

Abstract
A Bayesian two-stage phase I-II design is proposed for optimizing administration schedule and dose of an experimental agent based on the times to response and toxicity in the case where schedules are non-nested and qualitatively different. Sequentially adaptive decisions are based on the joint utility of the two event times. A utility function is constructed by partitioning the two-dimensional positive real quadrant of possible event time pairs into rectangles, eliciting a numerical utility for each rectangle, and fitting a smooth parametric function to the elicited values. We assume that each event time follows a gamma distribution with shape and scale parameters both modeled as functions of schedule and dose. A copula is assumed to obtain a bivariate distribution. To ensure an ethical trial, adaptive safety and efficacy acceptability conditions are imposed on the (schedule, dose) regimes. In stage 1 of the design, patients are randomized fairly among schedules and, within each schedule, a dose is chosen using a hybrid algorithm that either maximizes posterior mean utility or randomizes among acceptable doses. In stage 2, fair randomization among schedules is replaced by the hybrid algorithm. A modified version of this algorithm is used for nested schedules. Extensions of the model and utility function to accommodate death discontinuation of follow up are described. The method is illustrated by an autologous stem cell transplantation trial in multiple myeloma, including a simulation study.
Keywords: Adaptive decision making, Bayesian design, Phase I/II clinical trial, Stem cell transplantation, Utility
1 Introduction
In clinical trials involving cytotoxic or other potentially harmful agents, adverse events (toxicities) generally occur at random times after the start of treatment. Most phase I clinical trial designs determine an optimal dose, or a maximum tolerable dose (MTD), using a binary indicator of toxicity occurring by a predetermined time from the start of therapy. These designs include the continual reassessment method (CRM, O’Quigley, et al., 1990) and many others. To use more available information, improve logistics, and protect patients from late onset toxicities, phase I designs based on YT = time to toxicity have been proposed, including the time-to-event (TiTE) CRM (Cheung and Chappell, 2000), and the designs of Braun (2006) and Bekele, et al. (2008). Many phase I/II designs based on binary or categorical response and toxicity have been proposed (Thall and Russell, 1998; Braun, 2002; Thall and Cook, 2004; Zhang, et al., 2006). Phase I/II designs also may be based on YT and YR = time to response. Denoting and for m = R, T, with Y = (YR, YT), , and δ = (δR, δT), dose-finding may be based on (Y∘, δ) (cf. Yuan and Yin, 2009).
Most phase I and phase I/II designs focus on dose, but many agents have schedule-dependent effects. An example in oncology is a nucleoside analog for which the MTD of a 30-minute infusion is (i) 2100 mg/m2 if given once, (ii) 1000 mg/m2 if given weekly for three weeks with total dose 3000 mg/m2 over 21 days, and (iii) 300 mg/m2 if given twice in each of weeks 1, 3, and 5 for total dose 1800 mg/m2 over 35 days. An example of an unexpected increase in toxicity after changing the schedule of a preparative agent in stem cell transplantation (SCT) from (d/2, d/2) on days (−8, −3) to (d/3, d/3, d/3) on days (−8, −6, −3) is described by Thall (2010, Section 1.1). Braun, et al. (2005) proposed a Bayesian design to optimize the schedule of administration times, s = (s1, …, sk), based on ( , δT), with fixed per-administration dose (PAD), assuming nested schedules with each s corresponding to a number of cycles of therapy. Braun, et al. (2007) extended this to allow PAD to vary, and jointly optimized (s, PAD) by minimizing the absolute difference between a fixed target probability and the posterior mean probability of toxicity by a specified time, , similar to the TiTE CRM. Li, et al. (2008) proposed an approach to optimizing dose and schedule for the case of two nested schedules and bivariate binary outcomes. However, no designs currently exist that optimize either schedule or (schedule, dose) in the case of non-nested, qualitatively different schedules, or where the outcomes are bivariate event times.
We address the problem of sequential adaptive optimization of treatment regime τ = (s, d) in a phase I/II clinical trial where schedules may differ qualitatively or quantitatively, and the outcomes are possibly right-censored event times (Y∘, δ). The total dose is d, with fractions given at the successive administration times. No solution to this design problem currently exists. We propose an adaptive Bayesian method using a utility function, U(y), defined on the positive real quadrant [0, ∞)2 of possible Y values. We construct U(y) by partitioning a compact subset of [0, ∞)2 where Y pairs are likely to occur into rectangles, eliciting a numerical utility on each rectangle from the physicians planning the trial, and fitting a parametric function to the elicited values. For each Ym, m = R, T, we specify a gamma marginal with shape and scale parameters each modeled as functions of (s, d), and use a copula to obtain association.
The design has two stages, and only allows τ with both acceptable safety and efficacy to be assigned. In stage 1, patients are randomized fairly among schedules in blocks. Within each schedule, the acceptable dose with maximum posterior mean utility is chosen, unless the current sample size for the optimal dose is larger than all current sample sizes for the other doses. In that case, patients are randomized among the assigned schedule’s acceptable doses with probabilities proportional to their posterior mean utilities. In stage 2, the block randomization among schedules is unbalanced using similar criteria, with each schedule’s assignment probability proportional to the posterior mean utility of its optimal dose. We include randomization to reduce the chance of getting stuck at suboptimal τ, which may occur with a “greedy” algorithm that only maximizes posterior mean utility.
Our design differs from those of Braun et al. (2007) and Li et al. (2008) in that we (1) use both time-to-response and time-to-toxicity, (2) use utilities as decision criteria, (3) use unbalanced randomization to choose regimes, (4) assume a bivariate gamma regression model, and (5) allow non-nested schedules. We also consider the case where YR is evaluated at a sequence of times rather than continuously, hence is interval censored. We describe extensions of the model and utility to accommodate death or discontinuation of follow up at toxicity. Our methodology synthesizes the above and several other existing ideas, including use of a copula to obtain association, randomizing to avoid getting stuck at a suboptimal regime, and regression modeling of both first and second order parameters.
