

Abstract
Once considered an inevitable consequence of HIV treatment, drug resistance is declining. This decline supports the hypothesis that antiretroviral therapy can arrest replication and prevent the evolution of resistance. Further support comes from excellent clinical outcomes, the failure of treatment intensification to reduce residual viremia, the lack of viral evolution in patients on optimal therapy, pharmacodynamics studies explaining the extraordinarily high antiviral activity of modern regimens, and recent reports of potential cures. Evidence supporting ongoing replication includes higher rates of certain complications in treated patients and an increase in circular forms of the viral genome after intensification with integrase inhibitors. Recent studies also provide an explanation for the observation that some patients fail protease-inhibitor based regimens without evidence for resistance.
Introduction
Not long after the introduction of the nucleoside analogue reverse transcriptase inhibitor (NRTI) zidovudine (AZT) as the first antiretroviral drug for the treatment of HIV-1 infection, resistance to AZT was detected in treated patients [1]. Since that time, resistance has been reported for each new antiretroviral drug introduced [2], and the field has been haunted by the specter of widespread and inevitable drug resistance, necessitating the continued development of new classes of antiretroviral drugs. However, recent clinical experience suggests that HIV-1 drug resistance is actually declining, and it is now becoming clear that resistance is not an inevitable consequence of HIV-1 treatment, but rather a reflection of suboptimal treatment. Optimal therapy appears to prevent the evolution of resistance, even over long time periods. This review will explore the theoretical basis for this remarkable development.
The modern era of HIV-1 treatment began in 1997 when two new classes of antiretroviral drugs were introduced, the non-nucleoside reverse transcriptase inhibitors (NNRTIs) and the protease inhibitors (PIs). Three drug combinations consisting of an NNRTI or a PI and two drugs from the NRTI class were tested in clinical trials [3-5]. In these trials, it was shown for the first time that combination antiretroviral therapy (ART) could reduce viremia to clinically undetectable levels. When patients start on an appropriate combination of three antiretroviral drugs, plasma virus levels fall within a few months from pre-therapy levels on the order of 104-105 copies of genomic viral RNA/ml to below the limit of detection of clinical assays (50 copies/ml). Clinically, the disappearance of detectable viremia is associated with increases in or preservation of CD4+ T cell counts and the reversal or prevention of immunodeficiency. Combination ART rapidly became the standard of care for HIV-1 infection (Figure 1). Newly revised US treatment guidelines [6**] call for the treatment of all infected individuals with ART regimens consisting of two NRTIs and either an NNRTI, a PI or an integrase strand transfer inhibitor (InSTI) [7,8]. Several recent studies suggest that patients who start modern ART regimens early in the course of disease have a near normal lifespan expectancy, even in resource limited settings [9*].
Figure 1.
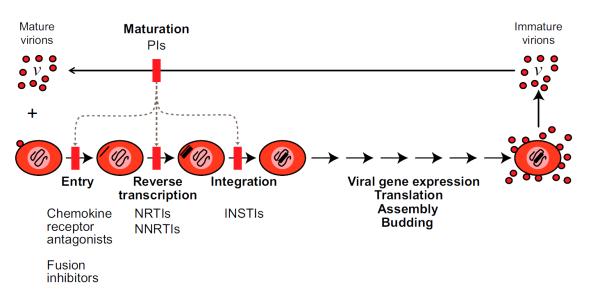
Antiretroviral therapy for HIV-1 infection. The steps in the life cycle blocked by different classes of antiretroviral drugs are indicated. Current ART regimens consist of two NRTIs and either an NNRTI, a PI, or an InSTI. Inhibitors of HIV-1 entry, chemokine receptor antagonists and fusion inhibitors, can be used. Note that all current antiretroviral drugs act to prevent new cells from becoming infected. They do not block the production of virus particles by a cell that already carries an integrated provirus. The PIs prevent virus particles from maturing to an infectious form. Immature virus particles show defects at multiple downstream steps in the virus life cycle (dotted lines), including entry, reverse transcription, and integration. See text for references.
Although ART can suppress viremia to clinically undetectable levels, there is inevitably a rebound in viremia within several weeks after discontinuation of therapy [10]. The ability of the virus to persist despite ART is due at least in part to is ability to establish a state of latent infection in resting memory CD4+ T cells [11,12]. This latent reservoir is extremely stable, even in patients on optimal ART [13-16], and is a likely source of viral rebound following interruption of therapy [17]. Another important indication of viral persistence during ART is the presence of trace levels of free virus in the plasma [18-21]. This residual viremia is detectable with RT-PCR assays that have single molecule sensitivity. All current antiretroviral drugs act by blocking new infection events rather than by blocking virus production by cells that already have an integrated provirus (Figure 1). Thus, the residual viremia may reflect the activation of latently infected cells or possibly the release of virus from other stable reservoirs [17] . The only exceptions to the rule of viral rebound after discontinuation of ART are rare cases of patients treated early in the course of infection who are able to control viral replication through unknown immunologic mechanisms [22,23**].
The decline in HIV-1 drug resistance
HIV-1 drug resistance is a result of random mutations introduced by the error prone HIV-1 reverse transcriptase when it converts the single stranded genomic viral RNA into double stranded DNA shortly after viral entry. On average, one mutation is introduced in the 10 kb genome for every three cycles of replication [24]. These mutations are typically base substitutions although insertions, duplications and recombination can also occur. Because resistance mutations arise during reverse transcription in newly infected cells, the evolution of resistance can in principle be arrested if new infection event are blocked by ART. Avoiding resistance has been a major guiding principle of ART, and recent studies suggest that this goal is actually achievable.
