

Abstract
TRH is a tripeptide amide that functions as a neurotransmitter but also serves as a neurohormone that has a critical role in the central regulation of the hypothalamic-pituitary-thyroid axis. Hypophysiotropic TRH neurons involved in this neuroendocrine process are located in the hypothalamic paraventricular nucleus and secrete TRH into the pericapillary space of the external zone of the median eminence for conveyance to anterior pituitary thyrotrophs. Under basal conditions, the activity of hypophysiotropic TRH neurons is regulated by the negative feedback effects of thyroid hormone to ensure stable, circulating, thyroid hormone concentrations, a mechanism that involves complex interactions between hypophysiotropic TRH neurons and the vascular system, cerebrospinal fluid, and specialized glial cells called tanycytes. Hypophysiotropic TRH neurons also integrate other humoral and neuronal inputs that can alter the setpoint for negative feedback regulation by thyroid hormone. This mechanism facilitates adaptation of the organism to changing environmental conditions, including the shortage of food and a cold environment. The thyroid axis is also affected by other adverse conditions such as infection, but the central mechanisms mediating suppression of hypophysiotropic TRH may be pathophysiological. In this review, we discuss current knowledge about the mechanisms that contribute to the regulation of hypophysiotropic TRH neurons under physiological and pathophysiological conditions.
Introduction
-
Organization of the Central Machinery Regulating the Hypothalamic-Pituitary-Thyroid Axis
Thyrotropin-releasing hormone (TRH) as central regulator of the HPT axis
Molecular characterization of the TRH gene
Processing of preproTRH
Inactivation of TRH
Anatomical characteristics of hypophysiotropic TRH neurons
Neuronal inputs of hypophysiotropic TRH neurons
Tanycytes as regulators of the HPT axis
Involvement of the autonomic nervous system in the regulation of the HPT axis
-
Negative Feedback Regulation of Hypophysiotropic TRH Neurons
Classical view of negative feedback regulation
Involvement of type 2 and type 3 deiodinases, thyroid hormone transporters, and pyroglutamyl-peptidase II in the negative feedback regulation of the hypophysiotropic TRH neurons
-
Central Regulation of the Hypothalamic-Pituitary-Thyroid Axis During Fasting
Role of the arcuato-paraventricular pathway in the regulation of the HPT axis during fasting
Direct action of leptin on hypophysiotropic TRH neurons
Involvement of tanycytes in the regulation of the HPT axis during fasting
Role of other neuronal pathways in the regulation of the HPT axis during fasting
Effects of Dehydration-Induced Anorexia on the Hypothalamic-Pituitary-Thyroid Axis
Regulation of the HPT Axis in High Fat Diet-Induced Obese Animals
-
Central Regulation of the HPT Axis During Infection and Prolonged Critical Illness
Role of neuronal pathways in the regulation of the hypophysiotropic TRH neurons during infection
Tanycytes as the key regulators of hypophysiotropic TRH neurons during infection
Regulation of hypophysiotropic TRH neurons during prolonged critical illness
Regulation of Hypophysiotropic TRH Neurons by Cold Exposure and Suckling
Translational Ramifications
Conclusions
I. Introduction
The hypothalamic-pituitary-thyroid (HPT) axis primarily functions to maintain normal, circulating levels of thyroid hormone that is essential for the biological function of all tissues, including brain development; regulation of cardiovascular, bone, and liver function; food intake; and energy expenditure among many others (1). Key to this regulatory system is a group of neurons that reside in the hypothalamic paraventricular nucleus (PVN), produce TRH, and integrate a wide variety of humoral and neuronal signals to regulate the HPT axis. In the present review, we will summarize current knowledge about the anatomy and physiology of these so called “hypophysiotropic” TRH neurons involved in the central regulation of the HPT axis under physiological and specific, pathophysiological conditions.
II. Organization of the Central Machinery Regulating the Hypothalamic-Pituitary-Thyroid Axis
A. Thyrotropin-releasing hormone (TRH) as central regulator of the HPT axis
TRH is a tripeptide amide (pGlu-His-ProNH2) (2) discovered simultaneously by the groups of Schally and Guillemin in 1969 (3, 4). In these pioneering studies, extracts from more than 250 000 porcine or sheep hypothalami containing only a few milligrams of TRH were shown to have TSH-releasing activity. The extracted material contained only three amino acids—glutamic acid, histidine, and proline (2)—and subsequently was shown to require cyclization of the glutamyl residue and amidation of the proline residue to achieve TSH-releasing activity (2).
TRH regulates the synthesis, release, and biological activity of TSH (5–7). This effect is mediated via the type 1 TRH receptor (8). At first, TRH stimulates the release of presynthesized TSH (6), and then it increases the synthesis of both TSH subunits, the α-glycoprotein hormone subunit, common to all three glycoprotein hormones of the anterior pituitary, and the TSH-specific β subunit (5). Binding of TRH to type 1 TRH receptor results in activation of phospholipase C, calcium mobilization, and activation of protein kinase C. This cascade leads to the synthesis of α-glycoprotein hormone subunit through effects on the pituitary LIM homeodomain factor, cAMP response element (CRE) binding protein (CREB), and CREB binding protein transcription factors (5). In contrast, the synthesis of the TSH-β subunit is mediated by the pituitary-specific transcription factor-1 and CREB binding protein transcription factors (5). TRH also has an important role in regulating the glycosylation of TSH by altering the oligosaccharide composition and structure of its three N-linked carbohydrate chains, important for the folding, assembly, secretion, metabolic clearance, and ultimately increasing the biological activity of TSH (6, 9–11). Indeed, TRH deficiency in both mouse models and man results in decreased TSH bioactivity and low peripheral thyroid hormone levels (12, 13).
B. Molecular characterization of the TRH gene
The first partial sequence of the preproTRH gene was cloned from frog skin by Richter et al (14), and then the nearly full-length cDNA of preproTRH was isolated from rat hypothalamic λgt11 library (15). A single copy of the preproTRH gene is present in the rat (chromosome 4), mouse (chromosome 6), and human (chromosome 3) genomes (16–19). The structure of the rat preproTRH gene is summarized in Figure 1. In all species, the gene contains three exons and two introns (16). In rats, the sizes of the introns are 750 and 450 bp, respectively (16). The first exon encodes the 5′ untranslated region of the mRNA, and the second exon encodes the signal peptide and a portion of the amino terminus of the proTRH peptide. The third exon encodes the remaining part of the amino-terminal peptide, five copies of the TRH sequence separated by non-TRH peptide sequences, and the carboxyl-terminal peptide followed by the 3′ untranslated region of the mRNA (16). The promoter of the gene contains a TATA box 28 bp upstream of the transcriptional initiation site (16). In addition, a series of regulatory elements has been identified in the proximal promoter of the gene. The human preproTRH gene contains three negative thyroid hormone response elements (TREs) (20). The so-called “site 4” is located −55 to −60 bp from the transcriptional initiation site. This thyroid hormone receptor (TR) binding half site binds both TR homodimers and TR-retinoid X receptor heterodimers (20–22). Two other functional TREs are present in the first exon between +14 to +19 and +37 to + 42, respectively (20). These sites can only bind TR monomers but are also necessary for the regulation of the gene by thyroid hormone (20). Site 4 of the human preproTRH gene is also thought to function as a CRE, resulting in competition between cAMP and TR for binding (23, 24). In the rat preproTRH gene, however, site 4 does not bind cAMP and functions exclusively as a TRE (21). A CRE-2 site identified 5′ to site 4 at −101 to −94 seems to function as the primary CRE in this gene (21). Glucocorticoid response element, activator protein-1, Krüppel-like factor 4, SP1, and signal transducers and activators of transcription-3 (STAT3) binding sites have also been identified in the proximal promoter of the preproTRH gene (21, 24, 25).
Figure 1.
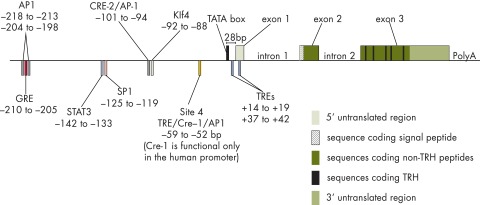
Schematic illustration of the organization of the rat preproTRH gene.
C. Processing of preproTRH
The 26-kDa rat preproTRH protein is composed of 255 amino acids (26). The N-terminal 25 amino acids comprise the signal peptide that directs the newly synthesized protein into the lumen of the rough endoplasmic reticulum after its synthesis on ribosomes (26). This sequence is cleaved during the passage into the rough endoplasmic reticulum (27), leaving a proTRH protein that contains five copies of TRH progenitor sequences, Gln-His-Pro-Gly, four non-TRH peptides located between the TRH progenitor sequences, and C-terminal and N-terminal flanking peptides (27). The proTRH protein of other species also contains multiple copies of TRH progenitor sequences (27), six copies in the human and seven copies in the frog (27). TRH progenitor peptides are flanked by paired basic amino acids, Lys-Arg or Arg-Arg, that serve as signals for endoprotease enzymes (27). The N-terminal flanking peptide is further cleaved at the pair of Arg amino acids located in the 51–52 position of the preproTRH molecule. Therefore, the processing of the rat proTRH results in five copies of TRH and seven non-TRH peptides (27). Because the non-TRH peptide located between the fourth and fifth TRH progenitors can be further cleaved and incomplete processing at Arg-Arg residues following the third and fourth TRH progenitor sequences can lead to C-terminally extended TRH peptides (27, 28), the actual number of peptides derived from proTRH processing can be even higher. In addition to mature TRH, other proTRH-derived peptides, such as proTRH 160–169 and proTRH 177–199, also have biological activity (27). Therefore, the large number of peptides derived from proTRH and evidence for differential processing of proTRH in different regions of the brain may serve to increase the diversity of how TRH neurons influence their targets.
The cleavage of proTRH at paired basic amino acid residues is primarily performed by two enzymes, prohormone convertase (PC) 1/3 and PC2 (27). However, both PC enzymes can cleave proTRH at multiple sites. In PC1/3 knockout (KO) mice, the concentration of the TRH tripeptide is reduced by almost 80%, but in PC2 KO mice it is reduced by only 44%. Therefore, PC1/3 may be more important in the processing of TRH (29). The lack of PC2 has more profound effects on the concentration of some of the non-TRH proTRH peptides (29). After cleavage, the basic amino acid residues are removed by carboxypeptidase E (27). The TRH progenitor, Gln-His-Pro-Gly, is then amidated by peptidylglycine α-amidating monooxygenase using the C-terminal glycine as amide donor (27). Finally, the N-terminal glycine is cyclized to pyroglutamate (27), catalyzed by N-glutaminyl cyclase (30), resulting in the mature form of TRH.
The processing of the proTRH protein takes place in the trans-Golgi network (TGN) and in the regulated secretory pathway (27). The first cleavage of proTRH occurs in the TGN (27) where PC1 processes the prohormone at the second or third TRH precursor, resulting in a 9.5- or 15-kDa N-terminal peptide and a 16.5- or 10-kDa C-terminal intermediate peptide (27). This initial cleavage is also critical for the targeting of proTRH-derived peptides into the regulated secretory pathway and the appropriate sorting of these peptides into secretory vesicles (31). After completion of the initial cleavage, the N- and C-terminal peptides of proTRH are sorted into different vesicles of the regulated secretory pathway (31). Prevention of the initial cleavage by mutation of the paired basic residues, however, directs proTRH protein into the constitutive secretory pathway (31). The C-terminal intermediate protein is further cleaved in the TGN at residues 201–202, but all other cleavage steps and the maturation of the TRH precursor take place in the immature and mature secretory vesicles (31).
D. Inactivation of TRH
Inactivation of secreted TRH in the brain is primarily catalyzed by a membrane-bound ectoenzyme, pyroglutamyl peptidase II (PPII) (32–34). PPII is a type II integral membrane protein comprised of a small, N-terminal, intracellular region and a large, extracellular domain containing the active site of the enzyme (32). PPII has stringent substrate specificity because it can only degrade peptides that are no longer than 4 amino acids with a pGlu-His-X structure, where X can be Pro, Ala, Trp, Pro-NH2, Pro-Gly, or Pro-β-NA (32). PPII produces the dipeptide His-ProNH2 from TRH, which is further degraded by dipeptidyl aminopeptidase IV, or spontaneously cyclizes to His-Pro diketopiperazine (32). PPII activity can be detected in most brain regions where the axons of TRH neurons terminate, but some mismatch is observed (32). PPII is primarily synthesized by neurons, but it is also produced by tanycytes, a specialized glial cell type, in the hypothalamus (32, 35). Inhibition of PPII activity markedly increases the amount of TRH released from brain tissue slices, supporting the importance of this peptidase in the metabolism of TRH (32).
In serum, TRH is degraded by a soluble enzyme that was formerly called thyroliberinase (35), but was subsequently shown to be a product of the PPII gene produced in the liver by proteolytic cleavage of membrane-bound PPII (36). Two broad-specificity cytosolic peptidases, pyroglutamyl peptidase I and prolyl endopeptidase, can also degrade TRH. However, because there is no evidence for the presence of these enzymes in the extracellular space and only a small proportion of the extracellular TRH is internalized, these enzymes do not play a major role in the inactivation of released TRH (32).
E. Anatomical characteristics of hypophysiotropic TRH neurons
TRH-synthesizing neurons are present in several brain regions, but only hypophysiotropic TRH neurons located in the PVN are involved in the central regulation of the HPT axis (37). This nucleus is a critical vegetative center of the hypothalamus and is located symmetrically at the upper third of the third ventricle.
The PVN contains a magnocellular and a parvocellular division. The magnocellular division houses oxytocin and vasopressin neurons that project to the posterior pituitary. The parvocellular division is further divided into anterior, periventricular, medial, ventral, dorsal, and lateral parvocellular subdivisions (38). In rats, TRH neurons are found in all parvocellular subdivisions (Figure 2, A–C) (39), but the hypophysiotropic TRH neurons are located only in the medial and periventricular subdivisions at the mid and caudal levels of the PVN (Figures 2, D–F, and 3, A–F) (40–42). In mice, hypophysiotropic TRH neurons are located only at the mid level of the PVN (Figure 3, G–L), intermingled with the magnocellular neurons (43, 44). The periventricular subdivision of the PVN does not contain TRH neurons, and the medial parvocellular subdivision at the caudal levels of the PVN houses nonhypophysiotropic TRH neurons (43, 44). In humans, the PVN also contains a large population of TRH neurons, especially in its medial part, but the location of hypophysiotropic TRH neurons is not yet known (45, 46).
Figure 2.

Distribution of TRH-synthesizing neurons in the rat PVN. A–C, Low-power micrographs illustrate the TRH neurons at three rostrocaudal levels of the PVN. D–F, Schematic drawings illustrate the subdivisions of the PVN where hypophysiotropic TRH neurons are localized (gray). AP, Anterior parvocellular subdivision; DP, dorsal parvocellular subdivision; LP, lateral parvocellular subdivision; MN, magnocellular part of PVN; MP, medial parvocellular subdivision, PV, periventricular parvocellular subdivision; VP, ventral parvocellular subdivision; III, third ventricle. [Reproduced from C. Fekete and R. M. Lechan: Negative feedback regulation of hypophysiotropic thyrotropin-releasing hormone (TRH) synthesizing neurons: role of neuronal afferents and type 2 deiodinase. Front Neuroendocrinol. 2007;28:97–114 (58), with permission. © Elsevier.]
Figure 3.

Darkfield photomicrographs showing proTRH mRNA expression in the anterior, mid, and posterior levels of the PVN in control (A–C) and hypothyroid (D–F) rats and in control (G–I) and hypothyroid (J–L) mice. Note the dramatic increase in silver grains denoting proTRH mRNA in the mid and caudal level of the hypothyroid rat PVN (E and F), whereas hypothyroidism increases proTRH mRNA only in midlevel neurons in mice (K). III, Third ventricle. [Panels G–L were reproduced from A. Kádár et al: Distribution of hypophysiotropic thyrotropin-releasing hormone (TRH)-synthesizing neurons in the hypothalamic paraventricular nucleus of the mouse. J Comp Neurol. 2010;518:3948–3961 (44), with permission. © Wiley-Liss Inc.]
Hypophysiotropic TRH neurons are functionally different from the nonhypophysiotropic TRH neurons in the PVN. Only hypophysiotropic TRH neurons project to the external zone of the median eminence (Figure 4), where their axon terminals release TRH into the extracellular space of this blood-brain barrier-free circumventricular organ (37). TRH is then conveyed to the anterior pituitary via the hypophysial portal circulation where TRH regulates the secretion of TSH from thyrotrophs and prolactin from lactotrophs (37, 47). In addition to TRH, hypophysiotropic neurons also express a second neuropeptide, cocaine and amphetamine-regulated transcript (CART) (42, 48). CART is simultaneously released into the hypophysial portal circulation and has been shown to inhibit the effect of TRH on prolactin secretion, but it has no effect on TRH-induced release of TSH (49). Hypophysiotropic TRH neurons also express the vesicular glutamate transporter 2, establishing the glutamatergic phenotype of these cells (50). Because TRH axon terminals in the median eminence contain a large number of small, clear vesicles (51), it is likely that glutamate is coreleased with TRH, but its physiological significance in the median eminence is currently unknown.
Figure 4.

Distribution of TRH-IR terminals in the mouse median eminence. TRH-IR axons densely innervate the external zone of the median eminence. III, Third ventricle; ME, median eminence.
In contrast to the hypophysiotropic TRH neurons, nonhypophysiotropic TRH-synthesizing neurons are widely distributed in the central nervous system (39). Currently, relatively little information is known about the projection fields and function of these neuronal groups, but some may be involved in the regulation of energy homeostasis. Nonhypophysiotropic TRH neurons in the anterior parvocellular subdivision of the PVN, for example, are densely innervated by inputs containing feeding-related peptides including axons containing agouti-related protein (AGRP), α-MSH, CART, galanin, and galanin-like peptide (42, 52, 53). In addition, these cells project to feeding-related nuclei such as the arcuate and dorsomedial nuclei and the amygdala (54). In the perifornical region, TRH is cosynthesized with another anorexigenic peptide, urocortin 3 (55). These neurons have a prominent projection field to the hypothalamic ventromedial nucleus (54, 55), known to be involved in the regulation of food intake (56). TRH neurons in the preoptic area influence energy homeostasis by regulating thermogenesis (57).
F. Neuronal inputs of hypophysiotropic TRH neurons
Hypophysiotropic TRH neurons are embedded in a dense network of neuronal axons in the PVN. These axons form numerous synaptic associations on the surface of TRH neurons and modulate the activity of these cells (58). Integration of these inputs together with humoral signals that can reach TRH neurons through the rich vascular supply of the median eminence or the PVN (59, 60) ensures fine tuning of the activity of the HPT axis and its adaptation to changing environmental conditions. Currently, three main neuronal groups are known to send synaptic inputs to the hypophysiotropic TRH neurons: the hypothalamic arcuate nucleus, the hypothalamic dorsomedial nucleus (DMN), and catecholamine-producing neurons in the brainstem (Figures 5 and 6) (58, 61).
Figure 5.
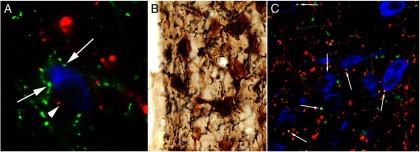
Innervation of the TRH neurons in the rat PVN by axons originating from the arcuate nucleus (A), DMN (B), and catecholaminergic neurons (C) in the brainstem. A, TRH neurons (blue) are contacted by axon terminals containing α-MSH (red; arrowhead) and AGRP (green; arrows). B, Axon varicosities containing the anterogradely transported marker protein, PHA-L (black) are juxtaposed to TRH-synthesizing neurons (brown) after iontophoretic administration of the tracer into the DMN. C, Both noradrenergic (red; open arrows) and adrenergic (yellow, white arrows) axons establish contacts with the TRH neurons. [Modified from C. Fekete et al: α-Melanocyte-stimulating hormone is contained in nerve terminals innervating thyrotropin-releasing hormone-synthesizing neurons in the hypothalamic paraventricular nucleus and prevents fasting-induced suppression of prothyrotropin-releasing hormone gene expression. J Neurosci. 2000;20:1550–1558 (52), with permission. © Society for Neuroscience. From E. Mihály et al: Hypothalamic dorsomedial nucleus neurons innervate thyrotropin-releasing hormone-synthesizing neurons in the paraventricular nucleus. Brain Res. 2001;891:20–31 (74), with permission. © Elsevier. And from T. Füzesi et al: Noradrenergic innervation of hypophysiotropic thyrotropin-releasing hormone-synthesizing neurons in rats. Brain Res. 2009;1294:38–44 (61), with permission. © Elsevier.]
Figure 6.
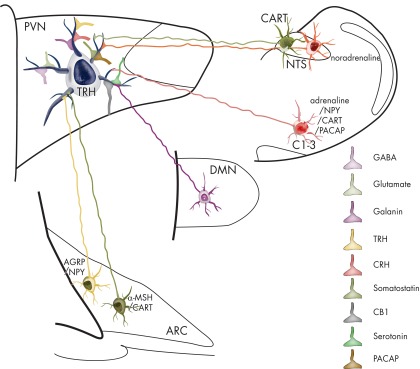
Schematic drawing summarizing known inputs to TRH neurons in the PVN. The inputs with identified origins are depicted by a neuron sending its axon to the TRH neuron. Inputs with currently unknown origins are labeled with axon terminals on the surface of the TRH neuron. PACAP, Pituitary adenylate cyclase-activating polypeptide.
