (HSC)70 (Enzo Life Sciences); slow myosin heavy chain (MyHC; Sigma-Aldrich, Poole, UK); nNOS, eNOS, iNOS, HSP10, HSP72, peroxiredoxin V (PRXV), Fast MyHC, and GAPDH (Abcam, Cambridge, UK) (28). Horseradish peroxidase–conjugated anti-mouse IgG, anti-rabbit IgG (Cell Signaling Technologies, Hitchin, UK) or anti-rat (Sigma-Aldrich) was used as the secondary antibody. Peroxidase activity was detected with an ECL kit (Amersham International, Cardiff, UK), and band intensities were analyzed by Quantity One Software (Bio-Rad Laboratories Ltd., Hemel Hempstead, UK). The specificity of the bands was identified in comparison with a sample that had not been exposed to the primary antibody, and the molecular mass was determined by using molecular mass markers. The NFκB subunits p50 and p65 were analyzed on nuclear protein extracts that were isolated from skeletal muscle (15). All protein contents were normalized to the GAPDH or β-actin content of the same sample.
Analyses of the 3-nitrotyrosine (3-NT) content of muscle proteins
Total cellular protein was isolated, and 30 μg was separated by SDS-PAGE followed by Western blot analysis, as describe above. The separated proteins were transferred onto polyvinylidene difluoride (PVDF) membranes. The content of 3-NT was analyzed by using a rabbit monoclonal antibody (Cell Biolabs, San Diego, CA, USA), according to manufacturer's instructions. The bands were visualized, and densitometric quantification was undertaken with a Chemi-Doc XRS System (Bio-Rad Laboratories, Hercules, CA, USA; ref. 14).
Determination of muscle structure
GTN muscles were cryosectioned at a thickness of 12 μm at −28°C through the midbelly, and fluorescent IHC staining was initiated the same day. Sections were rinsed with PBS and permeabilized in 0.2% Triton X-100 in PBS for 5 min. Wheat germ agglutinin (WGA) lectin AlexaFluor 488 (Molecular Probes) was used to identify extracellular matrix. Nuclei were identified with DAPI. Cross sections from 3–5 muscles/genotype were examined by observers in a blinded procedure to count the number of fibers with centrally located nuclei and to quantify individual fiber CSAs. For each of these analyses, 5 random fields/cross section with ∼300–400 fibers/field were counted for a total of 1500–2000 fibers/sample analyzed. ImageJ software (NIH, Bethesda, MD, USA) was used to measure individual fiber CSAs.
Real-time PCR analysis
Total RNA was isolated from GTN muscles by using Trizol reagent (Life Technologies, Gaithersburg, MD, USA) according to the manufacturer's instructions. cDNA was reverse transcribed, and gene expression was measured by SYBR Green assay and quantified by the 2−ΔΔCt method. The primer sequences are as follows: acetylcholine receptor α-subunit (AChRα), forward ACCTGGACCTATGACGGCTCT, reverse AGTTACTCAGGTCGGGCTGGT; and γ-actin, forward GCCGCCGCGTCCTT, reverse ATGACGAGTGCGGCGATT. The fold change in expression compared to WT values was calculated and plotted.
Statistical analysis
Data are presented as means ± sem for each experiment. Comparisons between genotypes were performed by 1-way ANOVA followed by the post hoc LSD test. Data were analyzed with SPSS 18 (SPSS Inc., Chicago, IL, USA). Values of P < 0.05 were considered statistically significant.
RESULTS
Generation of Sod1 neuron rescue Sod1−/− (SynTgSod1−/−) mice
As described in Materials and Methods and demonstrated by the data shown in Fig. 1, crossing of Sod1−/− mice with transgenic mice in which the human CuZnSOD protein was specifically expressed in neurons under control of the synapsin 1 promoter produced mice (SynTgSod1−/−) with neuron-specific Sod1 expression. To confirm the targeted rescue of the Sod1 gene in neurons, several tissues, including the AT (Fig. 1B) and GTN muscles (Fig. 1C), sciatic nerve (Fig. 1D, E), spinal cord (Fig. 1F), and liver (Fig. 1G) were dissected after the mice were euthanized, and the CuZnSOD protein content was examined. Expression of CuZnSOD protein in the SynTgSod1−/− mice was found in the sciatic nerve (Fig. 1D) and spinal cord (Fig. 1F), but not in the muscle tissue (Fig. 1A, B) or liver (Fig. 1G). The molecular masses of human and mouse CuZnSOD are 23 and 19 kDa, respectively, and this difference in molecular mass of the transgenic SOD1 can be seen in Fig. 1D. CuZnSOD protein content in sciatic nerve (Fig. 1D) and spinal cord (Fig. 1F) of the SynTgSod1−/− mice was on the order of 20–60% of the levels observed in these tissues in WT mice. In contrast, the SynTgSod1−/− mice exhibited ∼2-fold overexpression of hSOD1 in brain compared with endogenous Sod1 expression in the WT mice (data not shown).
Figure 1.

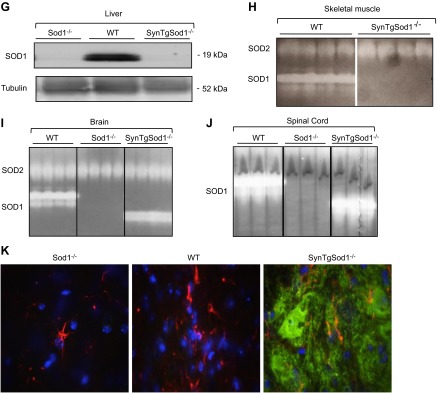
Generation of Sod1 neuron rescue Sod1−/− mice (SynTgSod1−/−). A) Schematic for generating Syn-hSOD1 transgenic mice and breeding to Sod1−/− mice to generate SynTgSod1−/− mice. B, C) Western blot analysis of SOD1 content of the AT (B) and GTN (C) muscles of Sod1−/−, WT, and SynTgSod1−/− mice. D, E) Western blot analysis of SOD1 (D) and SOD2 (E) content of sciatic nerve (SN) of Sod1−/−, WT, and SynTgSod1−/− mice and densitometric quantification of the blots. *P < 0.05 vs. WT. F) Western blot analysis of the SOD1 content of the spinal cord (SC) of WT, Sod1−/−, and SynTgSod1−/− mice and densitometric quantification of the blots. *P < 0.05 vs. WT. G) Representative Western blot of the SOD1 content of the liver of Sod1−/−, WT, and SynTgSod1−/− mice. H–J) Native gels stained for SOD1 and SOD2 enzyme activities in skeletal muscle (H), brain (I), and spinal cord (J) from WT, Sod1−/−, and SynTgSod1−/− mice. K) Transverse sections of lumbar spinal cord dorsal horn from WT, Sod1−/−, and SynTgSod1−/− mice labeled with human SOD1 (green) and GFAP (red), a marker of astrocytes. Nuclei (blue) were labeled with DAPI (×100).
