

Abstract
Since Hsp90 modulates all six hallmarks of cancer simultaneously, it has become an attractive target for the development of cancer chemotherapeutics. In an effort to develop more efficacious compounds for Hsp90 inhibition, novobiocin analogues were prepared by replacing the central coumarin core with naphthalene, quinolinone, and quinoline surrogates. These modifications allowed for modification of the 2-position, which was previously unexplored. Biological evaluation of these compounds suggests a hydrophobic pocket about the 2-position of novobiocin. Anti-proliferative activities of these analogues against multiple cancer cell lines identified 2-alkoxyquinoline derivatives to exhibit improved activity.
Introduction
Heat shock protein 90 (Hsp90) is one of the most abundant molecular chaperones in the cell, is highly conserved in both eukaryotes and prokaryotes, and plays a key role in protein homeostasis.1,2 Hsp90-dependent proteins are responsible for cellular growth, signaling, differentiation, survival and protection against misfolded proteins. More than 200 client proteins are dependent upon Hsp90 for activation and/or stability; a quarter of which are directly associated with cell growth or signaling.3 Several Hsp90-dependent clients are found in pathways that are commonly mutated or hijacked during oncogensis.4 Consequently, Hsp90 is overexpressed in cancer cells to maintain cellular homeostasis under such environments and to serve as a buffer within tumor cells.5,6 Since Hsp90 modulates multiple pathways simultaneously, its inhibition produces a mechanism to overcome resistance that results from the inhibition of a single protein.7–9 Therefore, inhibition of Hsp90 for the treatment of cancer represents a powerful paradigm for drug development. 10–12
Structurally, Hsp90 exists as a homodimer and possesses two nucleotide binding regions: The N-terminal ATP-binding pocket is required for ATPase activity and produces the requisite energy for client protein maturation.13–15 The C-terminal nucleotide binding pocket exhibits allosteric control over both substrates and the N-terminal ATP-binding site.16 The N-terminal ATP binding site is inhibited by the natural products radicicol and geldanamycicn.17 The C-terminal region contains a dimerization motif that modulates client protein release. N-Terminal inhibitors have been widely studied in both academia and the pharmaceutical industry, and some have advanced to clinical trials.18 In contrast, the C-terminal inhibitors have not advanced into clinical investigations thus far, and the most significant reason is likely the absence of a co-crystal structure bound to an inhibitor.19 The development of more efficacious C-terminal inhibitors is desired to better understand the ramifications of C-terminal inhibition and to probe the mechanism by which Hsp90 interacts with client proteins. As an anti-cancer agent, Hsp90 C-terminal inhibitors manifest a distinct advantage over N-terminal inhibitors, as they do not induce the pro-survival heat shock response, which is a deleterious consequence of N-terminal inhibition.20 Consequently, the development of the Hsp90 C-terminal inhibitors represents an intense area of research.21–26
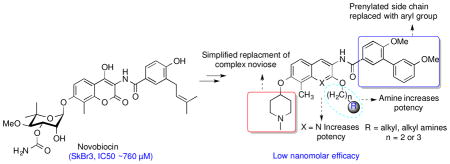
Novobiocin is a potent inhibitor of bacterial DNA gyrase and was also identified as the first Hsp90 C-terminal inhibitor alongside chlorobiocin and coumermycin (Figure 1).27–29 However, due to its low efficacy against cancer cells (IC50 ~ 700 μM), it was considered unsuitable for further evaluation as an Hsp90 inhibitor. Subsequent studies led to identification of some structure-activity relationships for novobiocin that resulted in KU-174 and DHN1, which manifest low micromolar activity. Additional studies led to compounds that exhibit greater potency and solubility than novobiocin and contained surrogates of the stereochemically complex noviose sugar as well as structural modifications to the benzamide side chain.23,30–33 The improved analogues exhibit mid nanomolar inhibitory activity against several cancer cell lines, while simultaneously manifesting increased solubility.25,26,34 In contrast to the sugar and amide termini of novobiocin, limited structure-activity relationship studies have been conducted on the central coumarin core.32 Therefore, structure-activity relationship studies for the coumarin core were pursued in an effort to develop more potent inhibitors and to explore this region for improved inhibitory activity.
Figure 1.

Hsp90 C-terminal inhibitors.
Construction of these novobiocin analogues were begun by replacing the central coumarin core with naphthalene, quinolinone and quinoline surrogates, Such motifs are abundant in natural products such as quinine, campothecin and cinchonidine, which manifest broad biological activities including anticancer, antimycobacterial, antimicrobial, anticonvulsant, anti-inflammatory and cardiovascular activities.35–41 The synthesis and structure-activity relationships of naphthalene, quinolinone, and quinolone replacements of the novobiocin coumarin core are presented in this article.
