

Abstract
The large diversity of viruses that exist in human populations are potentially excreted into sewage collection systems and concentrated in sewage sludge. In the US, the primary fate of processed sewage sludge (class B biosolids) is application to agricultural land as a soil amendment. To characterize and understand infectious risks associated with land application, and to describe the diversity of viruses in human populations, shotgun viral metagenomics was applied to 10 sewage sludge samples from 5 wastewater treatment plants throughout the continental U.S, each serving between 100,000 and 1,000,000 people. Nearly 330 million DNA sequences were produced and assembled, and annotation resulted in identifying 43 (26 DNA, 17 RNA) different types of human viruses in sewage sludge. Novel insights include the high abundance of newly emerging viruses (e.g. Coronavirus HKU1, Klassevirus, and Cosavirus) the strong representation of respiratory viruses, and the relatively minor abundance and occurrence of Enteroviruses. Viral metagenome sequence annotations were reproducible and independent PCR-based identification of selected viruses suggests that viral metagenomes were a conservative estimate of the true viral occurrence and diversity. These results represent the most complete description of human virus diversity in any wastewater sample to date, provide engineers and environmental scientists with critical information on important viral agents and routes of infection from exposure to wastewater and sewage sludge, and represent a significant leap forward in understanding the pathogen content of class B biosolids.
Keywords: biosolids, metagenome, viral metagenome, next generation DNA sequencing, virus, Klassevirus
Introduction
Despite the important global public health burden of environmental viral infections, methods have not been fully developed and applied for describing the broad diversity of human viruses in environmental samples. Over 140 different viral pathogen genotypes have the potential to exist in the environment1, and new pathogenic strains and species continue to be discovered2. Each viral type has unique infectious, structural, environmental transport, and environmental survival characteristics. Due in large part to culturability limitations and cost associated with identifying and quantifying human viruses through culture-based methods, environmental regulations and monitoring schemes typically focus on bacterial indicators, such as fecal coliforms3.
Shotgun metagenomic approaches enable simultaneous and target independent identification of viral pathogens, and provide a potential solution to the challenges associated with the traditionally culture-based methods for viral identification. By isolating virus-sized particles, extracting their nucleic acids, sequencing random short nucleic acid fragments, and assembling and classifying these short sequences, the full diversity of viral pathogens in an environmental sample can be revealed4, 5. Although the rapid reductions in the time and cost of DNA sequencing and the increasing set of sequenced genomes from human viral pathogens have enabled this metagenome approach for human viral pathogen detection4–6, viral metagenomic approaches have yet to be applied to highlight viral pathogen diversity, prevalence, and dominant genotypes in a large cross-section of environmental samples.
Sewage sludge, the solid waste stream resulting from wastewater treatment, represents an ideal matrix with which to apply metagenomic viral pathogen detection technologies. Sewage sludge has a high potential for pathogen diversity due to its source of concentrated human waste from thousands to millions of humans, and all known viral pathogens can be excreted by humans in to wastewater collection systems. The ultimate fate of particle-associated viruses contained in raw sewage and secondary wastewater clarifiers is sewage sludge. In the U.S., the majority of biosolids (stabilized sewage sludge) are applied to agricultural land and approximately 75% of these are of class B status7. While class B biosolids are known to contain pathogens, the full pathogen content has not been fully described, especially for viruses8, 9. The independent identification of viral pathogen diversity in sewage sludge would represent a significant advance in the science of sewage sludge management. More broadly, viral pathogen diversity data can assist in making rational regulatory and treatment design decisions in a variety of environmental scenarios including the assessment of human exposure routes to viruses in wastewater effluents, transport of reclaimed wastewater viruses through the subsurface, wastewater source tracking, and tracking the efficacy of virus removal from a variety of water and wastewater treatment schemes, including point of use devices10, 11.
We conducted a metagenomic analysis on ten sewage sludge samples (mesophilic anaerobic digester influent and effluent from five plants) collected from major wastewater treatment facilities located throughout the continental United States. DNA and RNA (cDNA) from virus-sized particles were isolated and sequenced using three separate runs of Illumina® HiSeq technology. These sequences were then quality trimmed, assembled, and annotated to identify the diversity of human DNA and RNA viruses in raw and stabilized sewage sludge. Replicated sample preparation and sequencing runs, and independent confirmation by polymerase chain reaction were employed to assess the quality of viral pathogen annotation. The ability to identify the full diversity of human viruses in environmental samples will assist in moving environmental science and engineering away from a strong reliance on indicator organisms, and closer to the analysis of the relevant infectious agents, thus enabling new approaches for determining the environmental fate of and exposure to viruses that are infectious to humans.