Sod1−/− mice showed no evidence of CuZnSOD expression in any tissues. MnSOD, the mitochondrial SOD isoform, was detected in the sciatic nerves from the Sod1−/−, WT, and SynTgSod1−/− mice, but no significant differences in content were seen between genotypes (Fig. 1E). The neuron-specific expression of CuZnSOD in the SynTgSod1−/− mice was further confirmed by enzymatic activity assays. As shown in Fig. 1H–J, CuZnSOD activity was evident only in the brain (Fig. 1I) and spinal cord (Fig. 1J) of the SynTgSod1−/− mice; the skeletal muscles from these mice showed no detectable CuZnSOD activity. The activity of MnSOD in muscle of the SynTgSod1−/− mice was unchanged compared with that in the WT mice, indicating that no compensatory up-regulation of MnSOD had occurred in response to the absence of CuZnSOD in the skeletal muscle of the SynTgSod1−/− mice (Fig. 1H). To further assess neuron-specific hSOD1 expression, we performed IHC studies of the spinal cords (Fig. 1K). hSOD1 was absent in the WT and Sod1−/− mice but was highly expressed in the SynTgSod1−/− mice. In addition, immunofluorescence imaging with double labeling for hSOD1 and GFAP, a well-characterized marker of astrocytes, indicated no colocalization of hSOD1 with astrocytes, supporting the specific expression of the Sod1 transgene under the synapsin promoter in neurons rather than in nonneuronal glial cells in the Sod1 neuron rescue Sod1−/− mice.
Changes in muscle mass and morphology in SynTgSod1−/− mice
We showed previously (13) that loss of skeletal muscle mass in the Sod1−/− mice occurs as early as 3 mo of age. To determine whether neuron-specific expression of CuZnSOD in the rescue SynTgSod1−/− mice would prevent the loss of muscle mass that occurs in Sod1−/− mice, body weight and the mass of several skeletal muscles were studied. No differences in body weight were observed between genotypes in this study (Table 1). Thus, absolute muscle masses are presented (Table 1). As previously reported, mass was substantially reduced in muscles of the Sod1−/− mice compared with that in WT muscles, with the sole exception of the SOL muscles, which were not different between the genotypes. In contrast to the Sod1−/− mice, which had lower muscle masses, the SynTgSod1−/− mice showed no evidence of atrophy in the AT, EDL, GTN, Quad, or PLT muscles (Table 1). To examine the structure of skeletal muscle, we obtained transverse sections of the GTN muscles from WT, Sod1−/−, and SynTgSod1−/− mice (Fig. 2A). Sections of muscles of the Sod1−/− mice showed variability in fiber size and the presence of a substantial number of fibers containing centrally located nuclei, in agreement with prior results (20). Fibers with centrally located nuclei were rarely detectable in the WT mice (Fig. 2A), but the presence of occasional central nuclei was observed in the GTN muscle fibers from the SynTgSod1−/− mice (Fig. 2A). Quantification of the number of fibers with centrally located nuclei in the SynTgSod1−/− mice indicated that, whereas the number of centrally nucleated fibers was greater in the muscles of the Sod1−/− mice than in those of the WT mice, the number was not significantly elevated in the SynTgSod1−/− mice compared with control values (Fig. 2B). As we have shown for Sod1−/− and WT mice at 8 and 20 mo of age (12), average fiber CSA for GTN muscles did not differ significantly in the WT, Sod1−/−, and SynTgSod1−/− mice (Fig. 2C). However, quantitative analysis of individual fiber CSAs revealed that the myofiber size distribution shifted to the left in the Sod1−/− mice, suggesting that smaller fibers were more prevalent in muscle of those mice (Fig. 2D). In addition, no differences were observed between the WT and SynTgSod1−/− mice, indicating that the absence of CuZnSOD in skeletal muscle of SynTgSod1−/− mice did not induce muscle fiber size changes (Fig. 2D). Similarly, no changes in slow (Fig. 2E) and fast (Fig. 2F) MyHC content was seen between muscles from the WT and SynTgSod1−/− mice, implying no changes in fiber type composition, a finding that was confirmed by IHC (data not shown). Previously, we showed that the absence of CuZnSOD in Sod1−/− mice alters the ultrastructure of myofibrils, as shown by abnormally organized sarcomeres and clustered mitochondria (11). In contrast, muscles of the SynTgSod1−/− mice showed no changes in myofibrillar organization or mitochondrial accumulation compared with muscles of the WT mice (Fig. 2G).
Table 1.
Body and skeletal muscle masses of WT, Sod1−/−, and SynTgSod1−/− mice
| Tissue | WT | Sod1−/− | SynTgSod1−/− |
|---|---|---|---|
| Body weight (g) | 30.9 ± 1.2 | 28.1 ± 1.7 | 29.8 ± 2.5 |
| AT (mg) | 51.3 ± 1.5 | 34.2 ± 2.9* | 50.9 ± 1.7 |
| EDL (mg) | 10.8 ± 0.2 | 8.4 ± 0.7* | 10.2 ± 0.5 |
| GTN (mg) | 146.1 ± 7.0 | 72.4 ± 9.3* | 139.7 ± 8.1 |
| SOL (mg) | 7.8 ± 0.5 | 8.4 ± 0.7 | 9.5 ± 0.5 |
| Quad (mg) | 246.1 ± 9.9 | 136.0 ± 13.3* | 228.5 ± 10.9 |
| PLT (mg) | 15.7 ± 1.1 | 7.3 ± 2.7* | 13.6 ± 1.0 |
Values are presented as means ± sem; n = 5/group.
