

Abstract
We are studying the projections from the entorhinal cortex to the hippocampal formation in the mouse. The dentate gyrus is innervated by the lateral entorhinal cortex (lateral perforant path) and medial entorhinal cortex (medial perforant path). The entorhinal cortex also projects to hippocampal areas CA3 and CA1, and to the subiculum. In young transgenic Alzheimer’s disease mouse models (before amyloid-β pathology), the connections are not different from normal mice. In Alzheimer’s disease mice with pathology, two changes occur: first, dystrophic axon endings appear near amyloid-β plaques, and second, there are sparse aberrant axon terminations not in the appropriate area or lamina of the hippocampus. Furthermore, MRI–diffusion tensor imaging analysis indicates a decrease in the quality of the white matter tracts connecting the hippocampus to the brain; in other words, the fimbria/fornix and perforant path. Similar changes in white matter integrity have been found in Alzheimer’s disease patients and could potentially be used as early indicators of disease onset.
Keywords: entorhinal cortex, hippocampus, limbic system, perforant path, tractography
The entorhinal cortex (EC) functions as the gateway to hippocampal formation because its output, through the perforant path, is the major cortical source of input to the hippocampus and, furthermore, together with the subiculum, it also provides the major output of the hippocampus [1]. Ramón y Cajal was the first to clearly describe fibers arising from the entorhinal area going to the fascia dentata and hippocampus proper (i.e., cornu Ammonis) [2]. More recently, modern tracing studies performed in many species have confirmed that the EC projects by way of the perforant path to the dentate gyrus and hippocampus [3–7]. It has been demonstrated by a large number of studies in several species that the superficial layers of the EC project to the hippocampus[4,5,7,8]. In the rat, the layer II cells of the EC have been shown to project primarily to the dentate gyrus [4,9–11]. The EC layer III cells have been shown to predominantly project to area CA1 of the hippocampus, and this projection to area CA1 is bilateral [4,7]. In early studies, it was concluded that a topography existed in the entorhinal–hippocampal projection [4,12]; in other words, the axons arising from the laterodorsal EC terminate in the septal (dorsal) and more ventral hippocampus, and medial bands of EC project to the temporal (ventral) hippocampus (Figure 1B). Similarly, we have shown that in the mouse, entorhinal–hippocampal projections are, for the most part, similarly organized to those in the rat; however, some differences between these two species have been shown to exist [7,13]. The current article describes the differences in projections from the EC to the hippocampal formation in the Alzheimer’s disease (AD) mouse model compared with the C57BL/6 mouse and we show that after the development of pathology, abnormalities are present in these connections [14]. We have studied the changes in plasticity in these connections in transgenic (Tg) AD model mice at different ages [15,16].
Figure 1. Overview of entorhinal–hippocampal connections.
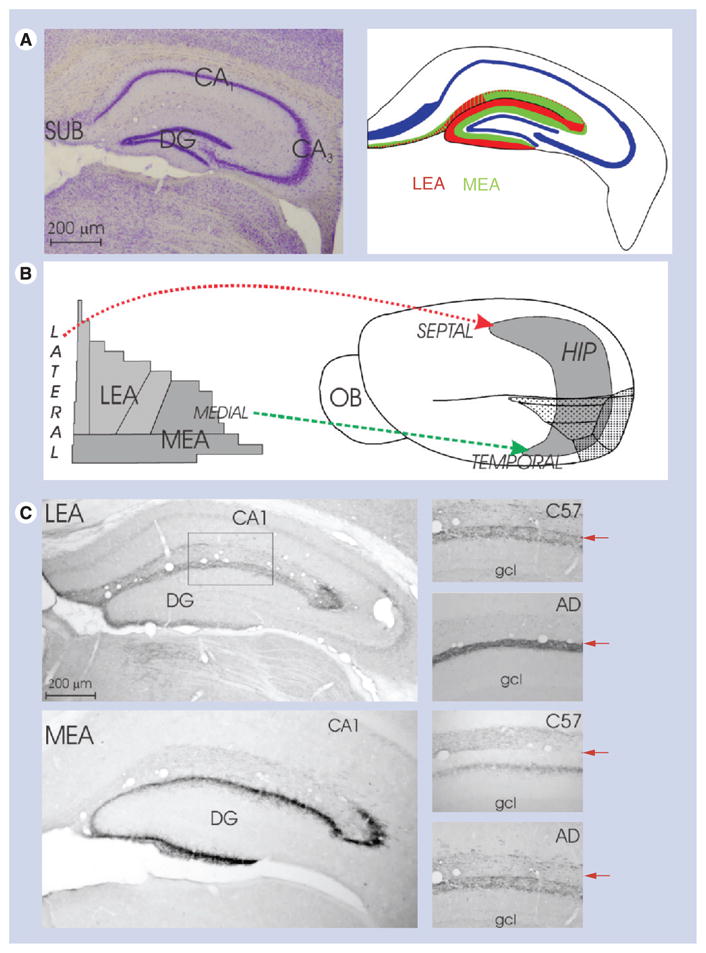
(A) Low-power photomicrograph of a coronal section of the septal hippocampal formation and 2D reconstruction map of the HIP to demonstrate the labeling pattern of the entorhinal cortex axons in the HIP; (B) 2D reconstruction maps of the entorhinal cortex (left) and mouse brain (right) to demonstrate the septotemporal distribution of the entorhinal cortex axons in the HIP; (C) left, two low-power photomicrographs of coronal sections through the septal hippocampal formation to demonstrate the labeling patttern of the entorhinal cortex axons in the HIP. Right, four higher-power images of the molecular layer of the DG showing the terminals of the LEA and MEA. Top image: C57BL mouse; bottom image: young (2 month) transgenic AD mouse model. The arrows indicate hippocampal fissure.
AD: Alzheimer’s disease; DG: Dentate gyrus; gcl: Granule cell layer; HIP: Hippocampus; LEA: Lateral entorhinal cortex; MEA: Medial entorhinal cortex; OB: Olfactory bulb; SUB: Subiculum.