MATERIALS AND METHODS
Summaries of the materials and methods for sample preparation, nucleic acid extraction and sequencing, and bioinformatic analyses are provided below. In-depth details of these methods are provided in the Supporting Information.
Sewage sludge sample preparation
The influent and effluent sludge from mesophilic anaerobic digesters was sampled from five domestic wastewater treatment plants within the continental United States (one each from the Southwest, Southeast and Midwest, and two from the Northeast). All influent samples were mixtures of primary and secondary sludge. Effluent sewage sludge samples were of a class B product, prior to dewatering. Samples were shipped on ice and fecal coliform and coliphage analysis plus virus extraction was performed within 24 hours of sampling. The sampled treatment plants served populations between 100,000 and 1,000,000 residents and utilized activated sludge processes.
Viruses were eluted from sewage sludge samples and then concentrated following a procedure adapted from Monpoeho and co-workers12 (see Supporting Information for details). The viral particles were then purified and concentrated further by overnight polyethylene glycol 8000 (Fisher Scientific, Massachusetts, USA) precipitation. The final product was suspended in 12 ml of sterile phosphate buffered saline (PBS, 0.13M sodium chloride, 0.003M potassium chloride, and 0.01M phosphate, pH=7.3) (Fisher Scientific, Massachusetts, USA) and used for subsequent coliphage culturing and nucleic acid extraction.
Somatic and male-specific coliphages in the samples were cultured using U.S. EPA method 160213 via the dual agar method (see Supporting Information for details). Fecal coliforms were enumerated by serial diluting sewage sludge in sterile PBS, spread plating on m-FC agar (BD Diagnostics, Maryland, USA), incubating at 44.5°C overnight, and visually enumerating colonies. Treatment plant digester characteristics and indicator organism concentrations are summarized in Table 1.
Table 1.
Anaerobic Digester Operation and Sludge Indicator Concentrations.
| Samplea |
Time and Temp. |
TSS |
Fecal Coliformsb |
Somatic Coliphagesc |
Male-Specific Coliphagesd |
||
|---|---|---|---|---|---|---|---|
| (%) | log(CFU/Dry Gram) | log(PFU/Dry Gram) | log(PFU/Dry Gram) | ||||
| A | INFLUENT | AI | >15 days, 37°C | 3.7 | 7.4 | 4.7 | 3.6 |
| EFFLUENT | AE | >15 days, 37°C | 1.7 | 5.9 | 3.8 | 2.1 | |
| B | INFLUENT | BI | >15 days, 37°C | 3.8 | 6.4 | 4.7 | 3.6 |
| EFFLUENT | BE | >15 days, 37°C | 1.2 | 5.2 | 4.0 | 2.2 | |
| C | INFLUENT | CI | >15 days, 37°C | 3.2 | 6.2 | 3.4 | 4.7 |
| EFFLUENT | CE | >15 days, 37°C | 2.0 | 4.1 | 1.1 | 3.5 | |
| D | INFLUENT | DI | >15 days, 37°C | 2.9 | 6.5 | 1.3 | 3.4 |
| EFFLUENT | DE | >15 days, 37°C | 1.4 | 5.1 | 1.1 | 3.0 | |
| E | INFLUENT | EI | >15 days, 37°C | 3.6 | 6.5 | 3.4 | 5.3 |
| EFFLUENT | EE | >15 days, 37°C | 2.5 | 6.0 | 2.3 | 4.8 |
Influent samples were mixtures of primary and secondary sludge,
average log10 reductions for fecal coliforms =1.3 ± 0.6 CFU/dry gram,
average log10 reductions for somatic coliphages = 1.0 ± 0.8 PFU/dry gram,
average log10 reductions for male-Specific coliphages = 1.0 ± 0.5 PFU/dry gram.
Nucleic acid extraction and DNA sequencing
RNA and DNA were recovered from the viral concentrate using a Qiagen Viral RNA extraction kit (Qiagen, California, USA) following manufacturer's instructions. To obtain a sufficient quantity of DNA and reverse transcribed RNA (cDNA) for sequencing, it was necessary to amplify the viral nucleic acids using a random transcription/amplification protocol as previously described6, 14. This nucleic acid kit and amplification method have previously been recognized to extract and amplify both genomic RNA and DNA5, 15. Recovered DNA and cDNA was sent to the Yale University Center for Genome Analysis where the viral genomic DNA and cDNA were fragmented to ~200 bp (base pair) lengths and sequenced using the Illumina® HiSeq 2000 platform in three separate runs, generating both paired-end (2 runs) and single-end (1 run) 76 bp reads (see Supporting Information for details).
