

Summary
T cells bearing receptors made up of α and β chains (TCRs) usually react with peptides bound to major histocompatibility complex proteins (MHC). This bias could be imposed by positive selection, the phenomenon that selects thymocytes to mature into T cells only if the TCRs they bear react with low but appreciable affinity with MHC + peptide combinations in the thymus cortex. However, it is also possible that the polypeptides of TCRs themselves do not have random specificities but rather are biased toward reaction with MHC. Evolution would therefore have selected for a collection of TCR variable elements that are prone to react with MHC. If this were to be so, positive selection would act on thymocytes bearing a pre biased collection of TCRs to pick out those that react to some extent, but not too well, with self MHC + self-peptides. A problem with studies of this evolutionary idea is the fact that there are many TCR variable elements and that these differ considerably in the amino acids with which they contact MHC. However, recent experiments by our group and others suggest that one group of TCR variable elements, those related to the mouse Vβ8 family, has amino acids in their CDR2 regions that consistently bind a particular site on an MHC α-helix. Other groups of variable elements may use different patterns of amino acids to achieve the same goal. Mutation of these amino acids reduces the ability of T cells and thymocytes to react with MHC. These amino acids are present in the variable regions of distantly related species such as sharks and human. Overall the data indicate that TCR elements have indeed been selected by evolution to react with MHC proteins. Many mysteries about TCRs remain to be solved, including the nature of auto-recognition, the basis of MHC allele specificity, and the very nature and complexity of TCRs on mature T cells.
Keywords: comparative immunology, T cells, T-cell receptors, thymus
Introduction
The major histocompatibility complex (MHC) was discovered in the first half of the 20th century because of its ability to raise very rapid and powerful immune responses (1, 2). The latter half of that same century was devoted to studies that eventually showed us how MHC proteins function in healthy and diseased individuals. Many people contributed to our understanding of what was going on (3–13), but one could argue that the culminating finding was the solution, using X-ray crystallography, of the structure of an MHC class I protein with some unidentified substance bound in a groove on its surface (14, 15). This unidentified substance turned out to be, of course, a peptide. This finding showed us the structural basis for what cellular immunologists had already established, that a single MHC protein could bind and present many different peptides to T cells (3, 12).
The T-cell receptors (TCRs) that engage MHC + peptide are heterodimers, made up of an α and β chain, each of which varies in sequence from one T cell to another (16–21). The genes coding for TCRs are created by rearrangement in the thymus with each thymocyte in mice, having a choice of one of 20–50 germline-encoded Vβs, one of 12Jβs, and one or two Dβs (to be read in any frame) to make up its TCRβ chain. Likewise TCR α chains are constructed from a choice of one of approximately 50–100 Vαs and one of about 50 Jαs (22–26). [The numbers of the germline-encoded variable elements vary from one species to another and even within species (IMGT database, http://www.imgt.org/)]. Furthermore, additional variability is conferred by removal of nucleotides or introduction of P nucleotides or non-germline-encoded bases at the joining points between the individual segments (27, 28). Simple arithmetic shows us that immense numbers of different TCRs can be constructed in this way.
With all this knowledge in hand, the next challenge was to find out how TCRs, with their many possible sequences, bind their ligand, MHC + peptide. Several ideas about the issue were suggested (29, 30). Eventually the answer, in the form of another X-ray cystallographically solved structure, turned out to be a compromise between some of the hypotheses (31). The TCR in that and many subsequently solved structures engages the MHC/peptide at a diagonal angle via 6 loops at the ends of some of its variable β strands. On the whole, the loops created by the germline-encoded Vα and Vβ regions (CDR1α, CDR2α and CDR1β, CDR2β) lie over the α helices of the α2/β1 and α1 domains of the MHC proteins, respectively. On the other hand, the loops created by the non-germline portions of the TCR, CDR3α, and CDR3β, tend to lie over and contact the peptide bound in the groove of the MHC protein (24, 32–42) (Fig. 1). There are interesting exceptions, however, as discussed below and elsewhere in this volume.
Fig. 1. Structure of a TCR + MHC + peptide complex.
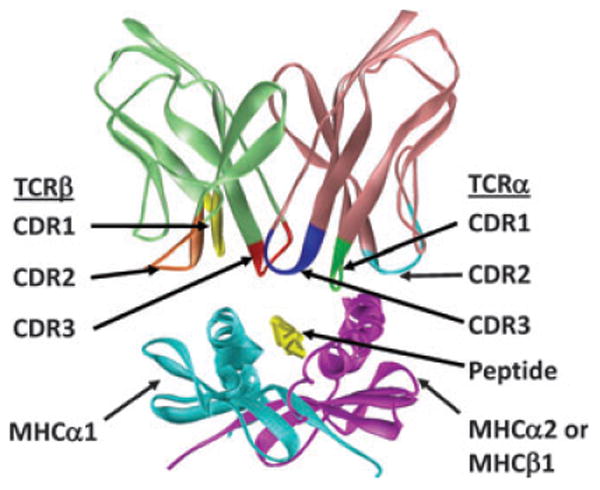
Shown is the X-ray crystallographically solved structure of the YAe62.8 TCR bound to IAb + the 3K peptide (33). The TCR V region loops (CDR1-3) of each of the TCR chains are highlighted in color.
Why are TCRs so obsessed with MHC?
Theoretically, T cells have about the same number of TCR sequences available to them as B cells have immunoglobulin sequences, and yet TCRs usually bind MHC + peptide, whereas immunoglobulins can bind an apparently limitless number of different structures. What accounts for the focus of TCRs on MHC? Neils Jerne (43) suggested, years before TCRs were discovered, that the germline-encoded versions of lymphocyte receptors must have evolved to bind MHC proteins and that to avoid reactivity with self-MHC, somatic mutation must muffle this germline reactivity. Because the germline genes coding for TCR components do not routinely mutate (44, 45), the second part of Jerne's hypothesis is not correct; however, the first portion of his idea could hold water. The portions of TCRs that are the most significant contributors to MHC engagement, CDR1α, CDR2α, CDR1β, and CDR2β, may have been selected evolutionarily to bind MHC.
Thymocytes are not allowed to escape the thymus unless the TCRs they bear can engage, with some low avidity/ affinity, an MHC + peptide combination that is present on cells in the thymus cortex (46, 47). Thus, this so-called positive selection allows cells to mature only if they can react with self-MHC. Positive selection all by itself can therefore account for the manifest obsession of the TCRs on mature T cells to react with MHC.
It is interesting that the Jerne hypothesis and positive selection, in isolation, can both account for TCR/MHC bias and tolerance to self from exactly opposite starting propositions. Jerne's hypothesis would argue that TCRs are evolutionarily selected to react well with MHC. During development, thymocytes bearing TCRs that continue to react well are destroyed, whereas those that mutate their TCR genes and thus achieve low level of reactivity with MHC are retained (along with, incidentally some thymocytes that lose MHC reactivity altogether). By contrast, the positive selection only hypothesis would argue that the TCR repertoire is intrinsically random and therefore only rare TCRs are MHC reactive. The thymocytes that bear these rare reactivities are allowed to mature, providing, of course, that they do not violate self-tolerance by reacting too well with self-MHC molecules.
We now know that thymocytes must go through positive selection. However, the existence of positive selection does not negate the first half of Jerne's hypothesis. The germline genes coding for TCRs may still give rise to TCRs that are likely to engage MHC and less likely to engage other ligands. Positive selection may act in various ways to refine the unselected TCR repertoire.
Evidence for and against the evolutionary hypothesis
Some years ago, several groups published data suggesting that the evolutionary hypothesis might be correct. The experiments studied the MHC reactivities of TCRs that could not have gone through positive selection. In one case, we constructed a T-cell hybridoma expressing a TCR using a TCRα chain and a TCRβ chain that had not been subject, as a pair, to positive selection. The hybridoma, when tested, responded to mouse MHC class II (MHCII) IAb plus some unknown peptide. We concluded that such random pairs reacted unexpectedly with MHC proteins (48). In other cases, TCRs from thymocytes that had not gone through positive selection were examined. Again, such TCRs turned out to react with MHC with unexpectedly high frequency (49, 50).
When the first structures of TCRs binding their MHC/peptide ligands began to appear many of us expected that some germline ‘rules of engagement’ would be evident. To some extent they were. For the vast majority of complexes that have been solved to date, the TCR usually adopts a diagonal orientation on MHC, using its CDR loops as described above, with the TCRα on one side of the MHC and the TCRβ on the other (reviewed in 51). However, no consistent use of a particular TCR amino acid side chain to engage MHC was evident. In retrospect it was obvious that this would be the case. The CDR1 and CDR2 loops of TCRs vary tremendously in sequence from one Vα or Vβ to another (22, 23). This is exemplified by the list of CDR2β sequences of the mouse Vβs, shown in Fig. 2A. Therefore, since the first sets of TCR/MHC/peptide structures involved TCRs with different Vαs and Vβs, no TCR amino acids should have been expected to be consistently present to illustrate the MHC-engaging job they might do.