Clinical experience and recent observational studies indicate that the incidence and prevalence of HIV-1 drug resistance is actually declining [25**,26*,27*,28*,29*]. A comprehensive study of essentially all infected individuals in Sweden found a dramatic decrease in the prevalence of resistance between 2003 and 2007 with slower decreases thereafter [25**]. This decrease coincided with the phase out of older drugs and the introduction of newer classes of antiretroviral drugs. Most mutations were found in patients with a history of suboptimal treatment. The only worrying trend was a very slight recent increase in the prevalence of NNRTI resistance mutations, which was attributed to the infection with resistant viruses in low to middle income countries where NNRTI regimens are very common and resistance monitoring is absent. A large multicohort European study found a decrease in the prevalence of drug resistance, particularly multiclass resistance, beginning around 2005 and continuing through the study end date in 2008 [26*]. In a large study of French patients with virologic failure (two plasma HIV-1 measurements >50 copies/ml), the fraction of samples with common RT resistance mutations (M184V/I, K103N) declined in the period between 2005 and 2010, despite continued use of drugs that select those mutations (lamivudine and emtricitabine for M184V/I and efavirenz for K103N) [27*]. The authors attribute this decline to the use of single pill, once a day regimens which promote improved adherence and prevent differential adherence to components of a regimen. Selection of the NRTI resistance mutation K65R has also been declining despite the widespread use of tenofovir, which selects this mutation [28*]. The prevalence of multidrug resistance has been declining in Portugal in the time between 2001 and 2013 [29*]. Overall, these studies suggest that improvements in ART, including convenient single pill regimens consisting of three relatively non-toxic drugs, have improved adherence and allowed the full potential of ART to become apparent.
The efficacy of modern ART regimens in suppressing viral replication and preventing the evolution of resistance is a major factor in recent change in HIV-1 treatment guidelines. The concern about resistance was often cited as a reason for delaying the initiation of ART. However, numerous studies have shown that early treatment is associated with better outcomes (reviewed in [6]), and with the high antiviral activity and improved toxicity profiles of modern regimens, there is no medical reason to delay treatment. Thus current US guidelines recommend treating all infected individuals with ART.
The debate over ongoing viral replication
The dramatic success of ART suggests that viral replication, and hence viral evolution, can be completely blocked in adherent patients. However, a vigorous debate over whether ART can completely inhibit viral replication has raged for several years. Table 1 lists the major arguments supporting and contradicting the hypothesis that ART can completely block HIV-1 replication and the evolution of resistance. Because of the large number of people currently being treated with ART, there is an enormous amount of clinical data that pertains to this question. Careful analysis by committees that set treatment guidelines has shown that early initiation of treatment is associated with the best outcomes in HIV-1 infection [6**]. There is no evidence that adherent patients spontaneously fail treatment with resistant virus. There is very strong evidence that in untreated patients, ongoing viral replication in each infected individual leads to diversification of a single infecting founder virus into a complex quasispecies that shows progressive divergence from the founder virus over time. The error prone nature of reverse transcriptase results in an inevitable linkage between viral replication and viral evolution. In response to the selective pressure exerted by the immune response or suboptimal drug treatment, escape or resistance mutations arise very rapidly (within weeks). The success of treatment together with the dramatic decline in resistance described above argue strongly that ART can halt clinically significant viral replication and hence viral evolution.
Table 1.
Arguments for and against the hypothesis that ART can completely block HIV-1 replication and evolution (please see text for references).
| For | Against |
|---|---|
| Success of ART in controlling viral replication and preventing immunodeficiency |
Evidence for ongoing immune activation and inflammatory state in treated pateints |
| Failure of intensification to reduce residual viremia |
Transient increase in 2LTR cirlecs with RAL intensification |
| Lack of evidence for sequence change over time in most patients |
Sequence evolution in a subset of pateints |
| Pharmacodynamic data indicating extremely high antiviral activity of NNRTIs and PIs |
Cell to cell spread reduces antiviral effect |
| Recent reports of cure |
Interventional studies involving treatment intensification also support the conclusion that ART stops viral replication. As discussed above, patients on ART who have suppression of viremia to below the limit of detection of clinical assays do have trace levels of free virus in the blood that can be detected with extremely sensitive “single copy” assays [18-21]. If the residual viremia is a reflection of ongoing viral replication not fully suppressed by ART, then the addition of a fourth antiretroviral drug from a different class to an optimal ART regimen should produce a decline in residual viremia detectable with such assays. If, on the other hand, residual viremia simply represents release of virus from stable reservoirs, then intensification should not decrease residual viremia. As shown in Figure 1, none of the current antiretroviral drugs block release of virus from cells that have an integrated provirus. Thus the activation of a small fraction of the resting CD4+ T cells that harbor latent HIV-1 could result in trace levels of viremia that would not be affected by treatment intensification. Over the last several years, numerous intensification studies have been carried out with different intensification drugs [30-33]. The results have been remarkable consistent. All studies show that intensification has no effect on residual viremia. This means that residual viremia is largely due to release of virus from stable reservoirs, and that if there is ongoing replication, it results in a level of viremia that is insignificant even compared to the trace levels detected with the single copy assay. A recent study has shown that intensification also fails to reduce trace levels of HIV-1 RNA in the cerebrospinal fluid [34].
Direct studies of viral evolution in patients on ART are complicated by the very low levels of virus present in the blood but have generally not shown any evidence for evolutionary change even over long time intervals. In some cohort studies, a small subset of the patients show evidence of viral evolution [35,36], but it cannot be excluded that these were patients with poor adherence. No study has documented ongoing viral evolution in the setting of adequate drug levels, and most patients show a striking lack of evolutionary change in viral sequences. The residual viremia is composed of drug sensitive viruses that do not evolve over time [37-41]. In acute infection, the gut associated lymphoid tissue (GALT) is a major site of viral replication [42], and high levels of HIV-1 DNA are found in CD4+ T cells in the GALT even in treated patients [43]. A recent study has shown that even in the GALT, there is no detectable viral evolution in patients on ART, while evolution is readily detectable in untreated patients [44**].