Tracing studies in Tg AD model mice
Deeply anesthetized adult, male mice were injected either with an anterogradely transported tracer in the EC or with retrogradely transported tracers in the hippocampal formation [7]. Together, these data give the full spectrum of the connections between the EC and hippocampus. Furthermore, we have performed MRI–diffusion tensor imaging (DTI) measurements, by creating respiratory-gated MRI images of the mouse brain. They were obtained with a Bruker Biospec® (Bruker Biosciences Corporation, MA, USA) 9.4-T/21-cm horizontal-bore magnet spectrometer with a 7.2-cm resonator for radiowave transmitting and an active-decoupled 2.0-cm surface coil for signal receiving. T2-weighted magnetic resonance images were first acquired using spin echo sequence (rapid acquisition with relaxation enhancement) with acquisition parameters: repetition time/echo time: 4000/40ms; four slices of 0.75-mm thickness without gap; 128× 128pixels matrix; and field of view: 16 × 16mm. DTI data were collected using a standard spin echo sequence with similar imaging parameters to those used in T2. DTI parameter maps were calculated using the vendor’s software to give fractional anisotropy (FA), mean diffusity, axial diffusivity and radial diffusivity. Our data indicate that in Tg AD model mice that have significant AD pathology; in other words, with large amounts of amyloid-β deposits, the FA of the white matter tract (fornix) leaving the hippocampus is reduced by 20%. We are currently investigating the changes in the perforant path in a Tg AD mouse model, but since this is a less organized fiber bundle, it is more difficult to obtain the data.
Overview of entorhinal hippocampal connections in mice
We have subdivided the EC into two main parts; in other words, the classical lateral entorhinal area (LEA) and the classical medial entorhinal area (MEA) (Figure 1) [17,18]. We use the division of the hippocampal formation into septal (i.e., dorsal) and temporal (i.e., ventral) poles of the hippocampus according to Blackstad (Figure 1) [19]. It should be noted that in the molecular layer of the dentate gyrus of the mouse, the two bands of the lateral and medial perforant path are wider than the inner band of labeling (which occupies approximately 17% of the total stratum moleculare) [20]. The inner band of the molecular layer contains the terminals of the associational and commissural axons.
Laminar organization of projection of the LEA & MEA
LEA axons travel via the lateral perforant path; in the dentate gyrus, terminals are present only in the outer a third of the molecular layer (Figure 1) [7], while in CA3 and CA1, terminals are in the stratum lacunosum-moleculare, and in the subiculum, in the superficial part of the stratum moleculare (Figure 1) [7]. MEA axons enter the medial perforant path; in the dentate gyrus, terminals are present in the middle a third of the molecular layer (Figure 1) [7], and in CA3 and CA1, terminals are in the stratum lacunosum-moleculare (Figure 1) [7]. In general, the mouse shows a laminar organization that is similar to that displayed in the rat [9,21] and other species, including monkeys [22–24]; LEA is the origin of the lateral perforant path that terminates in the outer a third of the molecular layer of the dentate gyrus, and MEA is the origin of the medial perforant path that ends in the middle a third of the molecular layer of the dentate gyrus [7,25]. The Tg AD model mouse has no significant differences in the origin and distribution of axons originating in the LEA or in MEA, but they do show a small amount of aberrant axon terminals in inappropriate layers (Figure 2A–D) [25]. The number of incorrectly ending axonal terminals from the EC in the hippocampal formation is less then 1% of all axonal terminals, suggesting that they probably do not play a big role in memory dysfunction in these AD model animals.
Figure 2. Four high-power photomicrographs of coronal sections through the dorsal hippocampal formation to demonstrate the labeling pattern in the hippocampus following injections of biotinylated dextran amine into the entorhinal cortex in transgenic Alzheimer’s disease mouse models that have Alzheimer’s disease pathology.

(A) Demonstrates labeling in the dentate gyrus following an injection into the lateral entorhinal cortex, while (B) shows the adjacent section stained for amyloid-β (WO-2 antibody). (C & D) Demonstrate labeling in the hippocampus following injections into the lateral entorhinal area and medial entorhinal area, respectively. The presence of plaques and dystrophic axon endings is highlighted by the arrows.
DG: Dentate gyrus.
Commissural connections
It is important to note that the entorhinal projections to the mouse hippocampus are bilateral to areas CA3 and CA1, but not to the dentate gyrus, where there is only a unilateral projection. This is different in rats where all projections are bilateral but, most likely, in humans, similar to monkeys [3,22], this projection is unilateral. In the mouse and rat, the hippocampus is strongly connected to the contralateral side of the brain; these connections arise from the the hilar cells and area CA3. However, the hippocampal commissural connections in monkeys (and most likely humans) are very limited; most intrinsic connections are unilateral [26,27]. In mice, both hippocampi are strongly interconnected [28,29], and the entorhinal cortices are connected.
Topographical organization
The projection of EC to the hippocampal formation is topographically organized, lateral parts of LEA project to the the dorsal, septal part of the hippocampal formation, whereas more medial parts of LEA terminate in the intermediate (septotemporal axis) part of the hippocampal formation. The most medial part of LEA terminates in the ventral, temporal part of the hippocampal formation (Figure 1B) [7]. MEA projections to the hippocampus display a similar septemporal distribution (Figure 1B) [7]. There are no significant differences present in the topographical organization between ‘normal’ and Tg AD model mice. Most species show a similar topographic organization of these connections [5,30].
Laminar organization in AD mice with plaques
Injections into LEA give rise to labeled axons in the lateral perforant path; in the dentate gyrus, labeled axons and terminals are primarily present in the outer a third of the molecular layer (Figure 3A) and in CA3 and CA,1 labeled axons and terminals are present in the stratum lacunosum-moleculare (Figure 3D) [25]. It should be noted that even with a significant amyloid-β load present in the molecular layer of the dentate gyrus (Figure 3B), the EC axonal endings are largely normal (Figure 3A). However, a few aberrant terminals can be observed in the inner molecular layer of the dentate gyrus (Figure 2A–D, arrows). Furthermore, at more advanced stages of pathology, the hippocampus shows shrinkage and the size of the dentate gyrus molecular layer is significantly decreased [31], probably indicating decreased synaptic innervation from the EC. Similarly, AD patients show shrinkage of the hippocampus [32–34].