Fig. 2. Amino acid sequences of the CDR2β regions of mouse Vβs illustrate residues shared between different Vβs.
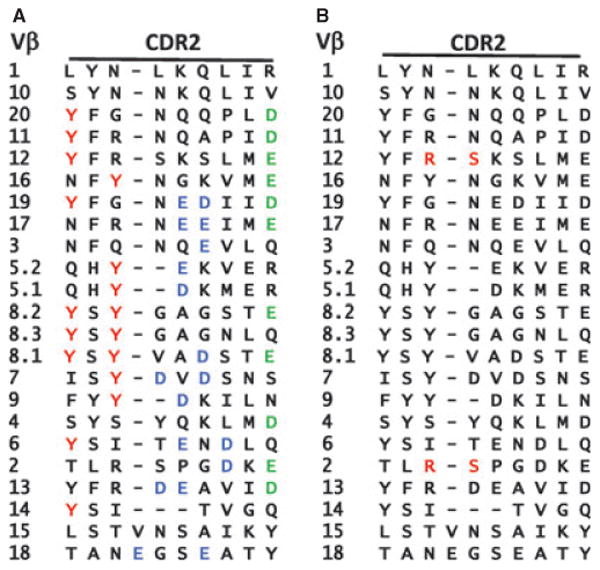
The amino acid sequences of the CDR2 regions of mouse Vβs are shown. (A) The Y46 Y48 tyrosine motif shared by relatives of the mouse Vβ8 family are highlighted in red. The acidic amino acids that often form salt bridges with basic amino acids in MHCII are colored green (D or E at position 54) or blue (other D or E in this region). (B) The R S motif that makes contact with MHC in two structures containing Vβ2 is colored red (39).
Thus, the ‘rules’ became evident only when a fair number of TCR/MHC/peptide structures involving identical and/or similar Vα and Vβ chains had been solved. At this point one could argue that the crystallographers had a lucky break. It turns out that some V regions allow TCRs to crystallize much more consistently than others. Chief among these are the TCRs that use relatives, in mouse and human, of the mouse Vβ8 family. Because TCRs that use these Vβ relatives often express better in E. coli or insect cells and because they fold more consistently, more structures have been solved with TCRs that include them than any other. Therefore, we can get a good idea of whether or not certain amino acids in these Vβs are used in a consistent way to bind MHC.
It turns out that they are. In agreement with many mutational studies, we and others have found that two amino acids in the CDR2 loops of these Vβ relatives, Y46 and Y48, called sometimes Y48 and Y50, very often engage the same surface of MHCII, whether the complex is human or mouse (31, 33, 42, 52–56). Such a conclusion is illustrated in Fig. 3, which shows the position of Y48 on a number of MHCII and MHC class I (MHCI) structures. Y48 is in almost exactly the same position in all the structures of TCRs that include this amino acid, bound to MHCII, regardless of the species source of the proteins or the peptide involved (Fig. 3A). LikewiseY48, when present, often engages MHCI, but in this case its binding site varies somewhat (Fig. 3B). Y46 (data not shown) and Y48 can move from one structure to another on the upper surface of MHCI. We do not know why the binding site of Y46 and Y48 is less consistent on MHCI than on MHCII. One possibility is that engagement of TCRs and MHCII is restricted by another feature, the fact that a third Vβ amino acid that is common to the family, E54, forms a salt bridge to MHCII αK39. αK39 is present in many alleles of MHCII and therefore this salt bridge, if formed, will tend to lock the TCR at a single location on the MHCII protein (see below).
Fig. 3. In different TCR + MHC + peptide complexes, Y48 of TCR Vβs that include it binds to the same site on MHCII but is more variably positioned on MHCI.
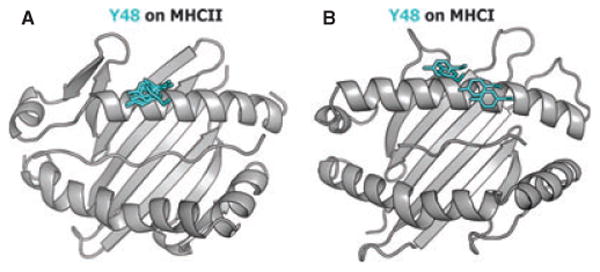
The positions of TCRβ Y48 in (A) 6 MHCII engaged structures and (B) 4 MHCI engaged structures is shown, with the tyrosine colored in cyan.
The roles of TCRβ Y46 and Y48 were most dramatically illustrated by two recent structures, which studied an unusual TCR that had two known ligands: one MHCII, IAb, plus the so-called 3K peptide, and another, MHCI, Kb, plus a different peptide. Y46 and Y48 are in almost exactly the same positions with respect to each other and with respect to the corresponding MHC α helices in the two structures (42). Y48 lies on the α helices of the two MHC proteins, pinned down to some extent by Y46.
Does mouse Vβ8 often use Y46 and Y48 to engage MHC, regardless of its TCRα partner?
The problem with structural studies is that they are laborious and, in the case of the variable TCRs, apply potentially only to the few structures that have been solved. A similar proposition applies also to mutational results. We therefore set up a system that would test the effects of loss of Y46 and Y48 on the ability of many TCRs to bind MHC. Our first experiments were done with retroviral transduction of a TCR wild-type (WT) or mutated (Y46A or Y48A) β chain into bone marrow stem cells that could not make any other TCRβ, but could make any TCRα. The stem cells were transferred into mice that lacked T cells and the ability of the different stem cells to create mature T cells measured (57). We have recently repeated these experiments in mice that are transgenic for the WT and mutant TCRβs, and, again, they were unable to make any other TCRβs, but were able to produce any TCRα. In both sets of experiments, the question to be addressed was whether the absence of Y46 or Y48 would inhibit recognition of MHC by TCRs expressing the mutated amino acid regardless of the TCRαs with which they were paired. That is, were the inhibitory effects of the mutations generalizable to many different TCRs? We decided to use efficiency of positive selection as a measure of how well the WT or mutated TCRβs could function, since MHC recognition is known to be needed for positive selection to occur.
The results of the retrogenic TCR experiments have been published (57). Results for the TCRβ transgenic mice were similar to those of the retrogenic mice. The thymi of the TCRβ transgenic mice contained about the same number of thymocytes, regardless of the TCR transgene they expressed. However, there was a dramatic reduction in the numbers of mature CD4+ and CD8+ T cells they contained, if the animals expressed A rather than Y at position 48. The same trend was apparent for CD4+ T cells in mice expressing A instead of Y at position 46, but the result did not apply for CD8+ cells in the same animals (Fig. 4). We interpret this result to indicate that loss of Y at position 48 of a Vβ8 related TCR leads to a reduction in positive selection of both CD4+ and CD8+ T cells, and this regardless of the fact that any possible TCRα was available to pair with the mutated TCRβ. We suggest that this occurred because of a lowered ability of the TCRs containing the mutant TCRβs to engage MHC. Apparently only a few TCRαs can compensate for the inadequacies of their TCRβ partner, as manifest by a shift in the TCRαs that are used by T cells in the mutant mice (57).
Fig. 4. TCRβ Y46 and Y438 adopt similar positions and configurations when engaging MHCII or MHCI.
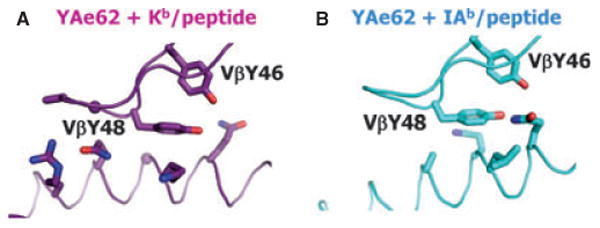
The YAe62 TCR can bind MHCII + the 3K peptide and MHCI + a different peptide. Shown are the positions of TCRβ Y46 and Y48 when bound to (A) IAb + 3K or (B) Kb + a different peptide. These data were previously shown in a different figure in Yin et al. (42).
These results indicate that the need for Y48 by Vβ8-related TCRs is quite generalizable and not a property of just a few TCRs. Nevertheless it could be and has been argued that these results apply only to the particular Vβ-CDR3β-Jβ sequence used to create the transduced stem cells and transgenic mice (58). Experiments are underway to test whether similar results will be obtained in a rearranging system, in which a WT and mutant Vβ can rearrange to a number of Jβs, creating many different CDR3β sequences as they do so.
Is VβY48 evolutionarily conserved?