Results and Discussion
Design of New Novobiocin Analogues
Modifications to three regions of novobiocin were pursued to reveal structure-activity relationships and to provide more efficacious compounds; 1) replacement of the central coumarin core with various heterocycles were investigated, 2) modifications to the 2-hydroxyl group of the corresponding 2-quinolinol tautomer ring system were prepared, and 3) modifications to the benzamide side chain were also pursued. Both, noviose (3) and the sugar surrogate N-methyl-4-piperidine, were chosen based upon prior studies with novobiocin. Similarly, the side chain was replaced with an optimized prenylated or biaryl side chain for further investigation of these new scaffolds. As shown in Figure 2, analogues containing the prenylated benzamide side chain were assembled via three components; 1) a central bicyclic aryl core (naphthalene or quinoline or quinolinone scaffold), 2) a prenylated benzoic acid 4, and 3) noviose. The prenylated acid (4) could be coupled with the amino group enlisting 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDCI) as the coupling agent. Addition of noviose could then be achieved via the coupling of the trichloroacetimidate of noviose carbonate with the phenol. The naphthalene core could be prepared from commercially available 6-bromonaphthalen-2-ol (9) and both the quinolinone or quinoline cores were envisioned for construction from commercially available 2-methyl-3-nitrophenol (8).
Figure 2.

Retrosynthetic analysis of novobiocin analogues
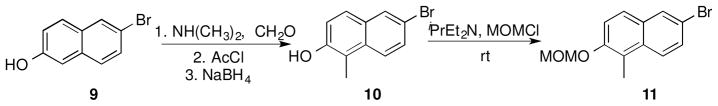
Synthesis of the naphthalene-containing analogues commenced by introduction of a methyl group at the 2-position of commercially available 6-bromonaphthalen-2-ol (9, Scheme 1) via a three-step procedure. Reductive amination of 9 with formaldehyde and N,N-dimethylamine followed by acylation of the phenol and subsequent reductive cleavage of N,N-dimethylamine furnished 6-bromo-1-methylnaphthalen-2-ol (10), in quantitative yield.42 The phenol was then protected as the methoxymethyl ether (MOM) to give compound 11 (Scheme 1) for use in subsequent steps.
Scheme 1.
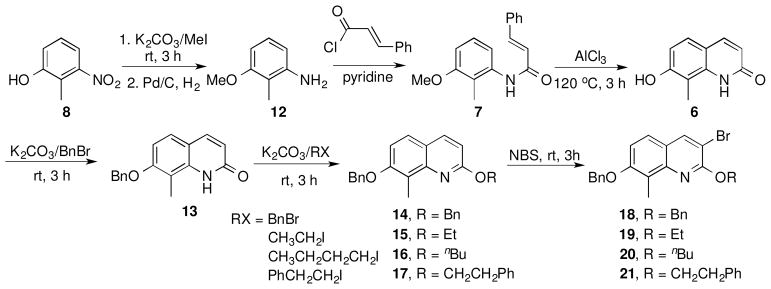
Construction of the quinolinone- and quinolone-containing analogues was accomplished via methylation of 2-hydroxy-3-nitrophenol using potassium carbonate and methyl iodide, followed by reduction of the nitro group to afford the corresponding aniline, 12 (Scheme 2). Treatment of 12 with trans-cinnamoyl chloride gave amide 7, which underwent intramolecular cyclization upon addition of aluminum chloride in chlorobenzene at 120 °C to give 8-methylquinoline-2,7-diol (6) in high yield.43 It is noteworthy that purification was not required until this point, as prior steps gave compounds in exceptional yields and purity. To incorporate alkyl ethers at the 2-position, the 7-phenol was selectively protected as the corresponding benzyl ether by the use of one equivalent of benzyl bromide and potassium carbonate. Taking advantage of the dynamic equilibrium that exists between the quinolone (6) and its quinolinol isomer, various hydrophobic groups were introduced at the 2-position. Treatment of compound 13 with alkyl halides in the presence of potassium carbonate afforded the corresponding alkyl ethers, 14–17, which were exposed to N-bromosuccinimide to give the brominated compounds, 18–21, in excellent yields (Scheme 2). The brominated compounds (11, 18–21) served as precursors for introduction of the amine and subsequent attachment of the benzamide side chain.
Scheme 2.
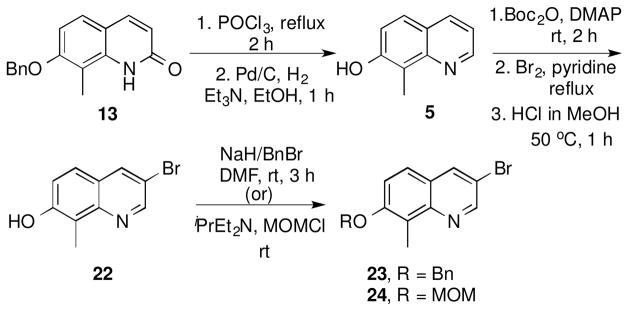
For direct comparison, quinoline analogues were also pursued. Preparation of quinoline precursors 23 and 24 began with quinolinol intermediate 13, which upon heating in the presence of phosphorous oxychloride, and subsequent hydrogenation gave the quinoline intermediate 5 as a yellow amorphous solid (Scheme 3).44 The phenolic moiety of 5 was then protected as the tert-butyl-carbonate to provide steric hindrance and enable regioselective bromination. In the event, bromination of 5 was achieved upon the addition of bromine to a solution of 5 in carbon tetrachloride and pyridine.32 Subsequent addition of methanolic HCl led to cleavage of the carbonate and formation of the corresponding phenol 22, which was then modified with methoxy methyl chloride or benzyl bromide to give intermediates 23 and 24, respectively.