Recent studies of HIV-1 pharmacodynamics have provided a quantitative basis for understanding the ability of ART to inhibit viral replication. Pharmacodynamics refers to the relationship between drug concentration and drug effect. The most commonly used parameter pharmacodynamic parameter is the IC50, the drug concentration at which there is 50% inhibition of some measure of viral replication. The fraction of infection events affected (inhibited) by a drug (fa) at a given concentration D is related to the IC50 by the median effect equation [45]:
where m is a measure of the slope or steepness of the dose response curve. This parameter has an exponential relationship to drug effect, and when the D > IC50 , slope values greater than 1 can produce extremely high levels of inhibition. The slope parameter is related to the Hill coefficient, a measure of cooperativity, and slope values >1 are typically associated with cooperative binding of ligands to a multivalent receptor. However, the molecular targets of most antiretroviral drugs are monovalent with respect to inhibitors, and thus high slope values were not expected for antiretroviral drugs. In 2008, Shen et al. showed that some classes of antiretroviral drugs, notably the NNRTIs and the PIs, show highly cooperative dose response curves with high slope values in the inhibition of infectivity [45]. These high slopes allowed clinical concentrations of these drugs to produce many logs of inhibition of viral replication. When the combined effects of multiple drugs used together in ART regimens were considered, it became clear that most ART regimens could produce levels of inhibition that were consistent with a complete block in ongoing replication [46*].
One particularly interesting argument supporting the efficacy of ART in blocking new infection involves recent reports of potential cures. The original report described the “Berlin patient”, an HIV-1-infected man who had sustained suppression of viremia on ART when he developed acute myeloid leukemia [47]. As part of the treatment for leukemia, he received two hematopoetic stem cell (HSC) transplants from a donor who was homozygous for a 32 base pair deletion in the HIV-1 co-receptor CCR5. ART was discontinued at the time of the initial transplant, and viremia never rebounded. A recent exhaustive study of multiple tissues from this patient five years after the initial transplant failed to uncover consistent evidence of viral persistence [48*]. In this case, donor cells were protected from infection by the absence of CCR5. Two additional cases of HSC transplantation have recently been reported, and in these cases the donors were wild type for CCR5 [49**]. In these cases, the donor cells were protected from HIV-1 infection by ART, which was continued uninterrupted throughout the entire transplant period. HIV-1 became undetectable at the time when 100% chimerism was documented. It remains to be determined whether these two new cases actually represent cures, but if so, they would dramatically illustrate the ability of ART to prevent otherwise susceptible donor cells from HIV-1 infection.
Although the preponderance of evidence suggests that modern ART regimens can arrest HIV-1 evolution in adherent patients, there is some evidence that trace amounts of replication continue. Patients on ART do experience higher levels of immune activation and higher rates of some non-infectious complications [50]. There is concern that some level of replication continuing in particular compartments could contribute to these complications and account for differences in life expectancy between patients and uninfected individuals. However, studies of this kind are complicated by differences in other risk factors in patient populations, legacy effects persisting from the time before treatment, and by drug toxicity.
Another line of evidence supporting some degree of ongoing replication is a recent study of the effects of intensification with the InSTI raltegravir [51]. Although levels of residual viremia do not change with intensification, there is a transient increase in the level of 2LTR circles, especially in patients on PI-based regimens. The circles form in the nucleus of infected cells when integration fails and the reverse transcribed HIV-1 DNA undergoes an end to end intermolecular ligation. The increase in these circles suggests that there were some new infection events in which virions released from an infected cell successfully matured and infected another cell in which reverse transcription then proceeded to completion. It is currently unclear how to reconcile this result with the evidence against ongoing replication presented above.
Finally, a scenario in which replication could continue despite ART has recently be described by Sigal and Baltimore [52**]. These investigators have shown that the local spread of HIV-1 from an infected cell to a neighboring cell can involve very high multiplicities of infection, which from a simple probability standpoint, reduces the effectiveness of antiretroviral drugs. This finding raises the possibility of local burst of replication of new infection, perhaps initiated by reactivation of latently infected cells.
Failure without resistance
An interesting development in the analysis of HIV-1 drug resistance is the phenomenon of treatment failure without resistance [53,54*]. Some patients on PI-based regimens develop detectable viremia but do not have resistance mutations in HIV-1 protease. Of course, non-adherence is one simple explanation, but in many cases the patients do show resistance to other drugs in the regimens. Two recent studies have offered potential explanations for PI failure without resistance (Figure 2). Rosenbloom et al. used pharmacokinetic and pharmacodynamics parameters and data on the fitness cost of resistance mutations to show that the time spent in the mutant selection window (MSW) is short for PIs [55*]. The MSW refers to the time following treatment interruption during which a drug resistant mutant has both a positive growth rate and a selective advantage over wild type virus. In evolutionary terms a positive growth rate is indicated by R0 > 1, where R0 is the basic reproductive ratio, the number of new cells that are infected by the virus released from a single infected cell. At very high drug concentrations, the resistant virus will be inhibited to a lesser extent than wild type virus but may still be unable to grow (R0 < 1). As the drug concentration falls with non-adherence, it will reach a level at which the mutant will have a positive growth rate (R0 > 1). This defines one boundary of the MSW (Figure 2A). Most drug resistance mutations cause a reduction in viral fitness in the absence of drug, and therefore the drug concentration will fall to a point where the wild type virus has a higher replication rate than the mutant virus. This defines the other boundary of the MSW. At drug concentrations within this window, the mutant virus will be selected. The time spent in the MSW after treatment is interrupted is a complex function of the drug half-life, and the effect of the resistance mutations on IC50, m, and viral fitness. Recent studies indicate that for PIs, the time spend in the mutant selection window is extremely short [54]. With falling PI concentrations, wild type virus rapidly gains a selective advantage, and thus patients can fail due to non-adherence without developing resistance.
Figure 2.