The hypothalamic arcuate nucleus plays a key role in the regulation of energy homeostasis by relaying humoral signals to second-order neuronal groups in the brain (56). Two major feeding-related neuronal populations are involved: the medially located, orexigenic neurons that synthesize neuropeptide Y (NPY), AGRP, and γ-amino butyric acid (GABA); and laterally located anorexigenic neurons that produce α-MSH and CART (56). Both neuronal groups are responsive to peripheral, feeding-related signals such as changes in leptin, ghrelin, insulin, and glucose (56), but are oppositely regulated (56).
Numerous AGRP- and NPY-immunoreactive (IR) axon varicosities form juxtaposition with virtually all TRH neurons in the PVN (Figure 5A) and establish a symmetric type of synaptic association with these cells, indicative of an inhibitory function (52, 62, 63). Because the neurons that synthesize AGRP are found only in the arcuate nucleus, the innervation of the TRH neurons by AGRP-IR axons must originate exclusively from this nucleus (63). NPY, however, is synthesized by a number of neuronal populations, but the two major sources for the NPY-IR innervation of the PVN are the arcuate nucleus and catecholaminergic neurons in the brainstem (64). The arcuate nucleus neurons provide approximately 75% of the NPY innervation to TRH neurons in the PVN (64, 65), whereas the adrenergic NPY neurons of the brainstem contribute the remaining 25% (64).
Anorexigenic α-MSH/CART neurons of the arcuate nucleus also innervate TRH neurons in the PVN (Figure 5A) (58) but establish fewer synapses than observed for axons containing AGRP/NPY (52). α-MSH is synthesized in two brain regions, the hypothalamic arcuate nucleus and the nucleus tractus solitarius (NTS) of the brainstem. Only α-MSH-containing neurons in the arcuate nucleus coexpress CART (42), and therefore, the colocalization of these two peptides can be used as a marker for α-MSH axons originating from the arcuate nucleus. Because all α-MSH-IR axon varicosities on the surface of the TRH neurons contain CART, the arcuate nucleus is the exclusive source of the α-MSH-IR innervation to TRH neurons in the PVN (42). Not all CART-IR axons in juxtaposition to the TRH neurons contain α-MSH, however, indicating that the arcuate nucleus is not the only source for the CART-IR innervation of these cells (42). Indeed, only a relatively small portion of the CART-IR varicosities on the surface of TRH neurons derive from the arcuate nucleus. CART-synthesizing neurons that innervate the PVN can also be found in the lateral hypothalamus, perifornical area, zona incerta, C1–3 adrenergic neuronal groups, and the medial subnucleus of the NTS (66). Because unilateral transection of the ascending brainstem pathways to the PVN results in an approximately 60% reduction of the CART innervation of hypophysiotropic TRH neurons, it would appear that the brainstem gives rise to most of the CART input to these cells (67).
TRH neurons in the PVN of the human hypothalamus also receive inputs from the infundibular nucleus, the analog of the rodent arcuate nucleus (46). Similar to the rodent, the human infundibular nucleus contains separate populations of AGRP/NPY and α-MSH-synthesizing neurons (46). In addition, TRH neurons in the human PVN are densely innervated by axons containing these peptides (46), suggesting that the arcuato (infundibulo)-paraventricular pathway is evolutionarily conserved, and thereby of importance in the regulation of hypophysiotropic TRH neurons (46). There is a major difference between the human and the rodent pathways, however. Although CART and α-MSH are coexpressed by the arcuate nucleus neurons in the rodent, CART is not present in α-MSH neurons of the human infundibulum (68). Moreover, CART can be detected in approximately 30% of AGRP/NPY neurons (68), indicating that CART may have a somewhat different role in the regulation of the thyroid axis in humans.
The DMN also plays an important role in the regulation of energy homeostasis and vegetative functions (69). Like the feeding-related neurons of the arcuate nucleus, DMN neurons sense circulating energy homeostasis-related hormones such as leptin (70), but these signals also influence the DMN indirectly via the arcuate nucleus (71). In addition, the DMN is a critical node in the circuit regulating food-entrainable circadian rhythms and the stress response (69, 72), integrating these signals and relaying the information to other neuronal populations such as the sympathetic nervous system and the PVN (69). The DMN also contributes to the regulation of the HPT axis because bilateral destruction of the DMN increases 24-hour release of T3 (73), suggesting a net inhibitory effect. Indeed, anterograde tract-tracing studies have demonstrated (Figure 5B) that the vast majority of the TRH neurons in the PVN receive input from the DMN, primarily establishing symmetric type synaptic associations with the TRH neurons characteristic of an inhibitory function (74). Little is known about how the DMN regulates hypophysiotropic TRH neurons, however, but it has been hypothesized that it may be involved in circadian regulation of the hypophysiotropic TRH neurons (58).
Brainstem catecholaminergic cell groups are involved in the regulation of a wide variety of physiological functions including attention, sleep/wakefulness, learning, memory, emotion, reproduction, neuroendocrine processes, and central responses to stress (75). These neurons can be subdivided into adrenergic and noradrenergic subtypes based on their transmitter content. Both neuronal populations produce dopamine-β hydroxylase, the noradrenaline-synthesizing enzyme, but only adrenergic neurons express phenylethanolamine N-methyltransferase, the enzyme that converts noradrenaline to adrenaline (76).
Hypophysiotropic TRH neurons receive a dense catecholaminergic innervation (77), perhaps comprising the largest input to these neurons. Catecholaminergic axons establish asymmetric-type synaptic specializations on the surface of TRH neurons (77), indicative of an activating effect and consistent with the observation that noradrenaline stimulates the transcription of TRH gene (78). It has been shown by triple-labeling immunofluorescence (Figure 5C) that approximately two-thirds of the catecholaminergic innervation of TRH neurons originate from adrenergic neurons, whereas the noradrenergic neuronal groups give rise to the remaining one-third (61). Adrenergic neurons are located exclusively in the C1–3 regions of the medulla, and because the axons of all three adrenergic regions have highly similar distribution patterns in the PVN (79), it is likely that each adrenergic group contributes to the innervation of hypophysiotropic TRH neurons (61).
In addition to the classical transmitters, subpopulations of the adrenergic neurons that innervate the TRH neurons in the PVN also synthesize peptidergic transmitters. Approximately 50% of the adrenergic innervation to TRH neurons cocontain CART (80), and more than 70% cocontain NPY (64). A large proportion of these adrenergic terminals contain pituitary adenylate cyclase-activating polypeptide (81). Currently, it is unknown how the corelease of multiple transmitters modulates the effect of adrenergic neurons on their targets, but it is likely that the capacity to release a large array of transmitters from the same terminals provides substantial flexibility for the ability of adrenergic neurons to differentially respond to diverse physiological and pathophysiological conditions.
The brainstem has six noradrenergic cells groups (A1–A6), but only the A1, A2, and A6 noradrenergic cell groups project to the PVN (82). The A1 noradrenergic cell group innervates primarily the magnocellular part of the PVN in rats (83), suggesting that this cell group may be only a minor source of the noradrenergic innervation to TRH neurons. The A2 and A6 noradrenergic cell groups, however, densely innervate the periventricular and medial parvocellular subdivisions of the PVN where the hypophysiotropic TRH neurons reside (83), making it likely that these two noradrenergic cell populations are the primary sources of the innervation of hypophysiotropic TRH neurons.
In addition to the above-mentioned inputs, the TRH neurons in the PVN receive galanin-, TRH-, CRH-, somatostatin-, and endocannabinoid receptor-containing inputs (50, 53, 81, 84–87). Nonsynaptic contacts between serotoninergic axons and the TRH neurons have also been described (88). However, very little is known about the involvement of these inputs in the regulation of hypophysiotropic TRH neurons. The known inputs of the hypophysiotropic TRH neurons are summarized in Figure 6.
G. Tanycytes as regulators of the HPT axis
Tanycytes are specialized glial cells lining the ventrolateral walls and the floor of the third ventricle between the rostral and caudal limits of the median eminence (Figure 7, A and B) (89). Characteristic of these cells are a small cell body located in the ependymal layer and a long, basal process that projects either into the median eminence or the arcuate, ventromedial, or dorsomedial nuclei (90). Based on their location, morphology, cytochemistry, and ultrastructure, tanycytes can be classified into four subtypes: α1 and α2 tanycytes that line the ventrolateral walls of the third ventricle, and β1 and β2 tanycytes that line the floor and lateral extensions of the third ventricle (Figure 7B) (89, 90). α1 Tanycytes are located approximately in the middle third of the ventricular wall and project into the ventromedial and dorsomedial nuclei where their end-feet processes terminate on neurons (89, 90). α2 Tanycytes are located ventral to α1 tanycytes and project their processes into the arcuate nucleus, terminating on neurons and around capillaries. The most ventrally located α2 tanycytes, however, send their processes to the most lateral portion of the tuberoinfundibular sulcus (89, 90).
Figure 7.
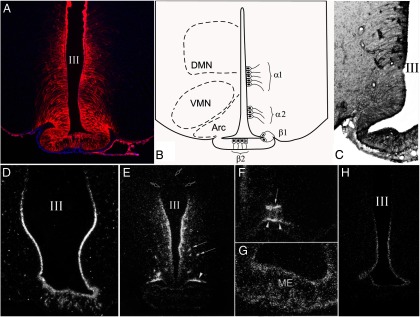
A and B, Organization of tanycyte subtypes in the MBH. A, Vimentin-immunolabeled (red) coronal section with DAPI counterstaining (blue) shows the distribution of tanycytes and their processes. B, The schematic diagram illustrates the location of tanycyte subtypes in the wall and floor of the third ventricle. C and D, All tanycyte subtypes synthesize MCT8 (C) and OATP1C1 (D) thyroid hormone transporters. E, Silver grains denoting D2 mRNA are accumulated over the cells lining the wall of the third ventricle, the tuberoinfundibular sulci (arrowheads), and around blood vessels in the arcuate nucleus (arrows). F and G, Higher power micrographs show the association of D2 mRNA with the tuberoinfundibular sulcus (arrowheads) and a blood vessel (arrows) in the arcuate nucleus (F), and in the external zone of the median eminence (G). H, Tanycyte expression of PPII mRNA. III, Third ventricle; Arc, arcuate nucleus; ME, median eminence; VMN, ventromedial nucleus. [Modified from E. Sánchez et al: Tanycyte pyroglutamyl peptidase II contributes to regulation of the hypothalamic-pituitary-thyroid axis through glial-axonal associations in the median eminence. Endocrinology. 2009;150:2283–2291 (35), with permission. © The Endocrine Society. From Kalló et al: A novel pathway regulates thyroid hormone availability in rat and human hypothalamic neurosecretory neurons. PLoS One. 2012;7:e37860 (51), with permission. © Public Library of Science. And from C. Fekete et al: DARPP-32 and CREB are present in type 2 iodothyronine deiodinase-producing tanycytes: implications for the regulation of type 2 deiodinase activity. Brain Res. 2000;862:154–161 (124), with permission. © Elsevier. Courtesy of Dr Gábor Wittmann.]
β1 Tanycytes line the lateral invaginations of the infundibular recess, and their processes arch toward the tuberoinfundibular sulcus and terminate on the surface of the pars tuberalis of the pituitary (89, 90). The β2 tanycytes line the floor of the infundibular recess, and their processes travel through the median eminence to terminate around the portal capillaries in the external zone of the median eminence (89, 90).
In addition to their distinct anatomical locations, the various tanycyte subtypes also differ in their chemical signatures, suggesting that they have independent functions. α and β1 Tanycytes express the glucose transporter-1, a blood-brain barrier marker (91). This observation is in keeping with the morphological observations that α tanycytes surround capillaries in the arcuate and ventromedial nuclei, and β1 tanycytes create the barrier that separates median eminence from the mediobasal hypothalamus (MBH) (91). However, α but not β tanycytes express somatostatin sst2a receptors, whereas β1 tanycytes are reactive for N-cadherin, while only α1 tanycytes express adenosine triphosphatase enzyme (90). In addition, β2 tanycytes lack the glucose transporter-1 but express Rab 4, a protein involved in vesicular transport (90, 91).
At the ultrastructural level, a common characteristic of tanycytes is the presence of both early and late endosomes near the apical surface of the cell bodies, suggesting that all types of tanycytes actively incorporate substances from the cerebrospinal fluid (CSF) (90). However, because tight junctions are absent between α and β1 tanycytes (90), substances from the CSF can also enter the neuropil directly through the ventrolateral portions of the third ventricular wall. In contrast, β2 tanycytes are bound together by both zonula adherens and tight junctions, forming an impermeable barrier between the CSF and the median eminence (90).
In addition to functioning as barrier cells, it is becoming increasingly clear that tanycytes are involved in neuroendocrine regulation (90). The role of tanycytes in the regulation of the hypothalamic-pituitary-gonadal axis has been long known (90). Changes in estrogen levels induce cytoskeletal remodeling of tanycytes, resulting in retraction of tanycyte end processes from the capillaries during the GnRH surge, allowing GnRH axons to secrete their products into the portal circulation (92). The role of tanycytes in the regulation of the HPT axis has been more recently recognized (89). Tanycytes express TRs (Figure 7, C and D), and changes in circulating thyroid hormone levels result in plastic remodeling of tanycyte end-feet processes (28, 93), perhaps also to regulate the entry of TRH released by the hypophysiotropic terminals into the portal circulation (93). In addition, the tanycytes express the TRH degrading enzyme, PPII (Figure 7H) (35), which is regulated in parallel to circulating thyroid hormone levels (35). Thus, hyperthyroidism results in up-regulation of tanycyte PPII, contributing to inhibition of TSH secretion by reducing the amount of TRH reaching the portal system. In support of this hypothesis, inhibition of PPII in the median eminence of hyperthyroid animals significantly increases the amount of secreted TRH (35).
Tanycytes are also thought to be involved in feedback regulation of the HPT axis through their expression of type 2 iodothyronine deiodinase (D2; Figure 7, E–G), an enzyme that catalyzes 5′ deiodination of T4 resulting in the generation of the active form of thyroid hormone, T3 (94). In most regions of the brain, D2 is expressed by astrocytes, but in the hypothalamus, D2 is primarily expressed by tanycytes (95–97). Most regions of the hypothalamus including the PVN are devoid of D2 activity (98).
Tanycyte D2 activity is precisely regulated at transcriptional and posttranslational levels (58, 99). Tanycytes synthesize ubiquitin ligase and deubiquitinase enzymes, WSB1 and USP33, respectively, that can quickly and reversibly regulate the activity of D2 (100). In addition, tanycytes are richly replete in the thyroid hormone transporters, MCT8 and OATP1C1 (Figure 7, C and D) (51, 101, 102), that facilitate the entry of T4 from the circulation or CSF and the release of the generated T3 into the neuropil or CSF. Under special conditions, tanycytes also express the thyroid hormone degrading enzyme, type 3 iodothyronine deiodinase enzyme (D3) (103). Precise transcriptional and posttranslational regulation of D2 and D3 in tanycytes provides a powerful mechanism to tightly control hypothalamic T3 availability that may contribute to regulation of the HPT axis.
H. Involvement of the autonomic nervous system in the regulation of the HPT axis
In addition to the stimulation of TSH secretion of the anterior pituitary by TRH, the central nervous system can also regulate thyroid function via the autonomic nervous system. The thyroid gland is innervated by both adrenergic nerve fibers of the sympathetic nervous system and the cholinergic axons originating from the vagus nerve (104, 105). Both sympathetic and parasympathetic nerves densely innervate the blood vessels of the thyroid gland, but axon terminals of these autonomic systems can also be found around the thyroid follicles (104, 105), indicating that not only the blood flow, but also the activity of thyroid follicles could be under direct control of autonomic inputs. Retrograde, virus-mediated tract-tracing studies has verified the existence of both the sympathetic and parasympathetic innervation of the thyroid gland, showing that 2 days after the injection of pseudorabies virus directly into the thyroid gland, sympathetic preautonomic neurons in the intermediolateral column of the spinal cord and the parasympathetic preautonomic neurons in the dorsal vagal complex were retrogradely labeled (106).
Relatively little data are available about how the autonomic inputs to the thyroid gland regulate thyroid function. However, the sympathetic input seems to have an inhibitory action because electrical stimulation of the cervical sympathetic trunk decreases thyroid blood flow (107). Noradrenaline also inhibits the stimulatory effect of TSH on the thyroid cells in vitro (108) and decreases thyroid hormone secretion in vivo (109).
In contrast, electric stimulation of the thyroid nerve, which carries parasympathetic inputs to the thyroid gland, results in increased thyroid blood flow that can be prevented by atropine pretreatment (107). In addition, transection of inferior laryngeal nerve that also carries parasympathetic input to the thyroid gland results in a fall of circulating T4 levels, supporting the stimulatory effect of the parasympathetic inputs on the activity of the thyroid gland (110).
In addition to the classical transmitters, the neuropeptides, NPY and vasoactive intestinal peptide are also present in axons innervating the thyroid gland (107). NPY is present in the sympathetic innervation of the thyroid gland and, similar to norepinephrine, inhibits thyroidal blood flow (111). In contrast, vasoactive intestinal peptide increases the thyroid blood flow and thyroid hormone secretion (107).
In addition to the primary preautonomic neuronal groups, multisynaptic connection of the suprachiasmatic nucleus and energy homeostasis-related neuronal groups of the hypothalamus including the PVN and the arcuate nucleus with the thyroid gland have also been demonstrated (106), suggesting that these hypothalamic cell groups may also be involved in the autonomic regulation of thyroid gland. Because central melanocortin and NPY signaling contributes to the metabolism of thyroid hormone in the liver by regulating sulfotransferases (112), it is conceivable that the autonomic nervous system is also involved in regulating the peripheral metabolism of thyroid hormones in addition to their synthesis, although the mechanism remains uncertain.
III. Negative Feedback Regulation of Hypophysiotropic TRH Neurons
A. Classical view of negative feedback regulation
Negative feedback regulation of hypophysiotropic TRH neurons is an important regulatory mechanism to ensure stability of circulating thyroid hormone levels (113). When circulating thyroid hormone levels are increased, TRH gene expression is decreased in hypophysiotropic neurons, whereas the converse is true in association with hypothyroidism (Figure 8) (58). Regulation of TRH transcription by thyroid hormone is relatively rapid because the exogenous administration of thyroid hormone can suppress transcription of the TRH gene in the PVN within 5 hours (114). This regulatory mechanism is a unique feature of hypophysiotropic TRH neurons because thyroid hormone does not regulate TRH gene expression in nonhypophysiotropic TRH neurons (113). Thyroid hormone is sensed directly by the hypophysiotropic TRH neurons because implantation of crystalline T3 immediately adjacent to the PVN in hypothyroid animals results in marked inhibition of TRH mRNA on that side but has no effect on TRH neurons on the contralateral side (115).
Figure 8.

In situ hybridization autoradiograms showing the effect of hypo- and hyperthyroidism on proTRH mRNA level in the medial parvocellular subdivision of the PVN. A substantial increase in silver grain accumulation is observed in the hypothyroid animal (A) compared to the fed control (B). In contrast, hyperthyroidism results in a marked reduction of proTRH mRNA level in the PVN (C). [Modified from E. M. Dyess et al: Triiodothyronine exerts direct cell-specific regulation of thyrotropin-releasing hormone gene expression in the hypothalamic paraventricular nucleus. Endocrinology. 1988;123:2291–2297 (115), with permission. © The Endocrine Society. And from C. Fekete and R. M. Lechan: Negative feedback regulation of hypophysiotropic thyrotropin-releasing hormone (TRH) synthesizing neurons: role of neuronal afferents and type 2 deiodinase. Front Neuroendocrinol. 2007;28:97–114 (58), with permission. © Elsevier.]
Hypophysiotropic TRH neurons express the TRα1, TRβ1, and TRβ2 isoforms of the TRs, although the TRβ1 isoform is present in relatively low abundance (116). The presence of all three TR isoforms in the same cell type may seem to be redundant. However, all isoforms have different roles in the regulation TRH gene expression in the hypophysiotropic neurons. In TRβ KO mice, in which TRα1 is the only functional TR, T3 treatment results in a significant increase of TRH gene expression in the PVN, whereas the lack of TRα1 in TRα KO mice enhances the T3-induced decrease in TRH mRNA in the PVN (117). These data suggest that T3 positively regulates the TRH gene via TRα1. This view is also supported by the stimulatory effect of T3 on the TRH gene expression in cell cultures of developing hypothalamic cells at the 12 division stage when the TRα1 is the predominant TR isoform (118). This TRα1-mediated stimulatory effect of T3 can be overridden by the TRβ-mediated inhibitory effect of T3 in wild-type mice and also in later stages of cultured hypothalamic cells (117, 118). Because a positive TRE has not been identified in the promoter of TRH gene, it is not clear whether the TRα1-mediated stimulatory effect exerted directly on the TRH promoter or through indirect effects (118). In contrast to the TRα KO mice, the T3-induced inhibition of TRH gene expression is completely absent in the PVN of both TRβ and TRβ2 KO mice (117, 119). Therefore, it has been suggested that negative feedback regulation of the TRH gene is mediated exclusively by the TRβ2 isoform (119). This view was challenged by Guissouma et al (120) using small interfering RNA (siRNA)-mediated knockdown of the different TRβ isoforms in the hypothalamus of mouse pups. The results of these experiments revealed that siRNA-mediated knockdown of either TRβ2 or TRβ1 prevents the T3-dependent inhibition of the activity of a TRH-luciferase construct transfected into the hypothalamus, suggesting that both TRβ1 and TRβ2 contribute to negative feedback regulation of the TRH gene. It is not clear, however, how the two TRβ isoforms interact in this regulatory process.