To provide a concrete frame of reference, we describe the illustrative SCT trial in Section 2. Section 3 describes a method for constructing a utility function from elicited values. Gamma regression models for [YR|s, d] and [YT|s, d], and likelihoods for both continuous and interval censored YR are given in Section 4. The design is presented in Section 5. Section 6 illustrates the method by application to the SCT trial, which has two schedules and three doses (six regimes), including a simulation study with comparison to two alternative designs, and evaluation of robustness and of sensitivity to the prior, cohort size, and sample size. We close with a discussion in Section 7.
2 Motivating Application
Melphalan is an alkalating agent commonly used as part of the preparative regimen for autologous SCT to treat multiple myeloma (MM), but there is no consensus for what (schedule, dose) combination is best in older patients. To address this, we designed a phase I/II trial to evaluate total doses d = 140, 180, or 200 mg/m2 of melphalan given either as a single 30-minute bolus infused on day −2 before SCT, or with the dose split into two equal boluses infused on days −3 and −2. Toxicity is defined as severe (grade 3 or 4) gastrointestinal toxicity or diarrhea, graft failure, or regimen-related death. Response is evaluated at 1, 3, 6, 9, and 12 months post transplant, so YR is interval censored while YT is observed continuously, which is common in early phase oncology trials. Response has three requirements, (i) normal bone marrow (< 5% myeloma cells), (ii) no new lytic lesions on bone X-ray, and (iii) absence of β2 microglobulin, a monoclonal protein characterizing MM in two consecutive tests.
Transforming pre-transplant administration days (−3, −2) to (0, 1), so transplant is on day 3 after the first administration, the six regimes in the MM trial are τ1 = {1, 140}, τ2 = {1, 180}, τ3 = {1, 200}, τ4 = {(0, 1), 140}, τ5 = {(0, 1), 180}, τ6 = {(0, 1), 200}. The subsets
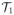 = {τ1, τ2, τ3} of one-day schedules and
= {τ1, τ2, τ3} of one-day schedules and
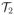 = {τ4, τ5, τ6} of two-day schedules have natural orderings, since the probabilities of toxicity and response each increase with d within each schedule. In contrast, for either response or toxicity, there is no obvious ordering among all six regimes in
= {τ4, τ5, τ6} of two-day schedules have natural orderings, since the probabilities of toxicity and response each increase with d within each schedule. In contrast, for either response or toxicity, there is no obvious ordering among all six regimes in
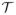 =
∪
. For example, although τ1 and τ4 deliver the same total dose, a 1-day or 2-day schedule may either be more toxic or have higher response rate. The terms “escalate” or “de-escalate” thus are meaningful when assigning doses within
or
. but not for assigning regimes within
. The additive hazard model of Braun et al. (2007) does not accommodate qualitatively different schedules with all administrations given early, or bivariate event time outcomes. We address this by treating schedule as qualitative and dose as quantitative, and defining a joint utility function for the two event times.
=
∪
. For example, although τ1 and τ4 deliver the same total dose, a 1-day or 2-day schedule may either be more toxic or have higher response rate. The terms “escalate” or “de-escalate” thus are meaningful when assigning doses within
or
. but not for assigning regimes within
. The additive hazard model of Braun et al. (2007) does not accommodate qualitatively different schedules with all administrations given early, or bivariate event time outcomes. We address this by treating schedule as qualitative and dose as quantitative, and defining a joint utility function for the two event times.
3 Utility Functions
We obtain a utility function, U(y), that represents the clinical desirability of each possible outcome pair. This utility may be used for decision-making with any sort of regime, and is not limited to dose-schedule optimization. Given follow up time Tmax, the physician is asked to partition the domains of YR and YT into subintervals that determine a grid of rectangular subsets partitioning [0, Tmax]2. A numerical utility is elicited for each rectangular subset, and nonlinear regression is used to fit a smooth surface by treating the midpoints of the rectangles in the Y domain as predictors and the corresponding elicited utilities as outcomes. The partition should be sufficiently refined to provide a discretization of Y in terms of the anticipated joint probability distribution that is realistic based on clinical experience, but sufficiently coarse that the elicitation is feasible. To facilitate refinement of the elicited numerical utilities or the grid, it is useful to show the physician a plot of the fitted surface, and iterate this process until an acceptable utility surface is obtained.
Since smaller yR and larger yT are more desirable, U(y) must decrease in yR and increase in yT, formally, ∂U(yR, yT)/∂yR < 0 < ∂U(yR, yT)/∂yT. We used the parametric function
| (1) |
To obtain 0 ≤ U(yR, yT) ≤ 100 with 0 corresponding to the worst and 100 to the best possible outcomes, we used the norming constants Umax = U∘(yR,min, yT,max) and Umin = U∘(yR,max, yT,min), denoting U∘(yR, yT) = b1 e−c1yR+ b2 e−c2yT+ b3 e−c1yR−c2yT. Any compact domain for U may be used, however. The inequalities c1, c2 > 0, b2 < 0 < b1, and b2 < −b3 < b1 are sufficient to ensure monotonicity of U(yR, yT) in each argument. Subject to these constraints, we solved for (c1, c2, b1, b2, b3) using nonlinear least squares with the midpoint of each rectangle as the X-variable and the elicited utilities U(e) on the rectangle as the Y-variable. For the autologous SCT trial design (Table 1) this gave estimates (ĉ1, ĉ2, b̂1, b̂2, b̂3) = (0.0631, 0.1088, 9.3557, −7.8677, 0.5301). Table 1 also gives the fitted utilities Û(y), and the surface obtained by plotting Û(y) on y is illustrated by Figure 1, where Tmax = 12 months for the SCT trial. For example, the rectangle defined by 1 < yR < 3 and 3 < yT < 6 has midpoint ymid = (2, 4.5) and elicited utility U(e) = 64.