Figure 3. Changes in terminal fields with Alzheimer’s disease.

(A–D) Four high-power photomicrographs to demonstrate improper localization of axons in the dentate gyrus in a transgenic Alzheimer’s disease mouse model with pathology. Arrows indicate incorrect location of axon endings. (E & F) High-power photomicrographs to demonstrate the pattern of labeled neurons in the entorhinal cortex following injections of retrogradely transported tracers into the dorsal hippocampus. (E) Demonstrates FluoroGold™-labeled (Fluorochrome, LLC, CO, USA) neurons in layer II of the entorhinal cortex following an injection into the dentate gyrus; (F) demonstrates labeled neurons in a transgenic Alzheimer’s disease mouse model with amyloid-β pathology.
Laminar origin
The entorhinal projection to the dentate gyrus predominantly originates from neurons in layer II (Figure 2) [4,7] and this projection is confined to the ipsilateral dentate gyrus, whereas the entorhinal projection to CA3, CA1 and the subiculum is bilateral, and it predominantly originates from neurons in layer III. No significant changes are present in Tg AD model mice (Figures 2 & 3). Importantly, it has been shown that the neurons in the superficial layers of the EC degenerate early in AD patients [35–37], thereby probably leading to decreased innervation of the hippocampus [38,39].
MRI–DTI measurements in mice & AD patients
To study the changes in connections in AD model mice compared with normal; in other words, C57BL mice, we performed preliminary MRI–DTI studies. The data indicate that small changes occur in hippocampal-related tracts in the Tg AD model mice that have developed significant pathology(Figure 4). The most easily identifiable changes occur in the fimbria/fornix; in the Tg AD model mice, there is a decrease in tract integrity (FA values increased by approximately 15%), as indicated by increased diffusion anisotropy, similar changes are observed in AD patients [40,41]. These changes in the integrity of myelin (white matter) correlate with observations of the white matter quality in the hippocampus of AD model mice by immunohistochemical methods [42]. In humans, it has been demonstrated that fornix FA both cross-sectionally correlated with and longitudinally predicted memory decline and progression to AD. Manually drawn regions of interest within the fornix show promise comparable with hippocampal volume as a predictive biomarker of progression [40]. Similarly, it has been demonstrated that the perforant path undergoes synaptic changes in the course of aging and dementia. Yassa et al. report direct evidence of age-related perforant path degradation in humans in vivo using ultra-high-resolution microstructural DTI [43]. They did not find evidence of white matter loss in a control pathway, the alveus, suggesting that these findings are not evidence for a global decline in white matter integrity. The extent of perforant path degradation correlated with performance on a word-list learning task sensitive to hippocampal deficits. They also show evidence for gray matter diffusion signals consistent with pyramidal dendrite orientation in the hippocampus and cerebral cortex [43]. Perforant path degradation is a unique biomarker that can be used in combination with traditional structural and functional neuroimaging methods to enhance detection of AD in its earliest stages.
Figure 4. Four MRI–diffusion tensor imaging images of the mouse brain.
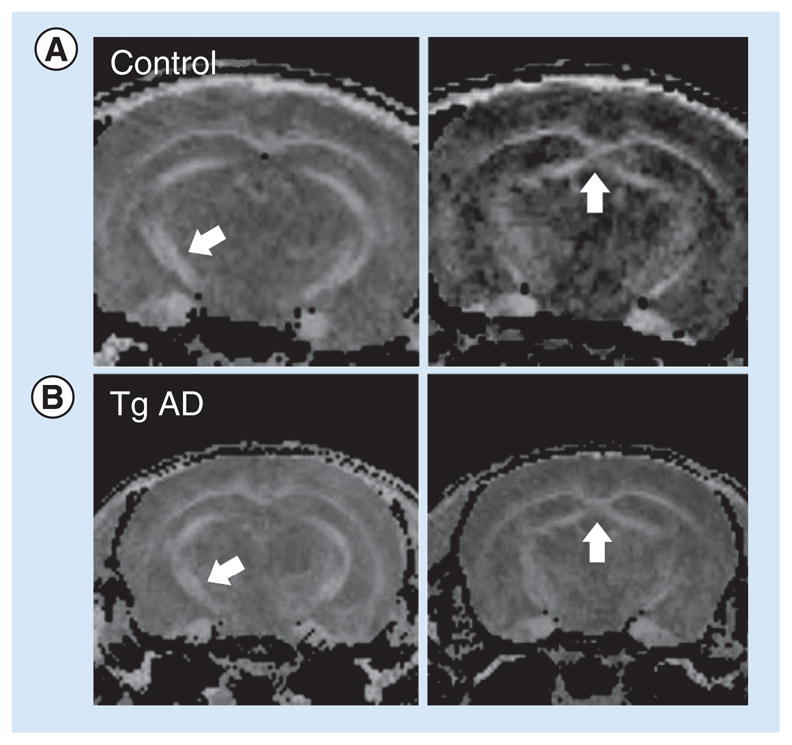
(A) Shows the normal mouse and (B) shows the Tg AD mouse model at 6 months of age. Arrows indicate areas with changes in fractional anisotropy between normal and Tg AD model mice; in other words, the fornix and perforant path. The fractional anisotropy has been reduced by 20% in the AD mice.
AD: Alzheimer’s disease; Tg: Transgenic.
Plasticity in connections
In general, the magnitude and significance of sprouting, both normal and aberrant, in AD have been underestimated [44,45]; most studies have been focused on degeneration in brain connections, especially on the entorhinal to hippocampal connections [38,46].