The two TRβ isoforms, however, unequivocally play different roles in ligand-independent stimulation of the TRH gene (117, 120). Although the TRH mRNA level is markedly increased in the PVN of TRβ2 KO mice independent of thyroid status (119), TRH expression is significantly decreased in both hypo- and euthyroid TRβ KO mice (117). These data suggest that the TRβ1 isoform is critical for ligand-independent stimulation of the TRH promoter and is supported by evidence that siRNA- mediated knockdown of the TRβ1 isoform significantly decreases activity of the TRH promoter in the mouse hypothalamus (120).
In addition to the regulation of TRH at the transcriptional level, thyroid hormone also influences posttranslational processing of proTRH. Increased levels of circulating thyroid hormone lead to the PVN selective down-regulation of two major proTRH-processing enzymes, PC1/3 and PC2 resulting in the accumulation of intermediate processing products of proTRH (121). Thyroid hormone may also affect expression of neuropeptide receptors in hypophysiotropic TRH neurons, such as the melanocortin 4 receptor (122), influencing the sensitivity of TRH neurons to their excitatory and inhibitory inputs.
B. Involvement of type 2 and type 3 deiodinases, thyroid hormone transporters, and pyroglutamyl-peptidase II in the negative feedback regulation of the hypophysiotropic TRH neurons
1. Role of deiodinases
The concept that the circulating level of T3 is solely responsible for negative feedback regulation of hypophysiotropic TRH by acting directly on these neurons was challenged by Kakucska et al (123), showing that restoration of circulating levels of T3 to normal levels in hypothyroid rats without the administration of T4 does not normalize TRH gene expression in the PVN (Figure 9). Only if very high hyperthyroid levels of T3 were achieved in the circulating blood was it possible to decrease TRH mRNA levels in the PVN into the normal, euthyroid range (123). These data indicate that in addition to T3, circulating T4 is also necessary for appropriate feedback control of hypophysiotropic TRH neurons. However, because T4 functions primarily as a prohormone, its conversion to T3 within the central nervous system must be an essential part of the feedback regulatory mechanism.
Figure 9.

In situ hybridization autoradiographs of proTRH mRNA in the PVN of hypothyroid (A), euthyroid (B), and hypothyroid (C and D) animals receiving a constant infusion of 0.5 μg (C) or 0.75 μg (D) of T3 per 100 g body weight per day. Mean plasma T3 levels (±SEM) are shown for each group at the bottom of the photomicrographs. Note that only the higher dose of T3 that raised plasma T3 levels into the supranormal range was capable of suppressing proTRH mRNA to euthyroid levels. E, Regression analysis of the above experiment. Interrupted line represents the mean ln(proTRH mRNA) for euthyroid animals, and its intercept with the regression line estimates the plasma T3 concentration required to suppress proTRH mRNA to euthyroid levels. Ninety-five percent confidence intervals for each intercept are bracketed. Open dots denote values for hypothyroid animals and hypothyroid animals infused with graded doses of T3. Closed dots denote values for euthyroid controls. [Modified from I Kakucska et al: Thyrotropin-releasing hormone gene expression in the hypothalamic paraventricular nucleus is dependent upon feedback regulation by both triiodothyronine and thyroxine. Endocrinology. 1992;130:2845–2850 (123), with permission. © The Endocrine Society. And from C. Fekete and R. M. Lechan: Negative feedback regulation of hypophysiotropic thyrotropin-releasing hormone (TRH) synthesizing neurons: role of neuronal afferents and type 2 deiodinase. Front Neuroendocrinol. 2007;28:97–114 (58), with permission. © Elsevier.]
As noted previously, the conversion of T4 to T3 in the brain is catalyzed by D2 (94), and in the hypothalamus, this enzyme is expressed primarily in tanycytes (95–98, 124). Although D2 is also present in less abundance in astrocytes in the median eminence and arcuate nucleus region (95), selective ablation of D2 from astrocytes in transgenic mice has no significant effect on feedback regulation of the hypophysiotropic TRH neurons (125), indicating that astrocytes have little or no role in the regulation of this response. Presumably, therefore, tanycyte D2 is responsible for generating the additional T3 required to maintain normal homeostasis in the thyroid axis, although this has not yet been directly tested experimentally. Under certain conditions, the thyroid hormone-inactivating enzyme, D3, is also present in tanycytes (126, 127). It is unknown, however, whether this enzyme contributes to the regulation of hypothalamic T3 concentrations.
In most brain regions, the primary role of D2 is to maintain local T3 concentrations if circulating levels of T4 and T3 decline (128). In the cortex, for example, hypothyroidism up-regulates D2 activity to produce more T3, whereas hyperthyroidism down-regulates D2 activity (94). Therefore, the local T3 concentration in the cortex is unchanged even if the circulating T4 concentrations vary over a relatively wide range (128). D2 is also regulated by thyroid hormone in tanycytes at the transcriptional level (97, 129); however, the increased gene expression is not accompanied by a rise of D2 activity in these cells (130). Although hypothyroidism results in a more than 4-fold increase of D2 activity in the cortex (131), it has no effect on D2 activity in the MBH (130). Similarly, no increase in D2 activity has been observed in the MBH in association with iodine deficiency, contrary to other regions in the brain (132). The posttranscriptional attenuation of thyroid hormone-induced regulation of D2 activity in the MBH suggests that the main role of D2 in this region is not to maintain a constant, local T3 concentration but rather to allow the hypothalamus to sense changes in peripheral thyroid hormone levels using T3 as a regulatory signal. This feature is important because stable hypothalamic T3 concentrations would reduce the sensitivity of the feedback regulation mechanism of TRH neurons in the PVN.
Despite all the above-mentioned data supporting the role of D2 expression of tanycytes in the feedback regulation of the hypophysiotropic TRH neurons, D2 KO mice are euthyroid, and their HPT axis has seemingly intact negative feedback regulation (133). These data could question the role of D2 in the feedback regulation of the HPT axis. However, it is important to note that during the embryonic period, thyroid hormone levels have a major influence on the development of the setpoint for feedback regulation of thyroid hormone on the HPT axis (134). In D2 KO animals, D2 activity is already absent from the tanycytes during the embryonic period. Therefore, it is likely that during development, compensatory mechanisms are brought into play that allow feedback regulation of the HPT axis to proceed normally, even in the absence of the thyroid hormone-activating capacity of tanycytes. This hypothesis is supported by the observation that thyroid hormone availability during the neonatal period can highly influence the setpoint for negative feedback regulation of the HPT axis (135). Namely, exogenous administration of T4 during the first 12 days after birth results in a permanent decrease in circulating TSH and thyroid hormone concentrations that persists into adulthood despite the normal TSH secretory capacity of animals in response to exogenous TRH administration (135). Were the setpoint of negative feedback regulation to be altered (lowered) during development by chronically low T3 levels in the hypothalamus of D2 KO mice, the HPT axis of the adult D2 KO mice would be expected to function normally despite low T3 availability in the hypothalamus of adult D2 KO mice. However, it is likely that ablation of D2 expression in adult animals would have a far more profound impact on the parameters of the HPT axis.
Some hypophysiotropic axon terminals in the median eminence contain D3 (51), indicating that these neurons can regulate T3 availability intracellularly, independent of the thyroid hormone concentration in the neuropil by degrading T3. Importantly, however, the vast majority of the TRH-containing hypophysiotropic terminals do not contain D3 and therefore do not have an internal mechanism for degrading T3 once taken up into the neuron (51).
2. Role of thyroid hormone transporters
Despite their lipophilic nature, the transport of thyroid hormone through cell membranes requires active transport (136). Currently, two main thyroid hormone transporters are known to be involved in thyroid hormone transport in the brain, OATP1C1 and MCT8, members of the organic anion-transporting polypeptide (OATP) and the monocarboxylate transporter (MCT) families, respectively (136). OATP1C1 has a similar high affinity for T4 and T3 and is abundantly expressed in endothelial cells of brain blood vessels, the choroid plexus, and tanycytes (102, 137). The activity of the HPT axis is not affected by the lack of OATP1C1 in KO mice (138), however, suggesting that this transporter does not play a crucial role in feedback regulation of TRH neurons. In contrast, the MCT8 transporter, which is preferentially expressed in neurons including hypophysiotropic TRH neurons (51, 139), and tanycytes (139), has preferential affinity for T3 (139). In MCT8 KO mice, TRH gene expression is increased in the PVN (140).
The location of tanycytes at the blood-brain and CSF-brain barriers and their high expression of thyroid hormone transporters place them in a strategic position to extract T4 from the bloodstream or CSF. The former could be accomplished through their end-feet processes terminating on portal capillaries or on blood vessels in the arcuate nucleus, and the latter via apical specializations after T4 has traversed the choroid plexus (101, 102, 141). Although increased circulating levels of T3 in MCT8 KO mice, and the high expression of this transporter in tanycytes implicate MCT8 as an essential component of the feedback regulation mechanism on hypophysiotropic TRH neurons, the considerable distance between tanycytes and the cell bodies of hypophysiotropic TRH neurons in the PVN raises questions as to how locally synthesized T3 can be transported from tanycytes to the perikarya of TRH neurons. It was initially hypothesized that T3 released from tanycytes into the CSF or neuropil can reach TRH neurons by diffusion through the brain extracellular space (97). This hypothesis, however, does not address how it is possible that only hypophysiotropic TRH neurons are regulated by the changes of peripheral thyroid hormone levels, whereas nonhypophysiotropic TRH neurons in the hypothalamus (including the PVN) that also express TRs are not affected (116).
Despite the relatively large distance between the tanycytes and the perikarya of the hypophysiotropic TRH neurons, there is one location where the two cell types are closely associated. This is the external zone of the median eminence where the axon terminals of hypophysiotropic TRH neurons are intertwined with end-feet processes of β2 tanycytes (142). MCT8 is present on the surface of practically all axon terminals in the external zone of the median eminence, including the terminals of hypophysiotropic TRH neurons (51). This observation makes it likely that T3 secreted from the tanycytes can be taken up by hypophysiotropic TRH terminals via MCT8 and then transported to the perikarya of TRH neurons by retrograde axonal transport. In support of this hypothesis, early studies by Dratman et al (143) presumed axonal transport of T3, based on the presence of radiolabeled T3 in neuronal processes and migration of autoradiographic signal among brain areas during the first 48 hours after iv administration of radiolabeled T3. In addition, rapid retrograde axonal transport of other bioactive molecules such as neurotrophins has already been proven (144, 145). To demonstrate that retrograde transport of T3 from the median eminence actually occurs and changes of peripheral thyroid hormone levels result in different intranuclear T3 concentration in the hypophysiotropic and nonhypophysiotropic TRH neurons, however, will require further study.
The median eminence is a unique brain region because its T3 content derives from at least two different sources. Being outside the blood-brain barrier (90), T3 circulating in the peripheral blood can readily enter the extracellular space of the median eminence through fenestrated portal capillaries without the need for a specific transport mechanism. However, tanycytes may also contribute to the T3 content in the median eminence given their high concentration of D2 (90) and, hence, ability to convert T4 to T3. Because nearly a twice normal circulating level of T3 is required to normalize the TRH mRNA level in the PVN if only T3 is administered to rats after inhibition of thyroid hormone synthesis (123), T3 derived from tanycytes may contribute substantially to the T3 content of the median eminence in euthyroid animals. Because only hypophysiotropic TRH neurons project to the median eminence, this hypothesis would also provide an explanation as to why hypophysiotropic and nonhypophysiotropic TRH neurons are differentially regulated by thyroid hormone. However, further studies are needed to demonstrate that the T3 content of hypophysiotropic and nonhypophysiotropic TRH neurons is differentially regulated by changes of peripheral thyroid hormone levels.
3. Role of PPII
In addition to modulating feedback regulation of TRH neurons by influencing T3 availability in the median eminence, tanycytes can also influence the amount of TRH that reaches portal capillaries in the median eminence. Tanycytes synthesize the TRH degrading enzyme, PPII, which is highly regulated by circulating levels of thyroid hormone (35). Peripheral administration of T4 markedly increases PPII mRNA synthesis and activity in tanycytes (35). Tanycyte end-feet processes surround the hypophysiotropic terminals in the external zone of the media eminence, and PPII is a membrane-bound protein with a large extracellular C-terminal region that contains the exopeptidase and catalytic motifs (146). Therefore, PPII synthesized by tanycytes is in an anatomical position to degrade TRH secreted from TRH-containing axon terminals into the extracellular space of the median eminence (35). In support of this hypothesis, inhibition of PPII by Hermodice carunculata protease inhibitor (HcPI) increases TRH release from median eminence explants, whereas in vivo, a single peripheral injection of HcPI results in a significant increase in circulating TSH levels in animals exposed to a cold environment compared to the vehicle-injected animals (35).
A summary of the mechanisms contributing to feedback regulation of hypophysiotropic TRH neurons is illustrated in Figure 10.
Figure 10.
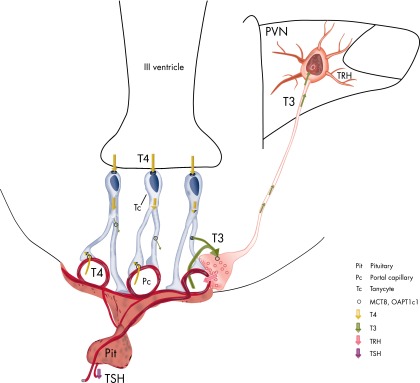
Schematic illustration of the machinery involved in negative feedback regulation of the HPT axis by thyroid hormone.
IV. Central Regulation of the Hypothalamic-Pituitary-Thyroid Axis During Fasting
The HPT axis plays a critical role in the regulation of energy expenditure by affecting basal metabolic rate and through the actions of thyroid hormone to stimulate mitochondrial oxygen consumption and increase thermogenesis (147). It is not surprising, therefore, that alternations in energy availability would be intimately linked to control of the HPT axis. During fasting, for example, circulating thyroid hormone levels decline, associated with low or normal TSH levels and inhibition of TRH gene expression in the PVN characteristic of central hypothyroidism (Figure 11, A and B) (148–150). Presumably, fasting-induced hypothyroidism is a homeostatic mechanism to conserve energy stores until food is once again available. The mechanism by which hypophysiotropic TRH neurons sense alterations in peripheral energy stores is orchestrated by leptin, a white adipose tissue-derived circulating hormone (148–150), because fasting-induced central hypothyroidism can be completely prevented by the exogenous administration of leptin (Figure 11C) (148). The primary target site for leptin that mediates its effect on the HPT axis is the arcuate nucleus, because ablation of this nucleus abolishes both fasting- and leptin-induced regulation of the HPT axis (151). The circuitry and peptide mediators involved are discussed in detail this section and are illustrated in Figure 12.
Figure 11.

Darkfield illumination photomicrographs of proTRH mRNA in the hypothalamic PVN in (A) fed, (B) fasted, and (C) fasted animals receiving leptin. Note the marked reduction in silver grains over neurons in the PVN in the fasted animals but restoration to normal in the fasted animals receiving leptin. III, Third ventricle. [Reproduced from C. Fekete and R. M. Lechan: Negative feedback regulation of hypophysiotropic thyrotropin-releasing hormone (TRH) synthesizing neurons: role of neuronal afferents and type 2 deiodinase. Front Neuroendocrinol. 2007;28:97–114 (58), with permission. © Elsevier.]
Figure 12.

Schematic drawing summarizing the regulation of TRH neurons by fasting.
A. Role of the arcuato-paraventricular pathway in the regulation of the HPT axis during fasting
Two antagonistic, neuronal populations in the arcuate nucleus, the orexigenic NPY/AGRP/GABA neurons and the anorexigenic α-MSH/CART neurons, are primarily responsible for sensing and relaying information to hypophysiotropic TRH neurons about the concentration of leptin in the bloodstream (56). Both leptin-sensitive neuronal groups directly target hypophysiotropic TRH neurons and establish synaptic specializations with their perikarya and dendrites in the PVN (58). Similar to leptin, central administration of α-MSH or CART to fasted animals completely prevents fasting-induced inhibition of TRH gene expression in the PVN (Figure 13) (42, 52) and can increase TRH release from hypothalamic explants (52, 152). α-MSH has also been shown to depolarize TRH neurons in the PVN and increase their firing rate (153). In contrast, central administration of NPY or AGRP to fed animals induces a state of central hypothyroidism similar to that observed in fasted animals (Figure 14), despite that the animals markedly increase their food intake secondary to the potent orexigenic effect of these peptides (152, 154, 155). NPY has also been shown to hyperpolarize the TRH neurons in the PVN and decrease the firing rate of these neurons (153).
Figure 13.

Darkfield illumination micrographs of proTRH mRNA in the medial and periventricular parvocellular subdivisions of the hypothalamic PVN in fed animals (A and D), fasted animals (B and E), and fasted animals receiving an intracerebroventricular infusion of either α-MSH (C) or CART (F) every 6 hours for 64 hours. Note the reduction in the accumulation of silver grains over the PVN in fasted animals compared with the fed controls. Both α-MSH and CART administration prevent the fasting-induced fall of proTRH mRNA. III, Third ventricle. [Modified from C. Fekete et al: Association of cocaine- and amphetamine-regulated transcript-immunoreactive elements with thyrotropin-releasing hormone-synthesizing neurons in the hypothalamic paraventricular nucleus and its role in the regulation of the hypothalamic-pituitary-thyroid axis during fasting. J Neurosci. 2000;20:9224–9234 (42), with permission. © Society for Neuroscience. From C. Fekete et al: α-Melanocyte-stimulating hormone is contained in nerve terminals innervating thyrotropin-releasing hormone-synthesizing neurons in the hypothalamic paraventricular nucleus and prevents fasting-induced suppression of prothyrotropin-releasing hormone gene expression. J Neurosci. 2000;20:1550–1558 (52), with permission. © Society for Neuroscience. And from C. Fekete and R. M. Lechan: Negative feedback regulation of hypophysiotropic thyrotropin-releasing hormone (TRH) synthesizing neurons: role of neuronal afferents and type 2 deiodinase. Front Neuroendocrinol. 2007;28:97–114 (58), with permission. © Elsevier.]
Figure 14.

Darkfield illumination photomicrographs of proTRH mRNA in the medial parvocellular subdivision of the PVN in control (A), AGRP-treated (B), and NPY-treated (C) animals. Note the marked reduction in silver grains over neurons in the PVN in both the AGRP- and NPY-infused groups. III, Third ventricle. [Modified from C. Fekete et al: Agouti-related protein (AGRP) has a central inhibitory action on the hypothalamic-pituitary-thyroid (HPT) axis; comparisons between the effect of AGRP and neuropeptide Y on energy homeostasis and the HPT axis. Endocrinology. 2002;143:3846–3853 (155), with permission. © The Endocrine Society. And from C. Fekete and R. M. Lechan: Negative feedback regulation of hypophysiotropic thyrotropin-releasing hormone (TRH) synthesizing neurons: role of neuronal afferents and type 2 deiodinase. Front Neuroendocrinol. 2007;28:97–114 (58), with permission. © Elsevier.]
The α-MSH/CART and NPY/AGRP/GABA arcuate neurons interact at multiple levels to regulate hypophysiotropic TRH neurons. First, both α-MSH and AGRP are ligands of the melanocortin 3 and 4 receptors (MC3R and MC4R) (156), with α-MSH functioning as an agonist and AGRP as a high-affinity antagonist (156). The MC4R is expressed by hypophysiotropic TRH neurons (24), and because the inhibitory effect of AGRP on TRH gene expression is abolished in MC4R KO mice (157), melanocortin signaling to TRH neurons must be mediated primarily by the MC4R. All TRH neurons that are innervated by α-MSH-containing axon varicosities are also innervated by AGRP-containing terminals (Figure 5A) (52), indicating direct, functional interactions between these two peptides. The interaction of the two peptides in the regulation of the TRH neurons is further supported by in vitro studies, demonstrating that AGRP prevents the stimulatory effect of α-MSH on TRH release (152).
A second mechanism may involve postreceptor effects arising from the interaction between α-MSH and NPY. The MC4R is coupled to Gs proteins and when activated by α-MSH, induces the adenylyl cyclase–protein kinase A cascade (158). The resultant increase in cAMP, and hence CREB phosphorylation, regulates TRH gene expression by the binding of phosphoCREB to CREs present in the TRH gene (21, 159). NPY also regulates cAMP synthesis, but because NPY receptors couple to Gi or G0 proteins, NPY inhibits adenylate cyclase, reducing cAMP accumulation (160). In this manner, NPY could inhibit α-MSH-induced CREB phosphorylation in hypophysiotropic TRH neurons by reducing the intracellular cAMP concentration. In fact, pretreating animals with centrally administered NPY markedly reduces the ability of intracerebroventricularly injected α-MSH to induce CREB phosphorylation in TRH neurons (161).
The inhibitory effects of NPY on hypophysiotropic TRH neurons are mediated primarily by Y1 and Y5 receptors because the central administration of either selective Y1 or Y5 receptor agonist is equally effective in inhibiting hypophysiotropic TRH gene expression and the HPT axis (162). In addition, Y1 receptor mRNA is expressed in TRH neurons in the PVN (163), and the Y5 receptor has been localized to the PVN (164), although its specific expression in hypophysiotropic TRH neurons has not been studied. Because the fasting-induced fall of leptin stimulates NPY/AGRP neurons and inhibits α-MSH/CART neurons (56), the increased inhibitory tone of orexigenic peptides and simultaneous decrease in the stimulatory input from anorexigenic neurons of arcuate nucleus origin play a critical role in the development of fasting-induced central hypothyroidism. This hypothesis is supported by evidence showing that fasting-induced central hypothyroidism is prevented in mice lacking both NPY and the MC4R (112).
Currently, little is known about the interactions between CART and melanocortins or NPY, partly hindered by the fact that the CART receptor(s) has not yet been identified. Central administration of CART increases CREB phosphorylation at least in the CRH neurons in the PVN, but phosphoCREB is absent from hypophysiotropic TRH neurons after this treatment (165). Therefore, it is unknown whether the effect of CART on the hypophysiotropic TRH neurons involves the interaction with the NPY and AGRP signaling at the level of second messengers or only at the level of the TRH promoter.