Table 1.
Utilities for rectangles of YR = time to response and YT = time to toxicity in the multiple myeloma autologous stem cell transplantation trial. For each (YR, YT) rectangle, the two tabled values are U(e) = the elicited utility and Û = the fitted parametric function evaluated at the rectangle’s midpoint.
| YT = Months to Toxicity | YR = Months to Response | ||||
|---|---|---|---|---|---|
| [0, 1) | [1, 3) | [3, 6) | [6, 9) | [9, 12) | |
|
|
|
||||
| [9, 12) | 95, 93.9 | 88, 86.0 | 74, 74.5 | 64, 62.8 | 54, 53.1 |
| [6, 9) | 85, 85.3 | 76, 77.4 | 63, 65.8 | 53, 54.0 | 43, 44.3 |
| [3, 6) | 75, 73.5 | 64, 65.5 | 52, 53.8 | 42, 41.9 | 32, 32.1 |
| [1, 3) | 62, 60.2 | 52, 52.1 | 41, 40.3 | 31, 28.3 | 21, 18.4 |
| [0, 1) | 50, 50.3 | 40, 42.2 | 30, 30.2 | 20, 18.1 | 10, 8.1 |
Figure 1.

Fitted utility surface for the times to response and toxicity in the multiple myeloma stem cell transplantation trial. Black and red dots show elicited values above and below the fitted surface, respectively.
Our criterion for choosing each cohort’s treatment regime is the posterior mean utility,
| (2) |
where we denote Ū(τ, θ) = EY|θ{U(Y) | τ, θ}, the mean over Y of the utility U(Y) of using regime τ for given θ. Another way to view u(τ, data) is obtained by applying the Fubini-Tonelli Theorem to switch the order of expectations in (2). Denoting the joint pdf of [Y | τ] by fR,T(y | τ, θ), this gives
The posterior expectation fR,T(y | τ, data) is the predictive distribution of Y, given the current data, for a patient treated with regime τ. Thus, u(τ, data) is the expected utility of τ for a newly enrolled patient. The design makes adaptive decisions based on the values of {u(τ, data), τ ∈
}, subject to safety and efficacy acceptability requirements.
4 Probability Model
4.1 Marginal Model
Our modeling strategy is to construct marginals for YR and YT that are functions of s and d, and use a bivariate copula (Nelsen, 1999) to obtain a joint distribution. For each outcome m = R, T and regime τ = (s, d), denote the pdf, cdf, and survivor function of Ym at time y > 0 by fm(y | τ, θm), Fm(y | τ, θm) = Pr(Ym ≤ y | τ, θm) and F̄m(y | τ, θm) = 1 − Fm(y | τ, θm), where θm is the marginal model parameter vector. The joint model parameter vector is θ = (θR, θT, ζ), where ζ is the copula’s association parameter.
We assume that, given s, larger d is associated with stochastically smaller YR and smaller YT. This says that, at any follow up time y, the probability of response, FR(y | (s, d), θR), and the probability of toxicity, FT (y | (s, d), θT), both increase in d for any s. The marginals are formulated so that these probabilities may either vary qualitatively with schedule or have monotone schedule effects. The utility function addresses the conflict between the goals to choose τ to make FR(yR|τ, θR) large while not allowing FT (yT |τ, θT) to become unacceptably large by quantifying the desirability of each possible (yR, yT) pair.
Let d1 < d2 < ··· < dJ denote the doses being considered. A practical difficulty when using u{(s, d), data} for decision making based on bivariate outcomes is that simply assuming FR(t | (s, d), θR) and FT (t | (s, d), θT) both are monotone in d may not distinguish adequately between different values of u{(s, dj), data} for doses dj near the optimum, in the case d1 < dj < dJ. A given change in an intermediate dj may produce changes of very different magnitudes in FR(t | (s, dj), θR) and FT (t | (s, dj), θT), which in turn may make it difficult to identify a middle dose for which (s, dj) has true maximum utility. To address this problem, for each outcome we define outcome-specific standardized doses,
denoting d̄ = (d1 + ··· + dJ)/J. The parameter λm controls the relative effects of doses that are not close to either d1 or dJ. Note that xR,1 = xT,1 = d1/d̄ and xR,J = xT,J = dJ /d̄, while all intermediate standardized doses for fm are parameterized by λm.
For brevity, hereafter we will index schedules by k = 1, ···, K and denote τ = (k, j) for the kth schedule and dose dj. To formulate flexible but reasonably parsimonious marginals for [Ym | τ], m = R, T, in preliminary simulations we explored the lognormal, Weibull, and gamma distributions across a diverse set of regime-outcome scenarios and true event time distributions. We chose the gamma, since it had the best overall performance and robustness of the three distributions. We used gamma marginals having the parametric form
where Γ(·) denotes the gamma function. The shape parameter ϕm,1 and scale parameter ϕm,2 both depend on dose and schedule as follows:
| (3) |
and
| (4) |
We require αm,1, αm,2, βm,1, βm,2 > 0, and assume the schedule effects, γm,1, ···, γm,K, have support [0, 2]. Different transformations are used for ϕm,1(k, j) and ϕm,2(k, j) because the shape and scale parameters play very different roles in determining the form of the gamma distribution, and we found that using a log transformation for ϕm,2{(k, j), θm} provided a well behaved dose-outcome model. For each outcome m = R, T and gamma shape (r = 1) or scale (r = 2) parameter, if dose is fixed and only schedule is varied, the right-hand sides of (3) and (4) reduce to βm,rγm,k, so there are K + 1 parameters for K effects. We thus define .