P < 0.05 vs. WT.
Figure 2.


Muscle morphology in SynTgSod1−/− mice. A) Transverse sections of GTN muscles from WT, Sod1−/−, and SynTgSod1−/− mice stained with WGA, to visualize extracellular matrix, and DAPI, to mark nuclei and show fiber sizes and presence of centrally located nuclei. B) Percentage of fibers showing centrally located nuclei. *P < 0.05 compared with values from WT mice. C) Average fiber CSA of GTN muscles from WT, Sod1−/−, and SynTgSod1−/− mice (n=4). D) Frequency distribution of fiber CSA of GTN muscle in WT, Sod1−/−, and SynTgSod1−/− mice (n=4). E, F) Representative Western blots of the slow (E) and fast (F) MyHC content of the GTN muscle of WT and SynTgSod1−/− mice. G) Electron micrographs of GTN muscles from WT and SynTgSod1−/− mice.
Muscle function is preserved in SynTgSod1−/− mice
To determine whether rescue of the Sod1 gene in neurons of the SynTgSod1−/− mice was sufficient to alter muscle function, we measured maximum isometric force generated by GTN muscles in situ in response to nerve stimulation. As shown in Fig. 3A, maximum isometric-specific force of GTN muscle was reduced by 30% in the Sod1−/− mice compared with the WT mice, whereas muscles from the SynTgSod1−/− mice showed no evidence of impaired muscle force generation. To determine the overall function of NMJs, maximum isometric contractions were also elicited by direct stimulation of the muscle tissue. No changes in maximum force generation were observed in muscles of the SynTgSod1−/− or WT mice (Fig. 3B), whereas a significant increase in force generation was found in the Sod1−/− mice after direct muscle stimulation. Thus, these data indicate that rescue of the CuZnSOD in neurons of Sod1−/− mice restores full functional innervation to the GTN muscle.
Figure 3.
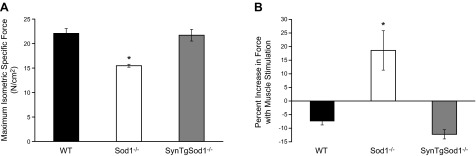
Muscle function in SynTgSod1−/− mice. A) Maximum isometric specific force normalized to total fiber CSA of GTN muscles from WT, Sod1−/−, and SynTgSod1−/− mice at 15 mo of age (n=5). B) Comparison of forces generated during direct muscle and nerve stimulation in GTN muscles of WT, Sod1−/−, and SynTgSod1−/− mice (n=5). Data represent the increase in force generated with direct muscle stimulation expressed as a percentage of the force generated with nerve stimulation. *P < 0.05 vs. WT.
Muscle redox status is not affected in skeletal muscles from SynTgSod1−/− mice deficient of CuZnSOD
To determine the effect of rescue of CuZnSOD in neurons on oxidative damage to SynTgSod1−/− mice, we measured the content of F2-isoprostanes, an indicator of lipid peroxidation, and found no difference in GTN muscle between the SynTgSod1−/− and WT mice (Fig. 4A). Similarly, no differences were observed in the contents of F2-isoprostanes in the brains from SynTgSod1−/− and WT mice. Both tissues (GTN and brain) from Sod1−/− mice showed a significant increase in F2-isoprostanes compared with age-matched WT mice (Fig. 4A) indicating an increase in lipid peroxidation. The level of protein nitration (Fig. 4B) and the protein content of PRXV, a peroxynitrite reductase, was also unchanged in skeletal muscle from the SynTgSod1−/− and WT mice (Fig. 4C), indicating no change in the overall redox status in the muscle of the SynTgSod1−/− mice. In contrast, skeletal muscle from the Sod1−/− mice showed a significant increase in both protein nitration (Fig. 4B) and PRXV expression (Fig. 4C), potentially reflecting an increase in peroxynitrite activity in skeletal muscle of the SOD1-null mice, as previously observed (14).
Figure 4.

Markers of oxidative damage in skeletal muscle and brain of SynTgSod1−/− mice. A) Lipid peroxidation assessed by F2-isoprostane content of skeletal muscle and brain of WT, Sod1−/−, and SynTgSod1−/− mice. B) Western blot analysis of changes in 3-NT content of skeletal muscles of Sod1−/−, WT, and SynTgSod1−/− mice and densitometric quantification of the blots. C) Representative Western blots of the content of PRXV, a peroxynitrite reductase, in skeletal muscle of Sod1−/−, WT, and SynTgSod1−/− mice and densitometric quantification of the blots. *P < 0.05 vs. WT.
No compensatory up-regulation in regulatory enzymes for RONS, stress responses, and adaptive signaling pathways in SynTgSod1−/− mice
The accelerated skeletal muscle-aging phenotype observed in Sod1−/− mice has been associated with increased oxidative damage and altered redox markers including an up-regulation in HSP expression and increased NFκB activation (15). To determine whether restoration of the neuron SOD1 in the absence of muscle SOD1 in SynTgSod1−/− mice might lead to a compensatory change in the expression or activity of proteins involved in antioxidant defense, we measured the expression of RONS regulatory enzymes and proteins in muscles. As shown in Fig. 5, no significant changes were detected in the RONS regulatory enzymes, including nNOS, eNOS, iNOS, and MnSOD, in muscle tissue of the SynTgSod1−/− mice compared with that of the WT mice. Muscle of the Sod1−/− mice showed a significant increase in the content of eNOS (Fig. 5B) and iNOS (Fig. 5C), compared with muscle from the WT mice. No significant difference was seen in the content of MnSOD in Sod1−/− muscle, compared with that in WT or SynTgSod1−/− muscle (Fig. 5D).