In addition to leptin, insulin and glucose also have anorexigenic effects when administered centrally (56). Circulating levels of these substances decrease during fasting and are sensed by the feeding-related neurons of the arcuate nucleus (56), raising the possibility that, similar to leptin, insulin and glucose are also involved in fasting-induced regulation of hypophysiotropic TRH neurons. However, whereas central administration of leptin completely prevents fasting-induced inhibition of the TRH gene expression, central administration of insulin or glucose has no effect on TRH gene expression in fasted rats (166). These observations suggest that changes in insulin and glucose levels do not have a critical role in the fasting-induced regulation of the HPT axis and indicate that leptin, insulin, and glucose have different effects on feeding-related neuronal groups in the arcuate nucleus. Indeed, whereas leptin can completely reverse fasting-induced changes in NPY, AGRP, proopiomelanocortin (POMC), and CART gene expression in arcuate nucleus neurons, centrally administered insulin only affects NPY and POMC gene expression, and glucose only affects NPY gene expression (166). Therefore, it would appear that all four feeding-related peptides of arcuate nucleus origin are necessary to appropriately regulate hypophysiotropic TRH neurons during fasting through the arcuato-paraventricular pathway (166).
Similar to the rodent arcuate nucleus, the human infundibular nucleus also contains neurons that coexpress AGRP and NPY and neurons that synthesize α-MSH (46). These neuronal populations give rise to axonal projections that directly innervate TRH neurons in the PVN (46). In contrast to the rodent, however, CART is not cosynthesized with α-MSH but is present in a subpopulation of the AGRP- and NPY-synthesizing neurons in the infundibular nucleus (68). The significance of this difference is uncertain, but it raises the possibility that CART may have a different role in the regulation of the HPT axis in the human brain compared to that observed in rodents (68).
B. Direct action of leptin on hypophysiotropic TRH neurons
Despite the observation that regulation of the hypophysiotropic TRH neurons by fasting and leptin is lost in arcuate nucleus-ablated animals (151), there has been an ongoing debate about whether leptin can exert a direct effect on hypophysiotropic TRH neurons (24, 37, 58, 153, 167). The presence of the leptin receptor in TRH neurons was shown in primary cultures of fetal rat hypothalamic cells (167), but the approach used did not differentiate between hypophysiotropic TRH neurons and the many other TRH-synthesizing neuronal populations present in the hypothalamus. In addition, it is not clear whether the expression of the leptin receptor observed in fetal cultures is sustained in adult animals or only exists transiently during development. Harris et al (24), however, have convincingly demonstrated that leptin administration induces the expression of suppressor of cytokine signaling 3 (SOCS3), an accepted marker of leptin receptor signaling, in a small subset (10%) of TRH neurons in the PVN. Furthermore, STAT3, a second messenger utilized by the leptin receptor, was shown to directly bind to the TRH promoter in mouse hypothalamus after peripheral administration of leptin (24). These data support the notion that a subgroup of hypophysiotropic TRH neurons in the PVN can be directly targeted by leptin, a view further supported by in vitro patch clamp electrophysiology data, demonstrating a direct excitatory effect of leptin on TRH neurons in the PVN (153).
However, it remains uncertain under what physiological conditions direct vs indirect leptin signaling to TRH neurons is utilized physiologically. During fasting, it seems clear that the arcuato-paraventricular pathway is the most important pathway that mediates the effect of leptin on hypophysiotropic TRH neurons. This is based on the observations that arcuate nucleus ablation prevents the effect of fasting and leptin administration on TRH gene expression in the PVN (148). In addition, whereas central administration of leptin results in both STAT3 and CREB phosphorylation in TRH neurons of the PVN (168), the administration of the melanocortin antagonist, SHU9119, completely prevents the stimulatory effect of centrally administered leptin on TRH release and TSH secretion (168), indicating that leptin influences hypophysiotropic TRH neurons primarily via the melanocortin system. Furthermore, in NPY/MC4R double KO mice lacking both NPY and melanocortin signaling, fasting-induced suppression of the HPT axis is completely prevented (112). These data, however, do not exclude the possibility that a direct effect of leptin on hypophysiotropic TRH neurons may be of secondary importance by increasing the expression of MC4R in these neurons (169), and hence their melanocortin sensitivity. In this manner, a direct effect of leptin may sensitize hypophysiotropic TRH neurons to the arcuate nucleus inputs, rather than directly influence the activity in these cells. A direct effect of leptin on hypophysiotropic TRH neurons, however, may be of importance in maintaining the activity of the HPT axis in association with obesity (170) and is discussed in greater detail in Section VI.
C. Involvement of tanycytes in the regulation of the HPT axis during fasting
In addition to neuronal groups projecting onto hypophysiotropic TRH neurons and a direct action of leptin, hypothalamic tanycytes may also participate in the central regulation of the HPT axis during fasting. Fasting results in an approximately 2-fold increase in D2 hybridization signal in the MBH (130) and a 1.6-fold increase of D2 activity (130). These changes are independent of the fasting-induced fall in peripheral thyroid hormone levels (130) because hypothyroidism has a relatively minimal effect on D2 activity in the MBH, and T4 treatment does not prevent the fasting-induced increase in MBH D2 activity (130). Changes of leptin or corticosterone levels alone have no effect on D2 activity in the MBH of fed animals (171), but either removal of the adrenal glands or preventing leptin levels from falling during fasting prevents the increase in D2 activity in the MBH in fasting animals (171). It is hypothesized, therefore, that the fall in leptin levels in fasted animals has a permissive role to enable the stimulatory effect of corticosterone on D2 activity (171). It has been further suggested that the fasting-induced increase of D2 activity in the MBH may contribute to inhibition of the HPT axis by increasing local tissue levels of thyroid hormone (172). This hypothesis is supported by a report that third ventricular administration of a deiodinase inhibitor, iopanoic acid, prevents the fasting-induced increase in hypothalamic T3 concentrations and blunts inhibition of TRH gene expression in the PVN (172). However, iopanoic acid has broad target specificity, inhibiting all deiodinase enzymes and blocking thyroid hormone transporters (173, 174). Therefore, it cannot be unequivocally stated that the observed effect of iopanoic acid treatment is due to inhibition of D2 activity in the MBH, especially because the fasting-induced fall in thyroid hormone levels is very similar in wild-type and D2 KO mice (175). An alternative hypothesis is that increased T3 production by tanycytes stimulates NPY neurons in the arcuate nucleus (175), indirectly inhibiting TRH neurons in the PVN. However, because thyroid hormone has no inhibitory effect on hypophysiotropic TRH neurons in TRβ2 KO mice, but the fasting-induced suppression of TRH mRNA in the PVN is completely intact in these animals (119), tanycyte-generated T3 during fasting likely has a minimal role in the regulation of the HPT axis. In addition, fasting inhibits TRH neurons in the PVN of Siberian hamsters, regardless of whether they are kept under short or long day photoperiods, although fasting has opposite effects on tanycyte D2 activity during long and short day photoperiods (176).
D. Role of other neuronal pathways in the regulation of the HPT axis during fasting
In addition to the arcuate nucleus, TRH neurons also receive inputs from other neuronal groups known to be involved in the regulation of energy homeostasis, including the DMN and neuronal groups in the brainstem (58). Axons arising from all subdivisions of the DMN densely innervate the vast majority of TRH neurons in the PVN (74). Approximately two-thirds of the PVN projecting DMN neurons are located in the medial part of ventral subdivision of the DMN, while only 10% of PVN projecting neurons are housed in the compact subdivision (71).
The physiological importance of the DMN in regulating hypophysiotropic TRH neurons remains uncertain but may be implicit in the knowledge that the DMN also has an important role in the regulation of energy homeostasis (177). Electrolytic and chemical (ibotenic acid) lesions of the DMN result in hypophagia due to an altered setpoint of body weight regulation (177). The DMN likely has both orexigenic and anorexigenic neuronal populations, however, because DMN lesions also increase food intake during the first hour of refeeding after a period of fasting and attenuate cholecystokinin-induced decrease in food intake (177).
Because the DMN contains leptin-, insulin-, and ghrelin-receptive and glucosensing neurons (70, 177–179), some of these neurons may function to transmit humoral signals to TRH neurons in the PVN. However, it is unlikely that the DMN functions independently of the arcuate nucleus in the regulation of hypophysiotropic TRH neurons, at least during fasting, because animals deficient in arcuate nucleus NPY and melanocortin peptides lose the ability to induce central hypothyroidism in response to fasting (112, 151).
Neurons of the DMN, however, receive feeding-related inputs from the arcuate nucleus and are densely innervated by both AGRP- and α-MSH-containing axons (71, 180). Furthermore, most DMN neurons that project to the PVN receive melanocortin input from the arcuate nucleus (71). This circuitry indicates that the DMN may serve as a relay station from the arcuate nucleus to the PVN, contributing to the effects of melanocortin signaling on the hypophysiotropic TRH neurons (71).
In addition to the regulation of food intake, the DMN is also involved in circadian regulation of hypothalamic systems (181). The DMN receives both direct and indirect inputs from the suprachiasmatic nucleus, and ablation of the DMN reduces the circadian rhythm of wakefulness, feeding, locomotor activity, and the hypothalamic-pituitary-adrenal axis (181). The DMN, therefore, may integrate feeding and circadian rhythm-related signals involved in the regulation of the HPT axis. In fact, the DMN is known to be a critical regulator of food-entrainable circadian rhythm of several hypothalamic systems (182). Fasting blunts the nocturnal increase of TSH secretion in humans (183), but it is unclear yet whether the DMN is involved in this process.
Hypophysiotropic TRH neurons also receive dense input from the brainstem that contains feeding-related peptides such as NPY and CART (64, 80). These inputs, however, do not seem to have a role in the fasting-induced regulation of TRH neurons because transection of ascending brainstem pathways does not affect fasting-induced inhibition of TRH gene expression in hypophysiotropic neurons (our unpublished personal observations).
V. Effects of Dehydration-Induced Anorexia on the Hypothalamic-Pituitary-Thyroid Axis
Dehydration induced by replacement of drinking water with 2.5% NaCl gradually decreases daily food intake by 80–90% and is associated with significant weight loss despite the fact that the animals have free access to food (184). This experimental model, commonly referred to as dehydration-induced anorexia (DIA), has been used by researchers to explore central mechanisms involved in the regulation of anorexia (184). Of interest, whereas food restriction inhibits all components of the HPT axis, DIA is accompanied by an increase in TRH gene expression in the PVN, elevated circulating TSH levels, yet unaltered T4 and decreased T3 concentrations in the peripheral blood (185). The increase in TSH concentration is likely secondary to increased TRH release, whereas the low T3 level may be secondary to increased peripheral T3 metabolism or clearance.
The effect of DIA on TRH neurons in the PVN, however, is not uniform (186). Similar to fasting, TRH mRNA is decreased in the mid level of the PVN (186), raising the possibility that these neurons are regulated by the arcuato-paraventricular pathway. Hypophysiotropic TRH neurons in the caudal part of the PVN, however, are stimulated by DIA but are inhibited in food-restricted animals (186), indicating that any inhibitory effect of the arcuato-paraventricular pathway is overridden by other factors. Because the TSH concentration is increased in DIA animals (185), it is likely that stimulation of the caudal TRH cell group and inhibition of the mid level TRH cell population in the PVN results in a net increase of TRH release into the hypophysial portal circulation.
One major difference between the peptide expression profile of the food-restricted and DIA animals is the increased CRH expression in lateral hypothalamic neurons in DIA animals (184). These CRH neurons are known to project to the PVN (184) and may contribute to the innervation of TRH neurons in the PVN (86). In support of this hypothesis, CRH stimulates TRH biosynthesis, and administration of a CRH-R2 antagonist prevents DIA-induced effects on TRH gene expression in the PVN (187).
In addition to the caudal part of the PVN, DIA also stimulates TRH neurons in the anterior parvocellular subdivision of the PVN (186). These neurons are not involved in the regulation of the HPT axis (58). Nevertheless, because the central administration of TRH inhibits food intake (188) and TRH neurons in the anterior PVN project to brain nuclei that are involved in the regulation of food intake (54), it is possible that the increased activity of TRH neurons in the anterior PVN contributes to the anorexigenic effect of DIA.
VI. Regulation of the HPT Axis in High-Fat Diet-Induced Obese Animals
Consumption of high-fat diet (HFD) results in increased caloric intake, increased weight gain, and also a stimulation of the HPT axis, characterized by increased TRH mRNA and protein levels in the PVN and increased TSH and thyroid hormone levels in the circulation (170).
Because the HFD-induced obese (DIO) animals have elevated leptin levels and the HFD does not stimulate the HPT axis in leptin-deficient ob/ob mice (170), the effect of a HFD on the HPT axis seems to be mediated by leptin (170). However, expression of the POMC (the precursor of α-MSH) gene in the arcuate nucleus of DIO rats remains unchanged (189), suggesting that a HFD decreases leptin sensitivity of anorexigenic neurons in this nucleus. This concept is supported by decreased STAT3 phosphorylation and increased suppressor of cytokine signaling 3 and PTP1B synthesis in the arcuate nucleus of HFD animals after leptin treatment (190, 191).
Therefore, in contrast to fasting, leptin would appear to regulate the HPT axis independently from POMC neurons. Because central leptin administration results in STAT3 phosphorylation in hypophysiotropic TRH neurons in DIO rats, it is hypothesized that the HPT axis is regulated by a direct action of leptin on these neurons (170). However, leptin-induced regulation of AGRP synthesis in the arcuate nucleus and the leptin sensitivity of neurons in the DMN remain intact in DIO animals (189, 190), necessitating additional studies to exclude the possibility that the effect of leptin on hypophysiotropic TRH neurons in DIO animals is mediated indirectly by these neuronal groups.
VII. Central Regulation of the HPT Axis During Infection and Prolonged Critical Illness
Similar to fasting, infection and other acute and chronic disorders can lead to inhibition of the HPT axis. In man, this condition is commonly referred to as the nonthyroidal illness syndrome (192), discussed in further detail in Section IX. Characteristic of this syndrome is a decline in circulating T3 and, in more severe cases, a general inhibition of the HPT axis due to the development of central hypothyroidism (192–194).
A. Role of neuronal pathways in the regulation of the hypophysiotropic TRH neurons during infection
A commonly used model for infection is the peripheral administration of bacterial lipopolysaccharide (LPS). However, whereas both fasting and LPS administration have a similar effect to induce central hypothyroidism, the mechanisms by which these two conditions influence the hypophysiotropic TRH neurons are different. As discussed previously, the fall in the circulating level of leptin orchestrates the inhibitory effect of fasting on hypophysiotropic TRH neurons via the arcuato-paraventricular pathway (37, 58). In contrast, LPS treatment increases circulating leptin levels (58, 195), stimulates POMC and CART gene expression in arcuate nucleus neurons, and does not increase NPY expression (195, 196)—changes that would predict activation of hypophysiotropic TRH neurons rather than suppression as observed (37, 58, 194, 197). Presumably, therefore, a more potent inhibitory mechanism becomes activated during infection that overrides any stimulatory action of leptin, α-MSH, or CART on hypophysiotropic TRH neurons.
One possibility to explain the effect of LPS on hypophysiotropic TRH neurons is activation of adrenergic neurons in the medulla because these neurons are known to mediate the effect of LPS on CRH neurons in the PVN and result in a marked increase in circulating levels of corticosterone (198). As described above, medullary adrenergic neurons directly innervate hypophysiotropic TRH neurons (77). Unilateral transection of ascending brainstem pathways or ablation of the catecholaminergic input of the PVN, which markedly attenuates LPS-induced c-Fos expression in the PVN and the increase in CRH gene expression (198, 199), however, results in an even more profound inhibition of TRH gene expression in the PVN (199). Thus, brainstem inputs would appear to stimulate rather than inhibit hypophysiotropic TRH neurons during infection, and in keeping with the activating effects of catecholamines on hypophysiotropic TRH (78).
B. Tanycytes as the key regulators of hypophysiotropic TRH neurons during infection
LPS has a potent effect on tanycyte D2 gene expression and enzymatic activity, resulting in an approximately 4-fold increase of D2 mRNA level and activity in the MBH within 12 hours of administration (Figure 15, A and B) (197, 200). It has been hypothesized, therefore, that LPS may inhibit hypophysiotropic TRH neurons by increasing local T3 levels in the median eminence, where the processes of β tanycytes and the axon terminals of the hypophysiotropic TRH neurons are closely associated (51, 99). In support of this hypothesis, LPS-induced inhibition of hypophysiotropic TRH neurons is abolished in the D2 KO mice (201). Although D2 activity rapidly increases in tanycytes and is maximal 9–12 hours after LPS treatment, D2 activity gradually increases in other responsive tissues, such as the cortex and the anterior pituitary, and becomes maximal at approximately 24 hours (99, 202). This difference in the D2 response favors distinct, tissue-specific mechanisms for activation of D2. The increase in D2 activity in the cortex and anterior pituitary is inversely related to peripheral thyroid hormone levels and can be prevented by clamping circulating T4 to euthyroid levels (99, 197). This indicates that in the cortex and anterior pituitary, D2 activation is secondary to LPS-induced reduction in peripheral thyroid hormone levels. The T4 clamp, however, has no effect on the LPS-induced increase in D2 activity in the tanycytes (197).
Figure 15.

A and B, Regulation of D2 gene expression in tanycytes by LPS. Low-power darkfield micrographs of the caudal part of the hypothalamus show the D2 expression in control animals (A) and 12 hours after LPS administration (B). Note marked increase in the density of silver grains, particularly in the external zone of the median eminence. C–E, Darkfield illumination photomicrographs of Iκ-Bα mRNA expression in the MBH in animals receiving saline (C) or LPS 3 hours (D) or 12 hours (E) before sacrifice. Note the marked accumulation of silver grains over the pars tuberalis in panel D 3 hours after the administration of LPS. Iκ-Bα mRNA is only seen in a subset of α tanycytes 12 hours after LPS administration (arrows in panel E). F and G (arrows), Expression of TSHβ mRNA in the pars tuberalis of animals receiving saline (F) or LPS (G) 9 hours before sacrifice. Note increased expression of TSHβ mRNA in the PT after LPS administration. Ventricular borders are demarcated with dotted lines. H, Double-labeled fluorescent image illustrates the presence of phospho-CREB (green and yellow, arrows) in the nucleus of tanycytes (red) 9 hours after administration of LPS. III, Third ventricle; ME, median eminence; PT, pars tuberalis. [Modified from C. Fekete et al: Lipopolysaccharide induces type 2 iodothyronine deiodinase in the mediobasal hypothalamus: implications for the nonthyroidal illness syndrome. Endocrinology. 2004;145:1649–1655 (99), with permission. © The Endocrine Society. And from E. Sánchez et al: Contribution of TNF-α and nuclear factor-κB signaling to type 2 iodothyronine deiodinase activation in the mediobasal hypothalamus after lipopolysaccharide administration. Endocrinology. 2010;151:3827–3835 (142), with permission. © The Endocrine Society.]
The mechanism by which LPS stimulates D2 activity in tanycytes remains uncertain. One attractive possibility includes a direct action of LPS on tanycytes via their expression of Toll-like receptor 4 (TLR4) (203) because the lack of TLR4 in TLR4 KO mice prevents the LPS-induced nonthyroidal illness syndrome (204). The main second messenger utilized by TLR4 and many of the cytokine receptors is the nuclear factor-κB (NF-κB) system (205), and the promoter of the D2 gene is highly responsive to NF-κB (99, 206). The human D2 gene contains two high-affinity NF-κB binding sites at positions −683 bp (called no. 2) and −198 bp (no. 5), 5′ to the transcriptional starting site (206). Although both sites have a similar binding affinity by EMSA assay, site-directed mutagenesis and promoter assay indicates that only site no. 5 possesses transactivation potency in the presence of the p65 subunit of NF-κB, increasing D2 promoter activity 150-fold (206). However, LPS-induced activation of IκBα mRNA, a marker of NF-κB signaling, is observed only in a subset of α tanycytes and only 12 hours after LPS administration (Figure 15, C–E), significantly later than the peak of the LPS-induced increase in D2 mRNA in these cells (142, 202). Thus, LPS-induced NF-κB activation via TLR4 or other cytokine receptors seems less likely as the initial stimulus that increases D2 activity in tanycytes in response to LPS treatment and rather may be involved in the maintenance of increased D2 (142).
IκBα mRNA, however, markedly increases in the pars tuberalis of the pituitary gland 3 hours after LPS treatment (Figure 15D) (142), a portion of the pituitary that is immediately adjacent to tanycyte end-feet processes in the external zone of the median eminence and in the sulcus tuberoinfundibularis (207). The pars tuberalis contains a large number of TSH-producing cells, but in contrast to thyrotropes in the anterior pituitary, TSH-producing cells of the pars tuberalis are not affected by TRH or thyroid hormone (207). Because the synthesis of the TSHβ-subunit increases significantly in these cells 9 hours after LPS treatment (Figure 15, F and G) (142), and the TSH receptor is expressed in tanycytes (103), TSH secretion from the pars tuberalis may be involved in mediating the effects of LPS on the tanycytes. Indeed, TSH has been shown to increase D2 activity in these cells via activation of cAMP (208). In addition, CREB phosphorylation, a marker of increased cAMP production, is evident in tanycytes 9 hours after LPS administration (Figure 15H) when D2 synthesis peaks, but absent from tanycytes at 6 hours after LPS administration when D2 synthesis is already increased (142).