The utility U(Y) reduces the two-dimensional outcome Y to a one-dimensional value, which in turn yields the posterior mean utility, u{(k, j), data}, that is used for decision making. In the models (3) for shape and (4) for scale, the relative magnitudes of the parametric contributions of k and xm,j must reflect their actual effects on u{(k, j), data}. In these models, βm,r may be thought of as the gamma’s usual shape (r = 1) or scale (r = 2) parameter, modified by the effects of dose and schedule. For each m = R, T, the same λm is used to define each standardized dose xm,j and, for each schedule k, the same parameter γm,k is used as a multiplicative effect on xm,j, for both ϕm,1 and ϕm,2.
4.2 Likelihood for Continuously Observed Response Times
Let t* denote study time, defined as the time from the start of the trial to the current time when a new patient is enrolled and an interim decision must be made. Let n* denote the number of patients accrued by t*. For the ith patient, i = 1, ···, n*, denote the treatment regime by τi and the outcome vectors evaluated at t* by and δi,t* = (δi,R,t*, δi,T,t*). For a patient with entry time ei < t*, the patient time at trial time t* is ti = t* − ei. Each patient’s outcome data change over time, starting at and δi,t* = (0,0) at accrual when ti = 0. Thereafter, each as long as δi,m,t* = 0, and achieves the final value Yi,m if and when the patient experiences event m, when δi,m,t* jumps from 0 to 1. That is, each ( , δi,t*) is a bivariate sample path of two step functions, jumping from 0 to 1 at their respective event times, with administrative right-censoring, from the time from that patient’s accrual to the most recent follow up time. Consequently, before computing posterior quantities used for making outcome-adaptive interim decisions at any study time t*, it is essential to update the trial data. We denote the interim data at trial time t* by .
Denote the joint cdf and survivor function of [Y | τ] by FR,T (y | τ, θ) and F̄R,T (y | τi, θ) = Pr(YR > yR, YT > yT | τi, θ). When both YR and YT are observed continuously, the likelihood for patient i at study time t* is
| (5) |
Once the marginals have been specified, a joint distribution of YR and YT may be defined in numerous ways. To obtain a parsimonious and tractable model, we use the bivariate Farlie-Gumbel-Morgenstern (FGM) copula (Nelsen, 1999). Hereafter, we will suppress t*, i, τi, and θ for brevity when no meaning is lost. The FGM copula is given in terms of the marginals and one association parameter ζ ∈ [−1, 1] by
| (6) |
To obtain the terms in (5) under the FGM copula, for (δR, δT) = (1,1) the joint pdf is
and F̄R,T (yR, yT) = FR,T (yR, yT) + F̄R(yR) + F̄T (yT) − 1. For (δR, δT) = (0,1) and a > 0,
and the term for (δR, δT) = (1,0) is obtained by symmetry. All likelihood contributions thus are determined by ζ and the marginal pdfs, with FR and FT and terms corresponding to administratively censored event times computed by numerical integration.
4.3 Likelihood for Interval Censored Response Times
To account for interval censoring when response is evaluated at successive times 0 = a0 < a1 < ··· < aL−1 < aL = ∞, rather than continuously, let Al = (al−1, al] denote the lth subinterval. If a response did not occur by al−1 but did occur by al, then YR ∈ Al. Let δ1,l denote this event. Given the partition {A1, ···, AL} of [0, ∞), the pair ( , δi,R) for continuously observed Yi,R are replaced by the vector of indicators δi,R = (δi,R,1, ···, δi,R,L), having one entry 1 and all other entries 0. At study time t*, the observed data of the ith patient are {δi,R(t*), , δi,T,t*}. When has been observed by study time t*, so that δi,T,t* = 1, the ith patient’s likelihood contribution is
| (7) |
denoting . Under the copula (6), this takes the form . Similarly, when patient i has not yet experienced toxicity, so δi,T,t* = 0 and Yi,T is censored at study time t*, the likelihood contribution is
| (8) |
denoting . Under the copula (6), this takes the form . Combining terms (7) and (8), if YR is interval censored the likelihood at trial time t* is
5 Trial Design
5.1 Treatment Regime Acceptability
While using utilities is a sensible way to combine efficacy and toxicity for optimizing treatment regimes, in practice some regimes may be excessively toxic or inefficacious. Such regimes should not be used to treat patients, and in the extreme case where all regimes are found to be either too toxic or inefficacious the trial should be terminated. We thus employ the following acceptability criteria, similar to those used by Thall and Cook (2004) and others for phase I/II trials. For m = R, T, let be a reference time from the start of therapy used to specify a limit on . Let π̄T be a fixed upper limit on and πR be a fixed lower limit on , both specified by the physician. Given upper probability cut-offs pT and pR, a regime τ is unacceptable if
| (9) |
and we denote the set of acceptable strategies by
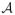 .
.
5.2 A Design for Non-Nested Schedules
The problem that a “greedy” sequential search algorithm, that always chooses the optimal action, may get stuck as a suboptimal action is well-known in optimization, but only recently has been addressed in dose-finding trials (Azriel, Mandel and Rinott, 2011; Thall and Nguyen, 2012). Our proposed design is a hybrid of a greedy design that always chooses τ = (k, j) to maximize posterior mean utility, and a nonadaptive, hence unethical design that simply randomizes patients fairly among regimes. The idea is avoid getting stuck at a suboptimal regime, but still conduct the trial ethically by using adaptive rules.