Scheme 3.
Introduction of the benzamide side chain was accomplished by utilization of the brominated derivatives, 11, 18–21, 23, 24 which were transformed into the corresponding anilines, 25–31, via two different conditions (Scheme 4).45, 46 Compounds 11, 18–21, 23, 24, were heated in a sealed tube with copper nanoparticles, azidotrimethyl silane, and amino ethanol in a solution of dimethyl acetamide to give anilines 25–31.46 The protecting groups on compounds 27–29 were exchanged from benzyl ethers to silyl ethers by hydrogenolysis on palladium/carbon and subsequent O-silylation with tert-butyldimethylsilyl chloride (TBSCl), which gave anilines 32–34. Anilines 25, 26, 31–34, were readily coupled with benzoic acid 4 enlisting N-(3-(dimethylamino)propyl)-N′-ethylcarbodiimide hydrochloride (EDCI) in a solution of pyridine and dichloromethane to give the corresponding amides, 35–40.47 Deprotection of the benzyl or methoxy methyl or tert-butyldimethyl silyl groups of 35–40 was readily achieved with freshly prepared lithium di-tert-butyl biphenyl (LiDBB) in THF, 4N HCl, or 1.0 M tetrabutylammonium fluoride, respectively. The free phenols of 41–46 were provided without modification of the olefinic side chain (Scheme 4).48, 49
Scheme 4.

Upon construction of the phenols, the noviose appendage was attached via published procedures.50 Phenols 41–46 were noviosylated with the trichloroacetimidate of noviose carbonate, 47, in the presence of boron trifluoride etherate to provide the corresponding carbonylated noviose products, which upon solvolysis with methanolic triethylamine afforded 48–53 in good yields (Scheme 5).
Scheme 5.
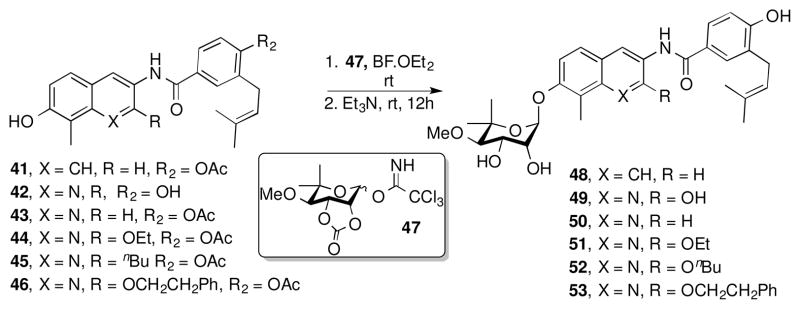
Biological evaluation of prenylated benzamide side chain containing coumarin surrogates
Upon construction of analogues 48–53, evaluation of anti-proliferative activity against SKBr3 (estrogen receptor negative, Her2 over-expressing breast cancer cells), MCF-7 (estrogen receptor positive breast cancer cells), LNCap (androgen-sensitive human prostate adenocarcinoma), 253B J-V (human bladder cancer), and PC3mm2 (androgen receptor insensitive prostate cancer) cell lines were investigated. Anti-proliferative activity of noviose containing analogues is shown in Table 1. Analogues lacking the coumarin core (49–50) manifested comparable activity to the corresponding coumarin-derived compounds, which confirms the lactone is not required for Hsp90 inhibition. These analogues also manifested comparable activities to DHN1 and DHN2 and manifested ~100-fold greater inhibitory activity than the natural product, novobiocin.31 Quinoline analogue 50 was found to be more efficacious (IC50 = 7.22 μ M against SKBr3 and IC50 = 0.26 μ M against PC3 cell lines) than the corresponding quinolinone (49) and naphthalene (48) analogues. Introduction of steric bulk at the 2-position of the quinolinol (51–53) revealed the ethoxy group (51) to be most active against both SKBr3 (IC50 = 1.94 μ M) and MCF7 (IC50 = 2.48 μM) cell lines. Given that analogue 51 (O-Et) manifested ~7-fold greater activity than analogue 53 (O-CH2CH2Ph) suggests that sterics are moderately tolerated.
Table 1.
Anti-proliferative activities of compounds 48–53.