Only a few studies have analyzed the effects of EC lesions on hippocampal plasticity in the mouse [31,47]. Our studies have shown that, surprisingly, even in Tg AD model animals with a high level of amyloid-β pathology, the response to an EC lesion is not significantly different from control age-matched mice [31]. We and others have shown that partial EC lesions lead to reinnervation of the denervated entorhinal–hippocampal pathway, both in control and Tg AD model mice. This, taken together with the use of Tg mice in studies on the involvement of the perforant path in AD [48] indicates that detailed anatomical information on the EC and its hippocampal connections in each species is important. Thus far, there is only one other study on the changes in the entorhinal–hippocampal connection in AD model mice [25]. Similar to our findings, their findings suggest that cerebral amyloid deposition has neurotropic effects and is the main cause of aberrant sprouting in the AD brain [49]. Furthermore, dystrophic axon terminals were found surrounding plaques; it should be noted that synaptic pathology is a major neurobiological substrate for cognitive dysfunction in AD [50].
Synaptic dysfunction is one of the first hallmarks of neurodegenerative disease. Spine pathology has been observed in association with many brain disorders, such as AD, Parkinson’s disease, prion diseases, schizophrenia, mental retardation and epilepsy. However, it is currently unclear how these phenotypes causally relate to disease progression. For example, in the vicinity of a cerebral infarct in mice, dendrites become exceptionally plastic, characterized by a long-lasting increase in the rate of spine turnover. These structural changes might provide a substrate for the long-term functional changes in the representational cortical maps that are observed after stroke models. Similarly, in mouse models of AD, the vicinity of amyloid plaques is characterized by highly dysmorphic neurites and spine turnover, causing a net loss of spines [51]. This phenotype could be caused by amyloid-β oligomers, which have been shown to block long-term potentiation and directly induce long-term depression, spine loss and memory loss [52].
AD & plasticity
The mechanisms that are involved in structural adaptive plasticity, allowing for the constant readjustment of connectivity providing the basis for ‘higher brain function’, are not very well understood. The integrative theory of neuro-plasticity suggests that no distinctions should be made between developmental, adaptive or restorative plasticity. It is thus reasonable to propose that the mechanism of reactive synaptic plasticity in the adult brain is identical to that involved in the natural turnover of synapses. The impact of experience on the CNS likely requires a lifelong high turnover of synapses that might, therefore, involve the same molecules as ‘reactive synaptogenesis’, as occurs, for example, after a lesion of the EC. Some of the proposed mechanisms are:
Neurotrophic factors such as NGF, BDNF, IGF-1, FGF or TGF-β;
Growth-associated proteins, such as GAP-43;
Neural cell-adhesion molecules, such as NCAM, and several synaptic proteins, such as synaptophysin and SNAP 25;
Cellular lipids and lipid carrier proteins, such as ApoE;
Changes in the expression and subcellular distrubution of microtubule-associated proteins and other cytoskeletal proteins [53,54].
In organs other than the brain, cell activation seems to increase ‘wear and tear’, for example, by increased free-radical formation, and thus cause an increased rate of aging. However, activation of nerve cells within the physiological range seems to lead to maintenance of neurons during aging and AD, possibly by preferentially stimulating the action of protective mechanisms, such as DNA repair. This ‘use it or lose it’ principle might explain why certain neurons degenerate in aging or AD, while others do not, and why recovery of various neuronal systems during aging has been obtained by restoration of the missing stimulus [55]. Consequently, neuronal activation may provide a means of prolonging neuronal function for the full length of our natural lifespan. There is a regional pattern of amyloid deposits and tangle pathology in AD brains that changes with disease duration [56]. Tangle density in AD shows systematic regional differences that can be observed both during aging and in AD [57]. The intensity of dendritic remodelling that can be observed during aging, as well as in AD, is regionally different and decreased in the following order: transentorhinal region > limbic areas (entorhinal region and hippocampus) > nonprimary association areas > primary sensory association areas > primary sensory and motor cortex [56]. These regional differences of neuronal plasticity follow the same pattern as the regional vulnerability to tangle formation in AD [57,58]. Furthermore, brain areas affected by AD pathology are primarily those structures that are involved in the regulation of ‘higher brain functions’ [59]. The functions these areas subserve, such as learning, memory, perception, self-awareness and consciousness, require a life-long adjusting of synaptic contacts that allows for the acquistion of new information, a process based on a particularly high degree of structural plasticity. Thus, there seems to be a relation between the development of AD pathology and disturbed neuronal plasticity [49,55].
AD & pathology spread
Many age-associated neurodegenerative diseases have, as the main pathology, the aggregation of specific proteins within the nervous system. For instance, in AD, the insidious pathogenic process begins many years before the symptoms emerge and the lesions that characterize the disease; in other words, senile plaques and neurofibrillary tangles are present throughout the brain. However, the pathology does not show up at random: a sequence of pathological events occurs [56,57]. There is clear evidence that both the amyloid-β and tau proteins, which aggregate to form senile plaques and neurofibrillary tangles, respectively, spread through the brain [48,60] following anatomical pathways. Recent data also indicate that the spread of these lesions from one site to another is mediated by the cellular uptake, transport and release of endogenous seeds formed by the cognate proteins; in other words, amyloid-β and tau [61,62].
AD & connections
It has been demonstrated that patients with AD experience a brain network breakdown [63], reflecting disconnection at both the structural and functional system levels [14,53,64]. Resting-state functional MRI studies demonstrated that the regional coherence of the functional MRI signal is significantly altered in patients with AD and amnestic mild cognitive impairment [65]. MRI–DTI has made it possible to track fiber bundle projections across the brain, revealing a substantially abnormal interplay of ‘critical’ white matter tracts in these conditions [66]. Regional cortical atrophy and cognitive function of AD patients have been shown to correlate with the structural changes of white matter. Synchronized structural changes of cingulum bundle and fornix, both of which are limbic tracts, were revealed. Widespread yet distinctive structural changes were demonstrated in limbic, commissural, association and projection tract groups between control and AD subjects [67,68]. In order to improve our understanding of the pathobiology of these findings, studies in Tg AD model mice are required [69].
Conclusion
Patients with AD demonstrate a brain network breakdown, reflecting disconnection at both the structural and functional system level. Our studies and many other studies are helping to establish the exciting potential of tract tracing as a neuropathological measure and as a biomarker of disease progression in AD.