The effect of LPS on the tanycytes, therefore, may be mediated by at least three factors. The first factor is responsible for the early increase in D2 but remains unknown. TSH released from the pars tuberalis may then contribute to enhancing D2 activation shortly thereafter, followed by other factors that maintain D2 activity by stimulating NF-κB signaling in tanycytes. Although changes in circulating levels of corticosterone have been suggested to have a role in the stimulation of D2 activity in fasted animals (171), a corticosterone clamp in adrenalectomized animals does not prevent the LPS-induced increase of D2 activity (202) or influence LPS-induced inhibition of TRH synthesis (194).
C. Regulation of hypophysiotropic TRH neurons during prolonged critical illness
The effect of prolonged critical illness on the HPT axis has been studied in a rabbit model using third-degree burn injury (209, 210). As in infection, prolonged critical illness results in low T4, T3, and TSH concentration in the circulation and decreased TRH mRNA level in the PVN (209, 210). In addition, it is associated with increased D2 gene expression in the MBH (210), indicating that similar to infection, burn injury may inhibit the HPT axis by inducing local hypothalamic hyperthyroidism caused by the increased thyroid hormone-activating capacity of tanycytes. Comparison of the T3 content in whole hypothalamic blocks of healthy and ill rabbits did not support this hypothesis, however, because T3 levels were not found to be higher in the experimental animals (210). Nevertheless, the approach did not take into account that burn-induced tissue hyperthyroidism may take place in only focal regions of the hypothalamus such as the median eminence and PVN, which may be sufficient to inhibit hypophysiotropic TRH neurons.
Prolonged illness is also accompanied by a nutritional deficit, raising the possibility that the decreased calorie intake also contributes to the development of central hypothyroidism during prolonged critical illness (209). Early parenteral nutrition, however, does not influence the illness-induced decrease in TRH gene expression and the decline in the circulating level of T4 (209). The expected decline in the circulating level of T3 was prevented, however, suggesting that the nutritional deficit may have its greatest influence on thyroid hormone metabolism in peripheral tissues (209).
VIII. Regulation of Hypophysiotropic TRH Neurons by Cold Exposure and Suckling
Thyroid hormone has a critical role in the regulation of both obligatory and adaptive thermogenesis (211). In the presence of hypothyroidism, obligatory thermogenesis, the heat dissipated due to the biochemical processes necessary for the maintenance of life, is reduced by approximately 30% (211). Hypothyroidism also interferes with adaptive thermogenesis, required to maintain normal body temperature in a cold environment (211). It is not surprising, therefore, that cold exposure serves as an important stimulus for TRH gene regulation in the PVN to increase thyroid hormone levels (212). The increase in TRH mRNA by cold exposure is transient, however, increasing by 100% within 1 hour of the cold exposure, followed by a decline to normal levels during the second hour (213). Peak TRH release from the median eminence also occurs 40 minutes after the cold exposure (213) and is facilitated by a cold-induced increase in the synthesis of PC enzymes, PC1/3 and PC2, involved in the processing of proTRH (214).
The physiological role of this early, transient increase in thyroid hormone levels is not completely clear. Cold exposure markedly increases D2 activity in brown adipose tissue (BAT) (94). As a result, TRs are fully occupied by T3 in the BAT after 4 hours of cold exposure (211) when the thyroid hormone levels have already normalized after a transient increase. In addition, administration of low doses of T4 to thyroidectomized rats can completely normalize the thermogenesis without normalizing the thyroid hormone concentration in the circulation (215), suggesting that even low circulating levels of thyroid hormone are sufficient for normal thermogenesis. We hypothesize, therefore, that the cold-induced transient increase in thyroid hormone may be necessary to initiate thermogenesis in BAT before D2 activity is sufficiently increased to maintain BAT thermogenesis, even without increased thyroid hormone levels in the circulation. This hypothesis, however, requires further clarification.
Because central administration of TRH stimulates thermogenesis in BAT via the activation of the sympathetic nervous system (57), the cold-induced increase in TRH gene expression in the PVN might be hypothesized to increase heat production in BAT simultaneously with stimulation of the HPT axis. Indeed, TRH has been shown to stimulate the BAT after its administration into the DMN, preoptic area, hypothalamic ventromedial nucleus, anterior hypothalamus, or hindbrain (57, 216). However, because none of these brain regions are innervated by hypophysiotropic TRH neurons (our unpublished observations), the effect of TRH on BAT is likely mediated by other groups of TRH-synthesizing neurons.
The effect of cold on the activity of hypophysiotropic TRH neurons is thought to be mediated primarily by the brainstem catecholaminergic neurons because noradrenaline stimulates TRH synthesis (78) and inhibition of noradrenaline prevents cold-induced activation of the HPT axis (217). In addition, the HPT axis does not respond to cold during the first 10 postnatal days in rodents, during which time catecholaminergic axons are still migrating to TRH neurons in the PVN (1). Furthermore, inhibition of adrenergic receptors blocks cold-induced TRH synthesis and release (214, 218).
Cold stimulates TRH gene expression, biosynthesis, and proteolytic processing via β adrenergic receptors and by increasing cAMP production (214). Both adrenergic and noradrenergic brainstem neurons densely innervate hypophysiotropic TRH neurons in the PVN (61), but the A2 noradrenergic cell group is the only catecholaminergic population in which cold-induced c-Fos activation can be observed. The release of TRH is also stimulated by cold via α-adrenergic receptors (214, 218). This effect is independent of the regulation of TRH synthesis (214, 218).
Similar to cold exposure, suckling also stimulates TRH gene expression (213, 219). TRH mRNA is increased in the PVN during the first days of lactation, but then declines after the eighth day (219). Acute suckling also results in a 2-fold increase in TRH mRNA in the PVN after 30 minutes of suckling, which gradually decreases to presuckling levels after 6 hours (213, 219). This response is associated with increased prolactin levels in the circulation, but surprisingly with unaltered TSH and thyroid hormone levels (219, 220). The mechanism for suckling-induced activation of hypophysiotropic TRH neurons is currently unclear, but the differential response of the anterior pituitary to TRH is likely mediated by several factors. Corticosterone may be important in the prevention of TRH-induced TSH secretion because corticosterone-clamped, adrenalectomized animals increase TSH in response to suckling (219). A decline in dopamine release in response to suckling may also contribute to the increased sensitivity of lactotrophs to TRH (47). CART, cosynthesized in hypophysiotropic TRH neurons, has been shown to modulate the effect of TRH on lactotrophs, preventing TRH from releasing prolactin without influencing TSH release during cold exposure and in association with hypothyroidism (220, 221). As opposed to cold exposure and hypothyroidism when CART is increased in hypophysiotropic neurons (220), CART remains unchanged during suckling in hypophysiotropic neurons, potentially favoring the release of prolactin (220).
IX. Translational Ramifications
Because of the similarities between the anatomical organization of the hypophysiotropic TRH-synthesizing neuronal system in experimental animals and man (see in Section II), it is reasonable to assume that regulation of the HPT axis in man may have similarities to that observed in rodents. In support of this hypothesis, the important role of TRH in the regulation of anterior pituitary TSH secretion in man is suggested by the presence of central hypothyroidism associated with mutations in the TRH receptor gene. Namely, individuals homozygous for TRH receptor mutations or compound heterozygotes demonstrate low levels of total and free T4 and have inappropriately low or normal TSH levels (222, 223). In addition, their TSH secretion is not influenced by exogenous administration of TRH (222, 223). Hence, although these individuals are able to produce TSH, the biological activity of their TSH is decreased, and they regulate TSH secretion at a lower setpoint, presumably due to deficiency in TRH signaling, resulting in lower than normal circulating levels of T4 (222, 223).
As in rodents, feedback regulation of the HPT axis in man would also appear to require the MCT8 thyroid hormone transporter. This is apparent in the Allan-Herndon-Dudley syndrome, secondary to mutations in MCT8, characterized by elevations in T3, a low T4 level, and a circulating TSH level in the upper normal range (224, 225). Although one might have anticipated a greater elevation in TSH if MCT8 has an important role in the transport of thyroid hormone to hypophysiotropic TRH neurons in man, these patients can have a blunted response to exogenously administered TRH (226), indicating that anterior pituitary thyrotropes may still retain the ability to transport T3 through other mechanisms, diminishing the direct stimulatory effect of TRH. Negative feedback inhibition of the HPT axis by thyroid hormone would also appear to be primarily mediated by TRβ in man because individuals with mutations in TRβ show major abnormalities in circulating thyroid hormone levels and have inappropriately elevated TSH levels (227). In contrast, whereas mutation of TRα is associated with clinical signs of severe hypothyroidism, it results in slightly low circulating levels of T4 and normal levels of T3 and TSH (228). This observation is also consistent with the concept that TRα1 may have an activating effect on the synthesis of TRH in hypophysiotropic TRH neurons (see Section III).
Leptin is also likely to have an important role in the regulation of the thyroid axis in man, as suggested by the close correlation between the circadian rhythm of leptin secretion and TSH (229). Circulating levels of both hormones are the highest between midnight and early morning and lowest in the early to midafternoon (229). In addition, subjects with congenital leptin deficiency, due to mutations in the leptin gene, have impaired thyroid function characterized by low levels of T4, inappropriately normal levels of TSH, dysregulation of TSH pulsatility and circadian rhythms, and/or an exaggerated response to exogenously administered TRH that is a characteristic feature of central hypothyroidism (229, 230). The thyroid phenotype, however, tends to be heterogeneous among the various families described (229, 230). In the Pakistani family with leptin deficiency reported by Montague et al (231), leptin replacement therapy significantly increased their T3 and T4 levels that were maintained at these higher levels with continued leptin treatment (232). Leptin also appears to orchestrate the suppression in circulating thyroid hormone levels during fasting and other disorders associated with a reduction in caloric intake in man such as hypothalamic amenorrhea and eating disorders. Leptin administration to normal individuals undergoing a 3-day fast restored fasting-induced effects on TSH pulsatility and increased T4 levels (233). In addition, leptin administration to individuals undergoing a more prolonged period of a less severe caloric restriction prevented the fall in T3 and T4 levels (234), although a similar response was not observed in a separate study working with obese patients (235). Leptin and thyroid hormone levels also tend to be low in individuals with hypothalamic amenorrhea induced by strenuous exercise and eating disorders (236). These alterations can be reversed with leptin replacement (237). Although there has been a reluctance to use leptin to treat patients with anorexia nervosa due to its effects on suppressing appetite and increasing energy expenditure, a correlation between low thyroid hormone and leptin levels is also well described in this disorder (238). Presumably, as in rodents, this response is homeostatic to reduce energy expenditure. Whether leptin is also responsible for increases in thyroid hormone levels observed in obese individuals is uncertain, but a correlation between increased leptin levels and higher T3 and TSH levels has been observed in obese patients (239).
A somewhat more complex issue is whether leptin is involved in the development of the so-called “nonthyroidal illness syndrome” or “euthyroid sick syndrome,” observed in approximately 40–70% of patients with severe illness in intensive care settings (240, 241). Low circulating levels of thyroid hormones but seemingly inappropriately low or normal TSH levels, diminished TSH pulsatility, and decreased TRH gene expression in the PVN are observed, suggesting the presence of central hypothyroidism, but the changes are completely reversible when the patient recovers (193, 240, 241). The decrease in thyroid hormone levels may be a homeostatic response to conserve energy stores under these adverse conditions and consistent with the physiology described above in experimental animals and human subjects associated with fasting. In support of this hypothesis, recent studies by Casaer et al (242) and Langouche et al (243) have demonstrated that early nutrient restoration to patients with critical illness in an intensive care unit setting is associated with greater complications and a more prolonged hospitalization. A very low T4 level, however, is predictive of poor outcomes in the intensive care unit setting, and perhaps an even better indicator of the severity of illness than the APACHE II score (244, 245). When T4 falls to less than 4 μg/dL, the risk of death rises to approximately 50%, and when T4 falls to < 2 μg/dL, mortality increases even more to approximately 80% (246–248). Thus, when T4 begins to decline, it remains unclear whether this is the progression of an adaptive response and does not require therapy, or whether it is indicative of a maladaptive response and should be vigorously treated to restore circulating thyroid hormone levels to normal. Evidence that the mechanism for central hypothyroidism associated with infection is vastly different than that elucidated for fasting, cited in Section VII, would give credence to the concern that the nonthyroidal illness syndrome associated with severe illness may not be a homeostatic mechanism mediated by leptin but is, in fact, maladaptive and pathophysiological (246). Namely, central hypothyroidism may be an epiphenomenon of infection-induced up-regulation of D2 in tanycytes causing hypothalamic hyperthyroidism and not part of an intended, homeostatic, physiological response to reduce metabolic rate and energy expenditure to promote survival.
A number of small clinical trials have attempted to determine whether thyroid hormone replacement in intensive care unit patients has any beneficial or detrimental effects on overall outcomes (246, 247, 249). Most of these studies have shown T4 or T3 to be safe and well tolerated (250, 251). Improvement in cardiac hemodynamic parameters including cardiac output, end diastolic volume, and stroke volume, and a reduction in peripheral arterial resistance have been observed in patients receiving T3 after coronary artery bypass surgery and/or in patients with dilated cardiomyopathy (249, 252–255), but at the expense of further suppression in circulating levels of TSH. However, other studies have not found a benefit, as summarized by Kaptein et al (256). It could be argued, however, that because of the associated rise in D1 and D3 in peripheral tissues in these patients (257), neither T4 nor T3 is appropriate therapy due to the effects of these enzymes to increase the conversion of T4 to rT3 (rather than T3) or degrade T3, respectively (246). The appropriate studies to determine whether correcting the HPT axis to normal in patients with severe illness, therefore, have not been conducted. A novel approach for the treatment of the nonthyroidal illness syndrome in critically ill patients has recently been suggested by Van den Berghe et al (258, 259). Using a continuous infusion of TRH together with a GH secretagogue restored thyroid hormone levels and TSH pulsatility in these patients and also improved catabolic parameters. However, carefully controlled studies are needed to determine whether this approach is associated with improved outcomes and reduced mortality.
X. Conclusion
Maintenance of circulating levels of thyroid hormones is controlled by what is classically called a “simple” negative feedback mechanism exerted on hypophysiotropic TRH neurons in the PVN. This regulatory mechanism, however, is vastly more complex, being modulated by glial-neuronal and neural-neural interactions as well as other circulating, humoral substances such as leptin. Given that hypophysiotropic TRH neurons are innervated by axons containing multiple peptides whose origin and physiological significance has not yet been determined (Figure 6), it is likely that the regulatory mechanisms involved are even more complex. Presumably, this finely tuned machinery provides flexibility to the HPT axis, enabling continuous adaptation to the constantly changing internal and external milieu. In most situations, alterations in the setpoint for negative feedback regulation would appear to be adaptive, contributing to the optimization of energy homeostasis. Elucidation of the basic mechanisms involved in central regulation of the thyroid axis, however, raise questions as to whether under certain circumstances, such as prolonged critical illness, regulation of the HPT axis may be maladaptive. Further research to elucidate the neurobiology involved in central regulation of hypophysiotropic TRH neurons will be instrumental in determining how these disorders give rise to alterations in the HPT axis and will facilitate the appropriate approach to therapy.
Acknowledgments
The authors give special thanks to Erzsébet Farkas for drawing the schematic illustrations.
This work was supported by grants from the Hungarian Science Foundation (OTKA K81845), Seventh EU Research Framework Programme (Health-F2–2010-259772), the Lendület Award of the Hungarian Academy of Sciences, the National Institutes of Health (DK-37021) and the Dr Gerald J. and Dorothy R. Friedman New York Foundation for Medical Research.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- AGRP
- agouti-related protein
- BAT
- brown adipose tissue
- CART
- cocaine and amphetamine-regulated transcript
- CRE
- cAMP response element
- CREB
- CRE binding protein
- CSF
- cerebrospinal fluid
- D2
- type 2 iodothyronine deiodinase
- D3
- type 3 iodothyronine deiodinase
- DIA
- dehydration-induced anorexia
- DIO
- HFD-induced obese
- DMN
- hypothalamic dorsomedial nucleus
- GABA
- γ-amino butyric acid
- HFD
- high-fat diet
- HPT
- hypothalamic-pituitary-thyroid
- IR
- immunoreactive
- KO
- knockout
- LPS
- lipopolysaccharide
- MBH
- mediobasal hypothalamus
- MC4R
- melanocortin 4 receptor
- MCT
- monocarboxylate transporter
- NF-κB
- nuclear factor-κB
- NPY
- neuropeptide Y
- NTS
- nucleus tractus solitarius
- OATP
- organic anion-transporting polypeptide
- PC
- prohormone convertase
- POMC
- proopiomelanocortin
- PPII
- pyroglutamyl peptidase II
- PVN
- hypothalamic paraventricular nucleus
- siRNA
- small interfering RNA
- STAT3
- signal transducers and activators of transcription-3
- TGN
- trans-Golgi network
- TLR4
- Toll-like receptor 4
- TR
- thyroid hormone receptor
- TRE
- thyroid hormone response element.