| |||||||
|---|---|---|---|---|---|---|---|
| Entry | X | R1 | SKBr3 (IC50, μ M) | MCF7 (IC50, μ M) | PC3 MM2 (IC50, μ M) | LN CaP (IC50, μ M) | 253JB-V (IC50, μ M) |
| 48 | CH | H | 10.21±0.84 | 12.57±1.4 | NT | NT | NT |
| 49 | N | OH | 11.13±0.66 | 8.04±1.17 | 4.91±.87 | NT | NT |
| 50 | N | H | 7.22±1.07 | 9.68±2.01 | 0.258±0.07 | NT | NT |
| 51 | N | OCH2CH3 | 1.94±0.44 | 2.48±0.03 | 5.75±0.34 | 5.16±0.66 | NT |
| 52 | N | OCH2(CH2)2CH3 | 10.26±2.26 | 3.98±0.71 | 8.48±2.62 | 7.37±0.53 | 8.48 |
| 53 | N | OCH2CH2Ph | 14.14±2.46 | 14.54±0.33 | 12.47±0.07 | 10.09±0.43 | 12.47 |
After evaluation of the analogues that contain the prenylated benzamide side chain, the previously optimized biaryl acid, 55, was incorporated to further investigate the activity of these scaffolds. Previous studies have shown that analogues possessing a biaryl benzamide side chain manifest anti-proliferation activities comparable to analogues containing the prenylated benzamide. As shown in Figure 3, the biaryl derivatives can be readily prepared by a Suzuki coupling reaction between commercially available boronic acids and the corresponding aryl iodide, followed by hydrolysis of the ester. Retrosynthesis of the biaryl amide analogues follows a similar method as that used to prepare prenylated benzamide derivatives. The biaryl amide analogues were assembled from three components, the central core containing naphthalene, quinoline or quinolinone scaffold, the biaryl acid (55), and the noviose sugar (3) or its surrogate 1-methylpiperidin-4-ol (54).
Figure 3.

Retrosynthetic analysis of biaryl amide-containing analogues
Synthesis of these biaryl analogues began with a N-(3-(dimethylamino)propyl)-N′-ethylcarbodiimide (EDCI)-mediated coupling of aniline intermediates 26–31 with biaryl acid 55 to give the corresponding amides, which upon cleavage of the benzyl or methoxy methyl groups gave the free phenols, 62–68 (Scheme 6). The resulting phenols were coupled with the trichloroacetimidate of noviose carbonate (48) in the presence of boron trifluoride etherate, followed by solvolysis of the cyclic carbonate to afford 69–74 (Scheme 7). 50
Scheme 6.
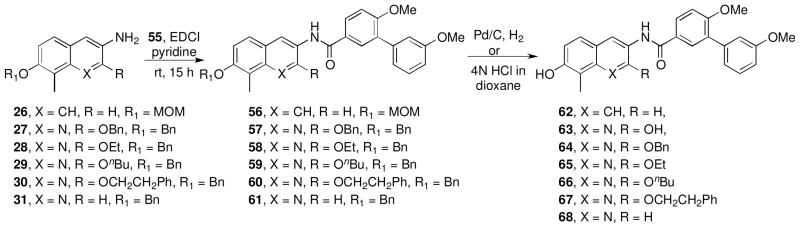
Scheme 7.
Evaluation of the anti-proliferative activities produced by analogues containing the biaryl benzamide moiety (69–74) against multiple cancer cell lines supported the nitrogen atom to be beneficial at the 1-position, as naphthalene analogues 69 and 70 were found to be 4- to 6-fold less active than corresponding quinoline analogue, 74 (Table 2). Increased steric bulk at the 2- position (71–73) was tolerated to some extent as both the ethyl and n-butyl ether manifested potency comparable to the unsubstituted analogue 74, however, introduction of bulkier groups (73) led to decreased potency.
Table 2.
Anti-proliferative Activities of compounds 69–74, 76–79, 81, 83, 85, 86.
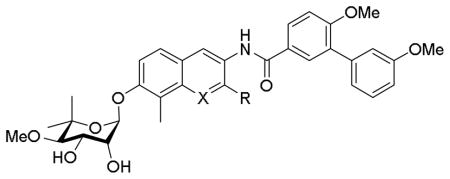
| |||||||
|---|---|---|---|---|---|---|---|
| Entry | X | R | SKBr3 (IC50, μ M) | MCF-7 (IC50, μ M) | PC3 MM2 (IC50, μ M) | LNCap (IC50, μ M) | 253JB-V (IC50, μ M) |
| 69 | CH | H | 38.1±2.2 | 25.7±3.7 | NT | NT | NT |
| 70 | N | OH | 56.8±10.1 | 11.87±0.72 | NT | NT | NT |
| 71 | N | OCH2CH3 | 13.5±1.13 | 13.8±0.82 | 6.05±1.9 | 4.71±2.4 | NT |
| 72 | N | OCH2(CH2)2CH3 | 8.174±0.22 | 11.27±0.53 | 5.487±1.1 | 3.768±0.2 | NT |
| 73 | N | OCH2CH2Ph | >100 | 77.41±5.4 | 29.76±2.5 | 22.52±8.9 | NT |
| 74 | N | H | 9.1±0.12 | 10.30±0.17 | NT | NT | NT |
| 76 | N | O-a | 1.13±0.02 | 2.09±0.31 | 3.95±0.04 | 1.798±0.0 | NT |
| 77 | N | O-d | 0.21±0.04 | 0.41±0.04 | 0.68±0.2 | 0.40±0.19 | NT |
| 78 | N | O-f | 1.02±0.21 | 1.41±0.03 | 1.41±0.01 | 1.67±0.07 | 1.12±0.40 |
| 79 | N | O-cyclohexyl | 26.40±4.2 | 44.14±2.7 | 14.12±1.3 | 13.20±0.0 | 7.42±0.1 |
| 81 | N | O-g | 0.54±0.24 | 0.72±0.41 | 1.78±0.4 | 1.88±0.16 | 1.79±0.07 |
| 83 | N | O-b | 4.58±0.34 | 5.45±1.1 | 2.08±0.2 | 5.49±5.6 | 0.73±0.7 |
| 85 | N | O-c | 0.12±0.04 | 0.56±0.12 | 1.33±0.36 | 1.26±0.07 | 0.99±0.01 |
| 86 | N | O-h | 0.56±0.21 | 1.21±0.39 | 1.54±0.31 | 2.39±0.77 | 1.89±0.09 |
Synthesis of noviose analogues containing alkyl or alkyl amine at the 2-position
Biological evaluation of quinoline containing analogues with the biaryl and prenylated side chains suggested that the binding pocket at the 2-position could be further probed for additional interactions, and perhaps greater inhibitory activity. Therefore, amines containing a 2- or 3- carbon linker consistent with the 2-ethoxy group were chosen for investigation (Figure 4). In addition, 2-bromoethoxy and the cyclohexyl ether of 2-phenol were prepared.