Future perspective
Together, the data presented indicate that deposition of cerebral amyloid-β leads to synaptic pathology, which causes the disruption of neuronal connectivity which, in turn, significantly contributes to AD dementia. Imaging provides a powerful quantitative measure of changes in structural connectivity measures derived from MRI–DTI methods. These methods offer additional markers of neuropathology arising from the secondary changes in axonal caliber and myelination that accompany decreased neuronal activity and neurodegeneration. DTI can especially be used for more finely mapping neurodegenerative changes in AD in patients and defining neuro-pathological changes in white matter. The differences between AD and control subjects, and those between mild cognitive impairment and control subjects indicate a progressive pattern of white matter disruption from limbic and commissural tracts to other tracts [67]. Together, the high correlation between FA, mean diffusity, and radial diffusivity measurements from limbic tracts and cortical atrophy suggests that the disruption of the limbic tracts is caused by neuronal damage [67], as is also shown by our anatomical studies [25].
Visualizing neuropathology can be enhanced by using more specific DTI measures and interpreting them relative to knowledge of local white matter anatomy in the healthy brain [68].
The available data suggest that AD risk is associated with a decline in white matter integrity in a subset of tracts. Specifically, AD risk has been associated with white matter integrity declines in tracts that connect gray matter structures associated with memory function [59]. These tracts include parahippocampal white matter, including the cingulum and the splenium of the corpus callosum. Some studies have indicated that AD risk declines are characterized by increases of radial diffusivity, raising the possibility that a myelin-related pathology may contribute to AD onset [67]. Together, these findings justify future research aimed at a more complete understanding of the neurobiological basis of DTI-based declines in AD. With continued refinement of imaging methods, DTI holds promise as a method to aid in the identification of presymptomatic AD [41,68,70–72].
Together, our and many other studies are helping to establish the exciting potential of tract tracing as a neuropathological measure and a biomarker of disease progression. The viability of these white matter tract integrity metrics as potential neuroimaging biomarkers of the earliest stages of AD and disease progression is unquestionable, but basic studies in Tg AD model mice are still urgently needed to validate these changes.
EXECUTIVE SUMMARY.
Lateral entorhinal area projects via the lateral perforant path to the dentate gyrus in the outer third of the molecular layer.
The Medial entorhinal area gives rise to labeled axons in the medial perforant path ending in the middle third of the molecular layer of the dentate gyrus.
The projection of the entorhinal cortex to the hippocampal formation is topographically organized. Neurons in lateral parts of the medial and lateral entorhinal cortex project to the dorsal, septal part of the hippocampal formation, medial parts of the entorhinal cortex project to the temporal, ventral hippocampal formation.
It is likely that cerebral amyloid deposition early on has neurotrophic effects and is the main cause of aberrant sprouting in the Alzheimer’s disease (AD) brain. Furthermore, dystrophic axon terminals were found surrounding plaques; it should be noted that synaptic pathology is the major neurobiological substrate for cognitive dysfunction in AD.
Small changes occur in hippocampal-related tracts in transgenic AD mouse models that have developed significant pathology.
The earliest and most easily identifiable changes occur in the fimbria/fornix, as indicated by increased diffusion anisotropy.
These changes are also observed in AD patients.
Patients with AD demonstrate a brain network breakdown, reflecting disconnection at both the structural and functional system level.
Together, our studies and many other studies are helping to establish the exciting potential of tract tracing as a neuropathological measure and biomarker of disease progression in AD.
Acknowledgments
The authors thank E von Schnier for his excellent comments on an earlier version of this manuscript.
Footnotes
For reprint orders, please contact: reprints@futuremedicine.com
Financial & competing interests disclosure
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
No writing assistance was utilized in the production of this manuscript.
References
Papers of special note have been highlighted as:
• of interest
•• of considerable interest
- 1.Witter MP, Groenewegen HJ, Lopes da Silva FH, Lohman AH. Functional organization of the extrinsic and intrinsic circuitry of the parahippocampal region. Prog Neurobiol. 1989;33(3):161–253. doi: 10.1016/0301-0082(89)90009-9. [DOI] [PubMed] [Google Scholar]
- 2.Ramon y Cajal S. Notes on the fine anatomy of the large brain. Z Wissensch Zool. 1893;56:615–672. [Google Scholar]
- 3.Van Hoesen GW, Pandya DN. Some connections of the entorhinal (area 28) and perirhinal (area 35) cortices of the rhesus monkey. III Efferent connections. Brain Res. 1975;95(1):39–59. doi: 10.1016/0006-8993(75)90206-1. [DOI] [PubMed] [Google Scholar]
- 4.Steward O, Scoville SA. Cells of origin of entorhinal cortical afferents to the hippocampus and fascia dentata of the rat. J Comp Neurol. 1976;169(3):347–370. doi: 10.1002/cne.901690306. [DOI] [PubMed] [Google Scholar]