References
- 1. Lechan RM, Hollenberg A, Fekete C. Hypothalamic-pituitary-thyroid axis: organization, neural/endocrine control of TRH. In: Squire LR, ed. Encyclopedia of Neuroscience. Oxford, UK: Academic Press; 2009:75–87 [Google Scholar]
- 2. Reichlin S. TRH: historical aspects. Ann NY Acad Sci. 1989;553:1–6 [DOI] [PubMed] [Google Scholar]
- 3. Boler J, Enzmann F, Folkers K, Bowers CY, Schally AV. The identity of chemical and hormonal properties of the thyrotropin releasing hormone and pyroglutamyl-histidyl-proline amide. Biochem Biophys Res Commun. 1969;37:705–710 [DOI] [PubMed] [Google Scholar]
- 4. Burgus R, Dunn TF, Desiderio D, Guillemin R. [Molecular structure of the hypothalamic hypophysiotropic TRF factor of ovine origin: mass spectrometry demonstration of the PCA-His-Pro-NH2 sequence]. C R Acad Sci Hebd Seances Acad Sci D. 1969;269:1870–1873 [PubMed] [Google Scholar]
- 5. Hashimoto K, Zanger K, Hollenberg AN, Cohen LE, Radovick S, Wondisford FE. cAMP response element-binding protein-binding protein mediates thyrotropin-releasing hormone signaling on thyrotropin subunit genes. J Biol Chem. 2000;275:33365–33372 [DOI] [PubMed] [Google Scholar]
- 6. Persani L. Hypothalamic thyrotropin-releasing hormone and thyrotropin biological activity. Thyroid. 1998;8:941–946 [DOI] [PubMed] [Google Scholar]
- 7. Harris AR, Christianson D, Smith MS, Fang SL, Braverman LE, Vagenakis AG. The physiological role of thyrotropin-releasing hormone in the regulation of thyroid-stimulating hormone and prolactin secretion in the rat. J Clin Invest. 1978;61:441–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rabeler R, Mittag J, Geffers L, et al. Generation of thyrotropin-releasing hormone receptor 1-deficient mice as an animal model of central hypothyroidism. Mol Endocrinol. 2004;18:1450–1460 [DOI] [PubMed] [Google Scholar]
- 9. Grossmann M, Szkudlinski MW, Tropea JE, et al. Expression of human thyrotropin in cell lines with different glycosylation patterns combined with mutagenesis of specific glycosylation sites. Characterization of a novel role for the oligosaccharides in the in vitro and in vivo bioactivity. J Biol Chem. 1995;270:29378–29385 [DOI] [PubMed] [Google Scholar]
- 10. Magner JA, Kane J, Chou ET. Intravenous thyrotropin (TSH)-releasing hormone releases human TSH that is structurally different from basal TSH. J Clin Endocrinol Metab. 1992;74:1306–1311 [DOI] [PubMed] [Google Scholar]
- 11. Taylor T, Weintraub BD. Altered thyrotropin (TSH) carbohydrate structures in hypothalamic hypothyroidism created by paraventricular nuclear lesions are corrected by in vivo TSH-releasing hormone administration. Endocrinology. 1989;125:2198–2203 [DOI] [PubMed] [Google Scholar]
- 12. Shibusawa N, Yamada M, Hirato J, Monden T, Satoh T, Mori M. Requirement of thyrotropin-releasing hormone for the postnatal functions of pituitary thyrotrophs: ontogeny study of congenital tertiary hypothyroidism in mice. Mol Endocrinol. 2000;14:137–146 [DOI] [PubMed] [Google Scholar]
- 13. Beck-Peccoz P, Amr S, Menezes-Ferreira MM, Faglia G, Weintraub BD. Decreased receptor binding of biologically inactive thyrotropin in central hypothyroidism. Effect of treatment with thyrotropin-releasing hormone. N Engl J Med. 1985;312:1085–1090 [DOI] [PubMed] [Google Scholar]
- 14. Richter K, Kawashima E, Egger R, Kreil G. Biosynthesis of thyrotropin releasing hormone in the skin of Xenopus laevis: partial sequence of the precursor deduced from cloned cDNA. EMBO J. 1984;3:617–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lechan RM, Wu P, Jackson IM, et al. Thyrotropin-releasing hormone precursor: characterization in rat brain. Science. 1986;231:159–161 [DOI] [PubMed] [Google Scholar]
- 16. Lee SL, Stewart K, Goodman RH. Structure of the gene encoding rat thyrotropin releasing hormone. J Biol Chem. 1988;263:16604–16609 [PubMed] [Google Scholar]
- 17. Yamada M, Satoh T, Monden T, Mori M. Assignment of the thyrotropin-releasing hormone gene (TRH) to human chromosome 3q13.3–>q21 by in situ hybridization. Cytogenet Cell Genet. 1999;87:275. [DOI] [PubMed] [Google Scholar]
- 18. Roller ML, Camper SA. Localization of the thyrotropin-releasing hormone gene, Trh, on mouse chromosome 6. Mamm Genome. 1995;6:443–444 [DOI] [PubMed] [Google Scholar]
- 19. Yamada M, Wondisford FE, Radovick S, Nakayama Y, Weintraub BD, Wilber JF. Assignment of human preprothyrotropin-releasing hormone (TRH) gene to chromosome 3. Somat Cell Mol Genet. 1991;17:97–100 [DOI] [PubMed] [Google Scholar]
- 20. Hollenberg AN, Monden T, Flynn TR, Boers ME, Cohen O, Wondisford FE. The human thyrotropin-releasing hormone gene is regulated by thyroid hormone through two distinct classes of negative thyroid hormone response elements. Mol Endocrinol. 1995;9:540–550 [DOI] [PubMed] [Google Scholar]
- 21. Díaz-Gallardo MY, Cote-Vélez A, Carreón-Rodríguez A, Charli JL, Joseph-Bravo P. Phosphorylated cyclic-AMP-response element-binding protein and thyroid hormone receptor have independent response elements in the rat thyrotropin-releasing hormone promoter: an analysis in hypothalamic cells. Neuroendocrinology. 2010;91:64–76 [DOI] [PubMed] [Google Scholar]
- 22. Satoh T, Yamada M, Iwasaki T, Mori M. Negative regulation of the gene for the preprothyrotropin-releasing hormone from the mouse by thyroid hormone requires additional factors in conjunction with thyroid hormone receptors. J Biol Chem. 1996;271:27919–27926 [DOI] [PubMed] [Google Scholar]
- 23. Wilber JF, Xu AH. The thyrotropin-releasing hormone gene 1998: cloning, characterization, and transcriptional regulation in the central nervous system, heart, and testis. Thyroid. 1998;8:897–901 [DOI] [PubMed] [Google Scholar]
- 24. Harris M, Aschkenasi C, Elias CF, et al. Transcriptional regulation of the thyrotropin-releasing hormone gene by leptin and melanocortin signaling. J Clin Invest. 2001;107:111–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pérez-Monter C, Martínez-Armenta M, Miquelajauregui A, et al. The Krüppel-like factor 4 controls biosynthesis of thyrotropin-releasing hormone during hypothalamus development. Mol Cell Endocrinol. 2011;333:127–133 [DOI] [PubMed] [Google Scholar]
- 26. Sevarino KA, Goodman RH, Spiess J, Jackson IM, Wu P. Thyrotropin-releasing hormone (TRH) precursor processing. Characterization of mature TRH and non-TRH peptides synthesized by transfected mammalian cells. J Biol Chem. 1989;264:21529–21535 [PubMed] [Google Scholar]
- 27. Nillni EA, Sevarino KA. The biology of pro-thyrotropin-releasing hormone-derived peptides. Endocr Rev. 1999;20:599–648 [DOI] [PubMed] [Google Scholar]
- 28. Lechan RM, Qi Y, Berrodin TJ, et al. Immunocytochemical delineation of thyroid hormone receptor β 2-like immunoreactivity in the rat central nervous system. Endocrinology. 1993;132:2461–2469 [DOI] [PubMed] [Google Scholar]
- 29. Cyr NE, Stuart RC, Zhu X, Steiner DF, Nillni EA. Biosynthesis of proTRH-derived peptides in prohormone convertase 1 and 2 knockout mice. Peptides. 2012;35:42–48 [DOI] [PubMed] [Google Scholar]
- 30. Schilling S, Kohlmann S, Bäuscher C, et al. Glutaminyl cyclase knock-out mice exhibit slight hypothyroidism but no hypogonadism: implications for enzyme function and drug development. J Biol Chem. 2011;286:14199–14208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Perello M, Stuart R, Nillni EA. Prothyrotropin-releasing hormone targets its processing products to different vesicles of the secretory pathway. J Biol Chem. 2008;283:19936–19947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Charli JL, Vargas MA, Cisneros M, et al. TRH inactivation in the extracellular compartment: role of pyroglutamyl peptidase II. Neurobiology (Bp). 1998;6:45–57 [PubMed] [Google Scholar]
- 33. Heuer H, Schäfer MK, Bauer K. The thyrotropin-releasing hormone-degrading ectoenzyme: the third element of the thyrotropin-releasing hormone-signaling system. Thyroid. 1998;8:915–920 [DOI] [PubMed] [Google Scholar]
- 34. Sanchez E, Charli JL, Lechan RM. Pyroglutamyl-peptidase II. In: Rawlings ND, Salvesen G, eds. Handbook of Proteolytic Enzymes. 3rd ed London, UK: Academic Press; 2013:414–419 [Google Scholar]
- 35. Sánchez E, Vargas MA, Singru PS, et al. Tanycyte pyroglutamyl peptidase II contributes to regulation of the hypothalamic-pituitary-thyroid axis through glial-axonal associations in the median eminence. Endocrinology. 2009;150:2283–2291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schmitmeier S, Thole H, Bader A, Bauer K. Purification and characterization of the thyrotropin-releasing hormone (TRH)-degrading serum enzyme and its identification as a product of liver origin. Eur J Biochem. 2002;269:1278–1286 [DOI] [PubMed] [Google Scholar]
- 37. Lechan RM, Fekete C. The TRH neuron: a hypothalamic integrator of energy metabolism. Prog Brain Res. 2006;153:209–235 [DOI] [PubMed] [Google Scholar]
- 38. Swanson LW, Sawchenko PE. Hypothalamic integration: organization of the paraventricular and supraoptic nuclei. Annu Rev Neurosci. 1983;6:269–324 [DOI] [PubMed] [Google Scholar]
- 39. Lechan RM, Segerson TP. Pro-TRH gene expression and precursor peptides in rat brain. Observations by hybridization analysis and immunocytochemistry. Ann NY Acad Sci. 1989;553:29–59 [DOI] [PubMed] [Google Scholar]
- 40. Ishikawa K, Taniguchi Y, Inoue K, Kurosumi K, Suzuki M. Immunocytochemical delineation of thyrotrophic area: origin of thyrotropin-releasing hormone in the median eminence. Neuroendocrinology. 1988;47:384–388 [DOI] [PubMed] [Google Scholar]
- 41. Merchenthaler I, Liposits Z. Mapping of thyrotropin-releasing hormone (TRH) neuronal systems of rat forebrain projecting to the median eminence and the OVLT. Immunocytochemistry combined with retrograde labeling at the light and electron microscopic levels. Acta Biol Hung. 1994;45:361–374 [PubMed] [Google Scholar]
- 42. Fekete C, Mihály E, Luo LG, et al. Association of cocaine- and amphetamine-regulated transcript-immunoreactive elements with thyrotropin-releasing hormone-synthesizing neurons in the hypothalamic paraventricular nucleus and its role in the regulation of the hypothalamic-pituitary-thyroid axis during fasting. J Neurosci. 2000;20:9224–9234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Biag J, Huang Y, Gou L, et al. Cyto- and chemoarchitecture of the hypothalamic paraventricular nucleus in the C57BL/6J male mouse: a study of immunostaining and multiple fluorescent tract tracing. J Comp Neurol. 2012;520:6–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kádár A, Sánchez E, Wittmann G, et al. Distribution of hypophysiotropic thyrotropin-releasing hormone (TRH)-synthesizing neurons in the hypothalamic paraventricular nucleus of the mouse. J Comp Neurol. 2010;518:3948–3961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Fliers E, Noppen NW, Wiersinga WM, Visser TJ, Swaab DF. Distribution of thyrotropin-releasing hormone (TRH)-containing cells and fibers in the human hypothalamus. J Comp Neurol. 1994;350:311–323 [DOI] [PubMed] [Google Scholar]
- 46. Mihály E, Fekete C, Tatro JB, Liposits Z, Stopa EG, Lechan RM. Hypophysiotropic thyrotropin-releasing hormone-synthesizing neurons in the human hypothalamus are innervated by neuropeptide Y, agouti-related protein, and α-melanocyte-stimulating hormone. J Clin Endocrinol Metab. 2000;85:2596–2603 [DOI] [PubMed] [Google Scholar]
- 47. Freeman ME, Kanyicska B, Lerant A, Nagy G. Prolactin: structure, function, and regulation of secretion. Physiol Rev. 2000;80:1523–1631 [DOI] [PubMed] [Google Scholar]
- 48. Broberger C. Hypothalamic cocaine- and amphetamine-regulated transcript (CART) neurons: histochemical relationship to thyrotropin-releasing hormone, melanin-concentrating hormone, orexin/hypocretin and neuropeptide Y. Brain Res. 1999;848:101–113 [DOI] [PubMed] [Google Scholar]
- 49. Fekete C, Lechan RM. Neuroendocrine implications for the association between cocaine- and amphetamine regulated transcript (CART) and hypophysiotropic thyrotropin-releasing hormone (TRH). Peptides. 2006;27:2012–2018 [DOI] [PubMed] [Google Scholar]
- 50. Hrabovszky E, Wittmann G, Turi GF, Liposits Z, Fekete C. Hypophysiotropic thyrotropin-releasing hormone and corticotropin-releasing hormone neurons of the rat contain vesicular glutamate transporter-2. Endocrinology. 2005;146:341–347 [DOI] [PubMed] [Google Scholar]
- 51. Kallo I, Mohacsik P, Vida B, et al. A novel pathway regulates thyroid hormone availability in rat and human hypothalamic neurosecretory neurons. PLoS One. 2012;7:e37860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Fekete C, Légrádi G, Mihály E, et al. α-Melanocyte-stimulating hormone is contained in nerve terminals innervating thyrotropin-releasing hormone-synthesizing neurons in the hypothalamic paraventricular nucleus and prevents fasting-induced suppression of prothyrotropin-releasing hormone gene expression. J Neurosci. 2000;20:1550–1558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wittmann G, Sarkar S, Hrabovszky E, Liposits Z, Lechan RM, Fekete C. Galanin- but not galanin-like peptide-containing axon terminals innervate hypophysiotropic TRH-synthesizing neurons in the hypothalamic paraventricular nucleus. Brain Res. 2004;1002:43–50 [DOI] [PubMed] [Google Scholar]
- 54. Wittmann G, Füzesi T, Singru PS, Liposits Z, Lechan RM, Fekete C. Efferent projections of thyrotropin-releasing hormone-synthesizing neurons residing in the anterior parvocellular subdivision of the hypothalamic paraventricular nucleus. J Comp Neurol. 2009;515:313–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wittmann G, Füzesi T, Liposits Z, Lechan RM, Fekete C. Distribution and axonal projections of neurons coexpressing thyrotropin-releasing hormone and urocortin 3 in the rat brain. J Comp Neurol. 2009;517:825–840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Schwartz MW, Woods SC, Porte D, Jr, Seeley RJ, Baskin DG. Central nervous system control of food intake. Nature. 2000;404:661–671 [DOI] [PubMed] [Google Scholar]
- 57. Shintani M, Tamura Y, Monden M, Shiomi H. Thyrotropin-releasing hormone induced thermogenesis in Syrian hamsters: site of action and receptor subtype. Brain Res. 2005;1039:22–29 [DOI] [PubMed] [Google Scholar]
- 58. Fekete C, Lechan RM. Negative feedback regulation of hypophysiotropic thyrotropin-releasing hormone (TRH) synthesizing neurons: role of neuronal afferents and type 2 deiodinase. Front Neuroendocrinol. 2007;28:97–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ambach G, Palkovits M. Blood supply of the rat hypothalamus. II. Nucleus paraventricularis. Acta Morphol Acad Sci Hung. 1974;22:311–320 [PubMed] [Google Scholar]
- 60. Ambach G, Palkovits M, Szentágothai J. Blood supply of the rat hypothalamus. IV. Retrochiasmatic area, median eminence, arcuate nucleus. Acta Morphol Acad Sci Hung. 1976;24:93–119 [PubMed] [Google Scholar]
- 61. Füzesi T, Wittmann G, Lechan RM, Liposits Z, Fekete C. Noradrenergic innervation of hypophysiotropic thyrotropin-releasing hormone-synthesizing neurons in rats. Brain Res. 2009;1294:38–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Toni R, Jackson IM, Lechan RM. Neuropeptide-Y-immunoreactive innervation of thyrotropin-releasing hormone-synthesizing neurons in the rat hypothalamic paraventricular nucleus. Endocrinology. 1990;126:2444–2453 [DOI] [PubMed] [Google Scholar]
- 63. Légrádi G, Lechan RM. Agouti-related protein containing nerve terminals innervate thyrotropin-releasing hormone neurons in the hypothalamic paraventricular nucleus. Endocrinology. 1999;140:3643–3652 [DOI] [PubMed] [Google Scholar]
- 64. Wittmann G, Liposits Z, Lechan RM, Fekete C. Medullary adrenergic neurons contribute to the neuropeptide Y-ergic innervation of hypophysiotropic thyrotropin-releasing hormone-synthesizing neurons in the rat. Neurosci Lett. 2002;324:69–73 [DOI] [PubMed] [Google Scholar]
- 65. Légrádi G, Lechan RM. The arcuate nucleus is the major source for neuropeptide Y-innervation of thyrotropin-releasing hormone neurons in the hypothalamic paraventricular nucleus. Endocrinology. 1998;139:3262–3270 [DOI] [PubMed] [Google Scholar]
- 66. Fekete C, Wittmann G, Liposits Z, Lechan RM. Origin of cocaine- and amphetamine-regulated transcript (CART)-immunoreactive innervation of the hypothalamic paraventricular nucleus. J Comp Neurol. 2004;469:340–350 [DOI] [PubMed] [Google Scholar]
- 67. Fekete C, Sarkar S, Lechan RM. Relative contribution of brainstem afferents to the cocaine- and amphetamine-regulated transcript (CART) innervation of thyrotropin-releasing hormone synthesizing neurons in the hypothalamic paraventricular nucleus (PVN). Brain Res. 2005;1032:171–175 [DOI] [PubMed] [Google Scholar]
- 68. Menyhért J, Wittmann G, Lechan RM, Keller E, Liposits Z, Fekete C. Cocaine- and amphetamine-regulated transcript (CART) is colocalized with the orexigenic neuropeptide Y and agouti-related protein and absent from the anorexigenic α-melanocyte-stimulating hormone neurons in the infundibular nucleus of the human hypothalamus. Endocrinology. 2007;148:4276–4281 [DOI] [PubMed] [Google Scholar]
- 69. Bernardis LL, Bellinger LL. The dorsomedial hypothalamic nucleus revisited: 1998 update. Proc Soc Exp Biol Med. 1998;218:284–306 [DOI] [PubMed] [Google Scholar]
- 70. Scott MM, Lachey JL, Sternson SM, et al. Leptin targets in the mouse brain. J Comp Neurol. 2009;514:518–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Singru PS, Fekete C, Lechan RM. Neuroanatomical evidence for participation of the hypothalamic dorsomedial nucleus (DMN) in regulation of the hypothalamic paraventricular nucleus (PVN) by α-melanocyte stimulating hormone. Brain Res. 2005;1064:42–51 [DOI] [PubMed] [Google Scholar]
- 72. Fuller PM, Lu J, Saper CB. Differential rescue of light- and food-entrainable circadian rhythms. Science. 2008;320:1074–1077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Bellinger LL, Bernardis LL, McCusker RH, Campion DR. Plasma hormone levels in growth-retarded rats with dorsomedial hypothalamic lesions. Physiol Behav. 1985;34:783–790 [DOI] [PubMed] [Google Scholar]
- 74. Mihály E, Fekete C, Légrádi G, Lechan RM. Hypothalamic dorsomedial nucleus neurons innervate thyrotropin-releasing hormone-synthesizing neurons in the paraventricular nucleus. Brain Res. 2001;891:20–31 [DOI] [PubMed] [Google Scholar]
- 75. Itoi K, Sugimoto N. The brainstem noradrenergic systems in stress, anxiety and depression. J Neuroendocrinol. 2010;22:355–361 [DOI] [PubMed] [Google Scholar]
- 76. Wittmann G. Regulation of hypophysiotrophic corticotrophin-releasing hormone- and thyrotrophin-releasing hormone-synthesising neurones by brainstem catecholaminergic neurones. J Neuroendocrinol. 2008;20:952–960 [DOI] [PubMed] [Google Scholar]
- 77. Liposits Z, Paull WK, Wu P, Jackson IM, Lechan RM. Hypophysiotrophic thyrotropin releasing hormone (TRH) synthesizing neurons. Ultrastructure, adrenergic innervation and putative transmitter action. Histochemistry. 1987;88:1–10 [DOI] [PubMed] [Google Scholar]
- 78. Cote-Vélez A, Pérez-Martínez L, Díaz-Gallardo MY, et al. Dexamethasone represses cAMP rapid upregulation of TRH gene transcription: identification of a composite glucocorticoid response element and a cAMP response element in TRH promoter. J Mol Endocrinol. 2005;34:177–197 [DOI] [PubMed] [Google Scholar]
- 79. Cunningham ET, Jr, Bohn MC, Sawchenko PE. Organization of adrenergic inputs to the paraventricular and supraoptic nuclei of the hypothalamus in the rat. J Comp Neurol. 1990;292:651–667 [DOI] [PubMed] [Google Scholar]
- 80. Wittmann G, Liposits Z, Lechan RM, Fekete C. Medullary adrenergic neurons contribute to the cocaine- and amphetamine-regulated transcript-immunoreactive innervation of thyrotropin-releasing hormone synthesizing neurons in the hypothalamic paraventricular nucleus. Brain Res. 2004;1006:1–7 [DOI] [PubMed] [Google Scholar]
- 81. Légrádi G, Hannibal J, Lechan RM. Association between pituitary adenylate cyclase-activating polypeptide and thyrotropin-releasing hormone in the rat hypothalamus. J Chem Neuroanat. 1997;13:265–279 [DOI] [PubMed] [Google Scholar]
- 82. Sawchenko PE, Swanson LW, Grzanna R, Howe PR, Bloom SR, Polak JM. Colocalization of neuropeptide Y immunoreactivity in brainstem catecholaminergic neurons that project to the paraventricular nucleus of the hypothalamus. J Comp Neurol. 1985;241:138–153 [DOI] [PubMed] [Google Scholar]
- 83. Cunningham ET, Jr, Sawchenko PE. Anatomical specificity of noradrenergic inputs to the paraventricular and supraoptic nuclei of the rat hypothalamus. J Comp Neurol. 1988;274:60–76 [DOI] [PubMed] [Google Scholar]
- 84. Fekete C, Wittmann G, Liposits Z, Lechan RM. GABA-ergic innervation of thyrotropin-releasing hormone-synthesizing neurons in the hypothalamic paraventricular nucleus of the rat. Brain Res. 2002;957:251–258 [DOI] [PubMed] [Google Scholar]