Bioinformatic analysis
The overall bioinformatic strategy included the following steps: (i) trim and clean sequencing reads, (ii) generate a master assembly of all sequence data, (iii) annotate the assembled contiguous sequences (contigs) through MG-RAST tBLASTx, and (iv) map sample specific reads onto the master assembly to determine sequence coverage (relative abundance). In total, 12 samples were sequenced; ten representing the influent (I) and effluent (E) of the five digesters (A, B, C, D, E) and an additional set of true biological replicates of the digester B samples (BI2 and BE2). Sequencing was also performed twice (technical replicates) for digesters B, C, D, and E. Biological replicates are defined as different nucleic acid extracts prepared from the same sample and viral elution. Technical replicates are defined as replicate sequencing runs from the same nucleic acid extracts. Short reads were assembled into contiguous sequences (contigs) and blasted against the NCBI viral genome database using tBLASTx with a maximum e-value of 0.001, which has previously been shown to minimize false negatives in metagenome annotation for human viral pathogen identification4. To determine sample-specific sequence presence and relative abundance, reads from each sample were mapped onto contigs generated from the master assembly. For each sample, this relative abundance is presented as the log10 of reads mapped to a specific viral pathogen contig divided by the total reads in that sample. Assembled contigs are available via MG-RAST accession number 4497937.3.
Finally, PCR of selected human viruses was used to validate metagenome annotation results. Viruses targeted by specific PCR primers (Table S1) included human strains of Adenovirus, Enterovirus, Parechovirus, and Norovirus GII. All PCR assays were performed on the nucleic acid extracts from the viral elution. The Supporting Information contains full details of the bioinformatic analysis and PCR validation experiments.
RESULTS
Viral metagenome sequencing and assembly results
Shotgun metagenome DNA and cDNA sequencing was done on three lanes of an Illumina® HiSeq 2000 generating 38.7 Gb (gigabases) of raw sequence data. Following trimming for quality scores, amplification adaptors, and read length, 21.6 Gb (~330 million reads) were used for assembling reads into contiguous sequences. The number of trimmed reads produced for each sample is summarized in Table S2.
The master assembly, which incorporated sequence reads from all samples, produced 412,654 contigs longer than 200 bp (Figure S1, Table S2). A contig length cutoff of 200 bp was chosen based on previous results4 that indicated contigs shorter than 200 bp are subject to increased annotation error. For the master assembly, the N50 (median contig size) was 512 bp, N90 was 231 bp, and the total assembly size was 186,505,658 bp. A histogram of contig sizes is shown in Figure S1.
Annotation results
Master assembly contigs were annotated through a tBLASTx comparison with the National Center for Biotechnology Information (NCBI) viral genome database. General annotation characteristics compiled by MG-RAST are shown in Figure 1A. Even though MG-RAST predicted that over 90% of all sequences coded for proteins, only 19.7% (81,265 contigs) had significant hits to known sequences in the MG RAST database (Figure 1A). The failure to annotate contigs demonstrates the limitations of viral diversity (especially for bacteriophages) represented in genome databases. Our experimental efforts to remove contaminating nucleic acids and also the low rRNA gene identifications in MG-RAST annotation (Figure 1A) suggest that contamination by non-viral nucleic acids was minimal. The majority of contigs annotated by tBLASTx searches (Figure 1B) were bacteriophages, followed by non-human eukaryotic viruses. Human pathogens comprised 0.1% of all contigs.
Figure 1.
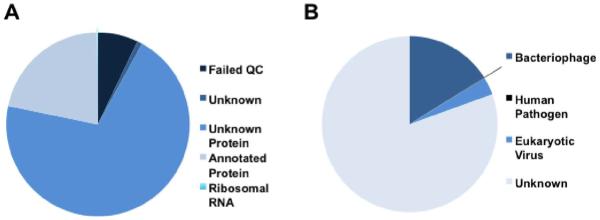
(A) Pie chart showing the MG-RAST subsystem annotation distribution for all contiguous sequences longer than 200 bp. (B) Pie chart demonstrating the distribution of tBLASTx virus contig annotations. Eukaryote pathogens do not include human viral pathogens. Human viral pathogen annotations represented ~0.1% of total contigs.
Viral pathogen identification
Of the 81,265 annotated contigs and 412,654 total contigs, 470 contigs (0.58% of annotated and 0.11% of total contigs) were tentatively identified as human viral pathogens. The N50 sequence length of these human pathogen contigs was 630 bp (Figure S1 inset). The most abundant potential human pathogen, with the majority of annotated contigs, belonged to the taxonomic family Herpesvirus. Of the other viral pathogen annotations, 32 contigs were identified as DNA viruses of non-Herpesvirus origin (Table S3) and 25 contigs were identified as RNA human viral pathogens (Table S4). DNA human viral pathogens included type strains of Papillomavirus, Adenovirus, Bocavirus, Parvovirus, and Torque Teno Virus (TTV). RNA pathogen identifications included type strains of Coronavirus, Cosavirus, Klassevirus, Rotavirus, Hepatitis C virus, Parechovirus, Sapovirus, Astrovirus, Coxsackievirus, Rhinovirus, T-lymphotropic virus, Human Immunodeficiency virus, Aichi virus, and Rubella virus. A full list of viruses identified to their highest taxonomic level are included Tables S3 and S4.