- 5.Witter MP, Groenewegen HJ. Laminar origin and septotemporal distribution of entorhinal and perirhinal projections to the hippocampus in the cat. J Comp Neurol. 1984;224(3):371–385. doi: 10.1002/cne.902240305. [DOI] [PubMed] [Google Scholar]
- 6.Amaral DG, Witter MP. The three-dimensional organization of the hippocampal formation: a review of anatomical data. Neuroscience. 1989;31(3):571–591. doi: 10.1016/0306-4522(89)90424-7. [DOI] [PubMed] [Google Scholar]
- 7.van Groen T, Miettinen P, Kadish I. The entorhinal cortex of the mouse: organization of the projection to the hippocampal formation. Hippocampus. 2003;13(1):133–149. doi: 10.1002/hipo.10037. [DOI] [PubMed] [Google Scholar]
- 8.Pohle W, Ott T, Muller-Welde P. Identification of neurons of origin providing the dopaminergic innervation of the hippocampus. J Hirnforsch. 1984;25(1):1–10. [PubMed] [Google Scholar]
- 9.Dolorfo CL, Amaral DG. Entorhinal cortex of the rat: topographic organization of the cells of origin of the perforant path projection to the dentate gyrus. J Comp Neurol. 1998;398(1):25–48. [PubMed] [Google Scholar]
- 10.Ruth RE, Collier TJ, Routtenberg A. Topography between the entorhinal cortex and the dentate septotemporal axis in rats: I. Medial and intermediate entorhinal projecting cells. J Comp Neurol. 1982;209(1):69–78. doi: 10.1002/cne.902090107. [DOI] [PubMed] [Google Scholar]
- 11.Ruth RE, Collier TJ, Routtenberg A. Topographical relationship between the entorhinal cortex and the septotemporal axis of the dentate gyrus in rats: II. Cells projecting from lateral entorhinal subdivisions. J Comp Neurol. 1988;270(4):506–516. doi: 10.1002/cne.902700404. [DOI] [PubMed] [Google Scholar]
- 12.Hjorth-Simonsen A, Jeune B. Origin and termination of the hippocampal perforant path in the rat studied by silver impregnation. J Comp Neurol. 1972;144(2):215–232. doi: 10.1002/cne.901440206. [DOI] [PubMed] [Google Scholar]
- 13.Deller T, Haas CA, Frotscher M. Sprouting in the hippocampus after entorhinal cortex lesion is layer-specific but not translaminar: which molecules may be involved? Restor Neurol Neurosci. 2001;19(3–4):159–167. [PubMed] [Google Scholar]
- 14.von Gunten A, Kovari E, Bussiere T, et al. Cognitive impact of neuronal pathology in the entorhinal cortex and CA1 field in Alzheimer’s disease. Neurobiol Aging. 2006;27(2):270–277. doi: 10.1016/j.neurobiolaging.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 15.Kadish I, Van Groen T. Low levels of estrogen significantly diminish axonal sprouting after entorhinal cortex lesions in the mouse. J Neurosci. 2002;22(10):4095–4102. doi: 10.1523/JNEUROSCI.22-10-04095.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Deller T, Haas CA, Frotscher M. Reorganization of the rat fascia dentata after a unilateral entorhinal cortex lesion. Role of the extracellular matrix. Ann NY Acad Sci. 2000;911:207–220. doi: 10.1111/j.1749-6632.2000.tb06728.x. [DOI] [PubMed] [Google Scholar]
- 17.van Groen T. Entorhinal cortex of the mouse: cytoarchitectonical organization. Hippocampus. 2001;11(4):397–407. doi: 10.1002/hipo.1054. [DOI] [PubMed] [Google Scholar]
- 18.Lorente De Nó R. Studies on the structure of the cerebral cortex. II Continuation of the study of the ammonic system. J Psychol Neurol. 1934;46:113–177. [Google Scholar]
- 19.Blackstad TW. Commissural connections of the hippocampal region in the rat, with special reference to their mode of termination. J Comp Neurol. 1956;105(3):417–537. doi: 10.1002/cne.901050305. [DOI] [PubMed] [Google Scholar]
- 20.West MJ, Andersen AH. An allometric study of the area dentata in the rat and mouse. Brain Res. 1980;2(3):317–348. doi: 10.1016/0165-0173(80)90012-0. [DOI] [PubMed] [Google Scholar]
- 21.van Groen T, Kadish I, Wyss JM. Species differences in the projections from the entorhinal cortex to the hippocampus. Brain Res Bull. 2002;57(3–4):553–556. doi: 10.1016/s0361-9230(01)00683-9. [DOI] [PubMed] [Google Scholar]
- 22.Suzuki WA, Amaral DG. Topographic organization of the reciprocal connections between the monkey entorhinal cortex and the perirhinal and parahippocampal cortices. J Neurosci. 1994;14(3 Pt 2):1856–1877. doi: 10.1523/JNEUROSCI.14-03-01856.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Witter MP, Amaral DG. Entorhinal cortex of the monkey: V. Projections to the dentate gyrus, hippocampus, and subicular complex. J Comp Neurol. 1991;307(3):437–459. doi: 10.1002/cne.903070308. [DOI] [PubMed] [Google Scholar]
- 24.van Groen T, van Haren FJ, Witter MP, Groenewegen HJ. The organization of the reciprocal connections between the subiculum and the entorhinal cortex in the cat: I. A neuroanatomical tracing study. J Comp Neurol. 1986;250(4):485–497. doi: 10.1002/cne.902500407. [DOI] [PubMed] [Google Scholar]
- 25••.Phinney AL, Deller T, Stalder M, et al. Cerebral amyloid induces aberrant axonal sprouting and ectopic terminal formation in amyloid precursor protein transgenic mice. J Neurosci. 1999;19(19):8552–8559. doi: 10.1523/JNEUROSCI.19-19-08552.1999. First paper to demonstrate changes in connections in transgenic Alzheimer’s disease model mice. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Amaral DG, Insausti R, Cowan WM. The commissural connections of the monkey hippocampal formation. J Comp Neurol. 1984;224(3):307–336. doi: 10.1002/cne.902240302. [DOI] [PubMed] [Google Scholar]