Although our data strongly supported the idea that Y46 and particularly Y48 residues are important for MHC interaction by a multitude of Vβ8-containing TCRs, they did not directly address whether these residues might have been evolutionary selected to fulfill this task. We tackled this issue by inspecting the Vβ sequences in species that are distantly related to mouse and human. We found Vβ sequences that include tyrosines at positions 46 and 48 even in sharks, members of the class Chondrichthyes, the most distant evolutionary class from mammals that is known to express an adaptive immune system (59–61). In spite of the presence of these two amino acids, the rest of the Vβ sequences that include them, in frogs, trout, and sharks, are only 25–30% identical to that of mouse Vβ8.2.
We set up experiments to find out if the xenogeneic Vβs can react with mouse MHC and whether, when they do so, their Y46 and Y48 residues are needed for the reaction. We exchanged the foreign Vβs or their mutants expressing Y46A or Y48A into a mouse TCR that reacts with IAd or IAb plus a peptide from ovalbumin (Ova). These genes were introduced into bone marrow stem cells that could not express any other TCRβs, but were competent for TCRα expression, and transferred the stem cells into T-cell deficient mice. The ability of the retrogenic TCRβs to react with mouse MHC was assessed, as above, by the efficiency with which mature CD4 and CD8 thymocytes appeared (57). Again, the thymi of all mice were comparable in cell number. The WT versions of the foreign Vβs gave rise to mature thymocytes quite efficiently whereas the mutant versions did not (62). Even more remarkably, the WT frog Vβ, when substituted into the mouse TCR that was specific for IAd + Ova, allowed recognition of that very combination, whereas its Y48A mutant did not.
These experiments suggest strongly, at least in the case of the Vβ8 analogs, that Y46 and Y48 have been present in some TCR Vβs for about 450 million years and that in TCRs, they probably perform a common function, engagement of MHC.
Do Vβs that are not related to the mouse Vβ8s have amino acids that bind in a consistent way to MHC?
A few years ago, we and Garcia et al. (53, 55) suggested that different Vβs might have different sets of amino acids to promote binding to MHC. Given the complexity of amino acid sequences of the CDR2 regions of Vβs in mice (Fig. 2A), this idea must be right, if V regions are to have any built-in propensity to engage MHC. Only about 30% of the mouse Vβs has Y46 and Y48 for example (Fig. 2), and this percentage is even lower for the human Vβs.
Unfortunately structural information for TCR/MHC combinations other than those that include Vβ8 relatives is sparse, due to expression and folding difficulties for such TCRs. There is the additional problem that now that the general rules for the engagement of TCRs and MHC are established, crystallographers have turned their attention to the unusual cases (see below). There is little incentive to solve yet more structures of TCRs that will obey the rules, even with different Vs that are already known. These two trends have contributed to our current inability to spot the crucial amino acids, if they exist, which routinely contribute to MHC recognition by other Vβs.
In spite of these difficulties, is there any evidence that there are motifs in Vβs that predispose toward MHC recognition, other than Y46 and Y48? To some extent, the answer to this question is yes. For example, we constructed retrogenic mice that expressed a single TCRβ chain that included Vβ6 with a tyrosine, the normal amino acid, or alanine at position 46. T cells were much less efficiently selected in mice expressing the mutant versus the WT TCRβ, suggesting that in Vβ6, Y46 contributes to MHC recognition (57). In a structural approach, Reiser et al. (39) compared MHC contacts made by two different TCRs containing mouse Vβ2. Two amino acids consistently made contact in the two complexes (Fig. 2B).
We and others have previously found that E54 in CDR2β of Vβ8-related TCRs often forms a salt bridge with a conserved K at position 39 of MHCII β1 helices (33, 53–55) (Fig. 5A). This interaction may serve to lock the TCR into its characteristic diagonal position in which the two accompanying Vβ8 tyrosines, Y46 and Y48, contact the MHCII β1 helix in a fixed position. It is worth noting that an acidic amino acid is often found at position 54 of mouse TCRβs (33) (Fig. 2A). A search of the PDB database reveals two mouse Vβ3-containing structures, involving the 2B4 and 226 TCRs that contain an E at a position 52, similar to that of the E54 mentioned above (56). However, in the 2B4 and 226 structures, E52 forms a salt bridge not with the conserved MHCII β1 K39, but instead with K67 on the same MHCII α helix (Fig. 5B).
Fig. 5. Replacement of TCRβ Y48 with an alanine in mice expressing a Vβ8-containing TCRβ as their only TCRβ chain severely reduces positive selection of T cells.
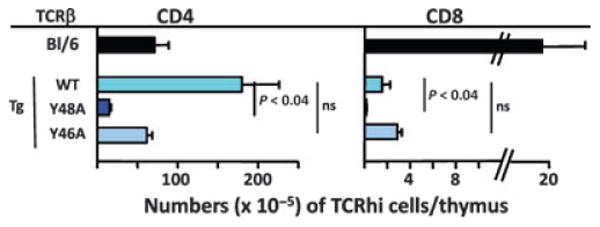
A TCRβ chain containing the WT DO11.10 TCRβ chain or the same chain with Y46 or Y48 replaced by alanines (Y46A or Y48A) was introduced into C57BL/6 embryos. The mice so generated were crossed to mice deficient in TCRβ (108) and intercrossed to produce mice that could make only the transgenic TCRβ and any TCRα. Thymi in these mice were analyzed for their content of mature CD4+ and CD8+ cells. Shown are the means and SEMs of at least three mice of each type.
To find out if the salt bridge involving K67 is found in other complexes, we investigated all the published TCR/pMHC II complex structures and found another two cases with such an interaction, but with interesting alternative patterns. Human Vβ2.1 does not contain an aspartic or glutamic acid at positions 52 or 54, but does have a glutamic acid residue at position 49. In the complex of a human Vβ2.1-containing TCR, MS2-3C8, recognizing DR4 (63), Vβ2.1 E49 forms a salt bridge with MHCII β1 K67 (Fig. 5C). Human Vβ3.1 has two acidic amino acids at or near CDR2, D49, and E54. Both form salt bridges to MHCII β1, VβD49 to MHCII β1 K67, and VβE54 to MHCII β1 K39 (64, 65) (Fig. 5D).
These data suggest a colorful spectrum of conserved salt bridges involving acidic amino acids near CDR2 of a number of Vβs and the complimentary K39 and/or K67 of MHCII. In agreement with this idea, about 80% of mouse Vβs and nearly 95% of all human TCR Vβs have acidic residues in or near CDR2β (Fig. 2A), all of which could be involved in salt bridges to MHCII β1 lysines. Perhaps the choice of bridging to K39 or K67 allows some flexibility, permitting accommodation of different Vβs and differences in the structures of CDR3 and peptide elements.
With respect to the ability of salt bridges to lock TCR positions on MHCII it is interesting to note that such salt bridges do not form when TCRs bind MHCI. Perhaps this is why the conserved TCR Y48 can move more freely from structure to structure on MHCI versus MHC II (Fig. 3).
Do Vαs contain amino acids that predispose toward MHC recognition?
As far as MHC binding is concerned, TCRαs have received much less attention than Vβs. This may be because in mice, at least, there are many more Vαs and Jαs than the corresponding TCRβ elements. Therefore, fewer TCR/MHC structures that involve the same Vα have been solved than for their Vβ counterparts and it is consequently more difficult to predict the rules. There is also the suspicion that the TCRα may play a different role in TCR/MHC recognition than TCRβ does (66–68) (see discussion below).
A few years ago we noticed that a tyrosine in the CDR1 region of some mouse and human Vαs often binds a particular site on the α2/β1 helix of MHC (33, 55). To find out whether or not this tyrosine plays a role in MHC engagement that is analogous to that of Vβ Y46 and Y48, we created transgenic mice expressing a single TCRα that contained Y29, or a mutant TCRα with an alanine at position 29. Along the lines of the Vβ8 experiments described above, the mice could express any TCRβ, but only a single TCRα, WT or Y29A. The effects of the Vα mutation on MHC binding when accompanied by any possible TCRβ were measured by counting the numbers of mature CD4+ or CD8+ T cells that appeared in the thymi of the transgenic mice. Thymi from both types of transgenic mice contained about the same number of thymocytes. However, there were significantly fewer mature CD4+ T cells in the thymi of mice expressing the mutant Y29A TCRα (Fig. 6).
Fig. 6. Acidic amino acids in CDR2β often make salt bridges with lysines in MHCII.

The X-ray crystallographically established positions of acidic amino acids in four TCRs engaged with MHCII + peptide, and their salt bridges with Ks in MHCII are shown. The data shown are from (A) Dai et al. (33), (B) Newell et al. (56), (C) Yin et al. (63), and (D) Hennecke et al. (64, 65).