Figure 5.
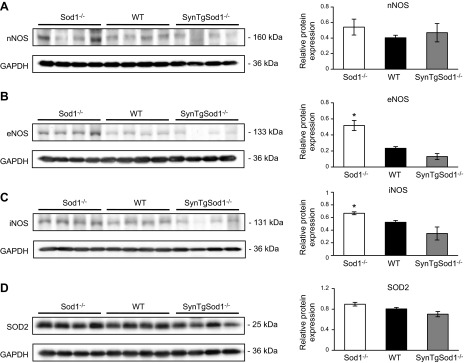
RONS regulatory protein contents in skeletal muscle of SynTgSod1−/− mice. Representative Western blots of nNOS (A), eNOS (B), iNOS (C), and SOD2 (D) protein content of skeletal muscles from Sod1−/−, WT, and SynTgSod1−/− mice and densitometric quantification of the blots. *P < 0.05 vs. WT.
HSPs have been shown to provide protection against an increase in RONS production (5), and we have shown elevated HSP content in muscle from Sod1−/− mice (15). We examined whether the content of various HSPs (Fig. 6) and markers of NFκB activation (Fig. 7) were altered in skeletal muscle of the SynTgSod1−/− mice in comparison with that of the Sod1−/− and WT mice. Protein expression of HSP10 (Fig. 6A), HSP25 (Fig. 6B), and HSC70 (Fig. 6E) was unchanged in the muscles of the SynTgSod1−/− mice, compared with expression in the WT muscle, but muscles from the Sod1−/− mice had an increased content of HSP25 (Fig. 6B), HSP60 (Fig. 6C), and HSP72 content (Fig. 6D). Interestingly, there was evidence of HSP60 down-regulation (Fig. 6C) in muscle from the SynTgSod1−/− mice compared with both other groups. Inducible HSP72 was not detected in skeletal muscle from the rescue and WT mice (Fig. 6D). Thus, rescue of neuron CuZnSOD in SynTgSod1−/− mice prevented the compensatory increases in HSPs that occurred in the whole-body SOD1-knockout mice. Previous data indicate that the NFκB signaling pathway is activated in muscle from Sod1−/− mice, but muscle from SynTgSod1−/− mice showed no evidence of this, as assessed by nuclear translocation of the NFκB cleavage products p50 and p65 (Fig. 7). Nuclear p50 (Fig. 7B) and p65 (Fig. 7C) contents were unchanged between the SynTgSod1−/− and WT mice.
Figure 6.

HSPs in skeletal muscle of SynTgSod1−/− mice. Representative Western blots of HSP10 (A), HSP25 (B), HSP60 (C), HSP72 (D), and HSC70 (E) protein content of skeletal muscle from Sod1−/−, WT, and SynTgSod1−/− mice and densitometric quantification of the blots. HSP72 was not detectable in muscles of WT and SynTgSod1−/− mice, and hence densitometry was not undertaken. *P < 0.05 vs. WT.
Figure 7.
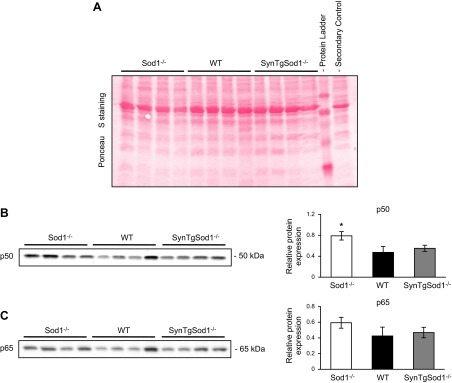
NFκB activation assessed by nuclear location of the p50 and p65 subunits of NFκB in skeletal muscle of SynTgSod1−/− mice. A) Ponceau S staining used to ensure equivalent loading of the samples, corresponding to the Western blots in panels B, C. B, C) Representative Western blots of the NFκB p50 (B) and p65 (C) subunits in nuclear fractions from skeletal muscle of Sod1−/−, WT, and SynTgSod1−/− mice and densitometric quantification of the blots. *P < 0.05 vs. WT.
Neuron-specific expression of CuZnSOD in SynTgSod1−/− mice preserves NMJ structure and function
Based on our observation that neuron-specific expression of CuZnSOD prevents the losses of muscle mass and function that occur in Sod1−/− knockout mice (Fig. 2 and 3), we hypothesized that rescue of CuZnSOD expression in motor neurons would also preserve motor neurons and NMJ structure and function. To test this hypothesis, we examined NMJ structure via immunofluorescent staining techniques (Fig. 8A). Motor neuron axons were stained with antibodies to neurofilaments and synaptotagmin 2 (NF/Syt2), whereas the motor end plates were identified with fluorophore-conjugated bungarotoxin, which stains the subunit of the postsynaptic nicotinic AChRs. As shown in Fig. 8A, immunohistologic examination of NMJs from the Sod1−/− mouse model exhibited abnormal axons and fragmented postsynaptic end plates (Fig. 8A). In comparison, NMJs in the SynTgSod1−/− mice showed no evidence of marked alterations, compared with those in the NMJs of the age-matched WT mice, indicating that rescue of CuZnSOD specifically in neurons restores both presynaptic and postsynaptic NMJ structure. Analysis of the content of mRNA for AChR-α in muscles was undertaken to examine the effect of restoration of neuronal CuZnSOD on AChR turnover in Sod1−/− mice. The AChR-α mRNA content was increased in muscle from the Sod1−/− mice, but was unchanged in the SynTgSod1−/− mice in comparison with that in the WT (Fig. 8B). Experiments to examine muscle electrophysiological properties indicated that expression of CuZnSOD exclusively in neurons in the Sod1−/− mice resulted in an electrophysiological phenotype similar to that of the WT animals (Fig. 8C). The amplitude of evoked EPPs and the frequency of spontaneous mEPPs in EDL muscle in the SynTgSod1−/− mice were substantially higher than those in the Sod1−/− mice (unpublished observations) and were comparable to those in the WT mice (SynTgSod1−/−, EPP: 30.2±1.5 mV, mEPP: 3.94±0.49 Hz, n=6; WT, EPP: 27.5±1.0 mV, mEPP: 3.77±0.28Hz, n=22), suggesting that synaptic vesicle release is fully restored by neuronal CuZnSOD expression. Figure 8D shows the effect of repetitive stimulation on the CMAP in the 3 groups of mice. At increased stimulation frequency, muscles from the Sod1−/− mice showed a decrease in CMAP, but this was not seen in the SynTgSod1−/− or WT mice, demonstrating that restoration of CuZnSOD in neurons rescues neurotransmission in SynTgSod1−/− mice.
