

Abstract
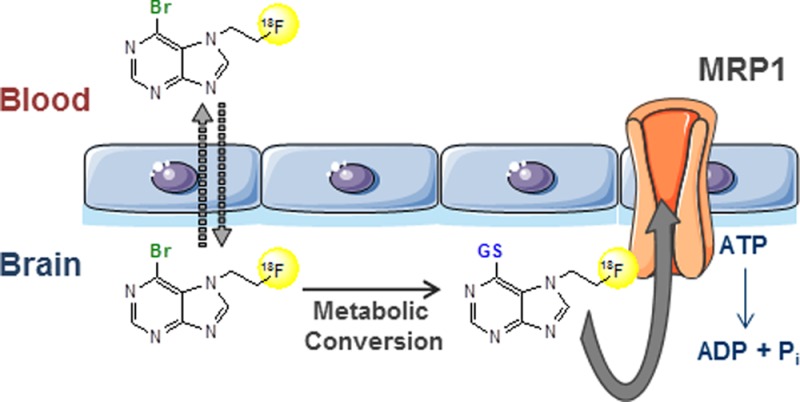
Multidrug resistance-associated protein 1 (MRP1) is a drug efflux transporter that has been implicated in the pathology of several neurological diseases and is associated with development of multidrug resistance. To enable measurement of MRP1 function in the living brain, a series of 6-halopurines decorated with fluorinated side chains have been synthesized and evaluated as putative pro-drug tracers. The tracers were designed to undergo conjugation with glutathione within the brain and hence form the corresponding MRP1 substrate tracers in situ. 6-Bromo-7-(2-[18F]fluoroethyl)purine showed good brain uptake and rapid metabolic conversion. Dynamic PET imaging demonstrated a marked difference in brain clearance rates between wild-type and mrp1 knockout mice, suggesting that the tracer can allow noninvasive assessment of MRP1 activity in vivo.
Introduction
Drug efflux transporters of the ATP-binding cassette (ABC) family of proteins mediate active transport of molecules across cell membranes and physiological barriers, and they have a major impact on the pharmacological behavior of most of the drugs in use today.1,2 ABC transporters have complementary substrate scope and expression patterns, and they maintain chemical homeostasis by extruding toxins and xenobiotics, including drugs, from cells and tissues.3−6 In particular, multidrug resistance-associated proteins (MRPs), P-glycoprotein (P-gp), and breast cancer-related protein (BCRP) are believed to play a key role in the development of multidrug resistance by extruding an array of therapeutic drugs, resulting in reduced access to their target sites, and hence rendering them ineffective.
MRP1, which is encoded by the ABCC1 gene, is ubiquitously expressed in the body but is found at elevated levels in the lungs, testis, kidneys, heart, and placenta, as well as at physiological barriers including the blood–brain barrier (BBB) and the blood–cerebrospinal fluid barrier.7−10 In contrast to P-gp, it is localized on the basolateral membranes in polarized cells. In the rat brain, MRP1 is present at higher levels in the astrocytes than in brain capillary endothelial cells.11 MRP1 functions mainly as a cotransporter of amphipathic organic anions. It can transport a variety of drugs, endogenous metabolites, xenobiotics, and hydrophobic drugs or other compounds that are conjugated to the anionic tripeptide glutathione (GSH), glucuronic acid, or sulfate.2,4,5 Because MRP1 also transports oxidized GSH, glutathione disulfide, it is believed to play additional roles in maintaining GSH homeostasis and regulating the redox state of cells.8,12 The role of MRPs in BBB permeability has been demonstrated by experiments in which inhibitors of MRPs, such as probenecid or MK-571, were shown to enhance drug penetration into the brain or to inhibit drug efflux from isolated brain endothelial cells.13
Upregulation of MRP1 is associated with drug resistance in a variety of solid tumors, including lung, breast, and prostate cancers,14 and has been shown to correlate with a poor response to treatment.10,15 Mice lacking intact mrp1 gene have altered response to inflammatory stimuli and show increased toxic response to the anticancer drug etoposide.16 Elevated MRP1 expression is also associated with refractory epilepsy,17,18 although the ability of MRP1 to transport antiepileptic drugs remains unclear.19,20 There is also evidence suggesting that MRP1 function may be impaired in patients with Alzheimer’s disease,21 and recent preclinical studies have provided compelling evidence for a role of this transporter in the clearance of amyloid-β (Aβ) peptides from the brain.22,23 Noninvasive imaging with positron emission tomography (PET) can enable assessment of MRP1 function in vivo and holds considerable potential as a tool to elucidate the role of MPR1 in human diseases, and to evaluate experimental treatments aimed at modulating transporter function.