Figure 4.
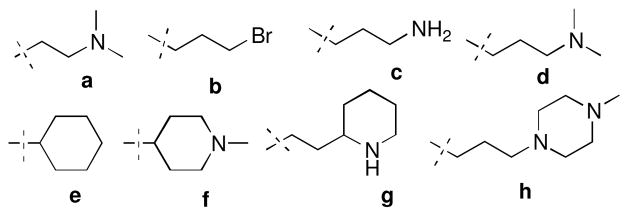
Selected alkyl amines and alkyl groups for probing the 2-position.
Analogues containing the alkyl side chains (a–h, Figure 4) at the 2-position were prepared by coupling the noviosylated intermediate 75 with various alcohols using standard Mitsunobu conditions followed by solvolysis of the carbonate to give coupled products, 76–80 (Scheme 8). Hydrogenolysis of the benzyl group on 80 afforded the secondary-amino analogue, 81. Treatment of 75 with dibromopropane in the presence of potassium carbonate, followed by solvolysis of the cyclic carbonate gave intermediate 83, which upon subsequent nucleophilic displacement with amines gave compounds 84 and 86, and subsequent hydrogenolysis gave 85.
Scheme 8.

Biological evaluation of analogues containing aminoalkyl substitutions at the 2-position revealed these substitutions to be beneficial as the majority of analogues manifested low micromolar to nanomolar activities against multiple cancer cell lines (Table 2). Their efficacy was greater than compounds containing the alkyl substituents (71–73 & 79). The two most potent analogues from this series were 77 (IC50 = 0.21±0.04 μM against MCF-7 cell line) and 85 (IC50 = 0.12±0.04 μM against MCF-7 cell line), which contain a 3-carbon linker between the quinolinol oxygen and the basic amine. Increased steric bulk as found in 86 resulted in decreased activity. Direct comparison of the 4-piperidine containing analogue, 78, with that containing a cyclohexane ring (79) suggests that the presence of charged group can provide improved activity. Overall, these results indicate that an amine attached to a 3-carbon linker is optimal at the 2-postion. Replacement of terminal primary-amine (76) with a tertiary-amine (77) resulted in increased efficacy against multiple cell lines. In summary, presence of hydrophilic amino group in the binding pocked enhances potency to a greater extant than increased lipophilicity.
Design and Synthesis of mono- and diamino substituted coumarin surrogate analogues
Encouraged by the efficacy manifested by noviose containing biaryl analogues that also contain an aminoalkyl substituent at the 2-position (76–78, 81, 83, 85, 86), compounds containing the noviose surrogate, N-methylpiperidine, were pursued (Scheme 9). N-Methylpiperidine is a readily available surrogate for the synthetically complex noviose sugar. 24, 32 Coupling of phenols 62, 63, 65–68 with 1-methylpiperidin-4-ol was achieved under Mitsunobu conditions employing diisopropyl azodicarboxylate (DIAD) and triphenylphosphine to give 87–92.26 Analogues containing aminoalkyl ethers at the 2-position were synthesized from diphenol 63, which upon treatment with five equivalents of piperidin-4-ol or 3-(dimethylamino)propan-1-ol gave analogues 93–94. Construction of 96 and 98 was accomplished from the phenol 64, which was obtained during partial hydrogenolysis of dibenzylated compound, 57. Treatment of 64 with dibromopropane in the presence of potassium carbonate, followed by coupling with 1-methylpiperazine gave 95. Subsequent hydrogenolysis of 95, followed by coupling of the corresponding phenol with 4 gave 96. Analogue 98 was synthesized from phenol 64 in a similar manner enlisting 3-(dimethylamino)propan-1-ol.
Scheme 9.