Figure 8.
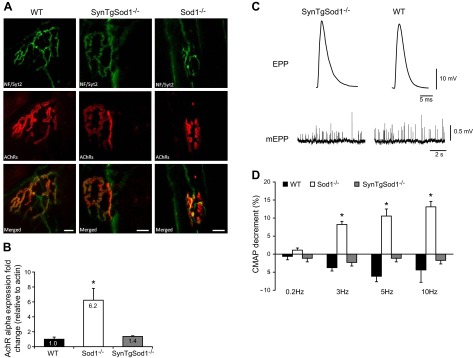
Structure and function of NMJs in SynTgSod1−/− mice. A) NMJ immunofluorescence images from AT muscles of WT, SynTgSod1−/−, and Sod1−/− mice at 8–12 mo of age. Top panels: morphology of presynaptic motor neurons stained with antibodies to neurofilaments and synaptotagmin-2 (green). Middle panels: morphology of postsynaptic AChRs labeled with AlexaFluor 594-conjugated bungarotoxin (red). Bottom panels: merged images of presynaptic motor neurons and AChRs. Scale bars = 10 μm. B) AChR-α mRNA expression in skeletal muscle of WT (n=9), Sod1−/− (n=8), and SynTgSod1−/− (n=6) mice measured by real-time PCR analysis, using γ-actin as the reference gene. Data are normalized to WT values and presented as fold change. C) Representative recordings of evoked EPPs and spontaneous mEPPs in the EDL muscles from WT and SynTgSod1−/− mice. D) Changes in neurotransmission in skeletal muscles (GTN), shown as the percentage decrement in muscle CMAP measured using EMG from WT (n=9), SynTgSod1−/− (n=9), and Sod1−/− (n=10) mice. *P < 0.05 vs. WT.
DISCUSSION
Considerable data have been presented examining the possibility that changes in oxidative stress or a failure of redox homeostasis play a role in aging processes. Although the area remains controversial (29, 30), substantial evidence indicates that age-related losses of skeletal muscle mass and function (sarcopenia) are associated with increased oxidative damage to the tissue and with activation of redox-regulated degenerative pathways (10, 31). Our previous studies have demonstrated that mice lacking CuZnSOD in all tissues (Sod1−/−) are characterized by an accelerated skeletal muscle–aging phenotype that includes many features of normal aging, such as loss of muscle fibers, weakness of the remaining fibers, and degeneration of NMJs (11–13). Muscles of Sod1−/− mice also show increased oxidative damage to lipids, proteins, and DNA, evidence of adaptations in stress responses to RONS, and defective redox signaling after contractile activity, all features that are also seen in aged WT mice (13, 15). We have therefore proposed that these mice can provide a mechanistic model to explore the processes by which oxidative damage or defective redox signaling can contribute to skeletal muscle aging.
Our group had previously refined the Sod1−/− mouse model to examine the role of a muscle-specific deletion of CuZnSOD in the accelerated muscle-aging phenotype and generated surprising results. Mice with muscle-specific deletion of the CuZnSOD gene maintained muscle masses at or above those of WT mice and thus did not reproduce the phenotype seen in whole-body Sod1−/− mice (21). Our aim in the current study was to examine the role of the deficit in CuZnSOD in motor neurons in the muscle atrophy phenotype exhibited by Sod1−/− mice. Our approach was to generate a neuron-specific transgenic Sod1−/− mouse model in which CuZnSOD was expressed in neurons under the control of the synapsin 1 promoter (SynTgSod1−/− mice). We hypothesized that if alterations in redox homeostasis within the motor neurons were the primary initiators of muscle atrophy, then targeted expression of CuZnSOD in the nerve of Sod1−/− mice would at least partially prevent the accelerated, age-related loss of muscle mass and function. SynTgSod1−/− mice showed no losses in muscle mass or maximum isometric specific force production up to at least 15 mo of age, a point in life when dramatic deficits in essentially all measures of neuromuscular function were seen in Sod1−/− mice (11–13). Increased oxidative damage and adaptations in stress responses observed in muscles from Sod1−/− mice (14, 15) were not present in SynTgSod1−/− mice. Finally, the accelerated degeneration in NMJ structure and function that occurred in Sod1−/− mice (11) was not seen in SynTgSod1−/− mice up to 15 mo of age.
Overall, the present findings support a potential role for changes in the redox status of neurons as a mechanism leading to loss of muscle mass and function during aging. Since muscles of SynTgSod1−/− mice lack CuZnSOD, the current results are also consistent with our previous observation that absence of CuZnSOD restricted to muscle is not sufficient to induce muscle atrophy or fiber loss (21). The partial rescue of CuZnSOD expression in motor neurons of the SynTgSod1−/− mice to ∼20% of WT levels dramatically reversed all aspects of the accelerated neuromuscular aging phenotype found in the Sod1−/− mice, including the multiple biochemical and physiological changes associated with the accelerated aging. Use of the synapsin 1 promoter in the transgenic mice allowed expression of CuZnSOD in all tissues of SynTgSod1−/− mice that contain synapses, including brain, spinal cord, and sensory and motor nerves, but excluded any role for other tissues and cell types that might be anticipated to play an essential role in maintenance of NMJs (e.g., Schwann cells) or muscles (e.g., satellite cells). Although we found no evidence of transgene expression in nonneuronal tissues, as indicated by immunoblot analysis and IHC and enzymatic studies, a comprehensive analysis of the localization of the hSOD1 expression in neurons has not been completed. Thus, we cannot rule out the possibility that CuZnSOD expression in other cell types within the nerve contributes to the protection from NMJ and muscle decline. The observation that the relatively low expression levels (∼20%) of CuZnSOD in the sciatic nerve of the SynTgSod1−/− mice were sufficient to rescue the accelerated muscle phenotype in the Sod1−/− is in accord with previous data showing that expression of ∼25% of the SOD1 protein in neurons of Sod1−/− mice rescues motor neuron neuropathy (32) and suggests that SOD1 in motor neurons of WT mice is in relative excess. We speculate that this reflects a specific requirement to protect neural tissue, in particular, against superoxide- or peroxynitrite-induced degeneration, since complete deletion of SOD1 in muscle fibers does not induce overt degenerative changes in that tissue (21).