To enable quantification of MRP1 function in the brain with PET, Okamura and co-workers recently applied a novel imaging concept, referred to as the metabolite extrusion method (MEM), which relies on a pro-drug/drug approach.24 The method was experimentally demonstrated using 6-bromo-7-[11C]methylpurine ([11C]7m6BP) as the pro-drug tracer.25 Following administration, [11C]7m6BP readily enters the brain and is subsequently converted to the corresponding GSH conjugate, which acts as a substrate for MRP1 (Figure 1). This enables MPR1 activity to be correlated directly to the efflux rate of radioactivity from the brain because the GSH conjugate S-[6-(7-[11C]methylpurinyl)]glutathione (PSG) is unable to cross the BBB by passive diffusion. While [11C]7m6BP enables imaging of MRP1 activity in rodents, the short half-life of carbon-11 (20.4 min) makes it difficult to determine the metabolic profile of the tracer in animal models. It also precludes dynamic imaging over prolonged periods of time and may be problematic for applications in higher species, such as primates and humans, which are likely to display lower rates of enzymatic GSH conjugation.26 To address these limitations, we have prepared and evaluated a series of fluorine-18 (half-life 110 min) labeled 6-halopurines as putative pro-drug tracers for dynamic imaging of MRP1 function in the brain.
Figure 1.

(a) Metabolic extrusion method (MEM): the pro-drug tracer crosses the BBB by passive diffusion (K1) and within the brain rapidly undergoes enzymatic conversion (k3) to a hydrophilic radioactive metabolite (tracer). If the passive diffusion of the tracer across the BBB is negligible, the activity of the drug efflux pump can be correlated to the clearance rate of the tracer (k4) from the brain. (b) Metabolic conversion of [11C]7m6BP to the MRP1 substrate S-[6-(7-[11C]methylpurinyl)]glutathione (PSG) by GSTs in the brain.
Results and Discussion
Chemistry
We explored two alternative strategies for preparation of fluorine-18 labeled 6-halopurines, namely, direct labeling of derivatives bearing fluoroalkyl chains or indirect labeling via click chemistry to give the resulting radiolabeled triazoles. The triazole-functionalized 6-halopurines 7–10 were synthesized by copper(I)-catalyzed cycloaddition of the corresponding alkynes (3–6) with 2-fluoroethyl azide as previously reported (Scheme 1).27 The N-fluoroalkylpurines 11–13 and the corresponding tosylate precursors 14–16 were prepared by base-promoted N-alkylation of 6-chloropurine (1) and 6-bromopurine (2) using a modified literature procedure.28 Alkylation provided the N7-isomers 11–16 in 4–13% yield, with the corresponding N9-isomers constituting the major products (40–65% yield). The increased polarity of the N7-isomers relative to their N9 alkylated counterparts allowed separation by flash chromatography on silica gel.29,30 Conjugation of the N7-fluorethyl analogue 11 with GSH under basic conditions24 provided S-[6-(7-(2-fluoroethyl)purinyl)]glutathione (17) in 84% yield as a nonradioactive reference compound for biological studies.
Scheme 1. Synthesis of Fluorinated 6-Halopurine Derivatives.

Reagents and conditions: (i) alkylating agent (1.3 equiv), NaH (1.1 equiv), DMF, overnight, rt; (ii) 2-fluoroethyl azide (1.3 equiv), CuSO4 (0.05 equiv), sodium ascorbate (0.1 equiv), DMF, 4 h, rt; (iii) K222/K2CO3 (30:15 mM), [18F]fluoride, 15 min, 80 °C.