Mature CD8+ thymocytes appeared with equal frequency whether the mice expressed the WT or mutant TCRα. We do not know why the mutation had a more profound effect on CD4+ rather than CD8+ T-cell selection and thus on MHCII versus MHCI recognition. A similar trend was observed for the TCRβ mutations described above (Fig. 4). In both cases, the WT TCRα and TCRβ chains used in the experiments came from CD4+ T cells. Perhaps a bias for MHCII reaction was present in some element of the individual TCR chains used, and this bias led to the mutations having a larger effect on recognition of the WT target. Alternatively, the rules of TCR/MHC engagement that we believe are being exposed in these experiments may apply more profoundly to MHCII rather than MHCI recognition.
There are difficulties with the approach used to study TCRα chains in the experiments described here. The transgenes were driven by a human CD2 promoter, a promoter that drives expression of the transgene during the double negative stage of thymocyte development (69). It is known that such early expression of TCRα leads to abnormalities in thymocyte development and numbers (70–72). Therefore, interpretation of the TCRα experiments must be considered with caution.
How do TCR/MHC combinations cope with variations in the lengths and amino acid sequences of TCR CDR3s and peptide ligands?
In theory, and to a lesser extent perhaps in practice, the CDR3 sequences of TCRαs and TCRβs can vary tremendously and are sometimes very long (73). Likewise, the upwardly pointing amino acids of the peptides bound to MHC can also differ enormously in sequence and, particularly for MHCI, in their ability to bulge upwards out of the groove (41). A priori it seems impossible that TCRs could engage MHC with rigid rules, in the way that other proteins bind their protein partners.
We suggest that evolution has coped with this problem by ‘designing’ the sites at which TCRs engage MHC in a fashion that allows flexibility. For example, Vβ Y48 binds the α1 helix of MHC at the same site in many different solved structures. However, Y48 does not approach the site in the same way from one structure to another. It can twist and tilt, still maintaining some van der Waals and polar interactions with the α carbon backbone and/or side chain amino acids lining the site (Fig. 7). The target of the Y48 on MHC is a shallow cradle that facilitates such flexibility.
Fig. 7. Replacement of TCRα Y29 with an alanine in mice expressing a Vα4-containing TCRα as their only TCRα chain reduces positive selection of T cells.
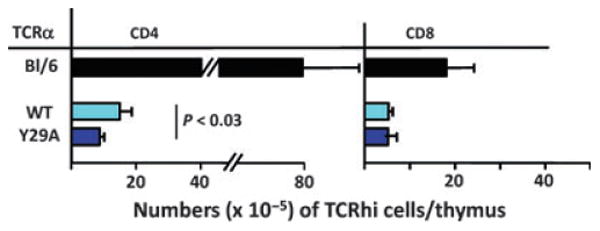
Mice were produced as described in Fig. 5 except that they expressed a single TCRα that of B3K508 (109) and any possible TCRβ. Thymi were analyzed for the numbers of CD4+ and CD8+ T cells they contained. Data shown are the means and SEMs of at least three mice of each type.
The variability thus allowed means that the engagement of MHC by other amino acids of the TCR can change from one structure to another, even when the structures involve exactly the same TCR. For example, in a recently published set of structures, Vβ Y46 and Y48 had almost exactly the same conformation with respect to MHC from one structure to the other, even though the MHC/peptide ligands in the two structures were different (74). However, other TCR amino acids, for example Q61, changed their rotamers considerably (74–76).
We have recently compared the structures of the same TCR bound to MHCI/peptide and MHCII/peptide ligands. Vβ Y46 and Y48 had exactly the same conformation with respect to MHCI and the analog MHCII in the two structures. However, when bound to MHCI, one of the β strands of TCRα had moved away from the strand containing Jα. This allowed the TCR structure to maintain stable binding of the α and β chains, but the rest of the TCRα to rotate and move away from TCRβ.
What about the TCR/MHC structures that do not use the built-in MHC-engaging TCR amino acids?
Apart from death and taxes, rules are not inviolate, and this remark certainly applies to the engagement of MHC by TCRs. Numerous examples have shown that some TCRs can bind ligands that include MHC without using the TCR amino acids that are predicted by the evolutionary hypothesis. For example, in the human TCR engaging an HIV peptide bound to HLA-A2 structure solved by the Wiley laboratory years ago, the TCR had a very long CDR3β sequence. In this complex, amino acids of the non-germ-line-encoded CDR3β loop were intimately involved in binding the MHC/peptide ligand, forcing the rest of the TCR well away from HLA-A2, thus prohibiting engagement of MHC by evolutionarily conserved residues, if they exist, of the TCR (73).
Sometimes peptides can bulge out of the MHC groove. Such is particularly evident in the case of peptides that bind to MHCI, which are locked by their charged N- and C-terminal residues in binding at each end of the MHCI groove. The bulged peptide again forces the TCR away from MHC, restricting engagement of conserved MHC-recognizing residues (41).
Such variations from the norm are particularly evident in structures of TCRs bound to MHC presenting autoantigens. Although relatively few examples of structures involving these interactions are currently available, it does seem that quite often, autoreactive TCRs approach their MHC + peptide ligands in unusual ways. For example, in the three available structures of TCRs from human multiple sclerosis patients binding to their MHC + autoantigen ligand the TCRs bind their ligands in an altered mode (40, 77, 78). In the most recently published structure, TCRα did not engage MHC at all, and only the CDR2β loop of the TCRβ chain engaged MHC. Most of the binding involved engagement of the peptide (40).
Although not all autoreactive TCRs bind their ligands in unusual ways (35, 63) there is often something unusual about the TCR, its ligand, or the interaction between the two. For example, in some complexes, the target peptide binds weakly to MHC (63, 79). In others, the peptide is truncated or modified (35, 80). In all cases, perhaps these features function to prevent negative selection of thymocytes bearing the relevant TCR in the thymus, thus allowing their escape to drive autoimmunity under the right circumstances.
What about TCRs that bind non-classical MHC ligands?
A few TCRs have been isolated that do not bind classical MHC/peptide ligands, but rather non-classical MHC. The best studied of these TCRs is the invariant natural killer T cell TCR (iNKT TCR), which, using a single TCRα sequence and a subset of TCRβ sequences, binds CD1d plus lipid ligands (81, 82). The properties and structures of these TCRs and their binding to CD1d + lipid have been extensively studied. The TCRβ partner of the conserved TCRα is often a relative of the mouse Vβ8 family. Although the invariant TCRα chain dominates the interaction with both the lipid and CD1d, the two Vβ tyrosines, Y46 and Y48, which we and others have implicated in engagement of classical MHC are also involved in binding CD1d, but at a completely different site (83, 84). The importance of these two tyrosines for the interaction of the iNKT TCR with CD1d and the making of iNKT cells was further emphasized by our experiments that swapped the CDR2 loop of Vβ8.2 into the Vβ6, a Vβ chain poorly represented within the iNKT repertoire. When we created retrogenic mice using a Vβ6 chain (that naturally lack the Y48 residue), no iNKT cells developed. However, when retrogenic mice were produced with the same Vβ6 chain in which the CDR2β loop had been swapped for the one of Vβ8.2, thereby introducing the Y48 residue, normal development of iNKT cell occurred (85, 86). Similarly, when inserted into an iNKT TCR the Y46 and Y48-containing Vβs from sharks, trout, and frogs can participate in and are required for CD1d recognition (62). This is quite an extraordinary result, since CD1d itself is not present in non-mammalian species. Therefore, when CD1d and the iNKT TCRs that bind it evolved, they apparently co-opted previously existing Vβs and their mode of MHC interaction.
What about TCRs that bind ligands that do not include MHC?
Some reports have described TCRs that react with ligands that do not include MHC. These include description of T cells that react with haptens related to fluorescein (87). Most notable, however, are two TCRs described in a recent article by Tikhonova et al. (58). These authors predicted that the bias of TCRs for reaction with MHC is imposed on a random TCR repertoire by positive selection in the thymus of thymocytes that bear CD4 and CD8. The authors suggested that because the tyrosine kinase Lck is needed for positive selection and because Lck is bound to CD4 and CD8 in thymocytes, positive selection requires simultaneous engagement of the same MHC + peptide complex by both the TCR and the appropriate costimulatory protein, CD4 or CD8. In the absence of CD4 and CD8 (and MHC proteins), Lck might be freed up in thymocytes undergoing positive selection, thereby allowing positive selection of thymocytes bearing TCR specificities for ligands other than MHC. The authors tested this hypothesis by examining the specificities of the few T cells that appear in mice that lack classical MHC and CD4 and CD8. Interestingly, two TCRs appeared in such mice that reacted with CD154, a non-MHC related protein. One of the TCRs was screened for amino acids involved in recognition of CD154 and TCRβ Y48 was among the amino acids of interest.