For each successive cohort of c patients, τ is chosen adaptively, as follows. Denote the regime maximizing u(τ, data*) among all τ ∈
by τopt. Denote the index of the optimal dose among acceptable regimes having schedule k by
Because the posterior mean utility u(τ, data*) is highly variable throughout much of the trial, randomizing among regimes with u{(k, j), data*} close to τopt is ethical, and reduces the risk of getting stuck at a suboptimal regime. The proposed hybrid design, Design 1, has two stages. Let n*(k, j) denote the number of patients up to trial time t* treated with τ = (k, j). Since only τ ∈
may be chosen, if
is empty then the trial is stopped and no τ is selected. If
is not empty, then for qualitatively different, non-nested schedules Design 1 proceeds as follows. Let N be the maximum overall sample size, and N1 the maximum stage 1 sample size, with N1 chosen to be a multiple of Kc reasonably close to N/2.
Stage 1
Randomize K cohorts of size c fairly among the schedules, restricted so that each schedule is assigned to exactly c patients. Repeat this until N1 patients have been treated. Within schedule k, starting at the lowest dose and not skipping an untried dose when escalating, treat the next patient at dose jopt(k), unless
| (10) |
where Δ1 is a small positive integer. If the inequality (10) holds, then within schedule k choose an acceptable dose randomly with probability proportional to u{(k, j), data*}.
Stage 2
For patients i = N1 + 1, ···, N, choose the schedule kopt with largest u{k, jopt(k)}, unless
| (11) |
where Δ2 is a small positive integer. If (11) holds, choose a schedule with probability proportional to u{(k, jopt(k)), data*}. Choose doses within schedules as in Stage 1.
The inequality (10) in Stage 1 says that the current sample size at the best acceptable dose within schedule k is at least Δ1 c larger than the current sample size at any other acceptable dose with that schedule. One may use Δ1 = Δ2 = 1, or slightly larger values, depending on c and possibly N, to control the amount of sample size imbalance between regimes. The randomization probabilities among doses within schedule k in Stage 1 at t* are
Similarly, the inequality (11) says that the current sample size at the best acceptable regime is at least Δ2 c larger than the current sample size at any other acceptable regime. The randomization probabilities among schedules in Stage 2 at t* are
Design 2, the “greedy” design, is a much simpler version Design 1 that chooses τ ∈
by simply maximizing u{(k, j), data*}, subject to the constraint that an untried dose may not be skipped when escalating within any schedule. With Design 2, schedules are chosen by fair randomization without replacement, as in the hybrid Design 1, but this is done throughout the trial, and within schedule k the current dose jopt(k) is chosen.
If schedules are nested, then γm,1 < γm,2 < ··· < γm,K for m = R, T, and consequently YR and YT are stochastically increasing in k as well as j, so the regime-finding algorithm must reflect this. Since in this case the word “escalate” pertains to both schedule and dose, i.e. to both k and j, the trial could be conducted by choosing (k, j) to maximize u{(k, j), data*} subject to a two-dimensional “do-not-skip” rule similar to that used by Braun et al. (2007), with escalation from (k, j) to optimize u{τ, data*} restricted to the three adjacent untried combinations (k +1, j), (k, j + 1), or (k + 1, j + 1). This could be elaborated, as in Design 1, to include randomization among regimes based on u{(k, j), data*}.
5.3 Accommodating Death During Follow Up
The model and utility may be modified to account for death during follow up, or discontinuation of follow up due to toxicity, possibly because the regime was changed at YT. This may be done parsimoniously using a semi-competing risks model, wherein we call either death or discontinuation of follow up at YT “fatal” toxicity, indicated by δTD, with δTA indicating “non-fatal” toxicity that allows follow up to continue for YR. Thus, δTD + δTA = δT, and (δTD, δTA) has possible values (1,0) or (0,1) if δT = 1 and (0,0) if δT = 0. If δTD = 1 and YT < YR then response will not occur. In this case, we define YR = +∞ and δR = 1, and extend the domain of (YR, YT) from E2 = [0, ∞)2 to . We do not assume that YR censors YT, however. Suppressing τ and θ, we define an extended distribution in terms of πTD = Pr(δTD = 1) and the conditional probabilities and , where πNR = Pr(YR > YT) is the probability of death before response if δTD = 1. It follows that is a probability distribution on , since .
To extend the likelihood (5) to this case, we first note that lines 2 and 4 of (5) are unchanged since in these cases YT is right-censored. The first line of (5) becomes
For line 3 of (5), if YR is censored and fatal toxicity occurs, then δTD = 1 and YR = +∞, a case already accounted for by line 1. If YR is censored and non-fatal toxicity occurs, this is accounted for by simply replacing δT with δTA.
The utility may be modified to accommodate death by considering the scaled original utility, U(yR, yT)/100, as a multiplicative discount factor for survival time on the follow-up interval [0, Tmax]. A utility function that does this is U +(yR, yT) = Tmax U (yR, yT)/100 if yT is the time of non-fatal toxicity and U +(yR, yT) = yT if yT is the time of death. Thus, 0 ≤ U+(yR, yT) ≤ Tmax, and the trial is conducted as described above. The model may be extended similarly if follow up is stopped at YR, although this is not commonly done if toxicity occurring over a prespecified follow up period of Tmax is considered important. One also might model πTD as a function of (s, d), if the death rate is sufficiently high to estimate the additional parameters, although this may be unlikely in practice.
6 Application to the SCT Trial
6.1 Prior and Design Parameters
We assumed that the positive real valued parameters αR,1, αR,2, βR,1, βR,2, λR, αT,1, αT,2, βT,1, βT,2, λT followed lognormal priors. The means were determined from the elicited values in Table 2 using a pseudo-sample based method similar to that described in Section 4.2 of Thall et al. (2011). Prior variances were calibrated to obtain a design with good performance across a broad range of scenarios. We assumed that ζ followed a beta distribution with parameters (1.1, 1.1), rescaled to have support on (−1, +1). Numerical values of the prior hyperparameters are given in Supplementary Table 1.
Table 2.