Results from the biological investigation of diamino analogues (87–94, 96 & 98) are shown in Table 3. These results confirm that the synthetically challenging noviose moiety can be replaced with commercially available amines without lowering potency as analogues 87–92 manifested IC50 values similar to compounds utilizing noviose (69–74). Biological evaluation of analogues containing alkyl amine substituents at the 2-position (93, 94, 96, 98) confirmed that N, N-dimethylpropylamine is an optimal substituent at the 2-position as analogue 108 manifested good potency against breast cancer (SKBr3; IC50 = 0.59±0.05 μM), prostate cancer (PC3MM2; IC50 = 0.409 μM) and, lung cancer (LNCaP; IC50 = 0.253 μM) cell lines. In contrast to noviose containing analogue 78, the corresponding N-methylpiperidine containing analogue 94 manifested better efficacy against the PC3MM2 and LNCaP cell lines. Replacement of N-methylpiperidine at the 7-position with acyclic N, N-dimethylpropylamine resulted in less activity.
Table 3.
Anti-proliferative activities of analogues 87–94, 104, 96, 98.

| ||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | X | R1 | R | SKBr3 (IC50, μ M) | MCF7 (IC50, μ M) | PC3 MM2 (IC50, μ M) | LN CaP (IC50, μ M) | 253JB-V (IC50, μ M) |
| 87 | CH | H | f | 2.32±0.61 | 1.39±0.1 | NT | NT | NT |
| 88 | N | OH | f | 1.30±0.08 | 1.43±0.19 | NT | NT | NT |
| 89 | N | OCH2CH3 | f | 1.86±0.27 | 1.97±0.17 | >100 | >100 | NT |
| 90 | N | OCH2(CH2)2CH3 | f | 1.06±0.02 | 1.72±0.33 | 2.23±0.02 | 2.06±0.06 | NT |
| 91 | N | OCH2CH2Ph | f | 1.89 ±0.4 | 3.19 ±0.6 | 2.46±0.37 | 2.37±0.04 | NT |
| 92 | N | H | f | 1.14±0.21 | 1.26±0.09 | 1.93±0.1 | NT | NT |
| 93 | N | O-d | d | 0.34±0.21 | 0.49±0.17 | 1.75±0.13 | 1.86±0.03 | 1.39±0.17 |
| 94 | N | O-f | f | 0.58±0.24 | 0.57±0.14 | 1.74±0.14 | 1.9±0.07 | NT |
| 96 | N | O-h | f | 0.64±0.31 | 0.67±0.42 | 1.75±0.1 | 1.79±0.2 | 1.30±0.15 |
| 98 | N | O-d | f | 0.59±0.05 | 1.58±0.16 | 2.4±0.12 | 2.6±1.10 | NT |
To confirm these analogues manifest inhibitory anti-proliferative activity through Hsp90 inhibition, western blot analyses of cell lysates resulting from the incubation of cells treated with the three most potent compounds 77, 85, and 94 for twenty four hours were performed. As shown in Figure 4, compound 77, 85 and, 94 induced degradation of Hsp90-dependent client proteins p-Akt and Her2. Whereas, actin levels remained unchanged. Actin is not an Hsp90-dependent substrate and used as a control. Absence of induction of heat shock response as indicated by constant levels of Hsp90 indicates that these compounds exhibit Hsp90 inhibitory activity through C-terminal inhibition.
In summary, the coumarin core of novobiocin has been replaced with a quinolinone, naphthalene, or quinolone ring system. These replacements resulted in the efficacious quinoline class of Hsp90 inhibitors that allowed probing at the 2-position to gain additional interactions with Hsp90. Addition of alkylamino substituents at the 2-position led to new analogues that exhibit increased activity against various cancer cell lines and analogue 87 was found to be the most potent, which manifested ~200 nm activity against the SKBr3 cell line. These results suggest that binding pocket surrounding the 2-phenolic position may be important for the development of more efficacious compounds.
Supplementary Material
Figure 5.
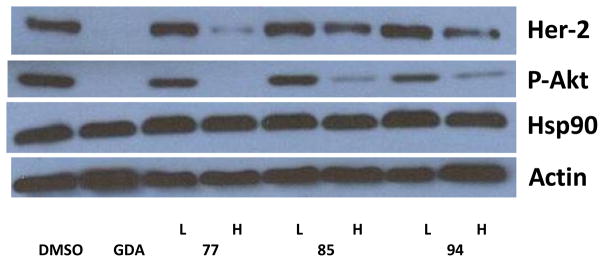
Western blot analyses of Hsp90 client protein degradation in MCF-7 breast cancer cell line. L represents a concentration ½ × IC50 value while H represents a concentration of 5 × IC50 value. GDA (500 nM) represents a positive control, while DMSO, vehicle, serves as the negative control.
Acknowledgments
The authors gratefully acknowledge the support of this project by NIH/NCI (CA120458 and CA167079) and frontier scientific for providing high quality boronic acids.