Explanations for PI failure without resistance mutations in the protease gene. (A) Pharmacodyamic properties of PIs restrict the evolution of resistance. During periods of non-adherence, resistant viruses emerge. As drug concentrations fall, there is a mutant selection window (MSW) in which the mutant virus has both a positive growth rate (R0 > 1) and a selective advantage over wild type virus. The length of the MSW depends on drug half-life, the properties of the dose-response curves for wild type (green) and resistant (red) viruses, and the fitness cost of the resistance mutation. For PIs, the time spent in the MSW is extremely short and with non-adherence, wild type virus rapidly emerges. Note that this form of treatment failure does not represent drug resistance, and suppression of viremia can be achieved by restoring adherence. (B) Mutations inside of (red) and outside of (*) the protease coding region can contribute to resistance. As shown in Figure 1, part of the inhibitory effect of PIs is due to effects on HIV-1 entry. Interactions between the uncleaved Gag precursor protein and the cytoplasmic domain of the Env protein inhibit entry, and thus PI-mediate inhibition of Gag cleavage results in inhibition of infectivity. Resistance to PIs may arise through mutations affecting the Gag-Env interaction. Mutations in the protease cleavage sites in Gag can also contribute to resistance. Current clinical assays for PI resistance examine only the protease gene and could miss other mutations contributing to resistance.
Recent studies have uncovered a second explanation for the phenomenon of failure without PI resistance [56*]. As discussed above, PIs are extremely effective antiretroviral drugs due to highly cooperative dose-response curves that are not fully explained by current pharmacodynamic theory [45,57]. The nature of these curves as well as the phenomenon of PI failure without resistance can both be explained through analysis of the effects of PIs on distinct steps in the life cycle (Figure 1). PIs do not affect virion release from infected cells but block entry, reverse transcription (RT), and post-RT steps [56]. The overall dose-response curves can be reconstructed by combining the curves for each step using the Bliss independence principle. Thus independent inhibition of multiple distinct steps in the life cycle generates the highly cooperative dose-response curves that make these drugs uniquely effective. Approximately half of the inhibitory potential of PIs is manifested at the entry step, likely reflecting interactions between the uncleaved Gag and the cytoplasmic tail (CT) of the Env protein [58**]. Sequence changes in Env, which are ignored in current clinical tests for PI resistance, can confer PI resistance, providing an explanation for PI failure without resistance. Mutations in the HIV-1 protease cleavage sites in the Gag polyprotein can also be selected for during PI failure [59**]. Thus some cases of PI failure without resistance mutations in the protease gene may actually represent the evolution of resistant viruses that have sequence changes in other genes that contribute to PI resistance. Because current clinical tests for PI resistance consider only the protease gene itself, this issue deserves additional exploration.
Conclusions
Recent clinical experience and experimental studies have clearly shown that HIV-1 infection can be successfully treated without the evolution of drug resistance. The underlying explanation for this development is the ability of current ART regimens to suppress viral replication to such a great extent that the evolution of drug resistance, which depends on new infection events, is essentially halted.
Highlights.
Recent studies suggest that the prevelance of HIV-1 drug resistance is declining.
This reflects the ability of antiretroviral therapy to halt viral evolution.
Recent studies explain treatment failure without protease inhibitor resistance.
Acknowledgements
This work was supported by NIH grant AI081600 and by the Howard Hughes Medical Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Larder BA, Darby G, Richman DD. HIV with reduced sensitivity to zidovudine (AZT) isolated during prolonged therapy. Science. 1989;243:1731–1734. doi: 10.1126/science.2467383. [DOI] [PubMed] [Google Scholar]
- 2*.Johnson VA, Calvez V, Gunthard HF, Paredes R, Pillay D, Shafer RW, Wensing AM, Richman DD. Update of the drug resistance mutations in HIV-1: March 2013. Top.Antivir Med. 2013;21:6–14. [PMC free article] [PubMed] [Google Scholar]
- This comprehensive summary of well characterized HIV-1 drug resistance mutations is compiled and updated by a panel of experts. Because of the close correlation between genotype and phenotype for HIV-1 drug resistance, the detection of these mutations by sequence analysis provides information that is extremely useful in patient management.
- 3.Gulick RM, Mellors JW, Havlir D, Eron JJ, Gonzalez C, McMahon D, Richman DD, Valentine FT, Jonas L, Meibohm A, et al. Treatment with indinavir, zidovudine, and lamivudine in adults with human immunodeficiency virus infection and prior antiretroviral therapy. N.Engl.J.Med. 1997;337:734–739. doi: 10.1056/NEJM199709113371102. [DOI] [PubMed] [Google Scholar]
- 4.Hammer SM, Squires KE, Hughes MD, Grimes JM, Demeter LM, Currier JS, Eron JJ, Jr, Feinberg JE, Balfour HH, Jr, Deyton LR, et al. A controlled trial of two nucleoside analogues plus indinavir in persons with human immunodeficiency virus infection and CD4 cell counts of 200 per cubic millimeter or less. AIDS Clinical Trials Group 320 Study Team. N.Engl.J.Med. 1997;337:725–733. doi: 10.1056/NEJM199709113371101. [DOI] [PubMed] [Google Scholar]
- 5.Perelson AS, Essunger P, Cao Y, Vesanen M, Hurley A, Saksela K, Markowitz M, Ho DD. Decay characteristics of HIV-1-infected compartments during combination therapy. Nature. 1997;387:188–191. doi: 10.1038/387188a0. [DOI] [PubMed] [Google Scholar]
- 6**.Department of Health and Human Services Panel on Antiretroviral Guidelines for Adults and Adolescents. . Guidelines for the use of antiretroviral agents in HIV-1-infected adults and adolescents. Available at http://aidsinfo.nih.gov/contentfiles/lvguidelines/AdultandAdolescentGL.pdf.
- Treatment recommendations of an expert US panel. Includes a detailed discussion of recent trends in HIV-1 treatment.