Taken together with our previous studies of muscle-specific CuZnSOD deletion (21), these data indicate that the increase in oxidative damage in muscle from Sod1−/− mice derives from changes in the redox status of neurons and hence potentially from a lack of appropriate innervation of the muscle. Isolated mitochondria from muscle of Sod1−/− mice have been shown to release increased amounts of hydrogen peroxide (11), and this process has been assumed to be the source of the increased ROS generation that leads to increased oxidative damage in Sod1−/− mice and also in aged WT mice (10, 31). Data from the current study provide further support for the conclusion that such changes in the muscle of Sod1−/− mice are secondary to neuronal deficits, because they did not occur in the SynTgSod1−/− mice where CuZnSOD was rescued only in the neurons. A potential mechanism for these changes comes from a study by Muller et al. (33), who demonstrated that transsection of the motor nerve induces large and sustained increases in the release of hydrogen peroxide and other ROS from mitochondria isolated from the denervated muscle. Thus, a logical hypothesis is that a primary effect of absence of CuZnSOD in neurons is to induce a functional denervation of muscle fibers that leads to an increase in ROS generation by muscle mitochondria, a consequent increase in oxidative damage in muscle, and an activation of adaptive responses to oxidative stress.
Clear evidence that the absence of CuZnSOD in neurons in Sod1−/− mice leads to functional denervation of muscle fibers, disruption of NMJs, and failure of neuromuscular transmission was provided by the observation that direct muscle stimulation showed an increase in maximum isometric force generation in muscle of the Sod1−/− mice that was not seen in muscles of the SynTgSod1−/− mice. In addition, both pre- and postsynaptic NMJ structures were disrupted in the Sod1−/− mice but not in the SynTgSod1−/− mice, and the electrophysiological properties, such as synaptic transmission at individual NMJs and muscle fiber recruitment, were maintained in the SynTgSod1−/− mice. In support of the hypothesis that maintenance of the integrity of NMJ structure is highly dependent on CuZnSOD expression in neurons, we also found no increase in the AChR-α mRNA content in muscle from the SynTgSod1−/− mice. Our previous observations of increased AChR mRNA levels in Sod1−/− mice with no increase in AChR protein level (11) was interpreted as a failed attempt to up-regulate the NMJ components in response to denervation in Sod1−/− mice. The lack of a compensatory increase observed in the present study in AChR mRNA in the SynTgSod1−/− mice suggests that there is no NMJ degeneration in this model. Thus, it appears that the absence of CuZnSOD in motor neurons specifically leads to disruption of motor neuron and NMJ structure and function. Several experimental models that lead to fragmentation and disruption of AChR and NMJs have been shown to induce a phenotype that resembles sarcopenia. These include depletion of the nerve-derived organizer of postsynaptic differentiation agrin (17), enhanced degradation of agrin through overexpression of neurotrypsin (18), and a reduction in the expression of the postsynaptic tyrosine kinase receptor B (TrkB; ref. 19). TrkB is a receptor for neurotrophic factors such as BDNF and neurotrophin-4 and -5. Although the latter model initially appears to be myocyte specific, all these models share the property of acting to disrupt communication from the motor neuron to the muscle, leading to fragmentation of the NMJ. It is tempting to speculate that this reflects a final common pathway that is also present in Sod1−/− mice, although there are currently no specific data in support of this notion.
Denervation and loss of NMJ integrity have been implicated in the loss of muscle fibers and failure of muscle force generation that occur with aging in humans and rodents (34–37). The model described herein indicates that deficits in redox homeostasis in motor nerves alone are sufficient to generate an aging phenotype in skeletal muscle and NMJs, with no independent contribution from muscle fibers, Schwann cells, or other associated cell types. It is currently unknown whether the age-related loss of muscle mass and function that occur in older humans and animals can similarly be attributed to deficits in motor neurons, or whether age-related deficits in skeletal muscles or other tissues play a role. This model potentially provides a powerful approach to help elucidate the roles of tissue-specific defects in redox status in this important area of muscle aging.
Acknowledgments
The authors thank Teresa M. Evans, Lauren K. Wood, Anne Erickson, and José Gomez for technical assistance.
This work was supported by U. S. National Institute on Aging/National Institutes of Health grants AG-020591 and T32 AG021890 (to M.V.I.).