Radiochemistry
The triazole-functionalized purines [18F]9 and [18F]10 were prepared in 40% and 55% isolated radiochemical yield (RCY), respectively, by ligand-accelerated copper(I)-catalyzed cycloaddition of the respective alkyne precursors 5 and 6 with [18F]fluoroethyl azide as reported elsewhere.27 Treatment of the tosylate precursors 14–16 with non-carrier-added (nca) [18F]fluoride in the presence of Kryptofix 222 (K222) and sodium carbonate provided the purines [18F]11–[18F]13 in 20–27% analytical RCY. The labeling reactions also yielded a lipophilic radioactive side product (up to 10% RCY), most likely due to competing substitution of the halide in the 6 position with [18F]fluoride. As expected, the formation of the radioactive side product was more pronounced for the brominated precursor 15 than for the 6-chloro analogues 14 and 16. The labeled compounds [18F]11–[18F]13 were obtained in 6–10% decay-corrected isolated RCY after formulation and sterile filtration, with a synthesis time of approximately 70 min. The chlorinated analogues [18F]11 and [18F]13 were isolated with a radiochemical purity of 99% after purification using a semipreparative monolithic HPLC column; however, in the case of 6-bromopurine [18F]12, this purification system gave a broad product peak that partly overlapped with an unknown radioactive side product. The use of a particle C18 column improved the separation and enabled us to obtain [18F]12 with 97% radiochemical purity. When this purification system and low levels of activity (1–1.5 GBq of [18F]fluoride) were used, the specific activity (SA) of [18F]12 was in a range of 5–7 GBq/μmol after formulation. Scale up of the reaction (∼5 GBq of [18F]fluoride) under optimized conditions improved the SA of [18F]12 to 180 GBq/μmol at end of synthesis.
Kinetic Studies
The reactivity of the purines 7–13 with GSH was evaluated by determining the pseudo-first-order nonenzymatic conjugation rates (knon, Table 1). To circumvent the need for radiolabeled analogues in this assay, we incubated compounds 7–13 at a concentration of 50 μM with an excess of GSH (10 mM) in phosphate buffer and used HPLC to analyze the composition of the reaction mixtures over time. Under