- 7.Markowitz M, Nguyen BY, Gotuzzo E, Mendo F, Ratanasuwan W, Kovacs C, Prada G, Morales-Ramirez JO, Crumpacker CS, Isaacs RD, et al. Rapid and durable antiretroviral effect of the HIV-1 Integrase inhibitor raltegravir as part of combination therapy in treatment-naive patients with HIV-1 infection: results of a 48-week controlled study. J.Acquir.Immune Defic.Syndr. 2007;46:125–133. doi: 10.1097/QAI.0b013e318157131c. [DOI] [PubMed] [Google Scholar]
- 8.Steigbigel RT, Cooper DA, Kumar PN, Eron JE, Schechter M, Markowitz M, Loutfy MR, Lennox JL, Gatell JM, Rockstroh JK, et al. Raltegravir with optimized background therapy for resistant HIV-1 infection. N.Engl.J.Med. 2008;359:339–354. doi: 10.1056/NEJMoa0708975. [DOI] [PubMed] [Google Scholar]
- 9*.Mills EJ, Bakanda C, Birungi J, Chan K, Ford N, Cooper CL, Nachega JB, Dybul M, Hogg RS. Life expectancy of persons receiving combination antiretroviral therapy in low-income countries: a cohort analysis from Uganda. Ann.Intern.Med. 2011;155:209–216. doi: 10.7326/0003-4819-155-4-201108160-00358. [DOI] [PubMed] [Google Scholar]
- A large study of 22,000 Ugandan patients initiating ART. Near normal life expectancy was seen, particularly in those initiating therapy with relatively high CD4 counts.
- 10.Davey RT, Jr, Bhat N, Yoder C, Chun TW, Metcalf JA, Dewar R, Natarajan V, Lempicki RA, Adelsberger JW, Miller KD, et al. HIV-1 and T cell dynamics after interruption of highly active antiretroviral therapy (HAART) in patients with a history of sustained viral suppression. Proc.Natl.Acad.Sci.U.S.A. 1999;96:15109–15114. doi: 10.1073/pnas.96.26.15109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chun TW, Finzi D, Margolick J, Chadwick K, Schwartz D, Siliciano RF. In vivo fate of HIV-1-infected T cells: quantitative analysis of the transition to stable latency. Nat.Med. 1995;1:1284–1290. doi: 10.1038/nm1295-1284. [DOI] [PubMed] [Google Scholar]
- 12.Chun TW, Carruth L, Finzi D, Shen X, DiGiuseppe JA, Taylor H, Hermankova M, Chadwick K, Margolick J, Quinn TC, et al. Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature. 1997;387:183–188. doi: 10.1038/387183a0. [DOI] [PubMed] [Google Scholar]
- 13.Finzi D, Hermankova M, Pierson T, Carruth LM, Buck C, Chaisson RE, Quinn TC, Chadwick K, Margolick J, Brookmeyer R, et al. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science. 1997;278:1295–1300. doi: 10.1126/science.278.5341.1295. [DOI] [PubMed] [Google Scholar]
- 14.Wong JK, Hezareh M, Gunthard HF, Havlir DV, Ignacio CC, Spina CA, Richman DD. Recovery of replication-competent HIV despite prolonged suppression of plasma viremia. Science. 1997;278:1291–1295. doi: 10.1126/science.278.5341.1291. [DOI] [PubMed] [Google Scholar]
- 15.Chun TW, Stuyver L, Mizell SB, Ehler LA, Mican JA, Baseler M, Lloyd AL, Nowak MA, Fauci AS. Presence of an inducible HIV-1 latent reservoir during highly active antiretroviral therapy. Proc.Natl.Acad.Sci.U.S.A. 1997;94:13193–13197. doi: 10.1073/pnas.94.24.13193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Siliciano JD, Kajdas J, Finzi D, Quinn TC, Chadwick K, Margolick JB, Kovacs C, Gange SJ, Siliciano RF. Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nat.Med. 2003;9:727–728. doi: 10.1038/nm880. [DOI] [PubMed] [Google Scholar]
- 17.Joos B, Fischer M, Kuster H, Pillai SK, Wong JK, Boni J, Hirschel B, Weber R, Trkola A, Gunthard HF, Swiss HIV Cohort Study HIV rebounds from latently infected cells, rather than from continuing low-level replication. Proc.Natl.Acad.Sci.U.S.A. 2008;105:16725–16730. doi: 10.1073/pnas.0804192105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dornadula G, Zhang H, VanUitert B, Stern J, Livornese L, Jr, Ingerman MJ, Witek J, Kedanis RJ, Natkin J, DeSimone J, Pomerantz RJ. Residual HIV-1 RNA in blood plasma of patients taking suppressive highly active antiretroviral therapy. JAMA. 1999;282:1627–1632. doi: 10.1001/jama.282.17.1627. [DOI] [PubMed] [Google Scholar]
- 19.Palmer S, Wiegand AP, Maldarelli F, Bazmi H, Mican JM, Polis M, Dewar RL, Planta A, Liu S, Metcalf JA, et al. New real-time reverse transcriptase-initiated PCR assay with single-copy sensitivity for human immunodeficiency virus type 1 RNA in plasma. J.Clin.Microbiol. 2003;41:4531–4536. doi: 10.1128/JCM.41.10.4531-4536.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maldarelli F, Palmer S, King MS, Wiegand A, Polis MA, Mican J, Kovacs JA, Davey RT, Rock-Kress D, Dewar R, et al. ART suppresses plasma HIV-1 RNA to a stable set point predicted by pretherapy viremia. PLoS Pathog. 2007;3:e46. doi: 10.1371/journal.ppat.0030046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Palmer S, Maldarelli F, Wiegand A, Bernstein B, Hanna GJ, Brun SC, Kempf DJ, Mellors JW, Coffin JM, King MS. Low-level viremia persists for at least 7 years in patients on suppressive antiretroviral therapy. Proc.Natl.Acad.Sci.U.S.A. 2008;105:3879–3884. doi: 10.1073/pnas.0800050105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Salgado M, Rabi SA, O'Connell KA, Buckheit RW, 3rd, Bailey JR, Chaudhry AA, Breaud AR, Marzinke MA, Clarke W, Margolick JB, et al. Prolonged control of replication-competent dual-tropic human immunodeficiency virus-1 following cessation of highly active antiretroviral therapy. Retrovirology. 2011;8 doi: 10.1186/1742-4690-8-97. 97-4690-897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23**.Saez-Cirion A, Bacchus C, Hocqueloux L, Avettand-Fenoel V, Girault I, Lecuroux C, Potard V, Versmisse P, Melard A, Prazuck T, et al. Post-treatment HIV-1 controllers with a long-term virological remission after the interruption of early initiated antiretroviral therapy ANRS VISCONTI Study. PLoS Pathog. 2013;9:e1003211. doi: 10.1371/journal.ppat.1003211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- This study identified 14 patients who were treated early in the course of infection and then were able to maintain control of viremia after treatment interruption. The mechanism of control is not clear but appears to be different from that observed in subset of patients known as elite supressors who typically have strong cytolytic T lymphocyte responses to HIV-1.