Footnotes
- 3-NT
- 3-nitrotyrosine
- AChR
- acetylcholine receptor
- AT
- anterior tibialis
- CMAP
- compound muscle action potential
- CSA
- cross-sectional area
- CuZnSOD
- copper-zinc superoxide dismutase
- EDL
- extensor digitorum longus
- EMG
- electromyography
- EPP
- end plate potential
- GFAP
- glial fibrillary acidic protein
- GTN
- gastrocnemius
- IHC
- immunohistochemistry
- Lf
- fiber length
- hSOD1
- human superoxide dismutase 1
- Lo
- optimal length
- HSC
- heat-shock cognate
- HSP
- heat-shock protein
- mEPP
- miniature end plate potential
- MnSOD
- manganese superoxide dismutase
- MyHC
- myosin heavy chain
- NMJ
- neuromuscular junction
- PFA
- paraformaldehyde
- PLT
- plantaris
- Po
- maximum isometric tetanic force
- PRXV
- peroxiredoxin V
- Quad
- quadriceps
- RONS
- reactive oxygen and nitrogen species
- SOD
- superoxide dismutase
- SOL
- soleus
- SPF
- specific pathogen-free
- TrkB
- tyrosine kinase receptor B
- UM
- University of Michigan
- UTHSCSA
- University of Texas Health Science Center at San Antonio
- WGA
- wheat germ agglutinin
- WT
- wild type
REFERENCES
- 1. Evans W. J. (2010) Skeletal muscle loss: cachexia, sarcopenia, and inactivity. Am. J. Clin. Nutr. 91, 1123S–1127S [DOI] [PubMed] [Google Scholar]
- 2. Larsson L. (1983) Histochemical characteristics of human skeletal muscle during aging. Acta Physiol. Scand. 117, 469–471 [DOI] [PubMed] [Google Scholar]
- 3. Porter M. M., Vandervoort A. A., Lexell J. (1995) Aging of human muscle: structure, function and adaptability. Scand. J. Med. Sci. Sports 5, 129–142 [DOI] [PubMed] [Google Scholar]
- 4. Lexell J. (1995) Human aging, muscle mass, and fiber type composition. J. Gerontol. A Biol. Sci. Med. Sci. 50(Spec. No.), 11–16 [DOI] [PubMed] [Google Scholar]
- 5. Broome C. S., Kayani A. C., Palomero J., Dillmann W. H., Mestril R., Jackson M. J., McArdle A. (2006) Effect of lifelong overexpression of HSP70 in skeletal muscle on age-related oxidative stress and adaptation after nondamaging contractile activity. FASEB J. 20, 1549–1551 [DOI] [PubMed] [Google Scholar]
- 6. Reid M. B., Durham W. J. (2002) Generation of reactive oxygen and nitrogen species in contracting skeletal muscle: potential impact on aging. Ann. N. Y. Acad. Sci. 959, 108–116 [DOI] [PubMed] [Google Scholar]
- 7. Vasilaki A., Simpson D., McArdle F., McLean L., Beynon R. J., Van Remmen H., Richardson A. G., McArdle A., Faulkner J. A., Jackson M. J. (2007) Formation of 3-nitrotyrosines in carbonic anhydrase III is a sensitive marker of oxidative stress in skeletal muscle. Proteomics Clin. Appl. 1, 362–372 [DOI] [PubMed] [Google Scholar]
- 8. Mecocci P., Fano G., Fulle S., MacGarvey U., Shinobu L., Polidori M. C., Cherubini A., Vecchiet J., Senin U., Beal M. F. (1999) Age-dependent increases in oxidative damage to DNA, lipids, and proteins in human skeletal muscle. Free Radic. Biol. Med. 26, 303–308 [DOI] [PubMed] [Google Scholar]
- 9. Palomero J., Vasilaki A., Pye D., McArdle A., Jackson M. J. (2013) Aging increases the oxidation of dichlorohydrofluorescein in single isolated skeletal muscle fibers at rest, but not during contractions. Am. J. Physiol. Regul. Integr. Comp. Physiol. 305, R351–R358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vasilaki A., Mansouri A., Remmen H., van der Meulen J. H., Larkin L., Richardson A. G., McArdle A., Faulkner J. A., Jackson M. J. (2006) Free radical generation by skeletal muscle of adult and old mice: effect of contractile activity. Aging Cell 5, 109–117 [DOI] [PubMed] [Google Scholar]
- 11. Jang Y. C., Lustgarten M. S., Liu Y., Muller F. L., Bhattacharya A., Liang H., Salmon A. B., Brooks S. V., Larkin L., Hayworth C. R., Richardson A., Van Remmen H. (2010) Increased superoxide in vivo accelerates age-associated muscle atrophy through mitochondrial dysfunction and neuromuscular junction degeneration. FASEB J. 24, 1376–1390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Larkin L. M., Davis C. S., Sims-Robinson C., Kostrominova T. Y., Remmen H. V., Richardson A., Feldman E. L., Brooks S. V. (2011) Skeletal muscle weakness due to deficiency of CuZn-superoxide dismutase is associated with loss of functional innervation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 301, R1400–R1407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Muller F. L., Song W., Liu Y., Chaudhuri A., Pieke-Dahl S., Strong R., Huang T. T., Epstein C. J., Roberts L. J., 2nd, Csete M., Faulkner J. A., Van Remmen H. (2006) Absence of CuZn superoxide dismutase leads to elevated oxidative stress and acceleration of age-dependent skeletal muscle atrophy. Free Radic. Biol. Med. 40, 1993–2004 [DOI] [PubMed] [Google Scholar]
- 14. Sakellariou G. K., Pye D., Vasilaki A., Zibrik L., Palomero J., Kabayo T., McArdle F., Van Remmen H., Richardson A., Tidball J. G., McArdle A., Jackson M. J. (2011) Role of superoxide-nitric oxide interactions in the accelerated age-related loss of muscle mass in mice lacking Cu,Zn superoxide dismutase. Aging Cell 10, 749–760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vasilaki A., van der Meulen J. H., Larkin L., Harrison D. C., Pearson T., Van Remmen H., Richardson A., Brooks S. V., Jackson M. J., McArdle A. (2010) The age-related failure of adaptive responses to contractile activity in skeletal muscle is mimicked in young mice by deletion of Cu,Zn superoxide dismutase. Aging Cell 9, 979–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Larsson J. E., Wahlstrom G. (1998) The influence of age and administration rate on the brain sensitivity to propofol in rats. Acta Anaesthesiol. Scand. 42, 987–994 [DOI] [PubMed] [Google Scholar]