- 27.Kondo H, Lavenex P, Amaral DG. Intrinsic connections of the macaque monkey hippocampal formation: II. CA3 connections. J Comp Neurol. 2009;515(3):349–377. doi: 10.1002/cne.22056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Swanson LW, Sawchenko PE, Cowan WM. Evidence for collateral projections by neurons in Ammon’s horn, the dentate gyrus, and the subiculum: a multiple retrograde labeling study in the rat. J Neurosci. 1981;1(5):548–559. doi: 10.1523/JNEUROSCI.01-05-00548.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Laurberg S. Commissural and intrinsic connections of the rat hippocampus. J Comp Neurol. 1979;184(4):685–708. doi: 10.1002/cne.901840405. [DOI] [PubMed] [Google Scholar]
- 30.Witter MP, Van Hoesen GW, Amaral DG. Topographical organization of the entorhinal projection to the dentate gyrus of the monkey. J Neurosci. 1989;9(1):216–228. doi: 10.1523/JNEUROSCI.09-01-00216.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kadish I, van Groen T. Lesion-induced hippocampal plasticity in transgenic Alzheimer’s disease mouse models: influences of age, genotype, and estrogen. J Alzheimers Dis. 2009;18(2):429–445. doi: 10.3233/JAD-2009-1157. [DOI] [PubMed] [Google Scholar]
- 32.de la Monte SM. Quantitation of cerebral atrophy in preclinical and end-stage Alzheimer’s disease. Ann Neurol. 1989;25(5):450–459. doi: 10.1002/ana.410250506. [DOI] [PubMed] [Google Scholar]
- 33.Laakso MP, Vaurio O, Savolainen L, et al. A volumetric MRI study of the hippocampus in type 1 and 2 alcoholism. Behav Brain Res. 2000;109(2):177–186. doi: 10.1016/s0166-4328(99)00172-2. [DOI] [PubMed] [Google Scholar]
- 34.Laakso MP, Frisoni GB, Kononen M, et al. Hippocampus and entorhinal cortex in frontotemporal dementia and Alzheimer’s disease: a morphometric MRI study. Biol Psychiatry. 2000;47(12):1056–1063. doi: 10.1016/s0006-3223(99)00306-6. [DOI] [PubMed] [Google Scholar]
- 35.Stranahan AM, Mattson MP. Selective vulnerability of neurons in layer II of the entorhinal cortex during aging and Alzheimer’s disease. Neural Plast. 2010;2010:108190. doi: 10.1155/2010/108190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kordower JH, Chu Y, Stebbins GT, et al. Loss and atrophy of layer II entorhinal cortex neurons in elderly people with mild cognitive impairment. Ann Neurol. 2001;49(2):202–213. [PubMed] [Google Scholar]
- 37.Gomez-Isla T, Price JL, Mckeel DW, Jr, Morris JC, Growdon JH, Hyman BT. Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer’s disease. J Neurosci. 1996;16(14):4491–4500. doi: 10.1523/JNEUROSCI.16-14-04491.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hyman BT, Van Hoesen GW, Damasio AR. Alzheimer’s disease: glutamate depletion in the hippocampal perforant pathway zone. Ann Neurol. 1987;22(1):37–40. doi: 10.1002/ana.410220110. [DOI] [PubMed] [Google Scholar]
- 39.Cabalka LM, Hyman BT, Goodlett CR, Ritchie TC, Van Hoesen GW. Alteration in the pattern of nerve terminal protein immunoreactivity in the perforant pathway in Alzheimer’s disease and in rats after entorhinal lesions. Neurobiol Aging. 1992;13(2):283–291. doi: 10.1016/0197-4580(92)90041-u. [DOI] [PubMed] [Google Scholar]
- 40.Mielke MM, Okonkwo OC, Oishi K, et al. Fornix integrity and hippocampal volume predict memory decline and progression to Alzheimer’s disease. Alzheimer’s Dement. 2012;8(2):105–113. doi: 10.1016/j.jalz.2011.05.2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Oishi K, Mielke MM, Albert M, Lyketsos CG, Mori S. The fornix sign: a potential sign for Alzheimer’s disease based on diffusion tensor imaging. J Neuroimaging. 2012;22(4):365–374. doi: 10.1111/j.1552-6569.2011.00633.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schmued LC, Raymick J, Paule MG, Dumas M, Sarkar S. Characterization of myelin pathology in the hippocampal complex of a transgenic mouse model of Alzheimer’s disease. Curr Alzheimers Res. 2013;10(1):30–37. doi: 10.2174/1567205011310010005. [DOI] [PubMed] [Google Scholar]
- 43.Yassa MA, Muftuler LT, Stark CE. Ultrahigh-resolution microstructural diffusion tensor imaging reveals perforant path degradation in aged humans in vivo. Proc Natl Acad Sci USA. 2010;107(28):12687–12691. doi: 10.1073/pnas.1002113107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hyman BT, Kromer LJ, Van Hoesen GW. Reinnervation of the hippocampal perforant pathway zone in Alzheimer’s disease. Ann Neurol. 1987;21(3):259–267. doi: 10.1002/ana.410210307. [DOI] [PubMed] [Google Scholar]
- 45•.Cotman CW, Anderson KJ. Synaptic plasticity and functional stabilization in the hippocampal formation: possible role in Alzheimer’s disease. Adv Neurol. 1988;47:313–335. First paper demonstrating very early pathology in layer II of the entorhinal cortex in Alzheimer’s disease. [PubMed] [Google Scholar]
- 46.Hyman BT, Van Hoesen GW, Kromer LJ, Damasio AR. Perforant pathway changes and the memory impairment of Alzheimer’s disease. Ann Neurol. 1986;20(4):472–481. doi: 10.1002/ana.410200406. [DOI] [PubMed] [Google Scholar]
- 47.Deller T, Del Turco D, Rappert A, Bechmann I. Structural reorganization of the dentate gyrus following entorhinal denervation: species differences between rat and mouse. Prog Brain Res. 2007;163:501–528. doi: 10.1016/S0079-6123(07)63027-1. [DOI] [PubMed] [Google Scholar]
- 48••.Van Groen T, Liu L, Ikonen S, Kadish I. Diffuse amyloid deposition, but not plaque number, is reduced in amyloid precursor protein/presenilin 1 double-transgenic mice by pathway lesions. Neuroscience. 2003;119(4):1185–1197. doi: 10.1016/s0306-4522(03)00215-x. Important review on the effects of plasticity on neural circuits in Alzheimer’s disease. [DOI] [PubMed] [Google Scholar]
- 49.Arendt T. Disturbance of neuronal plasticity is a critical pathogenetic event in Alzheimer’s disease. Int J Dev Neurosci. 2001;19(3):231–245. doi: 10.1016/s0736-5748(01)00007-7. [DOI] [PubMed] [Google Scholar]