This interesting result was interpreted to imply that since Y48 is a prominent amino acid on the tip of a CDR2 loop, it is almost bound to interact with any ligand for TCRs, including MHC and in the case the authors studied CD154 (58). The authors argued that the site-specific engagement of Vβ8 Y48 to MHC is caused by the arrangement of the trimolecular complexes of the TCR, the MHC, and CD4 or CD8. Positive selection then determines the bias of selected TCRs for MHC.
The authors (58) may be right in this conjecture. However, their ideas do not account for the results of the retrogenic and transgenic experiments described above. In those experiments, T-cell selection on MHC was severely reduced if the mice expressed a single TCRβ chain (Vβ8.2 or Vβ6) with a mutation at Y48 or Y46 versus the WT TCRβ sequence. The T cells in mice expressing a mutant TCRβ expressed a different TCRα repertoire (57), indicating that different TCRα chains could partner with the mutant TCRβs versus the WT TCRβs and allow successful positive selection to occur.
How then can one account for the finding that some TCRs can react with ligands that do not involve MHC at all? We think it is likely that because many TCR sequences are possible, some occasionally have the potential to bind unusual ligands, perhaps in unusual ways and perhaps in modes that are similar to those adopted by some antibodies. The idea that TCR V regions are biased toward reaction with MHC does not preclude their engagement of additional ligands. It is the general trend and bias that we would like to emphasize here, not an absolute rule. Ultimately, determining the frequency of MHC-reactive and non-MHC reactive out of completely random and unselected TCRs will answer the question of whether positive selection and/or evolutionary pressure is the most important force in imposing the reactivity of the TCR with MHC.
Finally in this category, we should mention superanti-gens. The staphylococcal enterotoxins bind MHCII and engage TCRs. However, they often but not always engage a portion of TCRs, the so-called CDR4β region (88, 89) that is not usually involved in binding MHC + peptide. In the two solved structures of TCR + MHC + superantigens, the TCR is lifted well away from the MHC, making no contacts with this protein at all (90, 91). Somehow microbes have used superantigen to bind TCRs and MHC without using at all the usual contacts between the two.
In 2012, what is to be gained from the study of more structures of TCRs bound to MHC proteins?
One could argue that we know all we need to know about the ways in which TCRs bind their ligands. Further investigations may reveal more about the fine details of the engagement, but will not help us understand or treat diseases, nor design better vaccines. There may be some truth to this argument, but we think not.
There are still some mysteries about T-cell specificity. Most obviously, we still do not understand autoimmunity. How do some T cells escape negative selection and regulatory control to attack their hosts? In rare cases, autoattack is caused by catastrophic mutations in genes, for example Fas or FoxP3 (92, 93), whose products are essential for proper control of the immune system. Conventional mapping and genome-wide association studies have revealed, however, that autoimmune diseases are most frequently caused by the presence in an individual of a combination of genetic polymorphisms (94–96). Of the implicated genes, the most common by far are those of the MHC (97), indicating that autoimmune diseases are most powerfully driven by some abnormality in T-cell reactivity to MHC plus, presumably, self-peptide(s). MHC association could be due to some MHC alleles presenting self-peptides too well. Alternatively and in our opinion more likely, MHC association of autoimmunity is a reflection of one of two phenomena. Some MHC alleles may present particular self-peptides poorly, allowing autoreactive T cells to escape negative selection in the thymus and thus become activated when exposed to higher concentrations of the MHC + relevant peptide in the periphery (63, 79). Some self-peptides may not be available in the thymus at all. Differences in protein cleavage or post-translational modifications, perhaps exclusive to peripheral tissues, may allow self-peptides to appear in the periphery and not at all in the thymus (80). In this latter case, the target peptides of autoreactive T cells may be, in effect, foreign peptides and treated as such by the T cells that recognize them. Further studies of the nature and structure of T-cell reaction with autoantigens are needed, for us to unravel these problems.
It has been argued that autoimmunity might sometimes arise because of a failure in control by regulatory T cells, as opposed to the active generation of T cells with autoreactive specificities. However, one should keep in mind that regulatory T cells also express a TCR, whose antigen specificity and MHC recognition is an inherent part of their development and most likely of their function (98–101). Clearly, not every TCR with any antigenic specificity can give rise to regulatory T cells, and there are still questions about how the affinity and nature of the TCR for self and foreign ligands affect the generation of regulatory T cells. Similar questions apply to the differentiation of T cells into other subtypes of cells, T-helper 1 (Th1), Th2, Th17, NKT, MAIT cell, etc. Here again, the TCR and its interaction with its ligands both initiate generation of the cells and potentially control their fate.
Positive selection does not just select for thymocytes bearing TCRs that react weakly with MHC. It also selects thymo-cytes bearing TCRs that turn out to react better with foreign peptides bound to MHC alleles that are self (expressed in the thymus cortex), rather than foreign. How can this be? How can TCRs distinguish between one allele of MHC and another? Our groups' experiments have suggested how TCRs have the ability to react with any MHC, in a generic way, but they have not yet tackled the issue of how TCRs can be MHC allele specific. Apart from the discovery that positive selection leads to a repertoire of TCRs biased toward recognition of self-MHC + foreign peptides, relatively few experiments have studied this issue. A flurry of articles in the 1990s from the Gascoigne group (66–68) suggested that MHC allele recognition was governed by Vα. A later article (102) suggested that the peptides involved in positive selection in the thymus governed the ability of TCRs to distinguish between self and foreign MHC. This finding would appear to be at odds with the Vα idea, given that peptides, self or foreign, are primarily recognized by the CDR3 regions of TCRs. The issue has yet to be resolved and is of some importance, since it has implications for our understanding of transplant rejection.
We do not properly understand the complexities of the T-cell repertoire. Recent deep sequencing experiments indicate that a very large number of different TCRs are expressed by naive T cells in mice and humans (103). However, not all estimates, including our own preliminary results, agree with this idea (104). Preliminary experiments from our laboratories suggest that TCR complexity, at least in mice, may not be as great as suggested. Moreover, the existence of public specificities, TCR sequences specific for particular MHC/peptides that have been found repeatedly in different individuals, indicates that the makeup of the TCR repertoire may be quite unexpectedly repetitive (105–107). Such uncertainties need to be resolved for us to properly understand and manipulate, perhaps via vaccines, our immune responses.
T cells are the underpinnings of the acquired immune system. Their recognition of antigen is the first and crucial step in their response to antigen. It behooves us to understand, at the most basic level, how antigen recognition by T cells is achieved.
Acknowledgments
The authors thank the many people who have contributed to our thoughts on the issues discussed here, particularly Dr. Eric Huseby, Dr. Brian Stadinski, Dr. Kira Rubtsova, Dr. Thierry Mallevaey, and Dr. Shaodong. The authors also thank Dean Becker, Frances Crawford, Desiree Garcia, Ella Kushnir, and Janice White, who have done much of the work that led to the ideas discussed in this article. This work was supported in part by USPHS grants AI-18785, AI-22295, AI-076463, and AI-078246. The authors have no conflicts of interest to declare.