- 24.Mansky LM, Temin HM. Lower in vivo mutation rate of human immunodeficiency virus type 1 than that predicted from the fidelity of purified reverse transcriptase. J.Virol. 1995;69:5087–5094. doi: 10.1128/jvi.69.8.5087-5094.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25**.Bontell I, Haggblom A, Bratt G, Albert J, Sonnerborg A. Trends in antiretroviral therapy and prevalence of HIV drug resistance mutations in Sweden 1997-2011. PLoS One. 2013;8:e59337. doi: 10.1371/journal.pone.0059337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- A thorough study involving records on essentially all known patients with HIV-1 infection in Sweden. The prevalence of HIV-1 drug resistance for all classes of antiretroviral drugs was tracked over time and shown to exhibit a dramatic decline.
- 26*.De Luca A, Dunn D, Zazzi M, Camacho R, Torti C, Fanti I, Kaiser R, Sonnerborg A, Codoner FM, Van Laethem K, et al. Declining prevalence of HIV-1 drug resistance in antiretroviral treatment-exposed individuals in Western Europe. J.Infect.Dis. 2013;207:1216–1220. doi: 10.1093/infdis/jit017. [DOI] [PubMed] [Google Scholar]
- Another study documenting the declining prevalence of HIV-1 drug resistance.
- 27*.Charpentier C, Lambert-Niclot S, Visseaux B, Morand-Joubert L, Storto A, Larrouy L, Landman R, Calvez V, Marcelin AG, Descamps D. Evolution of the K65R, K103N and M184V/I reverse transcriptase mutations in HIV-1-infected patients experiencing virological failure between 2005 and 2010. J.Antimicrob.Chemother. 2013 doi: 10.1093/jac/dkt184. [DOI] [PubMed] [Google Scholar]
- A study of over 9000 patients experience treatment failure between 2005 and 2010. The prevalence of important resistance mutations in reverse transcriptase declined over the study period, perhaps reflecting better adherence to newer regimens that combine three antiretroviral drugs into a single pill.
- Another study documenting the declining prevalence of HIV-1 drug resistance.
- 28*.Theys K, Snoeck J, Vercauteren J, Abecasis AB, Vandamme AM, Camacho RJ, Portuguese HIV-1 Resistance Study Group Decreasing population selection rates of resistance mutation K65R over time in HIV-1 patients receiving combination therapy including tenofovir. J.Antimicrob.Chemother. 2013;68:419–423. doi: 10.1093/jac/dks380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29*.Vercauteren J, Theys K, Carvalho AP, Valadas E, Duque LM, Teofilo E, Faria T, Faria D, Vera J, Aguas MJ, et al. The demise of multidrug-resistant HIV-1: the national time trend in Portugal. J.Antimicrob.Chemother. 2013;68:911–914. doi: 10.1093/jac/dks470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Another study documenting the declining prevalence of HIV-1 drug resistance.
- 30.Dinoso JB, Kim SY, Wiegand AM, Palmer SE, Gange SJ, Cranmer L, O'Shea A, Callender M, Spivak A, Brennan T, et al. Treatment intensification does not reduce residual HIV-1 viremia in patients on highly active antiretroviral therapy. Proc.Natl.Acad.Sci.U.S.A. 2009;106:9403–9408. doi: 10.1073/pnas.0903107106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yukl SA, Shergill AK, McQuaid K, Gianella S, Lampiris H, Hare CB, Pandori M, Sinclair E, Gunthard HF, Fischer M, et al. Effect of raltegravir-containing intensification on HIV burden and T-cell activation in multiple gut sites of HIV-positive adults on suppressive antiretroviral therapy. AIDS. 2010 doi: 10.1097/QAD.0b013e32833ef7bb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McMahon D, Jones J, Wiegand A, Gange SJ, Kearney M, Palmer S, McNulty S, Metcalf JA, Acosta E, Rehm C, et al. Short-course raltegravir intensification does not reduce persistent low-level viremia in patients with HIV-1 suppression during receipt of combination antiretroviral therapy. Clin.Infect.Dis. 2010;50:912–919. doi: 10.1086/650749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gandhi RT, Zheng L, Bosch RJ, Chan ES, Margolis DM, Read S, Kallungal B, Palmer S, Medvik K, Lederman MM, et al. The effect of raltegravir intensification on low-level residual viremia in HIV-infected patients on antiretroviral therapy: a randomized controlled trial. PLoS Med. 2010;7:e1000321. doi: 10.1371/journal.pmed.1000321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34*.Dahl V, Lee E, Peterson J, Spudich SS, Leppla I, Sinclair E, Fuchs D, Palmer S, Price RW. Raltegravir treatment intensification does not alter cerebrospinal fluid HIV-1 infection or immunoactivation in subjects on suppressive therapy. J.Infect.Dis. 2011;204:1936–1945. doi: 10.1093/infdis/jir667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Although previous studies had clearly documented that treatment intensification with the integrase inhibitor raltegravir did not reduce residual viremia, it was unclear whether intensification might affect the trace levels of virus present in the cerebrospinal fluid. The central nervous system (CNS)has been frequently discussed as a potential site of HIV-1 persistence, and not all antiretroviral drugs achieve optimal penetration of the CNS. In a small study of 14 patients, treatment intensification with the integrase inhibitor raltegravir did not reduce virus levels in the CSF.