Elicited prior means of FR(t | τ) and FT (t | τ) for the autologous stem cell transplantation trial to optimize (schedule, dose) of melphalan. For each total dose d, the prior means for the regimes τ = (−2, d) and τ = ((−3, −2), d) were identical.
| Days of Follow up | Prior Means of FR(t | τ)
|
Prior Means of FT (t | τ)
|
||||
|---|---|---|---|---|---|---|
| Total dose of Melphalan (mg/m2) | Total dose of Melphalan (mg/m2) | |||||
| d = 140 | d = 180 | d = 200 | d = 140 | d = 180 | d = 200 | |
|
|
|
|
||||
| t = 1 | – | – | – | .01 | .02 | .03 |
| t = 3 | – | – | – | .05 | .07 | .09 |
| t = 6 | – | – | – | .15 | .18 | .20 |
| t = 10 | – | – | – | .25 | .30 | .32 |
| t = 14 | – | – | – | .30 | .35 | .40 |
| t = 28 | – | – | – | .33 | .38 | .43 |
| t = 30 | .05 | .08 | .10 | – | – | – |
| t = 90 | .09 | .11 | .15 | – | – | – |
| t = 180 | .13 | .16 | .19 | .34 | .39 | .44 |
| t = 270 | .16 | .19 | .24 | – | – | – |
| t = 360 | .20 | .25 | .30 | .35 | .40 | .45 |
Since each schedule effect acts multiplicatively on the outcome-specific standardized doses, we found that γm,j ≤ 0.80 or ≥ 1.20 may have a large effect on xm,j. For example, γm,j = .80 would reduce some xm,j values by more than one full dose level. A more disperse prior on the γm,j’s also may cause the method to misinterpret a dose effect for a schedule effect in certain cases, especially those where a middle dose has highest utility. Consequently, we specified the priors of the γm,j’s to be highly concentrated beta distributions with domain [0, 2] and parameters (47.3, 47.3), which gives Pr[0.80 < γm,j < 1.20] = 0.95. Although these priors may appear to be overly informative, in fact small changes within the subdomain [0.80, 1.20] of [0, 2] allow the posterior mean utility to change substantively, so that one may detect true differences between schedules. In this case, the observed data easily have the necessary effect on the posterior distributions of the γm,j’s.
6.2 Simulation Study
We simulated the SCT trial with N1 = 36, N = 72, and c = 3, with YT monitored continuously and YR interval censored per the actual evaluation schedule at 1, 3, 6, 9, 12 months. We studied three competing designs: the hybrid design (Design 1), the greedy design (Design 2), and a randomized design with no interim decisions, restricting the randomization to treat exactly 12 patients at each of the six τ pairs, with the regime maximizing u(τ, data) selected at the end. We considered eight simulation scenarios (Supplementary Table 2). In Scenario 1, there is no schedule effect, toxicity is acceptable, and efficacy increases with dose. Scenario 2 also has no schedule effect, but toxicity is much higher, so the lowest dose has the highest utility. In Scenario 3, the 2-day schedule is superior due to higher efficacy. In Scenario 4, the 1-day schedule is superior. Scenario 5 has no schedule effect, but the middle dose is best. In Scenario 6, for both schedules, the utility is “V” shaped, lowest for the middle dose with the highest dose optimal. All regimes are unacceptably toxic in Scenario 7, and unacceptably inefficacious in Scenario 8. Each case was simulated 3000 times.
We use the statistic Rselect = {utrue(τselect) − umin}/(umax − umin), (cf. Thall and Nguyen, 2012) to quantify reliability of regime selection. This is the proportion of the difference between the utilities of the best and worst possible regimes achieved by τselect. A statistic quantifying the ethics of how well the method assigns regimes to patients in the trial is , where utrue(τ[i]) is the true utility of the regime given to patient i, and N is the final sample size.
The main simulation results are summarized in Table 3. In each of Scenarios 1 – 6, the hybrid design does a good job of selecting regimes with high true utilities, and is very likely to correctly stop early in both Scenarios 7 and 8. Table 4 compares the hybrid, greedy, and balanced designs in terms of Rtreat and Rselect. More detailed summaries of the simulations of the greedy and balanced non-adaptive designs are given in Supplementary Tables 3a and 3b, respectively. The main messages from Scenarios 1 – 6 in Table 3 are that (i) compared to the greedy design, the hybrid design has the same or higher Rselect while neither design is uniformly superior in terms of Rtreat; (ii) compared to the balanced design, the hybrid design has nearly identical Rselect but much higher values of Rtreat, so is much more ethical; and (iii) in Scenarios 7 and 8, both the hybrid and greedy designs correctly stop early with high probability, and both have much higher Rselect and Rtreat than the balanced design. In summary, the hybrid design has the best overall performance of the three designs and, as may be expected, the balanced design is ethically unacceptable.
Table 3.
The main simulation results using the hybrid algorithm with sample size 72 and cohort 3. The true time distribution is assumed to be lognormal.