The relative abundances and occurrence of viral pathogens are presented in Figure 2. The relative abundance of pathogen-annotated contigs in each specific sample represents the log10 transformed number of reads within a sample that map to a specific pathogen-annotated contig, normalized by the number of sequences in that sample. These values are not quantitative on an absolute (i.e. per mass) basis; thus, reliable estimates of pathogen removal through the digester cannot be performed. Figure 2 results demonstrate that the most abundant and ubiquitous viruses identified by metagenomics were the DNA viruses. Adenovirus, Herpesvirus, Papillomavirus, and Bocavirus were found in more than 90% of the samples. RNA viruses with occurrence greater than 80% of samples were Coronavirus, Klassevirus, and Rotavirus (Figure 2). Relative abundance was linked with occurrence, with the most abundant regions the figure (darkest shade) corresponding to highest occurrence. Variations of up to four orders of magnitude in normalized relative abundance were also observed in pathogen annotations between samples. To observe variations between influent and effluent viral populations for the different locations, viral pathogen populations are presented in a principle coordinate analysis using the Sorensen similarity index (Figure 3)16. The Sorensen similarity index is a pair-wise measure of sample similarity based on presence-absence data. Using this method of visualization, the normalizing impact of treatment was apparent, as effluent samples grouped with each other and were distinct from the influent samples.
Figure 2.

Heat map demonstrating the relative abundance and occurrence for human viral pathogens. Relative abundance is defined as the log10[reads mapped to a virus contig divided by the total reads in the sample]. The dashed box represents replicated samples. Tables S3 and S4 provide virus identification to the highest taxonomic level.
Figure 3.
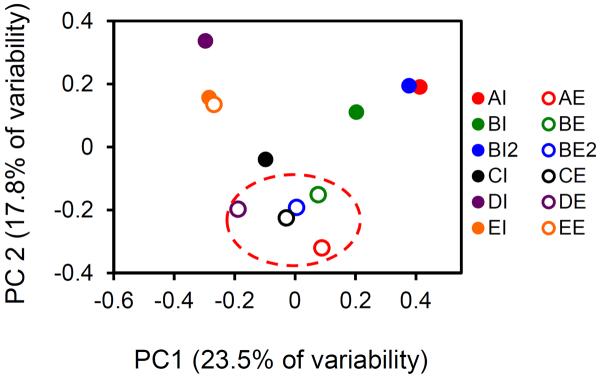
Principle component analysis of human viral pathogen population occurrence values. PCA plots were produced using the Sorensen similarity index.
Reproducibility and PCR confirmation
Two types of reproducibility, biological (replicates starting from DNA and RNA extraction) and technical (sequencing replicates), were investigated. Graphical representations comparing the duplicate relative abundances of annotated viral contiguous sequences are shown in Figure 4. It is both qualitatively and quantitatively evident from the scatter plots and line of best fit statistics that there is a high level of reproducibility (i.e. results are non-random) in metagenome annotation and sequencing. Technical reproducablity (average slope=0.90±0.03, average R2= 0.88±0.02) is higher (p<0.01) than biological reproducibility (average slope=0.79±0.2, average R2=0.62±0.07). Pathogen annotations for the true biological replicates were highly reproducible. Of the 40 virus data points presented in Figure 3 (see red box) for BI and BE, 37 agreed for presence and absence in the BI2 and BE2 samples.
Figure 4.

Comparison of the relative abundances of contigs, from replicate samples. Technical replicates refer to replicated sequencing of the same DNA/cDNA, while biological replicates refers to sequencing of DNA/cDNA extracted separately from the same sewage sludge sample. Slope and r2 values refer to data fitting to a straight line.
Additionally, PCR-based molecular assays were conducted for a suite of viruses that were expected to be present in the influent and effluent viral metagenomes. The results of these assays are presented in Table 2 and demonstrate the greater sensitivity of the targeted PCR methods. The sole DNA virus considered by targeted PCR, Adenovirus, was found in every sample by PCR and 92% of samples by metagenome annotation. Results for RNA viruses consistently showed higher occurrence by PCR than sequencing. Enteroviruses occurrence was 70% by PCR and 42% by sequence annotation (Coxsackievirus and Rhinovirus are included as Enterovirus), Parechoviruses occurrence of 100% by PCR and 58% by sequence annotation, and Norovirus GII occurrence of 80% by PCR, while not being identified by metagenome sequencing.