Footnotes
Supporting Information Available: Experimental procedures and characterization for all new compounds.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Chaudhury S, Welch TR, Blagg BS. Chem Med Chem. 2006;1:1331. doi: 10.1002/cmdc.200600112. [DOI] [PubMed] [Google Scholar]
- 2.Bishop SC, Burlison JA, Blagg BS. Curr Cancer Drug Targets. 2007;7:369. doi: 10.2174/156800907780809778. [DOI] [PubMed] [Google Scholar]
- 3.Powers MV, Workman P. Endocr-related cancer. 2006;13(Suppl 1):S125. doi: 10.1677/erc.1.01324. [DOI] [PubMed] [Google Scholar]
- 4.Workman P. Cancer Lett. 2004;206:149. doi: 10.1016/j.canlet.2003.08.032. [DOI] [PubMed] [Google Scholar]
- 5.Whitesell L, Lindquist SL. Nature Rev Cancer. 2005;5:761. doi: 10.1038/nrc1716. [DOI] [PubMed] [Google Scholar]
- 6.Dai C, Whitesell L. Future Oncol. 2005;1:529. doi: 10.2217/14796694.1.4.529. [DOI] [PubMed] [Google Scholar]
- 7.Xu W, Neckers L. Clin Can Res. 2007;13:1625. doi: 10.1158/1078-0432.CCR-06-2966. [DOI] [PubMed] [Google Scholar]
- 8.Tsuruo T, Naito M, Tomida A, Fujita N, Mashima T, Sakamoto H, Haga N. Cancer Sci. 2003;94:15. doi: 10.1111/j.1349-7006.2003.tb01345.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shimamura T, Li D, Ji H, Haringsma HJ, Liniker E, Borgman CL, Lowell AM, Minami Y, McNamara K, Perera SA, Zaghlul S, Thomas RK, Greulich H, Kobayashi S, Chirieac LR, Padera RF, Kubo S, Takahashi M, Tenen DG, Meyerson M, Wong KK, Shapiro GI. Cancer Res. 2008;68:5827. doi: 10.1158/0008-5472.CAN-07-5428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peterson LB, Blagg BS. Future Med Chem. 2009;1:267. doi: 10.4155/fmc.09.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maloney A, Workman P. Expert Opin Biol Thep. 2002;2:3. doi: 10.1517/14712598.2.1.3. [DOI] [PubMed] [Google Scholar]
- 12.Duerfeldt AS, Peterson LB, Maynard JC, Ng CL, Eletto D, Ostrovsky O, Shinogle HE, Moore DS, Argon Y, Nicchitta CV, Blagg BS. J Am Chem Soc. 2012;134:9796. doi: 10.1021/ja303477g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Prodromou C, Roe SM, O’Brien R, Ladbury JE, Piper PW, Pearl LH. Cell. 1997;90:65. doi: 10.1016/s0092-8674(00)80314-1. [DOI] [PubMed] [Google Scholar]
- 14.Chadli A, Bouhouche I, Sullivan W, Stensgard B, McMahon N, Catelli MG, Toft DO. Proc Natl Acad Sci USA. 2000;97:12524. doi: 10.1073/pnas.220430297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pearl LH, Prodromou C. Annu Rev Biochem. 2006;75:271. doi: 10.1146/annurev.biochem.75.103004.142738. [DOI] [PubMed] [Google Scholar]
- 16.Garnier C, Lafitte D, Tsvetkov PO, Barbier P, Leclerc-Devin J, Millot JM, Briand C, Makarov AA, Catelli MG, Peyrot V. The J Biol Chem. 2002;277:12208. doi: 10.1074/jbc.M111874200. [DOI] [PubMed] [Google Scholar]
- 17.Roe SM, Prodromou C, O’Brien R, Ladbury JE, Piper PW, Pearl LH. J Med Chem. 1999;42:260. doi: 10.1021/jm980403y. [DOI] [PubMed] [Google Scholar]
- 18.Neckers L, Workman P. Clin Can Res. 2012;18:64. doi: 10.1158/1078-0432.CCR-11-1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Matts RL, Dixit A, Peterson LB, Sun L, Voruganti S, Kalyanaraman P, Hartson SD, Verkhivker GM, Blagg BS. ACS Chem Biol. 2011;6:800. doi: 10.1021/cb200052x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bagatell R, Paine-Murrieta GD, Taylor CW, Pulcini EJ, Akinaga S, Benjamin IJ, Whitesell L. Clin Can Res. 2000;6:3312. [PubMed] [Google Scholar]
- 21.Khandelwal A, Hall JA, Blagg BS. J Org Chem. 2013;78:7859. doi: 10.1021/jo401027r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Donnelly A, Blagg BSJ. Curr Med Chem. 2008;15:2702. doi: 10.2174/092986708786242895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kusuma BR, Duerfeldt AS, Blagg BS. Bioorg Med Chem Lett. 2011;21:7170. doi: 10.1016/j.bmcl.2011.09.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kusuma BR, Peterson LB, Zhao H, Vielhauer G, Holzbeierlein J, Blagg BS. J Med Chem. 2011;54:6234. doi: 10.1021/jm200553w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao H, Donnelly AC, Kusuma BR, Brandt GEL, Brown D, Rajewski RA, Vielhauer G, Holzbeierlein J, Cohen MS, Blagg BSJ. J Med Chem. doi: 10.1021/jm200148p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhao H, Kusuma BR, Blagg BS. ACS Med Chem Lett. 2010;1:311. doi: 10.1021/ml100070r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marcu MG, Schulte TW, Neckers L. J Natl Cancer Inst. 2000;92:242. doi: 10.1093/jnci/92.3.242. [DOI] [PubMed] [Google Scholar]