| Scenario | 1-Day Schedule
|
2-Day Schedule
|
None |
Rselect Rtreat |
|||||
|---|---|---|---|---|---|---|---|---|---|
| Dose 1 | Dose 2 | Dose 3 | Dose 1 | Dose 2 | Dose 3 | ||||
| 1 | ūtrue(s, d) | 52.2 | 57.5 | 62.9 | 52.2 | 57.5 | 62.9 | ||
| % Sel | 5 | 8 | 38 | 5 | 8 | 35 | 1 | 0.82 | |
| # Pats | 11.6 | 9.8 | 14.5 | 11.6 | 9.5 | 14.4 | 0.54 | ||
|
| |||||||||
| 2 | ūtrue(s, d) | 59.0 | 53.7 | 48.1 | 59.0 | 53.7 | 48.1 | ||
| % Sel | 39 | 7 | 4 | 39 | 6 | 5 | 1 | 0.85 | |
| # Pats | 22.6 | 8.2 | 5.0 | 22.7 | 8.2 | 4.9 | 0.75 | ||
|
| |||||||||
| 3 | ūtrue(s, d) | 53.1 | 58.4 | 63.8 | 56.8 | 62.1 | 67.6 | ||
| % Sel | 3 | 5 | 16 | 6 | 12 | 58 | 1 | 0.81 | |
| # Pats | 11.1 | 9.1 | 12.9 | 11.6 | 10.5 | 16.5 | 0.54 | ||
|
| |||||||||
| 4 | ūtrue(s, d) | 58.6 | 54.6 | 49.7 | 55.4 | 51.4 | 46.5 | ||
| % Sel | 54 | 12 | 5 | 18 | 5 | 5 | 1 | 0.80 | |
| # Pats | 23.1 | 9.0 | 5.1 | 21.0 | 8.3 | 5.1 | 0.69 | ||
|
| |||||||||
| 5 | ūtrue(s, d) | 52.9 | 63.6 | 50.2 | 52.9 | 63.6 | 50.2 | ||
| % Sel | 8 | 34 | 6 | 9 | 36 | 6 | 1 | 0.74 | |
| # Pats | 13.0 | 16.7 | 0.5 | 12.8 | 16.7 | 6.2 | 0.54 | ||
|
| |||||||||
| 6 | ūtrue(s, d) | 53.5 | 48.1 | 56.5 | 53.5 | 48.1 | 56.5 | ||
| % Sel | 21 | 4 | 23 | 21 | 4 | 25 | 2 | 0.76 | |
| # Pats | 17.2 | 7.3 | 11.0 | 17.2 | 7.2 | 11.2 | 0.62 | ||
|
| |||||||||
| 7 | ūtrue(s, d) | 35.3 | 34.2 | 33.0 | 35.3 | 34.2 | 33.0 | ||
| % Sel | 0 | 0 | 0 | 0 | 0 | 0 | 100 | 0.87 | |
| # Pats | 5.0 | 1.6 | 0.5 | 5.0 | 1.6 | 0.5 | 0.81 | ||
|
| |||||||||
| 8 | ūtrue(s, d) | 39.9 | 37.8 | 35.6 | 39.9 | 37.8 | 35.6 | ||
| % Sel | 1 | 0 | 1 | 1 | 0 | 1 | 96 | 0.54 | |
| # Pats | 5.8 | 4.0 | 4.5 | 5.8 | 4.0 | 4.5 | 0.54 | ||
Table 4.
Summary statistics for the hybrid design, greedy design, and non-adaptive balanced allocation, for the (3-dose, 2-schedule) trial. The “balanced” method assigns 12 patients to each (schedule, dose) pair and does only one posterior computation, at the end of the trial. The number in parentheses after each Rselect is the percentage of times the trial is stopped with no (schedule, dose) selected. Because scenarios 7 (too toxic) and 8 (too inefficacious) have no acceptable treatments, the Rselect values are less relevant and thus are shown with a gray background.
| Scenario | Hybrid | Greedy | Balanced | |||
|---|---|---|---|---|---|---|
| Rselect | Rtreat | Rselect | Rtreat | Rselect | Rtreat | |
|
|
|
|
|
|||
| 1 | 0.82 (1) | 0.54 | 0.78 (1) | 0.45 | 0.85 (0) | 0.50 |
| 2 | 0.85 (1) | 0.75 | 0.85 (1) | 0.84 | 0.85 (0) | 0.51 |
| 3 | 0.81 (1) | 0.54 | 0.78 (1) | 0.48 | 0.83 (0) | 0.50 |
| 4 | 0.80 (1) | 0.69 | 0.80 (0) | 0.77 | 0.80 (0) | 0.51 |
| 5 | 0.74 (1) | 0.54 | 0.65 (1) | 0.48 | 0.77 (0) | 0.40 |
| 6 | 0.76 (2) | 0.62 | 0.75 (3) | 0.62 | 0.77 (0) | 0.55 |
| 7 | 0.87 (100) | 0.81 | 1.00 (100) | 0.83 | 0.94 (98) | 0.50 |
| 8 | 0.54 (96) | 0.54 | 0.54 (96) | 0.59 | 0.35 (72) | 0.50 |
In supplementary Table 4, we evaluate robustness of the hybrid design to the true event time distribution (lognormal, gamma, Weibull, or uniform). It shows that that (i) Rtreat is insensitive to the distributions studied, (ii) Rselect is insensitive to whether the true distribution is lognormal, gamma, or Weibull, but (iii) for a uniform distribution Rselect may be lower (Scenarios 1 and 3) or higher (Scenarios 2 and 4) than for the other distributions. Supplementary Table 5a shows that the hybrid design is insensitive to changes in prior hyperparameter σ̃ = 8 to 14, the assumed common prior sd of log(αm,l), log(βm,l), and log(λm) for all m = R, T and l = 1, 2. Supplementary Table 5b shows that the hybrid design is insensitive to changes in the 95% prior interval for the γm,j’s varying from (0.9, 1.1) to (0.6, 1.4), although Rselect and Rtreat both decrease slightly with the width of this interval in Scenario 5. This motivated our use of the 95% prior interval (0.8, 1.2). Supplementary Table 6 shows that Rselect is insensitive to c = 1, 2, or 3, and that Rtreat may increase or decrease slightly with c depending on the scenario. Supplementary Table 7 shows that, as N is increased from 48 to 360, both Rselect and Rtreat increase substantially.