Table 2.
PCR Versus Metagenome Sequencing Identification for Selected Human Viruses. Black Shading Represents Presence.
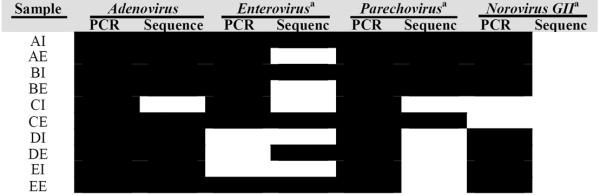
|
Quantitative PCR was performed for Enterovirus, Parechovirus, and Norovirus. Average concentrations and standard deviations in samples where detection occurred were 4.3±0.4 Enterovirus genomes/dry gram, 4.9±0.5 Parechovirus genomes/dry gram, and 4.8±0.2 Norovirus GII genomes/dry gram, when adjusted for sludge viral extraction efficiency. Viral extraction efficiency from sludge, as determined by extraction of spiked wild-type F+ coliphages from sterilized sewage sludge, was estimated to be 3%.
DISCUSSION
Two important, novel contributions can be drawn from this work's results. The first is the broad diversity of human viruses revealed in the sludge samples. In every sample surveyed, the degree of viral pathogen diversity is greater than had been previously demonstrated in any environmental or wastewater sample. The realistic implications of this diversity include the need to consider a broader selection of viruses in environmental fate and transport studies, and importance of considering multiple human exposure routes to sewage sludge and wastewater. For the second major contribution, this work demonstrates the utility of metagenomic approaches for viral pathogen identification. Metagenome results are highly reproducible, and verification of pathogen identifications highlights the conservative nature of metagenomic pathogen identifications.
Human virus diversity
DNA viruses causing latent infections, namely Herpesvirus and Papillomavirus, were the most ubiquitous and abundant viruses identified. Both viruses are highly prevalent in the general population. Papillomavirus infection has been previously documented in 68% of women (female-only cohort)17, and seroprevalence rates have been estimated at 85% for human Herpesvirus 6A and 6B18. This high prevalence coupled to the elevated relative abundances and 100% occurrence in the 12 influent and effluent samples considered here also demonstrate the potential for these viruses to be used as indicator or source tracking organisms for human waste. However, due to the presence of conserved genes between Herpesvirus and the human genome19, it has previously been recognized that Herpesvius will require additional confirmation before these applications can be developed20.
Beyond viruses causing latent infections, high occurrence and abundances were also observed for viruses associated with respiratory disease. Adenoviruses were highly occurring and abundant in samples considered in this study and were dominated type B and C respiratory strains, consistent with recent reports of Adenovrius diversity in sewage sludge21. By qPCR, Adenovirus has been observed at concentrations of 104 to 106 genome copies per dry gram in class B biosolids22. Bocavirus is frequently the cause of respiratory disease in children, has been recently detected in raw wastewater and occurred in 90% of the samples considered in this study23. The second most prevalent RNA virus was Coronavirus HKU1, a recently described agent associated with acute respiratory infections24. While the traditional concern with wastewater exposure has been ingestion, viruses that result in respiratory infections or are transmitted by the airborne route, droplet nuclei, or by fomite (Rhinovirus C, Bocavirus, Rubella virus, Coxsackievirus A16, Coronavirus HKU1, and respiratory Adenovirus B and C, and Parechovirus) were more abundant and prevalent than those which are transmitted by ingestion and result in gastrointestinal infection (Parvovirus, Rotavirus, Astrovirus, Sappovirus, Aichi virus, Parechovirus, and Adenovirus F). This load of respiratory viruses suggests the need for a broader view of human disease transmission modes due to wastewater exposure, and supports the concern over exposure to aerosols emitted during the land application of class B biosolids22, 25.
Among the most widely occurring and abundant RNA viruses were the emerging Picornaviruses including Parechovirus, Klassevirus and Cosavirus. Studies suggest Parechovirus seroprevalence in adults to be as high as 95%, and Parechoviruses are associated with both gastrointestinal and respiratory infections26. The infectious characteristics of Klassevirus, the most abundant and highly occurring RNA viruses identified, are currently unknown. Klassevirus has recently been found to be globally widespread in sewage27. Finally, Cosavirus, a novel genus in the Picornaviridae family that was present in 25% of sludge samples in this study, has been recently identified in children with non-poliovirus acute flaccid paralysis in Afghanistan and Pakistan, and more recently in a minority of raw sewage samples from U.S. and European wastewater treatment facilities23.