- 85. Toni R, Jackson IM, Lechan RM. Thyrotropin-releasing-hormone-immunoreactive innervation of thyrotropin-releasing-hormone-tuberoinfundibular neurons in rat hypothalamus: anatomical basis to suggest ultrashort feedback regulation. Neuroendocrinology. 1990;52:422–428 [DOI] [PubMed] [Google Scholar]
- 86. Liao N, Vaudry H, Pelletier G. Neuroanatomical connections between corticotropin-releasing factor (CRF) and somatostatin (SRIF) nerve endings and thyrotropin-releasing hormone (TRH) neurons in the paraventricular nucleus of rat hypothalamus. Peptides. 1992;13:677–680 [DOI] [PubMed] [Google Scholar]
- 87. Deli L, Wittmann G, Kalló I, et al. Type 1 cannabinoid receptor-containing axons innervate hypophysiotropic thyrotropin-releasing hormone-synthesizing neurons. Endocrinology. 2009;150:98–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Kiss J, Halász B. Ultrastructural analysis of the innervation of TRH-immunoreactive neuronal elements located in the periventricular subdivision of the paraventricular nucleus of the rat hypothalamus. Brain Res. 1990;532:107–114 [DOI] [PubMed] [Google Scholar]
- 89. Lechan RM, Fekete C. Infundibular tanycytes as modulators of neuroendocrine function: hypothetical role in the regulation of the thyroid and gonadal axis. Acta Biomed. 2007;78(suppl 1):84–98 [PubMed] [Google Scholar]
- 90. Rodríguez EM, Blázquez JL, Pastor FE, et al. Hypothalamic tanycytes: a key component of brain-endocrine interaction. Int Rev Cytol. 2005;247:89–164 [DOI] [PubMed] [Google Scholar]
- 91. Peruzzo B, Pastor FE, Blázquez JL, et al. A second look at the barriers of the medial basal hypothalamus. Exp Brain Res. 2000;132:10–26 [DOI] [PubMed] [Google Scholar]
- 92. Prevot V, Bellefontaine N, Baroncini M, et al. Gonadotrophin-releasing hormone nerve terminals, tanycytes and neurohaemal junction remodelling in the adult median eminence: functional consequences for reproduction and dynamic role of vascular endothelial cells. J Neuroendocrinol. 2010;22:639–649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Yamamura T, Yasuo S, Hirunagi K, Ebihara S, Yoshimura T. T(3) implantation mimics photoperiodically reduced encasement of nerve terminals by glial processes in the median eminence of Japanese quail. Cell Tissue Res. 2006;324:175–179 [DOI] [PubMed] [Google Scholar]
- 94. Bianco AC, Salvatore D, Gereben B, Berry MJ, Larsen PR. Biochemistry, cellular and molecular biology, and physiological roles of the iodothyronine selenodeiodinases. Endocr Rev. 2002;23:38–89 [DOI] [PubMed] [Google Scholar]
- 95. Diano S, Leonard JL, Meli R, Esposito E, Schiavo L. Hypothalamic type II iodothyronine deiodinase: a light and electron microscopic study. Brain Res. 2003;976:130–134 [DOI] [PubMed] [Google Scholar]
- 96. Guadaño-Ferraz A, Obregón MJ, St Germain DL, Bernal J. The type 2 iodothyronine deiodinase is expressed primarily in glial cells in the neonatal rat brain. Proc Natl Acad Sci USA. 1997;94:10391–10396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Tu HM, Kim SW, Salvatore D, et al. Regional distribution of type 2 thyroxine deiodinase messenger ribonucleic acid in rat hypothalamus and pituitary and its regulation by thyroid hormone. Endocrinology. 1997;138:3359–3368 [DOI] [PubMed] [Google Scholar]
- 98. Riskind PN, Kolodny JM, Larsen PR. The regional hypothalamic distribution of type II 5′-monodeiodinase in euthyroid and hypothyroid rats. Brain Res. 1987;420:194–198 [DOI] [PubMed] [Google Scholar]
- 99. Fekete C, Gereben B, Doleschall M, et al. Lipopolysaccharide induces type 2 iodothyronine deiodinase in the mediobasal hypothalamus: implications for the nonthyroidal illness syndrome. Endocrinology. 2004;145:1649–1655 [DOI] [PubMed] [Google Scholar]
- 100. Fekete C, Freitas BC, Zeöld A, et al. Expression patterns of WSB-1 and USP-33 underlie cell-specific posttranslational control of type 2 deiodinase in the rat brain. Endocrinology. 2007;148:4865–4874 [DOI] [PubMed] [Google Scholar]
- 101. Alkemade A, Friesema EC, Unmehopa UA, et al. Neuroanatomical pathways for thyroid hormone feedback in the human hypothalamus. J Clin Endocrinol Metab. 2005;90:4322–4334 [DOI] [PubMed] [Google Scholar]
- 102. Roberts LM, Woodford K, Zhou M, et al. Expression of the thyroid hormone transporters monocarboxylate transporter-8 (SLC16A2) and organic ion transporter-14 (SLCO1C1) at the blood-brain barrier. Endocrinology. 2008;149:6251–6261 [DOI] [PubMed] [Google Scholar]
- 103. Murphy M, Jethwa PH, Warner A, et al. Effects of manipulating hypothalamic triiodothyronine concentrations on seasonal body weight and torpor cycles in Siberian hamsters. Endocrinology. 2012;153:101–112 [DOI] [PubMed] [Google Scholar]
- 104. Amenta F, Caporuscio D, Ferrante F, Porcelli F, Zomparelli M. Cholinergic nerves in the thyroid gland. Cell Tissue Res. 1978;195:367–370 [DOI] [PubMed] [Google Scholar]
- 105. Melander A, Sundler F, Westgren U. Sympathetic innervation of the thyroid: variation with species and with age. Endocrinology. 1975;96:102–106 [DOI] [PubMed] [Google Scholar]
- 106. Kalsbeek A, Fliers E, Franke AN, Wortel J, Buijs RM. Functional connections between the suprachiasmatic nucleus and the thyroid gland as revealed by lesioning and viral tracing techniques in the rat. Endocrinology. 2000;141:3832–3841 [DOI] [PubMed] [Google Scholar]
- 107. Klieverik L, Kalsbeek A, Flier E. 2005 Autonomic innervation of the thyroid gland and its functional implications. Hot Thyroidology: European Thyroid Association. http://www.hotthyroidology.com/editorial_152.html
- 108. Juvenal GJ, Pregliasco LB, Krawiec L, Bocanera LV, Silberschmidt D, Pisarev MA. Long-term effect of norepinephrine on iodide uptake in FRTL-5 cells. Thyroid. 1997;7:795–800 [DOI] [PubMed] [Google Scholar]
- 109. Boado RJ, Romeo HE, Chuluyan HE, Cageao L, Cardinali DP, Zaninovich AA. Evidence suggesting that the sympathetic nervous system mediates thyroidal depression in turpentine-induced nonthyroidal illness syndrome. Neuroendocrinology. 1991;53:360–364 [DOI] [PubMed] [Google Scholar]
- 110. Romeo HE, Díaz MC, Ceppi J, Zaninovich AA, Cardinali DP. Effect of inferior laryngeal nerve section on thyroid function in rats. Endocrinology. 1988;122:2527–2532 [DOI] [PubMed] [Google Scholar]
- 111. Michalkiewicz M, Huffman LJ, Dey M, Hedge GA. Endogenous neuropeptide Y regulates thyroid blood flow. Am J Physiol. 1993;264:E699–E705 [DOI] [PubMed] [Google Scholar]
- 112. Vella KR, Ramadoss P, Lam FS, et al. NPY and MC4R signaling regulate thyroid hormone levels during fasting through both central and peripheral pathways. Cell Metab. 2011;14:780–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Segerson TP, Kauer J, Wolfe HC, et al. Thyroid hormone regulates TRH biosynthesis in the paraventricular nucleus of the rat hypothalamus. Science. 1987;238:78–80 [DOI] [PubMed] [Google Scholar]
- 114. Sugrue ML, Vella KR, Morales C, Lopez ME, Hollenberg AN. The thyrotropin-releasing hormone gene is regulated by thyroid hormone at the level of transcription in vivo. Endocrinology. 2010;151:793–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Dyess EM, Segerson TP, Liposits Z, et al. Triiodothyronine exerts direct cell-specific regulation of thyrotropin-releasing hormone gene expression in the hypothalamic paraventricular nucleus. Endocrinology. 1988;123:2291–2297 [DOI] [PubMed] [Google Scholar]
- 116. Lechan RM, Qi Y, Jackson IM, Mahdavi V. Identification of thyroid hormone receptor isoforms in thyrotropin-releasing hormone neurons of the hypothalamic paraventricular nucleus. Endocrinology. 1994;135:92–100 [DOI] [PubMed] [Google Scholar]
- 117. Dupré SM, Guissouma H, Flamant F, et al. Both thyroid hormone receptor (TR)β 1 and TR β 2 isoforms contribute to the regulation of hypothalamic thyrotropin-releasing hormone. Endocrinology. 2004;145:2337–2345 [DOI] [PubMed] [Google Scholar]
- 118. Carreón-Rodríguez A, Charli JL, Pérez-Martínez L. T3 differentially regulates TRH expression in developing hypothalamic neurons in vitro. Brain Res. 2009;1305:20–30 [DOI] [PubMed] [Google Scholar]
- 119. Abel ED, Ahima RS, Boers ME, Elmquist JK, Wondisford FE. Critical role for thyroid hormone receptor β2 in the regulation of paraventricular thyrotropin-releasing hormone neurons. J Clin Invest. 2001;107:1017–1023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Guissouma H, Froidevaux MS, Hassani Z, Demeneix BA. In vivo siRNA delivery to the mouse hypothalamus confirms distinct roles of TR β isoforms in regulating TRH transcription. Neurosci Lett. 2006;406:240–243 [DOI] [PubMed] [Google Scholar]
- 121. Perello M, Friedman T, Paez-Espinosa V, Shen X, Stuart RC, Nillni EA. Thyroid hormones selectively regulate the posttranslational processing of prothyrotropin-releasing hormone in the paraventricular nucleus of the hypothalamus. Endocrinology. 2006;147:2705–2716 [DOI] [PubMed] [Google Scholar]
- 122. Decherf S, Seugnet I, Kouidhi S, Lopez-Juarez A, Clerget-Froidevaux MS, Demeneix BA. Thyroid hormone exerts negative feedback on hypothalamic type 4 melanocortin receptor expression. Proc Natl Acad Sci USA. 2010;107:4471–4476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Kakucska I, Rand W, Lechan RM. Thyrotropin-releasing hormone gene expression in the hypothalamic paraventricular nucleus is dependent upon feedback regulation by both triiodothyronine and thyroxine. Endocrinology. 1992;130:2845–2850 [DOI] [PubMed] [Google Scholar]
- 124. Fekete C, Mihály E, Herscovici S, et al. DARPP-32 and CREB are present in type 2 iodothyronine deiodinase-producing tanycytes: implications for the regulation of type 2 deiodinase activity. Brain Res. 2000;862:154–161 [DOI] [PubMed] [Google Scholar]
- 125. Fonseca TL, Correa-Medina M, Campos MP, et al. Coordination of hypothalamic and pituitary T3 production regulates TSH expression. J Clin Invest. 2013;123:1492–1500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Barrett P, Ebling FJ, Schuhler S, et al. Hypothalamic thyroid hormone catabolism acts as a gatekeeper for the seasonal control of body weight and reproduction. Endocrinology. 2007;148:3608–3617 [DOI] [PubMed] [Google Scholar]
- 127. Ross AW, Helfer G, Russell L, Darras VM, Morgan PJ. Thyroid hormone signalling genes are regulated by photoperiod in the hypothalamus of F344 rats. PLoS One. 2011;6:e21351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Broedel O, Eravci M, Fuxius S, et al. Effects of hyper- and hypothyroidism on thyroid hormone concentrations in regions of the rat brain. Am J Physiol Endocrinol Metab. 2003;285:E470–E480 [DOI] [PubMed] [Google Scholar]
- 129. Anguiano B, Quintanar A, Luna M, et al. Neuroendocrine regulation of adrenal gland and hypothalamus 5′deiodinase activity. II. Effects of splanchnicotomy and hypophysectomy. Endocrinology. 1995;136:3346–3352 [DOI] [PubMed] [Google Scholar]
- 130. Diano S, Naftolin F, Goglia F, Horvath TL. Fasting-induced increase in type II iodothyronine deiodinase activity and messenger ribonucleic acid levels is not reversed by thyroxine in the rat hypothalamus. Endocrinology. 1998;139:2879–2884 [DOI] [PubMed] [Google Scholar]
- 131. Leonard JL, Kaplan MM, Visser TJ, Silva JE, Larsen PR. Cerebral cortex responds rapidly to thyroid hormones. Science. 1981;214:571–573 [DOI] [PubMed] [Google Scholar]
- 132. Serrano-Lozano A, Montiel M, Morell M, Morata P. 5′ Deiodinase activity in brain regions of adult rats: modifications in different situations of experimental hypothyroidism. Brain Res Bull. 1993;30:611–616 [DOI] [PubMed] [Google Scholar]
- 133. Schneider MJ, Fiering SN, Pallud SE, Parlow AF, St Germain DL, Galton VA. Targeted disruption of the type 2 selenodeiodinase gene (DIO2) results in a phenotype of pituitary resistance to T4. Mol Endocrinol. 2001;15:2137–2148 [DOI] [PubMed] [Google Scholar]
- 134. Alonso M, Goodwin C, Liao X, Page D, Refetoff S, Weiss RE. Effects of maternal levels of thyroid hormone (TH) on the hypothalamus-pituitary-thyroid set point: studies in TH receptor β knockout mice. Endocrinology. 2007;148:5305–5312 [DOI] [PubMed] [Google Scholar]
- 135. Dussault JH, Coulombe P, Walker P. Effects of neonatal hyperthyroidism on the development of the hypothalamic-pituitary-thyroid axis in the rat. Endocrinology. 1982;110:1037–1042 [DOI] [PubMed] [Google Scholar]
- 136. Jansen J, Friesema EC, Milici C, Visser TJ. Thyroid hormone transporters in health and disease. Thyroid. 2005;15:757–768 [DOI] [PubMed] [Google Scholar]
- 137. Sugiyama D, Kusuhara H, Taniguchi H, et al. Functional characterization of rat brain-specific organic anion transporter (Oatp14) at the blood-brain barrier: high affinity transporter for thyroxine. J Biol Chem. 2003;278:43489–43495 [DOI] [PubMed] [Google Scholar]
- 138. Mayerl S, Visser TJ, Darras VM, Horn S, Heuer H. Impact of Oatp1c1 deficiency on thyroid hormone metabolism and action in the mouse brain. Endocrinology. 2012;153:1528–1537 [DOI] [PubMed] [Google Scholar]
- 139. Heuer H, Maier MK, Iden S, et al. The monocarboxylate transporter 8 linked to human psychomotor retardation is highly expressed in thyroid hormone-sensitive neuron populations. Endocrinology. 2005;146:1701–1706 [DOI] [PubMed] [Google Scholar]
- 140. Trajkovic M, Visser TJ, Mittag J, et al. Abnormal thyroid hormone metabolism in mice lacking the monocarboxylate transporter 8. J Clin Invest. 2007;117:627–635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Southwell BR, Duan W, Alcorn D, et al. Thyroxine transport to the brain: role of protein synthesis by the choroid plexus. Endocrinology. 1993;133:2116–2126 [DOI] [PubMed] [Google Scholar]
- 142. Sánchez E, Singru PS, Wittmann G, et al. Contribution of TNF-α and nuclear factor-κB signaling to type 2 iodothyronine deiodinase activation in the mediobasal hypothalamus after lipopolysaccharide administration. Endocrinology. 2010;151:3827–3835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Dratman MB, Crutchfield FL, Futaesaku Y, Goldberger ME, Murray M. [125I] triiodothyronine in the rat brain: evidence for neural localization and axonal transport derived from thaw-mount film autoradiography. J Comp Neurol. 1987;260:392–408 [DOI] [PubMed] [Google Scholar]
- 144. von Bartheld CS, Williams R, Lefcort F, Clary DO, Reichardt LF, Bothwell M. Retrograde transport of neurotrophins from the eye to the brain in chick embryos: roles of the p75NTR and trkB receptors. J Neurosci. 1996;16:2995–3008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Watson FL, Heerssen HM, Moheban DB, et al. Rapid nuclear responses to target-derived neurotrophins require retrograde transport of ligand-receptor complex. J Neurosci. 1999;19:7889–7900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Chávez-Gutiérrez L, Matta-Camacho E, Osuna J, et al. Homology modeling and site-directed mutagenesis of pyroglutamyl peptidase II. Insights into omega-versus aminopeptidase specificity in the M1 family. J Biol Chem. 2006;281:18581–18590 [DOI] [PubMed] [Google Scholar]
- 147. Silva JE. The thermogenic effect of thyroid hormone and its clinical implications. Ann Intern Med. 2003;139:205–213 [PubMed] [Google Scholar]
- 148. Légrádi G, Emerson CH, Ahima RS, Flier JS, Lechan RM. Leptin prevents fasting-induced suppression of prothyrotropin-releasing hormone messenger ribonucleic acid in neurons of the hypothalamic paraventricular nucleus. Endocrinology. 1997;138:2569–2576 [DOI] [PubMed] [Google Scholar]
- 149. Rondeel JM, Heide R, de Greef WJ, et al. Effect of starvation and subsequent refeeding on thyroid function and release of hypothalamic thyrotropin-releasing hormone. Neuroendocrinology. 1992;56:348–353 [DOI] [PubMed] [Google Scholar]
- 150. van Haasteren GA, Linkels E, Klootwijk W, et al. Starvation-induced changes in the hypothalamic content of prothyrotrophin-releasing hormone (proTRH) mRNA and the hypothalamic release of proTRH-derived peptides: role of the adrenal gland. J Endocrinol. 1995;145:143–153 [DOI] [PubMed] [Google Scholar]
- 151. Legradi G, Emerson CH, Ahima RS, Rand WM, Flier JS, Lechan RM. Arcuate nucleus ablation prevents fasting-induced suppression of ProTRH mRNA in the hypothalamic paraventricular nucleus. Neuroendocrinology. 1998;68:89–97 [DOI] [PubMed] [Google Scholar]
- 152. Kim MS, Small CJ, Stanley SA, et al. The central melanocortin system affects the hypothalamo-pituitary thyroid axis and may mediate the effect of leptin. J Clin Invest. 2000;105:1005–1011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Ghamari-Langroudi M, Vella KR, Srisai D, Sugrue ML, Hollenberg AN, Cone RD. Regulation of thyrotropin-releasing hormone-expressing neurons in paraventricular nucleus of the hypothalamus by signals of adiposity. Mol Endocrinol. 2010;24:2366–2381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Fekete C, Kelly J, Mihály E, et al. Neuropeptide Y has a central inhibitory action on the hypothalamic-pituitary-thyroid axis. Endocrinology. 2001;142:2606–2613 [DOI] [PubMed] [Google Scholar]
- 155. Fekete C, Sarkar S, Rand WM, et al. Agouti-related protein (AGRP) has a central inhibitory action on the hypothalamic-pituitary-thyroid (HPT) axis; comparisons between the effect of AGRP and neuropeptide Y on energy homeostasis and the HPT axis. Endocrinology. 2002;143:3846–3853 [DOI] [PubMed] [Google Scholar]
- 156. Cone RD. Anatomy and regulation of the central melanocortin system. Nat Neurosci. 2005;8:571–578 [DOI] [PubMed] [Google Scholar]
- 157. Fekete C, Marks DL, Sarkar S, et al. Effect of Agouti-related protein in regulation of the hypothalamic-pituitary-thyroid axis in the melanocortin 4 receptor knockout mouse. Endocrinology. 2004;145:4816–4821 [DOI] [PubMed] [Google Scholar]
- 158. Jegou S, Cone RD, Eberle AN, Vaudry H. Melanocortins. In: Kastin AJ, ed. Handbook of Biologically Active Peptides. 2nd ed Amsterdam, The Netherlands: Academic Press; 2013:838–844 [Google Scholar]
- 159. Mayr B, Montminy M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat Rev Mol Cell Biol. 2001;2:599–609 [DOI] [PubMed] [Google Scholar]
- 160. Pedragosa-Badia X, Stichel J, Beck-Sickinger AG. Neuropeptide Y receptors: how to get subtype selectivity. Front Endocrinol (Lausanne). 2013;4:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Sarkar S, Lechan RM. Central administration of neuropeptide Y reduces α-melanocyte-stimulating hormone-induced cyclic adenosine 5′-monophosphate response element binding protein (CREB) phosphorylation in pro-thyrotropin-releasing hormone neurons and increases CREB phosphorylation in corticotropin-releasing hormone neurons in the hypothalamic paraventricular nucleus. Endocrinology. 2003;144:281–291 [DOI] [PubMed] [Google Scholar]
- 162. Fekete C, Sarkar S, Rand WM, et al. Neuropeptide Y1 and Y5 receptors mediate the effects of neuropeptide Y on the hypothalamic-pituitary-thyroid axis. Endocrinology. 2002;143:4513–4519 [DOI] [PubMed] [Google Scholar]
- 163. Kishi T, Aschkenasi CJ, Choi BJ, et al. Neuropeptide Y Y1 receptor mRNA in rodent brain: distribution and colocalization with melanocortin-4 receptor. J Comp Neurol. 2005;482:217–243 [DOI] [PubMed] [Google Scholar]
- 164. Morin SM, Gehlert DR. Distribution of NPY Y5-like immunoreactivity in the rat brain. J Mol Neurosci. 2006;29:109–114 [DOI] [PubMed] [Google Scholar]
- 165. Sarkar S, Wittmann G, Fekete C, Lechan RM. Central administration of cocaine- and amphetamine-regulated transcript increases phosphorylation of cAMP response element binding protein in corticotropin-releasing hormone-producing neurons but not in prothyrotropin-releasing hormone-producing neurons in the hypothalamic paraventricular nucleus. Brain Res. 2004;999:181–192 [DOI] [PubMed] [Google Scholar]
- 166. Fekete C, Singru PS, Sanchez E, et al. Differential effects of central leptin, insulin, or glucose administration during fasting on the hypothalamic-pituitary-thyroid axis and feeding-related neurons in the arcuate nucleus. Endocrinology. 2006;147:520–529 [DOI] [PubMed] [Google Scholar]
- 167. Nillni EA, Vaslet C, Harris M, Hollenberg A, Bjørbak C, Flier JS. Leptin regulates prothyrotropin-releasing hormone biosynthesis. Evidence for direct and indirect pathways. J Biol Chem. 2000;275:36124–36133 [DOI] [PubMed] [Google Scholar]
- 168. Perello M, Stuart RC, Nillni EA. The role of intracerebroventricular administration of leptin in the stimulation of prothyrotropin releasing hormone neurons in the hypothalamic paraventricular nucleus. Endocrinology. 2006;147:3296–3306 [DOI] [PubMed] [Google Scholar]
- 169. Ghamari-Langroudi M, Srisai D, Cone RD. Multinodal regulation of the arcuate/paraventricular nucleus circuit by leptin. Proc Natl Acad Sci USA. 2011;108:355–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Perello M, Cakir I, Cyr NE, et al. Maintenance of the thyroid axis during diet-induced obesity in rodents is controlled at the central level. Am J Physiol Endocrinol Metab. 2010;299:E976–E989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Coppola A, Meli R, Diano S. Inverse shift in circulating corticosterone and leptin levels elevates hypothalamic deiodinase type 2 in fasted rats. Endocrinology. 2005;146:2827–2833 [DOI] [PubMed] [Google Scholar]