References
- 1.The Nobel Lectures in Immunology. The Nobel Prize for Physiology or Medicine, 1980 awarded to Baruj Benacerraf, Jean Daussett & George D. Snell. Scand J Immunol. 1992;35:373–398. [PubMed] [Google Scholar]
- 2.Gorer P. The antigenic structure of tumors. Adv Immunol. 1961;1:345. [Google Scholar]
- 3.Babbitt BP, Allen PM, Matsueda G, Haber E, Unanue ER. Binding of immunogenic peptides to Ia histocompatibility molecules. Nature. 1985;317:359–361. doi: 10.1038/317359a0. [DOI] [PubMed] [Google Scholar]
- 4.Benacerraf B, McDevitt HO. Histocompatibility-linked immune response genes. Science. 1972;175:273–279. doi: 10.1126/science.175.4019.273. [DOI] [PubMed] [Google Scholar]
- 5.Bevan MJ. Interaction antigens detected by cytotoxic T cells with the major histocompatibility complex as modifier. Nature. 1975;256:419–421. doi: 10.1038/256419a0. [DOI] [PubMed] [Google Scholar]
- 6.Kappler JW, Marrack PC. Helper T cells recognise antigen and macrophage surface components simultaneously. Nature. 1976;262:797–799. doi: 10.1038/262797a0. [DOI] [PubMed] [Google Scholar]
- 7.Kappler JW, Skidmore B, White J, Marrack P. Antigen-inducible, H-2-restricted, interleukin-2-producing T cell hybridomas. Lack of independent antigen and H-2 recognition. J Exp Med. 1981;153:1198–1214. doi: 10.1084/jem.153.5.1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Katz DH, Hamaoka T, Dorf ME, Benacerraf B. Cell interactions between histoincompatible T and B lymphocytes. V. Failure of histoincompatible T cells to interfere with physiologic cooperation between syngeneic T and B lymphocytes. J Immunol. 1974;112:855–857. [PubMed] [Google Scholar]
- 9.Schwartz RH, Yano A, Paul WE. Interaction between antigen-presenting cells and primed T lymphocytes: an assessment of Ir gene expression in the antigen-presenting cell. Immunol Rev. 1978;40:153–180. doi: 10.1111/j.1600-065x.1978.tb00405.x. [DOI] [PubMed] [Google Scholar]
- 10.Shearer GM, Lozner EC, Rehn TG, Schmitt-Verhulst AM. Mixed lymphocyte reactivity and cell-mediated lympholysis to trinitrophenyl-modified autologous lymphocytes in C57BL/10 congenic and B10-A recombinant mouse strains. J Exp Med. 1975;141:930–934. [PMC free article] [PubMed] [Google Scholar]
- 11.Shevach EM, Green I, Paul WE. Alloantiserum-induced inhibition of immune response gene product function. II. Genetic analysis of target antigens. J Exp Med. 1974;139:679–695. doi: 10.1084/jem.139.3.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shimonkevitz R, Kappler J, Marrack P, Grey H. Antigen recognition by H-2-restricted T cells. I. Cell-free antigen processing. J Exp Med. 1983;158:303–316. doi: 10.1084/jem.158.2.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zinkernagel RM, Doherty PC. Immunological surveillance against altered self components by sensitised T lymphocytes in lymphocytic choriomeningitis. Nature. 1974;251:547–548. doi: 10.1038/251547a0. [DOI] [PubMed] [Google Scholar]
- 14.Bjorkman PJ, Saper MA, Samraoui B, Bennett WS, Strominger JL, Wiley DC. The foreign antigen binding site and T cell recognition regions of class I histocompatibility antigens. Nature. 1987;329:512–518. doi: 10.1038/329512a0. [DOI] [PubMed] [Google Scholar]
- 15.Bjorkman PJ, Strominger JL, Wiley DC. Crystallization and X-ray diffraction studies on the histocompatibility antigens HLA-A2 and HLA-A28 from human cell membranes. J Mol Biol. 1985;186:205–210. doi: 10.1016/0022-2836(85)90271-2. [DOI] [PubMed] [Google Scholar]
- 16.Allison JP, McIntyre BW, Bloch D. Tumor-specific antigen of murine T-lymphoma defined with monoclonal antibody. J Immunol. 1982;129:2293–2300. [PubMed] [Google Scholar]
- 17.Chien Y, Becker DM, Lindsten T, Okamura M, Cohen DI, Davis MM. A third type of murine T-cell receptor gene. Nature. 1984;312:31–35. doi: 10.1038/312031a0. [DOI] [PubMed] [Google Scholar]
- 18.Haskins K, Kubo R, White J, Pigeon M, Kappler J, Marrack P. The major histocompatibility complex-restricted antigen receptor on T cells. I. Isolation with a monoclonal antibody. J Exp Med. 1983;157:1149–1169. doi: 10.1084/jem.157.4.1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hedrick SM, Cohen DI, Nielsen EA, Davis MM. Isolation of cDNA clones encoding T cell-specific membrane-associated proteins. Nature. 1984;308:149–153. doi: 10.1038/308149a0. [DOI] [PubMed] [Google Scholar]
- 20.Meuer SC, Fitzgerald KA, Hussey RE, Hodgdon JC, Schlossman SF, Reinherz EL. Clonotypic structures involved in antigen-specific human T cell function. Relationship to the T3 molecular complex. J Exp Med. 1983;157:705–719. doi: 10.1084/jem.157.2.705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yanagi Y, Yoshikai Y, Leggett K, Clark SP, Aleksander I, Mak TW. A human T cell-specific cDNA clone encodes a protein having extensive homology to immunoglobulin chains. Nature. 1984;308:145–149. doi: 10.1038/308145a0. [DOI] [PubMed] [Google Scholar]
- 22.Arden B, Clark SP, Kabelitz D, Mak TW. Human T-cell receptor variable gene segment families. Immunogenetics. 1995;42:455–500. doi: 10.1007/BF00172176. [DOI] [PubMed] [Google Scholar]
- 23.Arden B, Clark SP, Kabelitz D, Mak TW. Mouse T-cell receptor variable gene segment families. Immunogenetics. 1995;42:501–530. doi: 10.1007/BF00172177. [DOI] [PubMed] [Google Scholar]
- 24.Concannon P, et al. Human T-cell receptor genes: organization, diversity, and polymorphism. Cold Spring Harb Symp Quant Biol. 1986;51:785–789. doi: 10.1101/sqb.1986.051.01.091. [DOI] [PubMed] [Google Scholar]
- 25.Glusman G, et al. Comparative genomics of the human and mouse T cell receptor loci. Immunity. 2001;15:337–349. doi: 10.1016/s1074-7613(01)00200-x. [DOI] [PubMed] [Google Scholar]
- 26.Hayday AC, et al. Unusual organization and diversity of T-cell receptor alpha-chain genes. Nature. 1985;316:828–832. doi: 10.1038/316828a0. [DOI] [PubMed] [Google Scholar]
- 27.Hagiya M, Davis DD, Shultz LD, Sakano H. Non-germ-line elements (NGE) are present in the T cell receptor beta-chain genes isolated from the mutant mouse, motheaten (me/me) J Immunol. 1986;136:2697–2700. [PubMed] [Google Scholar]
- 28.Leiden JM, Strominger JL. Generation of diversity of the beta chain of the human T-lymphocyte receptor for antigen. Proc Natl Acad Sci USA. 1986;83:4456–4460. doi: 10.1073/pnas.83.12.4456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Davis MM, Chien Y. Topology and affinity of T-cell receptor mediated recognition of peptide-MHC complexes. Curr Opin Immunol. 1993;5:45–49. doi: 10.1016/0952-7915(93)90079-8. [DOI] [PubMed] [Google Scholar]
- 30.Sant'Angelo DB, et al. The specificity and orientation of a TCR to its peptide-MHC class II ligands. Immunity. 1996;4:367–376. doi: 10.1016/s1074-7613(00)80250-2. [DOI] [PubMed] [Google Scholar]
- 31.Garcia KC, et al. Structural basis of plasticity in T cell receptor recognition of a self peptide-MHC antigen. Science. 1998;279:1166–1172. doi: 10.1126/science.279.5354.1166. [DOI] [PubMed] [Google Scholar]
- 32.Colf LA, et al. How a single T cell receptor recognizes both self and foreign MHC. Cell. 2007;129:135–146. doi: 10.1016/j.cell.2007.01.048. [DOI] [PubMed] [Google Scholar]
- 33.Dai S, et al. Crossreactive T Cells spotlight the germline rules for alphabeta T cell-receptor interactions with MHC molecules. Immunity. 2008;28:324–334. doi: 10.1016/j.immuni.2008.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Garboczi DN, Ghosh P, Utz U, Fan QR, Biddison WE, Wiley DC. Structure of the complex between human T-cell receptor, viral peptide and HLA-A2. Nature. 1996;384:134–141. doi: 10.1038/384134a0. [DOI] [PubMed] [Google Scholar]
- 35.Maynard J, et al. Structure of an autoimmune T cell receptor complexed with class II peptide-MHC: insights into MHC bias and antigen specificity. Immunity. 2005;22:81–92. doi: 10.1016/j.immuni.2004.11.015. [DOI] [PubMed] [Google Scholar]
- 36.McBeth C, et al. A new twist in TCR diversity revealed by a forbidden alphabeta TCR. J Mol Biol. 2008;375:1306–1319. doi: 10.1016/j.jmb.2007.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reinherz EL, et al. The crystal structure of a T cell receptor in complex with peptide and MHC class II. Science. 1999;286:1913–1921. doi: 10.1126/science.286.5446.1913. [DOI] [PubMed] [Google Scholar]
- 38.Reiser JB, et al. Crystal structure of a T cell receptor bound to an allogeneic MHC molecule. Nat Immunol. 2000;1:291–297. doi: 10.1038/79728. [DOI] [PubMed] [Google Scholar]