We also evaluated the hybrid design for an extended version of the SCT trial, with 4 doses and 3 schedules (12 regimes), obtained by interpolating the elicited priors and scenarios of the original 6-regime design. The three doses of the original trial are mapped into the first, third and fourth doses of the extended trial, with a new, second lowest dose corresponding to d = 160 mg/m2 added. Elicited prior and scenario probabilities of the original first two doses were interpolated to obtain values for the new dose. A new third schedule was obtained by averaging the prior and scenario probabilities of the two original schedules. Results for this 12-regime setting are given in Supplementary Tables 8 – 12.
Supplementary Table 8a gives a hypothetical utility that places greater weight on quick responses. Either for the 6 regime case (Supplementary Table 8b) or 12 regime case (Supplementary Table 15), the hybrid design’s behavior for this different utility, compared to the actual utility, has an equally high probability of correctly stopping the trial early in Scenarios 7 and 8, and in Scenarios 1 – 6 is better in three cases and worse in three cases. This is desirable, since otherwise there would be little point in using an elicited, subjective utility as an objective function.
7 Discussion
We have proposed an adaptive Bayesian method for jointly optimizing schedule of administration and dose in phase I-II trials based on event times for efficacy and toxicity. We modeled schedules qualitatively because either of the two outcome events may occur long after administration. This is very different from the additive hazard model, with a component for each administration, used by Braun et al. (2007), who dealt with time to toxicity occurring over a much shorter time frame. For regimes administered over a period longer than a few days, our methodology could be extended to allow each patient’s initial dose to be changed adaptively based on interim events or new data from other patients.
Our design uses a regime assignment algorithm that is a hybrid of a greedy algorithm and adaptive randomization. Extensive simulations show that, for a maximum sample size of 72, the proposed model and method provide a design that is reliable, safe, and robust, and that it works well in the cases of either six or 12 regimes.
Supplementary Material
Acknowledgments
The research of PT and HN was supported NCI grant RO1 CA 83932
Footnotes
Web Appendix 1 referenced in Section 6 is available with this paper at the Biometrics website on Wiley Online Library.
References
- 1.Azriel D, Mandel M, Rinott Y. The treatment versus experimentation dilemma in dose-finding studies. Journal of Statistical Planning and Inference. 2011;141:2759–2758. [Google Scholar]
- 2.Bekele BN, Ji Y, Shen Y, Thall PF. Monitoring late onset toxicities in phase I trials using predicted risks. Biostatistics. 2008;9:442–457. doi: 10.1093/biostatistics/kxm044. 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Braun TM. The bivariate continual reassessment method: extending the CRM to phase I trials of two competing outcomes. Contemporary Clinical Trials. 2002;23:240–256. doi: 10.1016/s0197-2456(01)00205-7. [DOI] [PubMed] [Google Scholar]
- 4.Braun TM, Yuan Z, Thall PF. Determining a maximum tolerated schedule of a cytotoxic agent. Biometrics. 2005;61:335–343. doi: 10.1111/j.1541-0420.2005.00312.x. [DOI] [PubMed] [Google Scholar]
- 5.Braun TM, Thall PF, Nguyen H, de Lima M. Simultaneously optimizing dose and schedule of a new cytotoxic agent. Clinical Trials. 2007;4:113–124. doi: 10.1177/1740774507076934. [DOI] [PubMed] [Google Scholar]
- 6.Braun TM. Generalizing the TITE-CRM to adapt for early- and late-onset toxicities. Statistics in Medicine. 2010;25:2071–2083. doi: 10.1002/sim.2337. [DOI] [PubMed] [Google Scholar]
- 7.Cheung Y, Chappell R. Sequential designs for Phase I clinical trials with late-onset toxicities. Biometrics. 2000;56:1177–1182. doi: 10.1111/j.0006-341x.2000.01177.x. [DOI] [PubMed] [Google Scholar]
- 8.Li Y, Bekele BN, Ji Y, Cook JD. Dose-schedule finding in phase I/II clinical trials using a Bayesian isotonic transformation. Statistics In Medicine. 2008;27:4895–4913. doi: 10.1002/sim.3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu CA, Braun TM. Identifying optimal treatment schedules via parametric non-mixture cure models. Journal of the Royal Statistical Society, C (Applied Statistics) 2009;58:225–236. doi: 10.1111/j.1467-9876.2008.00660.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nelsen RB. An Introduction to Copulas. Springer-Verlag; 1999. [Google Scholar]
- 11.Thall PF. Bayesian models and decision algorithms for complex early phase clinical trials. Statistical Science. 2010;25:227–244. doi: 10.1214/09-STS315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thall PF, Cook JD. Dose-finding based on efficacy-toxicity trade-offs. Biometrics. 2004;60:684–693. doi: 10.1111/j.0006-341X.2004.00218.x. [DOI] [PubMed] [Google Scholar]
- 13.Thall PF, Nguyen HQ. Adaptive randomization to improve utility-based dose-finding with bivariate ordinal outcomes. Journal of Bio-pharmaceutical Statistics. 2012;22:785–801. doi: 10.1080/10543406.2012.676586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thall PF, Russell KT. A strategy for dose finding and safety monitoring based on efficacy and adverse outcomes in phase I/II clinical trials. Biometrics. 1998;54:251–264. [PubMed] [Google Scholar]
- 15.Thall PF, Szabo A, Nguyen HQ, Amlie-Lefond CM, Zaidat OO. Optimizing the concentration and bolus of a drug delivered by continuous infusion. Biometrics. 2011;67:1638–1646. doi: 10.1111/j.1541-0420.2011.01580.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yuan Y, Yin G. Bayesian dose-finding by jointly modeling toxicity and efficacy as time-to-event outcomes. Journal of the Royal Statistical Society, Series C. 2009;58:954–968. [Google Scholar]
- 17.Zhang W, Sargent DJ, Mandrekar S. An adaptive dose-finding design incorporating both efficacy and toxicity. Statistics In Medicine. 2006;25:2365–2383. doi: 10.1002/sim.2325. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.