- 35.Frenkel LM, Wang Y, Learn GH, McKernan JL, Ellis GM, Mohan KM, Holte SE, De Vange SM, Pawluk DM, Melvin AJ, et al. Multiple viral genetic analyses detect low-level human immunodeficiency virus type 1 replication during effective highly active antiretroviral therapy. J.Virol. 2003;77:5721–5730. doi: 10.1128/JVI.77.10.5721-5730.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tobin NH, Learn GH, Holte SE, Wang Y, Melvin AJ, McKernan JL, Pawluk DM, Mohan KM, Lewis PF, Mullins JI, Frenkel LM. Evidence that low-level viremias during effective highly active antiretroviral therapy result from two processes: expression of archival virus and replication of virus. J.Virol. 2005;79:9625–9634. doi: 10.1128/JVI.79.15.9625-9634.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hermankova M, Ray SC, Ruff C, Powell-Davis M, Ingersoll R, D'Aquila RT, Quinn TC, Siliciano JD, Siliciano RF, Persaud D. HIV-1 drug resistance profiles in children and adults with viral load of <50 copies/ml receiving combination therapy. JAMA. 2001;286:196–207. doi: 10.1001/jama.286.2.196. [DOI] [PubMed] [Google Scholar]
- 38.Kieffer TL, Finucane MM, Nettles RE, Quinn TC, Broman KW, Ray SC, Persaud D, Siliciano RF. Genotypic analysis of HIV-1 drug resistance at the limit of detection: virus production without evolution in treated adults with undetectable HIV loads. J.Infect.Dis. 2004;189:1452–1465. doi: 10.1086/382488. [DOI] [PubMed] [Google Scholar]
- 39.Persaud D, Siberry GK, Ahonkhai A, Kajdas J, Monie D, Hutton N, Watson DC, Quinn TC, Ray SC, Siliciano RF. Continued production of drug-sensitive human immunodeficiency virus type 1 in children on combination antiretroviral therapy who have undetectable viral loads. J.Virol. 2004;78:968–979. doi: 10.1128/JVI.78.2.968-979.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nettles RE, Kieffer TL, Kwon P, Monie D, Han Y, Parsons T, Cofrancesco J, Jr, Gallant JE, Quinn TC, Jackson B, et al. Intermittent HIV-1 viremia (Blips) and drug resistance in patients receiving HAART. JAMA. 2005;293:817–829. doi: 10.1001/jama.293.7.817. [DOI] [PubMed] [Google Scholar]
- 41.Bailey JR, Sedaghat AR, Kieffer T, Brennan T, Lee PK, Wind-Rotolo M, Haggerty CM, Kamireddi AR, Liu Y, Lee J, et al. Residual human immunodeficiency virus type 1 viremia in some patients on antiretroviral therapy is dominated by a small number of invariant clones rarely found in circulating CD4+ T cells. J.Virol. 2006;80:6441–6457. doi: 10.1128/JVI.00591-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Veazey RS, DeMaria M, Chalifoux LV, Shvetz DE, Pauley DR, Knight HL, Rosenzweig M, Johnson RP, Desrosiers RC, Lackner AA. Gastrointestinal tract as a major site of CD4+ T cell depletion and viral replication in SIV infection. Science. 1998;280:427–431. doi: 10.1126/science.280.5362.427. [DOI] [PubMed] [Google Scholar]
- 43.Chun TW, Nickle DC, Justement JS, Meyers JH, Roby G, Hallahan CW, Kottilil S, Moir S, Mican JM, Mullins JI, et al. Persistence of HIV in gut-associated lymphoid tissue despite long-term antiretroviral therapy. J.Infect.Dis. 2008;197:714–720. doi: 10.1086/527324. [DOI] [PubMed] [Google Scholar]
- 44**.Evering TH, Mehandru S, Racz P, Tenner-Racz K, Poles MA, Figueroa A, Mohri H, Markowitz M. Absence of HIV-1 evolution in the gut-associated lymphoid tissue from patients on combination antiviral therapy initiated during primary infection. PLoS Pathog. 2012;8:e1002506. doi: 10.1371/journal.ppat.1002506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- A careful study which fails to detect any sequence changes in HIV-1 DNA in cells from the GALT of patients on ART. The GALT has a high level of infection and is frequently cited as a possible site of ongoing viral replication during ART. This study contradicts that idea.
- 45.Shen L, Peterson S, Sedaghat AR, McMahon MA, Callender M, Zhang H, Zhou Y, Pitt E, Anderson KS, Acosta EP, Siliciano RF. Dose-response curve slope sets class-specific limits on inhibitory potential of anti-HIV drugs. Nat.Med. 2008;14:762–766. doi: 10.1038/nm1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46*.Jilek BL, Zarr M, Sampah ME, Rabi SA, Bullen CK, Lai J, Shen L, Siliciano RF. A quantitative basis for antiretroviral therapy for HIV-1 infection. Nat.Med. 2012 doi: 10.1038/nm.2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- This study analyzes the inhibitory potential of a very large number of ART regimens, taking into account the complex and, in some cases, cooperative nature of the dose response curves for individuals drugs as well as their modes of interaction. The authors conclude that many ART regimens have sufficient inhibitory potential to fully suppress replication in all drug accessible compartments.