- 39.Reiser JB, et al. A T cell receptor CDR3beta loop undergoes conformational changes of unprecedented magnitude upon binding to a peptide/MHC class I complex. Immunity. 2002;16:345–354. doi: 10.1016/s1074-7613(02)00288-1. [DOI] [PubMed] [Google Scholar]
- 40.Sethi DK, et al. A highly tilted binding mode by a self-reactive T cell receptor results in altered engagement of peptide and MHC. J Exp Med. 2011;208:91–102. doi: 10.1084/jem.20100725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tynan FE, et al. A T cell receptor flattens a bulged antigenic peptide presented by a major histocompatibility complex class I molecule. Nat Immunol. 2007;8:268–276. doi: 10.1038/ni1432. [DOI] [PubMed] [Google Scholar]
- 42.Yin L, et al. A single T cell receptor bound to major histocompatibility complex class I and class II glycoproteins reveals switchable TCR conformers. Immunity. 2011;35:23–33. doi: 10.1016/j.immuni.2011.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jerne NK. The somatic generation of immune recognition. Eur J Immunol. 1971;1:1–9. doi: 10.1002/eji.1830010102. [DOI] [PubMed] [Google Scholar]
- 44.Ikuta K, Ogura T, Shimizu A, Honjo T. Low frequency of somatic mutation in beta-chain variable region genes of human T-cell receptors. Proc Natl Acad Sci USA. 1985;82:7701–7705. doi: 10.1073/pnas.82.22.7701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Marrack P, Shimonkevitz R, Hannum C, Haskins K, Kappler J. The major histocompatibility complex-restricted antigen receptor on T cells. IV. An antiidiotypic antibody predicts both antigen and I-specificity. J Exp Med. 1983;158:1635–1646. doi: 10.1084/jem.158.5.1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fink PJ, Bevan MJ. H-2 antigens of the thymus determine lymphocyte specificity. J Exp Med. 1978;148:766–775. doi: 10.1084/jem.148.3.766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zinkernagel RM, Callahan GN, Althage A, Cooper S, Klein PA, Klein J. On the thymus in the differentiation of “H-2 self-recognition” by T cells: evidence for dual recognition? J Exp Med. 1978;147:882–896. doi: 10.1084/jem.147.3.882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Blackman M, et al. The T cell repertoire may be biased in favor of MHC recognition. Cell. 1986;47:349–357. doi: 10.1016/0092-8674(86)90591-x. [DOI] [PubMed] [Google Scholar]
- 49.Merkenschlager M, Graf D, Lovatt M, Bommhardt U, Zamoyska R, Fisher AG. How many thymocytes audition for selection? J Exp Med. 1997;186:1149–1158. doi: 10.1084/jem.186.7.1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zerrahn J, Held W, Raulet DH. The MHC reactivity of the T cell repertoire prior to positive and negative selection. Cell. 1997;88:627–636. doi: 10.1016/s0092-8674(00)81905-4. [DOI] [PubMed] [Google Scholar]
- 51.Rudolph MG, Stanfield RL, Wilson IA. How TCRs bind MHCs, peptides, and coreceptors. Annu Rev Immunol. 2006;24:419–466. doi: 10.1146/annurev.immunol.23.021704.115658. [DOI] [PubMed] [Google Scholar]
- 52.Feng D, Bond CJ, Ely LK, Maynard J, Garcia KC. Structural evidence for a germline-encoded T cell receptor-major histocompatibility complex interaction ‘codon’. Nat Immunol. 2007;8:975–983. doi: 10.1038/ni1502. [DOI] [PubMed] [Google Scholar]
- 53.Garcia KC, Adams JJ, Feng D, Ely LK. The molecular basis of TCR germline bias for MHC is surprisingly simple. Nat Immunol. 2009;10:143–147. doi: 10.1038/ni.f.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Garcia KC, Teyton L, Wilson IA. Structural basis of T cell recognition. Annu Rev Immunol. 1999;17:369–397. doi: 10.1146/annurev.immunol.17.1.369. [DOI] [PubMed] [Google Scholar]
- 55.Marrack P, Scott-Browne JP, Dai S, Gapin L, Kappler JW. Evolutionarily conserved amino acids that control TCR-MHC interaction. Annu Rev Immunol. 2008;26:171–203. doi: 10.1146/annurev.immunol.26.021607.090421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Newell EW, et al. Structural basis of specificity and cross-reactivity in T cell receptors specific for cytochrome c-I-E(k) J Immunol. 2011;186:5823–5832. doi: 10.4049/jimmunol.1100197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Scott-Browne JP, White J, Kappler JW, Gapin L, Marrack P. Germline-encoded amino acids in the alphabeta T-cell receptor control thymic selection. Nature. 2009;458:1043–1046. doi: 10.1038/nature07812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tikhonova AN, et al. alphabeta T cell receptors that do not undergo major histocompatibility complex-specific thymic selection possess antibody-like recognition specificities. Immunity. 2012;36:79–91. doi: 10.1016/j.immuni.2011.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Criscitiello MF, Ohta Y, Graham MD, Eubanks JO, Chen PL, Flajnik MF. Shark class II invariant chain reveals ancient conserved relationships with cathepsins and MHC class II. Dev Comp Immunol. 2012;36:521–533. doi: 10.1016/j.dci.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dooley H, Flajnik MF. Shark immunity bites back: affinity maturation and memory response in the nurse shark, Ginglymostoma cirratum. Eur J Immunol. 2005;35:936–945. doi: 10.1002/eji.200425760. [DOI] [PubMed] [Google Scholar]
- 61.Sunyer JO. Evolutionary and functional relationships of B cells from fish and mammals: insights into their novel roles in phagocytosis and presentation of particulate antigen. Infect Disord Drug Targets. 2012;12:200–212. doi: 10.2174/187152612800564419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Scott-Browne JP, Crawford F, Young MH, Kappler JW, Marrack P, Gapin L. Evolutionarily conserved features contribute to alphabeta T cell receptor specificity. Immunity. 2011;35:526–535. doi: 10.1016/j.immuni.2011.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yin Y, Li Y, Kerzic MC, Martin R, Mariuzza RA. Structure of a TCR with high affinity for self-antigen reveals basis for escape from negative selection. EMBO J. 2011;30:1137–1148. doi: 10.1038/emboj.2011.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hennecke J, Carfi A, Wiley DC. Structure of a covalently stabilized complex of a human alphabeta T-cell receptor, influenza HA peptide and MHC class II molecule, HLA-DR1. EMBO J. 2000;19:5611–5624. doi: 10.1093/emboj/19.21.5611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hennecke J, Wiley DC. Structure of a complex of the human alpha/beta T cell receptor (TCR) HA1.7, influenza hemagglutinin peptide, and major histocompatibility complex class II molecule, HLA-DR4 (DRA*0101 and DRB1*0401): insight into TCR cross-restriction and alloreactivity. J Exp Med. 2002;195:571–581. doi: 10.1084/jem.20011194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sim BC, Gascoigne NR. Reciprocal expression in CD4 or CD8 subsets of different members of the V alpha 11 gene family correlates with sequence polymorphism. J Immunol. 1999;162:3153–3159. [PubMed] [Google Scholar]
- 67.Sim BC, Wung JL, Gascoigne NR. Polymorphism within a TCRAV family influences the repertoire through class I/II restriction. J Immunol. 1998;160:1204–1211. [PubMed] [Google Scholar]
- 68.Sim BC, Zerva L, Greene MI, Gascoigne NR. Control of MHC restriction by TCR Valpha CDR1 and CDR2. Science. 1996;273:963–966. doi: 10.1126/science.273.5277.963. [DOI] [PubMed] [Google Scholar]
- 69.Zhumabekov T, Corbella P, Tolaini M, Kioussis D. Improved version of a human CD2 minigene based vector for T cell-specific expression in transgenic mice. J Immunol Methods. 1995;185:133–140. doi: 10.1016/0022-1759(95)00124-s. [DOI] [PubMed] [Google Scholar]
- 70.Brabb T, Rubicz R, Mannikko V, Goverman J. Separately expressed T cell receptor alpha and beta chain transgenes exert opposite effects on T cell differentiation and neoplastic transformation. Eur J Immunol. 1997;27:3039–3048. doi: 10.1002/eji.1830271142. [DOI] [PubMed] [Google Scholar]
- 71.Erman B, Feigenbaum L, Coligan JE, Singer A. Early TCRalpha expression generates TCRalphagamma complexes that signal the DN-to-DP transition and impair development. Nat Immunol. 2002;3:564–569. doi: 10.1038/ni800. [DOI] [PubMed] [Google Scholar]
- 72.Huang CY, Kanagawa O. Impact of early expression of TCR alpha chain on thymocyte development. Eur J Immunol. 2004;34:1532–1541. doi: 10.1002/eji.200424870. [DOI] [PubMed] [Google Scholar]