- 50.Arendt T. Synaptic degeneration in Alzheimer’s disease. Acta Neuropathol. 2009;118(1):167–179. doi: 10.1007/s00401-009-0536-x. [DOI] [PubMed] [Google Scholar]
- 51••.Spires-Jones TL, Meyer-Luehmann M, Osetek JD, et al. Impaired spine stability underlies plaque-related spine loss in an Alzheimer’s disease mouse model. Am J Pathol. 2007;171(4):1304–1311. doi: 10.2353/ajpath.2007.070055. First important paper demonstrating the stages of pathology in the development of Alzheimer’s disease. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shankar GM, Li S, Mehta TH, et al. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med. 2008;14(8):837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Arendt T. Alzheimer’s disease as a disorder of mechanisms underlying structural brain self-organization. Neuroscience. 2001;102(4):723–765. doi: 10.1016/s0306-4522(00)00516-9. [DOI] [PubMed] [Google Scholar]
- 54.Holtmaat A, Svoboda K. Experience-dependent structural synaptic plasticity in the mammalian brain. Nat Rev Neurosci. 2009;10(9):647–658. doi: 10.1038/nrn2699. [DOI] [PubMed] [Google Scholar]
- 55.Swaab DF. Brain aging and Alzheimer’s disease, ‘wear and tear’ versus ‘use it or lose it’. Neurobiol Aging. 1991;12(4):317–324. doi: 10.1016/0197-4580(91)90008-8. [DOI] [PubMed] [Google Scholar]
- 56•.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82(4):239–259. doi: 10.1007/BF00308809. Good review on spreading of pathology in neurodegenerative diseases. [DOI] [PubMed] [Google Scholar]
- 57.Braak H, Braak E. Staging of Alzheimer’s disease-related neurofibrillary changes. Neurobiol Aging. 1995;16(3):271–278. doi: 10.1016/0197-4580(95)00021-6. discussion 278–284. [DOI] [PubMed] [Google Scholar]
- 58.Arendt T, Bruckner MK, Gertz HJ, Marcova L. Cortical distribution of neurofibrillary tangles in Alzheimer’s disease matches the pattern of neurons that retain their capacity of plastic remodelling in the adult brain. Neuroscience. 1998;83(4):991–1002. doi: 10.1016/s0306-4522(97)00509-5. [DOI] [PubMed] [Google Scholar]
- 59.Small SA, Schobel SA, Buxton RB, Witter MP, Barnes CA. A pathophysiological framework of hippocampal dysfunction in ageing and disease. Nat Rev Neurosci. 2011;12(10):585–601. doi: 10.1038/nrn3085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hardy J, Revesz T. The spread of neurodegenerative disease. N Engl J Med. 2012;366(22):2126–2128. doi: 10.1056/NEJMcibr1202401. [DOI] [PubMed] [Google Scholar]
- 61.Walker LC, Diamond MI, Duff KE, Hyman BT. Mechanisms of protein seeding in neurodegenerative diseases. JAMA Neurol. 2013;70(3):304–310. doi: 10.1001/jamaneurol.2013.1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wu JW, Herman M, Liu L, et al. Small misfolded tau species are internalized via bulk endocytosis and anterogradely and retrogradely transported in neurons. J Biol Chem. 2013;288(3):1856–1870. doi: 10.1074/jbc.M112.394528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Van Hoesen GW, Hyman BT. Hippocampal formation: anatomy and the patterns of pathology in Alzheimer’s disease. Prog Brain Res. 1990;83:445–457. doi: 10.1016/s0079-6123(08)61268-6. [DOI] [PubMed] [Google Scholar]
- 64.Matthews PM, Filippini N, Douaud G. Brain structural and functional connectivity and the progression of neuropathology in Alzheimer’s disease. J Alzheimers Dis. 2013;33(Suppl 1):S163–S172. doi: 10.3233/JAD-2012-129012. [DOI] [PubMed] [Google Scholar]
- 65.Agosta F, Pievani M, Geroldi C, Copetti M, Frisoni GB, Filippi M. Resting state fMRI in Alzheimer’s disease: beyond the default mode network. Neurobiol Aging. 2012;33(8):1564–1578. doi: 10.1016/j.neurobiolaging.2011.06.007. [DOI] [PubMed] [Google Scholar]
- 66.Filippi M, Agosta F. Structural and functional network connectivity breakdown in Alzheimer’s disease studied with magnetic resonance imaging techniques. J Alzheimers Dis. 2011;24(3):455–474. doi: 10.3233/JAD-2011-101854. [DOI] [PubMed] [Google Scholar]
- 67.Huang H, Fan X, Weiner M, et al. Distinctive disruption patterns of white matter tracts in Alzheimer’s disease with full diffusion tensor characterization. Neurobiol Aging. 2012;33(9):2029–2045. doi: 10.1016/j.neurobiolaging.2011.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fieremans E, Benitez A, Jensen JH, et al. Novel white matter tract integrity metrics sensitive to Alzheimer disease progression. AJNR Am J Neuroradiol. 2013;34(11):2105–2112. doi: 10.3174/ajnr.A3553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zerbi V, Kleinnijenhuis M, Fang X, et al. Gray and white matter degeneration revealed by diffusion in an Alzheimer mouse model. Neurobiol Aging. 2013;34(5):1440–1450. doi: 10.1016/j.neurobiolaging.2012.11.017. [DOI] [PubMed] [Google Scholar]
- 70.Johnson DK, Barrow W, Anderson R, et al. Diagnostic utility of cerebral white matter integrity in early Alzheimer’s disease. Int J Neurosci. 2010;120(8):544–550. doi: 10.3109/00207454.2010.494788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chao LL, Decarli C, Kriger S, et al. Associations between white matter hyperintensities and beta amyloid on integrity of projection, association, and limbic tensor MRI. PLoS ONE. 2013;8(6):e65175. doi: 10.1371/journal.pone.0065175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Clerx L, Visser PJ, Verhey F, Aalten P. New MRI markers for Alzheimer’s disease: a meta-analysis of diffusion tensor imaging and a comparison with medial temporal lobe measurements. J Alzheimers Dis. 2012;29(2):405–429. doi: 10.3233/JAD-2011-110797. [DOI] [PubMed] [Google Scholar]