- 47.Hutter G, Nowak D, Mossner M, Ganepola S, Mussig A, Allers K, Schneider T, Hofmann J, Kucherer C, Blau O, et al. Long-term control of HIV by CCR5 Delta32/Delta32 stem-cell transplantation. N.Engl.J.Med. 2009;360:692–698. doi: 10.1056/NEJMoa0802905. [DOI] [PubMed] [Google Scholar]
- 48*.Yukl SA, Boritz E, Busch M, Bentsen C, Chun TW, Douek D, Eisele E, Haase A, Ho YC, Hutter G, et al. Challenges in detecting HIV persistence during potentially curative interventions: a study of the Berlin patient. PLoS Pathog. 2013;9:e1003347. doi: 10.1371/journal.ppat.1003347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- A detailed, collaborative search for persistent HIV-1 in the Berlin patient. Except for sporadic PCR signals at the limit of detection, no consistent evidence supporting HIV-1 persistence in this patient was uncovered, supporting the conclusion that he is the first patient cured of HIV-1 infection.
- 49*.Henrich TJ, Hu Z, Li JZ, Sciaranghella G, Busch MP, Keating SM, Gallien S, Lin NH, Giguel FF, Lavoie L, et al. Long-term reduction in peripheral blood HIV type 1 reservoirs following reduced-intensity conditioning allogeneic stem cell transplantation. J.Infect.Dis. 2013;207:1694–1702. doi: 10.1093/infdis/jit086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- This study described two patients with well controlled HIV-1 infection who received HSC transplants for malignancies. During HSC, ART is often stopped due to complex drug interactions and side effects of the treatments. However, in these cases ART was continued throughout the transplant period and appears to have protected the donor cells, which were from individuals with wild type CCR5 alleles, from infection until 100% chimerism was achieved. If viremia does not rebound after treatment interruption, these may represent additional cases of cure and certainly illustrate the remarkable ability of ART to stop new infection events.
- 50.Deeks SG. HIV infection, inflammation, immunosenescence, and aging. Annu.Rev.Med. 2011;62:141–155. doi: 10.1146/annurev-med-042909-093756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Buzon MJ, Massanella M, Llibre JM, Esteve A, Dahl V, Puertas MC, Gatell JM, Domingo P, Paredes R, Sharkey M, et al. HIV-1 replication and immune dynamics are affected by raltegravir intensification of HAART-suppressed subjects. Nat.Med. 2010;16:460–465. doi: 10.1038/nm.2111. [DOI] [PubMed] [Google Scholar]
- 52**.Sigal A, Kim JT, Balazs AB, Dekel E, Mayo A, Milo R, Baltimore D. Cell-to-cell spread of HIV permits ongoing replication despite antiretroviral therapy. Nature. 2011;477:95–98. doi: 10.1038/nature10347. [DOI] [PubMed] [Google Scholar]
- This study provides in vitro evidence that high multiplicity infection that might occur in the local environment of a productively infected cell is more difficult to block with antiretroviral drugs.
- 53.Havlir DV, Hellmann NS, Petropoulos CJ, Whitcomb JM, Collier AC, Hirsch MS, Tebas P, Sommadossi JP, Richman DD. Drug susceptibility in HIV infection after viral rebound in patients receiving indinavir-containing regimens. JAMA. 2000;283:229–234. doi: 10.1001/jama.283.2.229. [DOI] [PubMed] [Google Scholar]
- 54*.Dolling DI, Dunn DT, Sutherland KA, Pillay D, Mbisa JL, Parry CM, Post FA, Sabin CA, Cane PA. Low frequency of genotypic resistance in HIV-1-infected patients failing an atazanavir-containing regimen: a clinical cohort study. J.Antimicrob.Chemother. 2013 doi: 10.1093/jac/dkt199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- A recent study of 2528 patients on regimens including the PI atazanavir. Among the 16% of patients who failed therapy, atazanavir resistance mutations were rare.
- 55*.Rosenbloom DI, Hill AL, Rabi SA, Siliciano RF, Nowak MA. Antiretroviral dynamics determines HIV evolution and predicts therapy outcome. Nat.Med. 2012;18:1378–1385. doi: 10.1038/nm.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- A recent study providing a pharmacodynamic analysis of treatment failure for different antiretroviral drugs. During periods of non-adherence to PIs, the time during which the mutant virus can grow and has a growth advantage over wild type virus is very short. Thus patients fail with virus that has no mutations in protease. This only occurs if adherence is suboptimal.
- 56*.Rabi SA, Laird GM, Durand CM, Laskey S, Shan L, Bailey JR, Chioma S, Moore RD, Siliciano RF. Multi-step inhibition explains HIV-1 protease inhibitor pharmacodynamics and resistance. J. Clin. Invest. 2013 doi: 10.1172/JCI67399. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- A recent study which explains the steep dose response curves for PIs in terms of effects on multiple downstream steps in the virus life cycle including virus entry. The study raises the possibility that mutations in the Env protein may contribute to PI resistance.
- 57.Shen L, Rabi SA, Sedaghat AR, Shan L, Lai J, Xing S, Siliciano RF. A critical subset model provides a conceptual basis for the high antiviral activity of major HIV drugs. Sci.Transl.Med. 2011;3:91ra63. doi: 10.1126/scitranslmed.3002304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58**.Chojnacki J, Staudt T, Glass B, Bingen P, Engelhardt J, Anders M, Schneider J, Muller B, Hell SW, Krausslich HG. Maturation-dependent HIV-1 surface protein redistribution revealed by fluorescence nanoscopy. Science. 2012;338:524–528. doi: 10.1126/science.1226359. [DOI] [PubMed] [Google Scholar]
- An elegant imaging study demonstrating differential localization of Env trimers on the surfaces of immature and mature virus particles. The authors suggest that Gag cleavage by HIV-1 protease is necessary to allow Env trimers to cluster in a configuration that promotes the entry process [Google Scholar]
- 59**.Fun A, Wensing AM, Verheyen J, Nijhuis M. Human Immunodeficiency Virus Gag and protease: partners in resistance. Retrovirology. 2012;9 doi: 10.1186/1742-4690-9-63. 63-4690-9-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- A comprehensive review of the interactions between HIV-1 protease and its cleavage sites in Gag as pertains to the evolution of drug resistance [Google Scholar]