The presence of some emerging Picornaviruses in sewage may be potentially captured by Enterovirus culture-based monitoring. Plaque formation on BGM cell lines has been documented for Parechoviruses but not Klassevirus or Cosavirus28. However, these emerging Picornaviruses may have risk implications that are independent of typical Enteroviruses. Overall, the human virus content in sewage sludge samples is diverse. The occurrence and relative abundance values in this study reveal the gaps in approaches such as using fecal coliforms as an indicator of pathogen content in biosolids. As an example, limitations in the indicator approach can be viewed in a comparison of sample BE versus EE, where fecal coliforms are greater in the EE sample than the BE sample, but occurrence of viruses (Figure 2) was 40% for EE versus 75% for the BE sample.
Reproducibility and confirmation
Although not common in metagenome studies, scientific investigations are dependent upon, and assume the reproducibility of results. Both biological and technical replicates performed in this study indicated that metagenome sequencing results were non-random and reproducible. The average coefficient of determination value (r2) for biological replicates was 0.62 and the average r2 value for technical sequencing replicates was 0.88. For biological replicates, human virus annotation was highly reproducible with only 3 disagreements in 40 pathogen annotations. These small differences are likely a factor of both inherent variability in the subset of nucleic acids prepared from the sample, laboratory variation in sample preparation, and the inclusion of technical sequencing variability in biological replicates. For all replicates, a bias was shown for sequences to be more readily annotated in one sequencing run over the other, as demonstrated by a slope of less than one and a non-zero intercept (Figure 4), and pointing to factors such as inconsistencies in sample loading onto the sequencing instrument or variation between instrument performance.
A selected group of metagenome viral pathogen identifications were further validated by PCR-based assays. For all viruses considered in this validation, metagenome sequencing under-represented human viruses compared to PCR. A lower degree of under-representation was associated with the higher relative abundances of human viruses identified through metagenomics, suggesting that these disagreement are likely due to insufficient sequencing depth, which ultimately results in higher method detection limits for human viruses. Adenovirus and Norovirus are notable examples. While PCR measures absolute presence/absence, regardless of the abundance of non-target nucleic acids such as those from bacteriophages or other sewage-derived viruses, metagenomics measures only a relative abundance, meaning that even in cases when a pathogen has a high absolute abundance, the identification may be obscured by even higher concentrations of other viruses (bacteriophages or non-human eukaryote viruses). Based on the virus extraction efficiency and fraction of eluted RNA or DNA used in the PCR reaction, detection levels were in the range of 1.9×103 to 5.7×103 genome copies/dry gram. Previously reported concentrations of Enterovirus, Adenovirus, and Norovirus GII genomes in biosolids have averaged 1.4×104, 1.3×105, and 3.2×104 genome copies/dry gram, respectively22 While the detection level is unknown for metagenomic approaches and partially dependent on the amount of sequence information generated (38.7 Gb generated here), the results in Table 2 illustrate that it is most certainly greater than the detection level for that of PCR. The greater sensitivity by PCR highlights the role of metagenomics as a method to reveal overall pathogen diversity, rather than a method to quantify concentration, and suggests that for biosolids, which are rich in bacteriophages and non-pathogenic eukaryote viruses, a greater amount of sequencing data will result in revealing an even larger diversity of human pathogens. The continuing trend of declining DNA sequencing costs should continue to enable and strengthen viral metagenomic approaches for revealing viral diversity29.
Additional limitations to uncovering the full human virus diversity exist. Of the more than 108 unique viral genotypes estimated to exist in the world30, only 4,159 genomes were included in the amended NCBI viral genome database when this study was conducted. Additionally exacerbating this problem, sequenced viral genomes are biased towards those that have a culturable host (bacterial culture or eukaryotic cell line). The vast majority of viruses are unculturable in the laboratory, thus challenging efforts to improve our limited view of viral diversity. While this problem is less severe for human pathogens due to high medical interest and also because genomes are now being produced for viruses that are not culturable (e.g. Klassevirus and Norovirus), there is still likely human pathogen diversity that is not represented in the viral genome database. Large percentages of illnesses are currently undiagnosed. Indeed, efforts to understand the etiological agent in diarrheal and respiratory illness in humans have consistently resulted in no identified agent in more than 40% of cases27, 31.
The results of this study serve to expand our view on the type, occurrence and abundance of viral pathogens in raw sewage sludge and class B biosolids. These results strongly suggest that current regulations for pathogens in sewage sludge that focus on fecal coliform indicators or the presence of Enterovirus, do not capture the full degree of pathogen diversity to which the public may be exposed during biosolids land application. Emerging viruses including Parechovirus, Klassevirus, Bocavirus, and Coronavirus HKU1, were abundantly identified, highlighting previously undemonstrated pathogen diversity in sewage sludge. These identifications demonstrate the need for developing an improved suite of indicator pathogens, and at a minimum, both DNA and RNA viruses, rather than just the RNA virus Enterovirus, must be considered in any future updates to sewage sludge regulation. More broadly, the viral metagenomics approaches applied here are reproducible and are applicable to any environmental sample where viral pathogen diversity may be a concern. However, viral metagenomics should be considered as an approach for revealing diversity, and should act as a basis for, rather than a substitute to, qPCR and culture-based monitoring.