- 73.Ding YH, Smith KJ, Garboczi DN, Utz U, Biddison WE, Wiley DC. Two human T cell receptors bind in a similar diagonal mode to the HLA-A2/Tax peptide complex using different TCR amino acids. Immunity. 1998;8:403–411. doi: 10.1016/s1074-7613(00)80546-4. [DOI] [PubMed] [Google Scholar]
- 74.Stadinski BD, et al. A role for differential variable gene pairing in creating T cell receptors specific for unique major histocompatibility ligands. Immunity. 2011;35:694–6704. doi: 10.1016/j.immuni.2011.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Turner SJ, Rossjohn J. alphabeta T cell receptors come out swinging. Immunity. 2011;35:660–662. doi: 10.1016/j.immuni.2011.11.002. [DOI] [PubMed] [Google Scholar]
- 76.Garcia K, et al. A closer look at TCR germline recognition. Immunity. 2012;36:887–887. doi: 10.1016/j.immuni.2012.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Li Y, Huang Y, Lue J, Quandt JA, Martin R, Mariuzza RA. Structure of a human autoimmune TCR bound to a myelin basic protein self-peptide and a multiple sclerosis-associated MHC class II molecule. EMBO J. 2005;24:2968–2979. doi: 10.1038/sj.emboj.7600771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hahn M, Nicholson MJ, Pyrdol J, Wucherpfennig KW. Unconventional topology of self peptide-major histocompatibility complex binding by a human autoimmune T cell receptor. Nat Immunol. 2005;6:490–496. doi: 10.1038/ni1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Stadinski BD, Zhang L, Crawford F, Marrack P, Eisenbarth GS, Kappler JW. Diabetogenic T cells recognize insulin bound to IAg7 in an unexpected, weakly binding register. Proc Natl Acad Sci USA. 2010;107:10978–10983. doi: 10.1073/pnas.1006545107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Stadinski BD, et al. Chromogranin A is an autoantigen in type 1 diabetes. Nat Immunol. 2010;11:225–231. doi: 10.1038/ni.1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cohen NR, Garg S, Brenner MB. Antigen presentation by CD1 lipids, T cells, and NKT cells in microbial immunity. Adv Immunol. 2009;102:1–94. doi: 10.1016/S0065-2776(09)01201-2. [DOI] [PubMed] [Google Scholar]
- 82.Matsuda JL, Mallevaey T, Scott-Browne J, Gapin L. CD1d-restricted iNKT cells, the ‘Swiss-Army knife’ of the immune system. Curr Opin Immunol. 2008;20:358–368. doi: 10.1016/j.coi.2008.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Borg NA, et al. CD1d-lipid-antigen recognition by the semi-invariant NKT T-cell receptor. Nature. 2007;448:44–49. doi: 10.1038/nature05907. [DOI] [PubMed] [Google Scholar]
- 84.Li Y, et al. The Valpha14 invariant natural killer T cell TCR forces microbial glycolipids and CD1d into a conserved binding mode. J Exp Med. 2010;207:2383–2393. doi: 10.1084/jem.20101335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mallevaey T, et al. A molecular basis for NKT cell recognition of CD1d-self-antigen. Immunity. 2011;34:315–326. doi: 10.1016/j.immuni.2011.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Scott-Browne JP, et al. Germline-encoded recognition of diverse glycolipids by natural killer T cells. Nat Immunol. 2007;8:1105–1113. doi: 10.1038/ni1510. [DOI] [PubMed] [Google Scholar]
- 87.Diamond DJ, et al. Major histocompatibility complex independent T cell receptor-antigen interaction: functional analysis using fluorescein derivatives. J Exp Med. 1991;174:229–241. doi: 10.1084/jem.174.1.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pullen AM, Bill J, Kubo RT, Marrack P, Kappler JW. Analysis of the interaction site for the self superantigen Mls-1a on T cell receptor V beta. J Exp Med. 1991;173:1183–1192. doi: 10.1084/jem.173.5.1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Choi YW, Herman A, DiGiusto D, Wade T, Marrack P, Kappler J. Residues of the variable region of the T-cell-receptor beta-chain that interact with S. aureus toxin superantigens. Nature. 1990;346:471–473. doi: 10.1038/346471a0. [DOI] [PubMed] [Google Scholar]
- 90.Wang L, et al. Crystal structure of a complete ternary complex of TCR, superantigen and peptide-MHC. Nat Struct Mol Biol. 2007;14:169–171. doi: 10.1038/nsmb1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Saline M, Rodstrom KE, Fischer G, Orekhov VY, Karlsson BG, Lindkvist-Petersson K. The structure of superantigen complexed with TCR and MHC reveals novel insights into superantigenic T cell activation. Nat Commun. 2010;1:119. doi: 10.1038/ncomms1117. [DOI] [PubMed] [Google Scholar]
- 92.Bennett CL, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet. 2001;27:20–21. doi: 10.1038/83713. [DOI] [PubMed] [Google Scholar]
- 93.Chun HJ, Lenardo MJ. Autoimmune lymphoproliferative syndrome: types I, II and beyond. Adv Exp Med Biol. 2001;490:49–57. doi: 10.1007/978-1-4615-1243-1_6. [DOI] [PubMed] [Google Scholar]
- 94.Bogdanos DP, et al. Twin studies in autoimmune disease: genetics, gender and environment. J Autoimmun. 2012;38:J156–J169. doi: 10.1016/j.jaut.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 95.Invernizzi P, Gershwin ME. The genetics of human autoimmune disease. J Autoimmun. 2009;33:290–299. doi: 10.1016/j.jaut.2009.07.008. [DOI] [PubMed] [Google Scholar]
- 96.Schaschl H, Aitman TJ, Vyse TJ. Copy number variation in the human genome and its implication in autoimmunity. Clin Exp Immunol. 2009;156:12–16. doi: 10.1111/j.1365-2249.2008.03865.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.McDevitt HO, Bodmer WF. HL-A, immune-response genes, and disease. Lancet. 1974;1:1269–1275. doi: 10.1016/s0140-6736(74)90021-x. [DOI] [PubMed] [Google Scholar]
- 98.Gabrysova L, Wraith DC. Antigenic strength controls the generation of antigen-specific IL-10-secreting T regulatory cells. Eur J Immunol. 2010;40:1386–1395. doi: 10.1002/eji.200940151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Gottschalk RA, Corse E, Allison JP. TCR ligand density and affinity determine peripheral induction of Foxp3 in vivo. J Exp Med. 2010;207:1701–1711. doi: 10.1084/jem.20091999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Moran AE, et al. T cell receptor signal strength in Treg and iNKT cell development demonstrated by a novel fluorescent reporter mouse. J Exp Med. 2011;208:1279–1289. doi: 10.1084/jem.20110308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Turner MS, Kane LP, Morel PA. Dominant role of antigen dose in CD4+Foxp3+ regulatory T cell induction and expansion. J Immunol. 2009;183:4895–4903. doi: 10.4049/jimmunol.0901459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Nikolic-Zugic J, Bevan MJ. Role of self-peptides in positively selecting the T-cell repertoire. Nature. 1990;344:65–67. doi: 10.1038/344065a0. [DOI] [PubMed] [Google Scholar]
- 103.Genolet R, Stevenson BJ, Farinelli L, Osteras M, Luescher IF. Highly diverse TCRalpha chain repertoire of pre-immune CD8(+) T cells reveals new insights in gene recombination. EMBO J. 2012;31:1666–1678. doi: 10.1038/emboj.2012.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Robins HS, et al. Overlap and effective size of the human CD8+ T cell receptor repertoire. Sci Transl Med. 2010;2:47ra64. doi: 10.1126/scitranslmed.3001442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Quigley MF, et al. Convergent recombination shapes the clonotypic landscape of the naive T-cell repertoire. Proc Natl Acad Sci USA. 2010;107:19414–19419. doi: 10.1073/pnas.1010586107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.van Bockel DJ, et al. Persistent survival of prevalent clonotypes within an immunodominant HIV gag-specific CD8+ T cell response. J Immunol. 2010;186:359–371. doi: 10.4049/jimmunol.1001807. [DOI] [PubMed] [Google Scholar]
- 107.Venturi V, et al. A mechanism for TCR sharing between T cell subsets and individuals revealed by pyrosequencing. J Immunol. 2011;186:4285–4294. doi: 10.4049/jimmunol.1003898. [DOI] [PubMed] [Google Scholar]
- 108.Mombaerts P, Clarke AR, Hooper ML, Tonegawa S. Creation of a large genomic deletion at the T-cell antigen receptor beta-subunit locus in mouse embryonic stem cells by gene targeting. Proc Natl Acad Sci USA. 1991;88:3084–3087. doi: 10.1073/pnas.88.8.3084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Huseby ES, et al. How the T cell repertoire becomes peptide and MHC specific. Cell. 2005;122:247–260. doi: 10.1016/j.cell.2005.05.013. [DOI] [PubMed] [Google Scholar]