- 17. Samuel M. A., Valdez G., Tapia J. C., Lichtman J. W., Sanes J. R. (2012) Agrin and synaptic laminin are required to maintain adult neuromuscular junctions. PLoS ONE 7, e46663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Butikofer L., Zurlinden A., Bolliger M. F., Kunz B., Sonderegger P. (2011) Destabilization of the neuromuscular junction by proteolytic cleavage of agrin results in precocious sarcopenia. FASEB J. 25, 4378–4393 [DOI] [PubMed] [Google Scholar]
- 19. Kulakowski S. A., Parker S. D., Personius K. E. (2011) Reduced TrkB expression results in precocious age-like changes in neuromuscular structure, neurotransmission, and muscle function. J. Appl. Physiol. 111, 844–852 [DOI] [PubMed] [Google Scholar]
- 20. Fischer L. R., Igoudjil A., Magrane J., Li Y., Hansen J. M., Manfredi G., Glass J. D. (2011) SOD1 targeted to the mitochondrial intermembrane space prevents motor neuropathy in the Sod1 knockout mouse. Brain 134, 196–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang Y., Davis C., Sakellariou G. K., Shi Y., Kayani A. C., Pulliam D., Bhattacharya A., Richardson A., Jackson M. J., McArdle A., Brooks S. V., Van Remmen H. (2013) CuZnSOD gene deletion targeted to skeletal muscle leads to loss of contractile force but does not cause muscle atrophy in adult mice. FASEB J. 27, 3536–3548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Elchuri S., Oberley T. D., Qi W., Eisenstein R. S., Jackson Roberts L., Van Remmen H., Epstein C. J., Huang T. T. (2005) CuZnSOD deficiency leads to persistent and widespread oxidative damage and hepatocarcinogenesis later in life. Oncogene 24, 367–380 [DOI] [PubMed] [Google Scholar]
- 23. Huang T. T., Yasunami M., Carlson E. J., Gillespie A. M., Reaume A. G., Hoffman E. K., Chan P. H., Scott R. W., Epstein C. J. (1997) Superoxide-mediated cytotoxicity in superoxide dismutase-deficient fetal fibroblasts. Arch. Biochem. Biophys. 344, 424–432 [DOI] [PubMed] [Google Scholar]
- 24. Kaja S., van de Ven R. C., van Dijk J. G., Verschuuren J. J., Arahata K., Frants R. R., Ferrari M. D., van den Maagdenberg A. M., Plomp J. J. (2007) Severely impaired neuromuscular synaptic transmission causes muscle weakness in the Cacna1a-mutant mouse rolling Nagoya. Eur. J. Neurosci. 25, 2009–2020 [DOI] [PubMed] [Google Scholar]
- 25. Van Remmen H., Salvador C., Yang H., Huang T. T., Epstein C. J., Richardson A. (1999) Characterization of the antioxidant status of the heterozygous manganese superoxide dismutase knockout mouse. Arch. Biochem. Biophys. 363, 91–97 [DOI] [PubMed] [Google Scholar]
- 26. Ward W. F., Qi W., Van Remmen H., Zackert W. E., Roberts L. J., 2nd, Richardson A. (2005) Effects of age and caloric restriction on lipid peroxidation: measurement of oxidative stress by F2-isoprostane levels. J. Gerontol. A Biol. Sci. Med. Sci. 60, 847–851 [DOI] [PubMed] [Google Scholar]
- 27. Morrow J. D., Roberts L. J., 2nd (1999) Mass spectrometric quantification of F2-isoprostanes in biological fluids and tissues as measure of oxidant stress. Methods Enzymol. 300, 3–12 [DOI] [PubMed] [Google Scholar]
- 28. Sakellariou G. K., Vasilaki A., Palomero J., Kayani A., Zibrik L., McArdle A., Jackson M. J. (2013) Studies of mitochondrial and nonmitochondrial sources implicate nicotinamide adenine dinucleotide phosphate oxidase(s) in the increased skeletal muscle superoxide generation that occurs during contractile activity. Antioxid. Redox Signal. 18, 603–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Perez V. I., Bokov A., Van Remmen H., Mele J., Ran Q., Ikeno Y., Richardson A. (2009) Is the oxidative stress theory of aging dead? Biochim. Biophys. Acta 1790, 628–638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gems D., Doonan R. (2009) Antioxidant defense and aging in C. elegans: is the oxidative damage theory of aging wrong? Cell Cycle 8, 1681–1687 [DOI] [PubMed] [Google Scholar]
- 31. Van Remmen H., Richardson A. (2001) Oxidative damage to mitochondria and aging. Exp. Gerontol. 36, 957–968 [DOI] [PubMed] [Google Scholar]
- 32. Flood D. G., Reaume A. G., Gruner J. A., Hoffman E. K., Hirsch J. D., Lin Y. G., Dorfman K. S., Scott R. W. (1999) Hindlimb motor neurons require Cu/Zn superoxide dismutase for maintenance of neuromuscular junctions. Am. J. Pathol. 155, 663–672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Muller F. L., Song W., Jang Y. C., Liu Y., Sabia M., Richardson A., Van Remmen H. (2007) Denervation-induced skeletal muscle atrophy is associated with increased mitochondrial ROS production. Am. J. Physiol. Regul. Integr. Comp. Physiol. 293, R1159–R1168 [DOI] [PubMed] [Google Scholar]
- 34. Campbell M. J., McComas A. J., Petito F. (1973) Physiological changes in ageing muscles. J. Neurol. Neurosurg. Psychiatry 36, 174–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rowan S. L., Rygiel K., Purves-Smith F. M., Solbak N. M., Turnbull D. M., Hepple R. T. (2012) Denervation causes fiber atrophy and myosin heavy chain co-expression in senescent skeletal muscle. PLoS ONE 7, e29082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Larsson L., Yu F., Hook P., Ramamurthy B., Marx J. O., Pircher P. (2001) Effects of aging on regulation of muscle contraction at the motor unit, muscle cell, and molecular levels. Int. J. Sports Nutr. Exerc. Metab. 11(Suppl.), S28–S43 [DOI] [PubMed] [Google Scholar]
- 37. Chai R. J., Vukovic J., Dunlop S., Grounds M. D., Shavlakadze T. (2011) Striking denervation of neuromuscular junctions without lumbar motoneuron loss in geriatric mouse muscle. PLoS ONE 6, e28090. [DOI] [PMC free article] [PubMed] [Google Scholar]