Supplementary Material
ACKNOWLEDGEMENTS
Kyle Bibby was supported by a fellowship from the Environmental Research and Education Foundation and STAR fellowship Assistance Agreement no. FP917115 awarded by the US Environmental Protection Agency (EPA). This article has not been formally reviewed by the EPA. The views expressed in this article are solely those of the authors, and the EPA does not endorse any products or commercial services mentioned in this article. Computing was carried out at the Yale University Center for High Performance Computation in Biology and Biomedicine (HPC) which is supported by NIH grant RR19895.
Footnotes
SUPPORTING INFORMATION AVAILABLE Additional materials and methods, metagenome assembly data, and detailed tables of identified DNA and RNA human viruses are available in the Supporting Information. This information is available free of charge via the Internet at http://pubs.acs.org/.
REFERENCES
- 1.Gerba CP, Gramos DM, Nwachuku N. Comparative inactivation of Enteroviruses and Adenovirus 2 by UV Light. Applied and Environmental Microbiology. 2002;68:5167–5169. doi: 10.1128/AEM.68.10.5167-5169.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Greninger A, Runckel C, Chiu C, Haggerty T, Parsonnet J, Ganem D, DeRisi J. The complete genome of Klassevirus - a novel picornavirus in pediatric stool. Virology Journal. 2009;6:82. doi: 10.1186/1743-422X-6-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.USEPA . Ambient Water Quality Criteria for Bacteria. 1986. EPA440/5-84-002. [Google Scholar]
- 4.Bibby K, Viau E, Peccia J. Viral metagenome analysis to guide human pathogen monitoring in environmental samples. Letters in Applied Microbiology. 2011;52:386–392. doi: 10.1111/j.1472-765X.2011.03014.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rosario K, Nilsson C, Lim YW, Ruan Y, Breitbart M. Metagenomic analysis of viruses in reclaimed water. Environmental Microbiology. 2009;11:2806–2820. doi: 10.1111/j.1462-2920.2009.01964.x. [DOI] [PubMed] [Google Scholar]
- 6.Cantalupo PG, Calgua B, Zhao G, Hundesa A, Wier AD, Katz JP, Grabe M, Hendrix RW, Girones R, Wang D, Pipas JM. Raw sewage harbors diverse viral Populations. mBio. 2011;2(5) doi: 10.1128/mBio.00180-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beecher N, Crawford K, Goldstein N, Lono-Batura M, Dziezyk E. A national biosolids regulation, quality, end use, and disposal survey. Northeast Biosolids and Residuals Association; Tamworth, NH: 2007. http://www.nebiosolids.org/uploads/pdf/NtlBiosolidsReport-20July07.pdf. [Google Scholar]
- 8.Viau E, Peccia J. Survey of wastewater indicators and human pathogen genomes in biosolids produced by class A and class B stabilization treatments. Applied and Environmental Microbiology. 2009;75:164–174. doi: 10.1128/AEM.01331-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wong K, Onan BM, Xagoraraki I. Quantification of enteric viruses, pathogen indicators, and Salmonella bacteria in class B anaerobically digested biosolids by culture and molecular methods. Applied and Environmental Microbiology. 2010;76:6441–6448. doi: 10.1128/AEM.02685-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McLennan SD, Peterson LA, Rose JB. Comparison of point-of-use technologies for emergency disinfection of sewage-contaminated drinking water. Applied and Environmental Microbiology. 2009;75:7283–7286. doi: 10.1128/AEM.00968-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Santo Domingo JW, Bambic DG, Edge TA, Wuertz S. Quo vadis source tracking? Towards a strategic framework for environmental monitoring of fecal pollution. Water Research. 2007;41:3539–3552. doi: 10.1016/j.watres.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 12.Monpoeho S, Maul A, Mignotte-Cadiergues B, Schwartzbrod L, Billaudel S, Ferro V. Best viral elution method available for quantification of Enteroviruses in sludge by both cell culture and reverse transcription-PCR. Applied and Environmental Microbiology. 2001;67:2484–2488. doi: 10.1128/AEM.67.6.2484-2488.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.USEPA . Method 1602: Male-specific (F+) and Somatic Coliphage in Water by Single Agar Layer (SAL) Procedure. 2001. EPA 821-R-01-029. [Google Scholar]