- 28.Lewis RJ, Singh OM, Smith CV, Skarzynski T, Maxwell A, Wonacott AJ, Wigley DB. EMBO J. 1996;15:1412. [PMC free article] [PubMed] [Google Scholar]
- 29.Marcu MG, Chadli A, Bouhouche I, Catelli M, Neckers LM. J Biol Chem. 2000;275:37181. doi: 10.1074/jbc.M003701200. [DOI] [PubMed] [Google Scholar]
- 30.Yu XM, Shen G, Neckers L, Blake H, Holzbeierlein J, Cronk B, Blagg BS. J Am Chem Soc. 2005;127:12778. doi: 10.1021/ja0535864. [DOI] [PubMed] [Google Scholar]
- 31.Burlison JA, Neckers L, Smith AB, Maxwell A, Blagg BS. J Am Chem Soc. 2006;128:15529. doi: 10.1021/ja065793p. [DOI] [PubMed] [Google Scholar]
- 32.Donnelly AC, Mays JR, Burlison JA, Nelson JT, Vielhauer G, Holzbeierlein J, Blagg BS. J Org Chem. 2008;73:8901. doi: 10.1021/jo801312r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Peterson LB, Blagg BS. Bioorg Med Chem Lett. 2010;20:3957. doi: 10.1016/j.bmcl.2010.04.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao H, Yan B, Peterson LB, Blagg BS. ACS Med Chem Lett. 2012;3:327. doi: 10.1021/ml300018e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Borras J, Scozzafava A, Menabuoni L, Mincione F, Briganti F, Mincione G, Supuran CT. Bioorg Med Chem. 1999;7:2397. doi: 10.1016/s0968-0896(99)00190-x. [DOI] [PubMed] [Google Scholar]
- 36.Musiol R, Jampilek J, Buchta V, Silva L, Niedbala H, Podeszwa B, Palka A, Majerz-Maniecka K, Oleksyn B, Polanski J. Bioorg Med Chem. 2006;14:3592. doi: 10.1016/j.bmc.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 37.Graves PR, Kwiek JJ, Fadden P, Ray R, Hardeman K, Coley AM, Foley M, Haystead TA. Mol Pharmcol. 2002;62:1364. doi: 10.1124/mol.62.6.1364. [DOI] [PubMed] [Google Scholar]
- 38.Michael JP. Nat Prod Rep. 2005;22:627. doi: 10.1039/b413750g. [DOI] [PubMed] [Google Scholar]
- 39.Perzyna A, Marty C, Facompre M, Goossens JF, Pommery N, Colson P, Houssier C, Houssin R, Henichart JP, Bailly C. J Med Chem. 2002;45:5809. doi: 10.1021/jm020235g. [DOI] [PubMed] [Google Scholar]
- 40.Kljun J, Bratsos I, Alessio E, Psomas G, Repnik U, Butinar M, Turk B, Turel I. Inorg Chem. 2013;52:9039. doi: 10.1021/ic401220x. [DOI] [PubMed] [Google Scholar]
- 41.Villemagne B, Crauste C, Flipo M, Baulard AR, Deprez B, Willand N. Eur J Med Chem. 2012;51:1. doi: 10.1016/j.ejmech.2012.02.033. [DOI] [PubMed] [Google Scholar]
- 42.Rodriguez R, Moses JE, Adlington RM, Baldwin JE. Org Biomol Chem. 2005;3:3488. doi: 10.1039/b508972g. [DOI] [PubMed] [Google Scholar]
- 43.Elliott MC, Wordingham SV. Synlett. 2004;2004:0898. [Google Scholar]
- 44.Deng J, Feng E, Ma S, Zhang Y, Liu X, Li H, Huang H, Zhu J, Zhu W, Shen X, Miao L, Liu H, Jiang H, Li J. J Med Chem. 2011;54:4508. doi: 10.1021/jm200161c. [DOI] [PubMed] [Google Scholar]
- 45.Messaoudi S, Brion JD, Alami M. Adv Synth Catal. 2010;352:1677. [Google Scholar]
- 46.Maejima T, Shimoda Y, Nozaki K, Mori S, Sawama Y, Monguchi Y, Sajiki H. Tetrahedron. 2012;68:1712. [Google Scholar]
- 47.Burlison JA, Avila C, Vielhauer G, Lubbers DJ, Holzbeierlein J, Blagg BSJ. The J Org Chem. 2008;73:2130. doi: 10.1021/jo702191a. [DOI] [PubMed] [Google Scholar]
- 48.Yang SH, Clark GR, Caprio V. Org Biomol Chem. 2009;7:2981. doi: 10.1039/b903904j. [DOI] [PubMed] [Google Scholar]
- 49.Choy N, Shin Y, Nguyen PQ, Curran DP, Balachandran R, Madiraju C, Day BW. J Med Chem. 2003;46:2846. doi: 10.1021/jm0204136. [DOI] [PubMed] [Google Scholar]
- 50.Shen G, Yu XM, Blagg BS. Bioorg Med Chem Lett. 2004;14:5903. doi: 10.1016/j.bmcl.2004.09.017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.