- 172. Coppola A, Hughes J, Esposito E, Schiavo L, Meli R, Diano S. Suppression of hypothalamic deiodinase type II activity blunts TRH mRNA decline during fasting. FEBS Lett. 2005;579:4654–4658 [DOI] [PubMed] [Google Scholar]
- 173. St Germain DL. The effects and interactions of substrates, inhibitors, and the cellular thiol-disulfide balance on the regulation of type II iodothyronine 5′-deiodinase. Endocrinology. 1988;122:1860–1868 [DOI] [PubMed] [Google Scholar]
- 174. Westholm DE, Stenehjem DD, Rumbley JN, Drewes LR, Anderson GW. Competitive inhibition of organic anion transporting polypeptide 1c1-mediated thyroxine transport by the fenamate class of nonsteroidal antiinflammatory drugs. Endocrinology. 2009;150:1025–1032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Coppola A, Liu ZW, Andrews ZB, et al. A central thermogenic-like mechanism in feeding regulation: an interplay between arcuate nucleus T3 and UCP2. Cell Metab. 2007;5:21–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Herwig A, Wilson D, Logie TJ, et al. Photoperiod and acute energy deficits interact on components of the thyroid hormone system in hypothalamic tanycytes of the Siberian hamster. Am J Physiol Regul Integr Comp Physiol. 2009;296:R1307–R1315 [DOI] [PubMed] [Google Scholar]
- 177. Bellinger LL, Bernardis LL. The dorsomedial hypothalamic nucleus and its role in ingestive behavior and body weight regulation: lessons learned from lesioning studies. Physiol Behav. 2002;76:431–442 [DOI] [PubMed] [Google Scholar]
- 178. Zigman JM, Jones JE, Lee CE, Saper CB, Elmquist JK. Expression of ghrelin receptor mRNA in the rat and the mouse brain. J Comp Neurol. 2006;494:528–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Wilcox BJ, Corp ES, Dorsa DM, et al. Insulin binding in the hypothalamus of lean and genetically obese Zucker rats. Peptides. 1989;10:1159–1164 [DOI] [PubMed] [Google Scholar]
- 180. Broberger C, Johansen J, Johansson C, Schalling M, Hökfelt T. The neuropeptide Y/agouti gene-related protein (AGRP) brain circuitry in normal, anorectic, and monosodium glutamate-treated mice. Proc Natl Acad Sci USA. 1998;95:15043–15048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Chou TC, Scammell TE, Gooley JJ, Gaus SE, Saper CB, Lu J. Critical role of dorsomedial hypothalamic nucleus in a wide range of behavioral circadian rhythms. J Neurosci. 2003;23:10691–10702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Gooley JJ, Schomer A, Saper CB. The dorsomedial hypothalamic nucleus is critical for the expression of food-entrainable circadian rhythms. Nat Neurosci. 2006;9:398–407 [DOI] [PubMed] [Google Scholar]
- 183. Romijn JA, Adriaanse R, Brabant G, Prank K, Endert E, Wiersinga WM. Pulsatile secretion of thyrotropin during fasting: a decrease of thyrotropin pulse amplitude. J Clin Endocrinol Metab. 1990;70:1631–1636 [DOI] [PubMed] [Google Scholar]
- 184. Watts AG, Sanchez-Watts G, Kelly AB. Distinct patterns of neuropeptide gene expression in the lateral hypothalamic area and arcuate nucleus are associated with dehydration-induced anorexia. J Neurosci. 1999;19:6111–6121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Jaimes-Hoy L, Joseph-Bravo P, de Gortari P. Differential response of TRHergic neurons of the hypothalamic paraventricular nucleus (PVN) in female animals submitted to food-restriction or dehydration-induced anorexia and cold exposure. Horm Behav. 2008;53:366–377 [DOI] [PubMed] [Google Scholar]
- 186. Alvarez-Salas E, Aceves C, Anguiano B, et al. Food-restricted and dehydrated-induced anorexic rats present differential TRH expression in anterior and caudal PVN. Role of type 2 deiodinase and pyroglutamyl aminopeptidase II. Endocrinology. 2012;153:4067–4076 [DOI] [PubMed] [Google Scholar]
- 187. de Gortari P, Mancera K, Cote-Vélez A, et al. Involvement of CRH-R2 receptor in eating behavior and in the response of the HPT axis in rats subjected to dehydration-induced anorexia. Psychoneuroendocrinology. 2009;34:259–272 [DOI] [PubMed] [Google Scholar]
- 188. Vijayan E, McCann SM. Suppression of feeding and drinking activity in rats following intraventricular injection of thyrotropin releasing hormone (TRH). Endocrinology. 1977;100:1727–1730 [DOI] [PubMed] [Google Scholar]
- 189. van den Heuvel JK, van Rozen AJ, Adan RA, la Fleur SE. An overview on how components of the melanocortin system respond to different high energy diets. Eur J Pharmacol. 2011;660:207–212 [DOI] [PubMed] [Google Scholar]
- 190. Münzberg H, Flier JS, Bjørbaek C. Region-specific leptin resistance within the hypothalamus of diet-induced obese mice. Endocrinology. 2004;145:4880–4889 [DOI] [PubMed] [Google Scholar]
- 191. White CL, Whittington A, Barnes MJ, Wang Z, Bray GA, Morrison CD. HF diets increase hypothalamic PTP1B and induce leptin resistance through both leptin-dependent and -independent mechanisms. Am J Physiol Endocrinol Metab. 2009;296:E291–E299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. De Groot LJ. Dangerous dogmas in medicine: the nonthyroidal illness syndrome. J Clin Endocrinol Metab. 1999;84:151–164 [DOI] [PubMed] [Google Scholar]
- 193. Fliers E, Guldenaar SE, Wiersinga WM, Swaab DF. Decreased hypothalamic thyrotropin-releasing hormone gene expression in patients with nonthyroidal illness. J Clin Endocrinol Metab. 1997;82:4032–4036 [DOI] [PubMed] [Google Scholar]
- 194. Kondo K, Harbuz MS, Levy A, Lightman SL. Inhibition of the hypothalamic-pituitary-thyroid axis in response to lipopolysaccharide is independent of changes in circulating corticosteroids. Neuroimmunomodulation. 1997;4:188–194 [DOI] [PubMed] [Google Scholar]
- 195. Kim YW, Kim KH, Ahn DK, et al. Time-course changes of hormones and cytokines by lipopolysaccharide and its relation with anorexia. J Physiol Sci. 2007;57:159–165 [DOI] [PubMed] [Google Scholar]
- 196. Sergeyev V, Broberger C, Hökfelt T. Effect of LPS administration on the expression of POMC, NPY, galanin, CART and MCH mRNAs in the rat hypothalamus. Brain Res Mol Brain Res. 2001;90:93–100 [DOI] [PubMed] [Google Scholar]
- 197. Fekete C, Sarkar S, Christoffolete MA, Emerson CH, Bianco AC, Lechan RM. Bacterial lipopolysaccharide (LPS)-induced type 2 iodothyronine deiodinase (D2) activation in the mediobasal hypothalamus (MBH) is independent of the LPS-induced fall in serum thyroid hormone levels. Brain Res. 2005;1056:97–99 [DOI] [PubMed] [Google Scholar]
- 198. Schiltz JC, Sawchenko PE. Specificity and generality of the involvement of catecholaminergic afferents in hypothalamic responses to immune insults. J Comp Neurol. 2007;502:455–467 [DOI] [PubMed] [Google Scholar]
- 199. Fekete C, Singru PS, Sarkar S, Rand WM, Lechan RM. Ascending brainstem pathways are not involved in lipopolysaccharide-induced suppression of thyrotropin-releasing hormone gene expression in the hypothalamic paraventricular nucleus. Endocrinology. 2005;146:1357–1363 [DOI] [PubMed] [Google Scholar]
- 200. Boelen A, Kwakkel J, Thijssen-Timmer DC, Alkemade A, Fliers E, Wiersinga WM. Simultaneous changes in central and peripheral components of the hypothalamus-pituitary-thyroid axis in lipopolysaccharide-induced acute illness in mice. J Endocrinol. 2004;182:315–323 [DOI] [PubMed] [Google Scholar]
- 201. Freitas BC, Gereben B, Castillo M, et al. Paracrine signaling by glial cell-derived triiodothyronine activates neuronal gene expression in the rodent brain and human cells. J Clin Invest. 2010;120:2206–2217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Sánchez E, Singru PS, Fekete C, Lechan RM. Induction of type 2 iodothyronine deiodinase in the mediobasal hypothalamus by bacterial lipopolysaccharide: role of corticosterone. Endocrinology. 2008;149:2484–2493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Hoshino K, Takeuchi O, Kawai T, et al. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J Immunol. 1999;162:3749–3752 [PubMed] [Google Scholar]
- 204. Rocchi R, Kimura H, Tzou SC, et al. Toll-like receptor-MyD88 and Fc receptor pathways of mast cells mediate the thyroid dysfunctions observed during nonthyroidal illness. Proc Natl Acad Sci USA. 2007;104:6019–6024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Zhang G, Ghosh S. Toll-like receptor-mediated NF-κB activation: a phylogenetically conserved paradigm in innate immunity. J Clin Invest. 2001;107:13–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Zeöld A, Doleschall M, Haffner MC, et al. Characterization of the nuclear factor-κ B responsiveness of the human dio2 gene. Endocrinology. 2006;147:4419–4429 [DOI] [PubMed] [Google Scholar]
- 207. Bockmann J, Böckers TM, Winter C, et al. Thyrotropin expression in hypophyseal pars tuberalis-specific cells is 3,5,3′-triiodothyronine, thyrotropin-releasing hormone, and pit-1 independent. Endocrinology. 1997;138:1019–1028 [DOI] [PubMed] [Google Scholar]
- 208. Nakao N, Ono H, Yamamura T, et al. Thyrotrophin in the pars tuberalis triggers photoperiodic response. Nature. 2008;452:317–322 [DOI] [PubMed] [Google Scholar]
- 209. Mebis L, Eerdekens A, Güiza F, et al. Contribution of nutritional deficit to the pathogenesis of the nonthyroidal illness syndrome in critical illness: a rabbit model study. Endocrinology. 2012;153:973–984 [DOI] [PubMed] [Google Scholar]
- 210. Mebis L, Debaveye Y, Ellger B, et al. Changes in the central component of the hypothalamus-pituitary-thyroid axis in a rabbit model of prolonged critical illness. Crit Care. 2009;13:R147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Silva JE. Thyroid hormone control of thermogenesis and energy balance. Thyroid. 1995;5:481–492 [DOI] [PubMed] [Google Scholar]
- 212. Zoeller RT, Kabeer N, Albers HE. Cold exposure elevates cellular levels of messenger ribonucleic acid encoding thyrotropin-releasing hormone in paraventricular nucleus despite elevated levels of thyroid hormones. Endocrinology. 1990;127:2955–2962 [DOI] [PubMed] [Google Scholar]
- 213. Uribe RM, Redondo JL, Charli JL, Joseph-Bravo P. Suckling and cold stress rapidly and transiently increase TRH mRNA in the paraventricular nucleus. Neuroendocrinology. 1993;58:140–145 [DOI] [PubMed] [Google Scholar]
- 214. Perello M, Stuart RC, Vaslet CA, Nillni EA. Cold exposure increases the biosynthesis and proteolytic processing of prothyrotropin-releasing hormone in the hypothalamic paraventricular nucleus via β-adrenoreceptors. Endocrinology. 2007;148:4952–4964 [DOI] [PubMed] [Google Scholar]
- 215. Bianco AC, Silva JE. Intracellular conversion of thyroxine to triiodothyronine is required for the optimal thermogenic function of brown adipose tissue. J Clin Invest. 1987;79:295–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Rogers RC, Barnes MJ, Hermann GE. Leptin “gates” thermogenic action of thyrotropin-releasing hormone in the hindbrain. Brain Res. 2009;1295:135–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Krulich L, Giachetti A, Marchlewska-Koj A, Hefco E, Jameson HE. On the role of the central noradrenergic and dopaminergic systems in the regulation of TSH secretion in the rat. Endocrinology. 1977;100:496–505 [DOI] [PubMed] [Google Scholar]
- 218. Arancibia S, Tapia-Arancibia L, Astier H, Assenmacher I. Physiological evidence for α 1-adrenergic facilitatory control of the cold-induced TRH release in the rat, obtained by push-pull cannulation of the median eminence. Neurosci Lett. 1989;100:169–174 [DOI] [PubMed] [Google Scholar]
- 219. van Haasteren GA, van Toor H, Klootwijk W, et al. Studies on the role of TRH and corticosterone in the regulation of prolactin and thyrotrophin secretion during lactation. J Endocrinol. 1996;148:325–336 [DOI] [PubMed] [Google Scholar]
- 220. Sánchez E, Fekete C, Lechan RM, Joseph-Bravo P. Cocaine- and amphetamine-regulated transcript (CART) expression is differentially regulated in the hypothalamic paraventricular nucleus of lactating rats exposed to suckling or cold stimulation. Brain Res. 2007;1132:120–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Raptis S, Fekete C, Sarkar S, et al. Cocaine- and amphetamine-regulated transcript co-contained in thyrotropin-releasing hormone (TRH) neurons of the hypothalamic paraventricular nucleus modulates TRH-induced prolactin secretion. Endocrinology. 2004;145:1695–1699 [DOI] [PubMed] [Google Scholar]
- 222. Collu R, Tang J, Castagné J, et al. A novel mechanism for isolated central hypothyroidism: inactivating mutations in the thyrotropin-releasing hormone receptor gene. J Clin Endocrinol Metab. 1997;82:1561–1565 [DOI] [PubMed] [Google Scholar]
- 223. Bonomi M, Busnelli M, Beck-Peccoz P, et al. A family with complete resistance to thyrotropin-releasing hormone. N Engl J Med. 2009;360:731–734 [DOI] [PubMed] [Google Scholar]
- 224. Dumitrescu AM, Liao XH, Best TB, Brockmann K, Refetoff S. A novel syndrome combining thyroid and neurological abnormalities is associated with mutations in a monocarboxylate transporter gene. Am J Hum Genet. 2004;74:168–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Friesema EC, Grueters A, Biebermann H, et al. Association between mutations in a thyroid hormone transporter and severe X-linked psychomotor retardation. Lancet. 2004;364:1435–1437 [DOI] [PubMed] [Google Scholar]
- 226. Boccone L, Mariotti S, Dessì V, Pruna D, Meloni A, Loudianos G. Allan-Herndon-Dudley syndrome (AHDS) caused by a novel SLC16A2 gene mutation showing severe neurologic features and unexpectedly low TRH-stimulated serum TSH. Eur J Med Genet. 2010;53:392–395 [DOI] [PubMed] [Google Scholar]
- 227. Refetoff S, Weiss RE, Usala SJ. The syndromes of resistance to thyroid hormone. Endocr Rev. 1993;14:348–399 [DOI] [PubMed] [Google Scholar]
- 228. Bochukova E, Schoenmakers N, Agostini M, et al. A mutation in the thyroid hormone receptor α gene. N Engl J Med. 2012;366:243–249 [DOI] [PubMed] [Google Scholar]
- 229. Mantzoros CS, Magkos F, Brinkoetter M, et al. Leptin in human physiology and pathophysiology. Am J Physiol Endocrinol Metab. 2011;301:E567–E584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Paz-Filho G, Wong ML, Licinio J. Ten years of leptin replacement therapy. Obes Rev. 2011;12:e315–e323 [DOI] [PubMed] [Google Scholar]
- 231. Montague CT, Farooqi IS, Whitehead JP, et al. Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature. 1997;387:903–908 [DOI] [PubMed] [Google Scholar]
- 232. Farooqi IS, Matarese G, Lord GM, et al. Beneficial effects of leptin on obesity, T cell hyporesponsiveness, and neuroendocrine/metabolic dysfunction of human congenital leptin deficiency. J Clin Invest. 2002;110:1093–1103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Chan JL, Heist K, DePaoli AM, Veldhuis JD, Mantzoros CS. The role of falling leptin levels in the neuroendocrine and metabolic adaptation to short-term starvation in healthy men. J Clin Invest. 2003;111:1409–1421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Rosenbaum M, Murphy EM, Heymsfield SB, Matthews DE, Leibel RL. Low dose leptin administration reverses effects of sustained weight-reduction on energy expenditure and circulating concentrations of thyroid hormones. J Clin Endocrinol Metab. 2002;87:2391–2394 [DOI] [PubMed] [Google Scholar]
- 235. Shetty GK, Matarese G, Magkos F, et al. Leptin administration to overweight and obese subjects for 6 months increases free leptin concentrations but does not alter circulating hormones of the thyroid and IGF axes during weight loss induced by a mild hypocaloric diet. Eur J Endocrinol. 2011;165:249–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Bruni V, Dei M, Morelli C, Schettino MT, Balzi D, Nuvolone D. Body composition variables and leptin levels in functional hypothalamic amenorrhea and amenorrhea related to eating disorders. J Pediatr Adolesc Gynecol. 2011;24:347–352 [DOI] [PubMed] [Google Scholar]
- 237. Chou SH, Chamberland JP, Liu X, et al. Leptin is an effective treatment for hypothalamic amenorrhea. Proc Natl Acad Sci USA. 2011;108:6585–6590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Misra M, Klibanski A. Neuroendocrine consequences of anorexia nervosa in adolescents. Endocr Dev. 2010;17:197–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Reinehr T, Isa A, de Sousa G, Dieffenbach R, Andler W. Thyroid hormones and their relation to weight status. Horm Res. 2008;70:51–57 [DOI] [PubMed] [Google Scholar]
- 240. Plikat K, Langgartner J, Buettner R, et al. Frequency and outcome of patients with nonthyroidal illness syndrome in a medical intensive care unit. Metabolism. 2007;56:239–244 [DOI] [PubMed] [Google Scholar]
- 241. Bello G, Ceaichisciuc I, Silva S, Antonelli M. The role of thyroid dysfunction in the critically ill: a review of the literature. Minerva Anestesiol. 2010;76:919–928 [PubMed] [Google Scholar]
- 242. Casaer MP, Wilmer A, Hermans G, Wouters PJ, Mesotten D, Van den Berghe G. Role of disease and macronutrient dose in the randomized controlled EPaNIC trial: a post hoc analysis. Am J Respir Crit Care Med. 2013;187:247–255 [DOI] [PubMed] [Google Scholar]
- 243. Langouche L, Vander Perre S, Marques M, et al. Impact of early nutrient restriction during critical illness on the nonthyroidal illness syndrome and its relation with outcome: a randomized, controlled clinical study. J Clin Endocrinol Metab. 2013;98:1006–1013 [DOI] [PubMed] [Google Scholar]
- 244. Rothwell PM, Lawler PG. Prediction of outcome in intensive care patients using endocrine parameters. Crit Care Med. 1995;23:78–83 [DOI] [PubMed] [Google Scholar]
- 245. Chinga-Alayo E, Villena J, Evans AT, Zimic M. Thyroid hormone levels improve the prediction of mortality among patients admitted to the intensive care unit. Intensive Care Med. 2005;31:1356–1361 [DOI] [PubMed] [Google Scholar]
- 246. De Groot LJ. Non-thyroidal illness syndrome is a manifestation of hypothalamic-pituitary dysfunction, and in view of current evidence, should be treated with appropriate replacement therapies. Crit Care Clin. 2006;22:57–86, vi [DOI] [PubMed] [Google Scholar]
- 247. Adler SM, Wartofsky L. The nonthyroidal illness syndrome. Endocrinol Metab Clin North Am. 2007;36:657–672, vi [DOI] [PubMed] [Google Scholar]
- 248. Angelousi AG, Karageorgopoulos DE, Kapaskelis AM, Falagas ME. Association between thyroid function tests at baseline and the outcome of patients with sepsis or septic shock: a systematic review. Eur J Endocrinol. 2011;164:147–155 [DOI] [PubMed] [Google Scholar]
- 249. Haas NA, Camphausen CK, Kececioglu D. Clinical review: thyroid hormone replacement in children after cardiac surgery–is it worth a try? Crit Care. 2006;10:213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250. Brent GA, Hershman JM. Thyroxine therapy in patients with severe nonthyroidal illnesses and low serum thyroxine concentration. J Clin Endocrinol Metab. 1986;63:1–8 [DOI] [PubMed] [Google Scholar]
- 251. Becker RA, Vaughan GM, Ziegler MG, et al. Hypermetabolic low triiodothyronine syndrome of burn injury. Crit Care Med. 1982;10:870–875 [DOI] [PubMed] [Google Scholar]
- 252. Klemperer JD, Klein I, Gomez M, et al. Thyroid hormone treatment after coronary-artery bypass surgery. N Engl J Med. 1995;333:1522–1527 [DOI] [PubMed] [Google Scholar]
- 253. Spratt DI, Frohnauer M, Cyr-Alves H, et al. Physiological effects of nonthyroidal illness syndrome in patients after cardiac surgery. Am J Physiol Endocrinol Metab. 2007;293:E310–E315 [DOI] [PubMed] [Google Scholar]
- 254. Pingitore A, Galli E, Barison A, et al. Acute effects of triiodothyronine (T3) replacement therapy in patients with chronic heart failure and low-T3 syndrome: a randomized, placebo-controlled study. J Clin Endocrinol Metab. 2008;93:1351–1358 [DOI] [PubMed] [Google Scholar]
- 255. Mullis-Jansson SL, Argenziano M, Corwin S, et al. A randomized double-blind study of the effect of triiodothyronine on cardiac function and morbidity after coronary bypass surgery. J Thorac Cardiovasc Surg. 1999;117:1128–1134 [DOI] [PubMed] [Google Scholar]
- 256. Kaptein EM, Sanchez A, Beale E, Chan LS. Clinical review: thyroid hormone therapy for postoperative nonthyroidal illnesses: a systematic review and synthesis. J Clin Endocrinol Metab. 2010;95:4526–4534 [DOI] [PubMed] [Google Scholar]
- 257. Boelen A, Kwakkel J, Fliers E. Beyond low plasma T3: local thyroid hormone metabolism during inflammation and infection. Endocr Rev. 2011;32:670–693 [DOI] [PubMed] [Google Scholar]
- 258. Van den Berghe G, Baxter RC, Weekers F, et al. The combined administration of GH-releasing peptide-2 (GHRP-2), TRH and GnRH to men with prolonged critical illness evokes superior endocrine and metabolic effects compared to treatment with GHRP-2 alone. Clin Endocrinol (Oxf). 2002;56:655–669 [DOI] [PubMed] [Google Scholar]
- 259. Van den Berghe G, Wouters P, Weekers F, et al. Reactivation of pituitary hormone release and metabolic improvement by infusion of growth hormone-releasing peptide and thyrotropin-releasing hormone in patients with protracted critical illness. J Clin Endocrinol Metab. 1999;84:1311–1323 [DOI] [PubMed] [Google Scholar]