- 14.Wang D, Coscoy L, Zylberberg M, Avila PC, Boushey HA, Ganem D, DeRisi JL. Microarray-based detection and genotyping of viral pathogens. Proceedings of the National Academy of Sciences. 2002;99:15687–15692. doi: 10.1073/pnas.242579699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wylie KM, Mihindukulasuriya KA, Sodergren E, Weinstock GM, Storch GA. Sequence analysis of the human virome in febrile and afebrile children. PLoS ONE. 2012;7:e27735. doi: 10.1371/journal.pone.0027735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sorensen T. Biologiske Skrifter. 1948. A method of establishing groups of equal amplitude in plant sociology based on similarity of species content. [Google Scholar]
- 17.J K, S R, Y J, B H, A N, Zimet GD. Rates of human Papillomavirus vaccination, attitudes about vaccination, and human Papillomavirus prevalence in young women. Obstetrics & Gynecology. 2008;111:8. doi: 10.1097/AOG.0b013e31817051fa. [DOI] [PubMed] [Google Scholar]
- 18.Levy JA, Ferro F, Greenspan D, Lennette ET. Frequent isolation of HHV-6 from saliva and high seroprevalence of the virus in the population. The Lancet. 1990;335:1047–1050. doi: 10.1016/0140-6736(90)92628-u. [DOI] [PubMed] [Google Scholar]
- 19.Arbuckle JH, Medveczky MM, Luka J, Hadley SH, Luegmayr A, Ablashi D, Lund TC, Tolar J, De Meirleir K, Montoya JG, Komaroff AL, Ambros PF, Medveczky PG. The latent human herpesvirus-6A genome specifically integrates in telomeres of human chromosomes in vivo and in vitro. Proceedings of the National Academy of Sciences. 2010;107:5563–5568. doi: 10.1073/pnas.0913586107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pipas J. Personal Communication. 2012.
- 21.Bibby K, Peccia J. Environmental Science: Processes & Impacts. 2013. Prevalence of respiratory adenovirus species B and C in sewage sludge. DOI: 10.1039/C2EM30831B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Viau E, Bibby K, Paez-Rubio T, Peccia J. Toward a consensus view on the infectious risks associated with land application of sewage sludge. Environmental Science & Technology. 2011;45:5459–5469. doi: 10.1021/es200566f. [DOI] [PubMed] [Google Scholar]
- 23.Blinkova O, Rosario K, Li L, Kapoor A, Slikas B, Bernardin F, Breitbart M, Delwart E. Frequent detection of highly diverse variants of Cardiovirus, Cosavirus, Bocavirus, and Circovirus in sewage samples collected in the United States. Journal of Clinical Microbiology. 2009;47:3507–3513. doi: 10.1128/JCM.01062-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Woo P, Lau S, Yip C, Huang Y, Yuen K-Y. More and more Coronaviruses: Human Coronavirus HKU1. Viruses. 2009;1:57–71. doi: 10.3390/v1010057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lewis D, Gattie DK. Pathogen risks from applying sewage sludge to land. Environmental Science and Technology. 2002;36:286A–293A. doi: 10.1021/es0223426. [DOI] [PubMed] [Google Scholar]
- 26.Joki-Korpela P, Hyypia T. Parechoviruses, a novel group of human picornaviruses. Annals of Medicine. 2001;33:466–471. doi: 10.3109/07853890109002095. [DOI] [PubMed] [Google Scholar]
- 27.Holtz L, Finkbeiner S, Zhao G, Kirkwood C, Girones R, Pipas J, Wang D. Klassevirus 1, a previously undescribed member of the family Picornaviridae, is globally widespread. Virology Journal. 2009;6:86. doi: 10.1186/1743-422X-6-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sedmak G, Nix WA, Jentzen J, Haupt TE, Davis JP, Bhattacharyya S, Pallansch MA, Oberste MS. Infant deaths associated with human Parechovirus infection in Wisconsin. Clinical Infectious Diseases. 2010;50:357–361. doi: 10.1086/649863. [DOI] [PubMed] [Google Scholar]
- 29.Shendure J, Ji H. Next-generation DNA sequencing. Nat Biotech. 2008;26:1135–1145. doi: 10.1038/nbt1486. [DOI] [PubMed] [Google Scholar]
- 30.Rohwer F. Global Phage Diversity. Cell. 2003;113:141. doi: 10.1016/s0092-8674(03)00276-9. [DOI] [PubMed] [Google Scholar]
- 31.Sloots TP, Whiley DM, Lambert SB, Nissen MD. Emerging respiratory agents: New viruses for old diseases? Journal of Clinical Virology. 2008;42:233–243. doi: 10.1016/j.jcv.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.