these conditions, knon of 7m6BP (0.49 ± 0.001 h–1) was comparable to the rate previously reported for [14C]7m6BP (0.28 ± 0.006 h–1) in the presence of physiological concentration of GSH (2 mM).26 Purines with bromine in the 6-position showed 3–4 fold higher conjugation rates than the corresponding chlorine analogues, with the highest rate observed for the triazole 8 (0.80 ± 0.02 h–1). The fluoroethyl derivative 12 was equally reactive to 7m6BP, whereas analogues of 8 and 11 with extended side chains showed a marked drop in reactivity.
Table 1. Nonenzymatic Reaction Rates of 6-Halopurine Analogues with GSH.
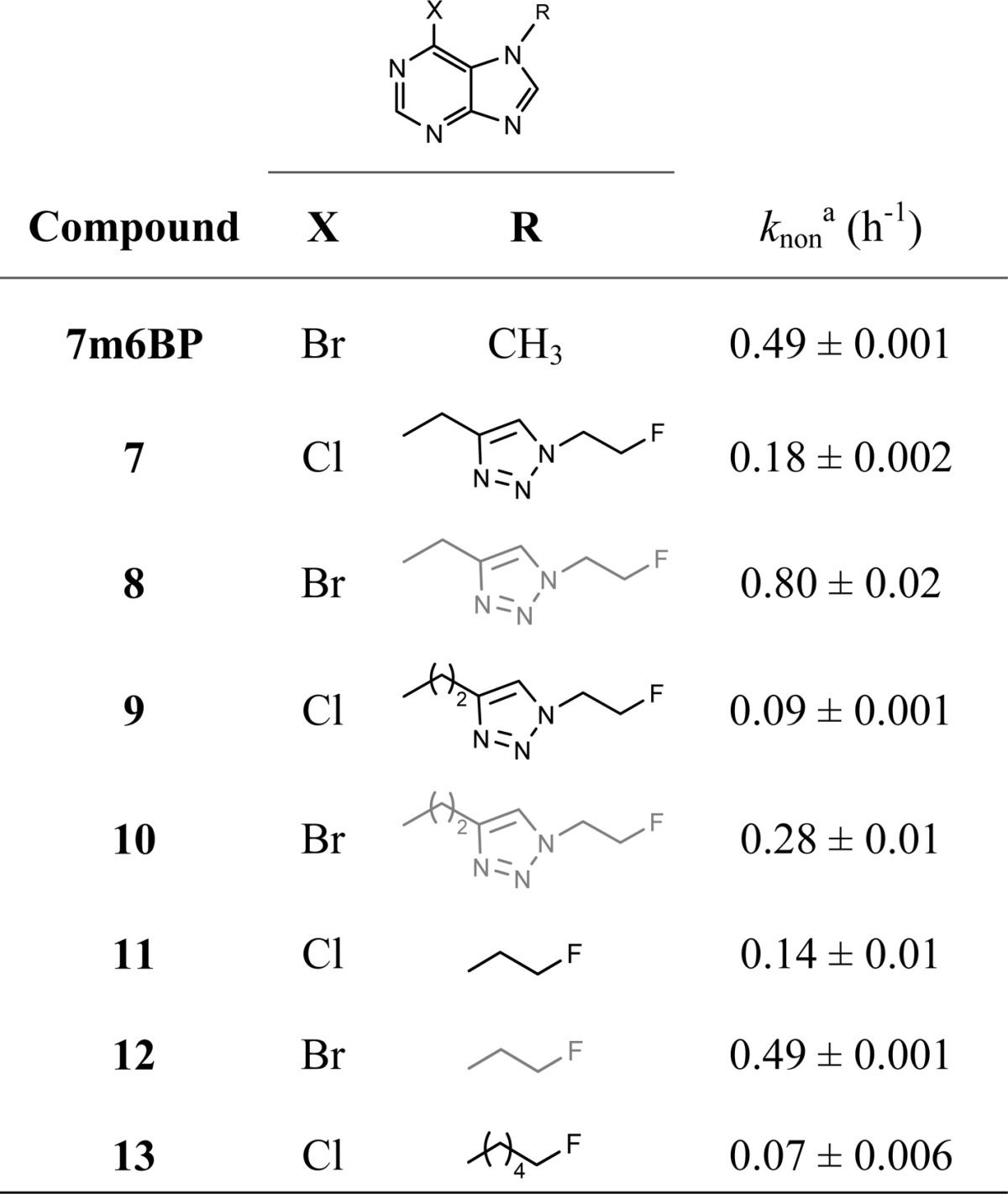
Reaction rates of 6-halopurine analogues (50 μM) in phosphate buffer (0.1 M, pH 7.4) in the presence of GSH (10 mM). Measurements were made in duplicate, and values are represented as mean ± standard deviation.
Biodistribution Studies
Motivated by the ease of radiolabeling,27 we initially selected the triazole [18F]10 for biological evaluation in wild-type BALB/c mice. The more reactive analogue 8 was excluded from biological evaluation because it was found to gradually hydrolyze in water. However, [18F]10 showed a negligible brain uptake at 5 min postinjection, and hence we decided not to further pursue evaluation of purines bearing triazole side chains. 6-Bromo-7-(2-fluoroethyl)purine (12), which showed the highest nonenzymatic GSH conjugation rate of the fluoroalkylated analogues, gave an initial brain uptake of 3.9% ± 0.5% injected dose per gram bodyweight (ID/g) at 5 min postinjection (p.i.), with a gradual clearance of radioactivity from the brain observed over a period from 15 to 90 min p.i. (Figure 2a). Intriguingly, the clearance of radioactivity from blood, as well as most of the other tissues examined (heart, liver, lungs, and spleen) mirrored that of the brain. A high initial uptake (45% ID/g at 5 min p.i.) and rapid elimination from the kidneys pointed to renal clearance as the main route of excretion. The uptake of radioactivity in bone tissue remained low throughout the time course of the experiment (∼1% ID/g in femur at 90 min p.i.). The biodistribution of the chlorinated analogue [18F]11 (Figure 2b) was practically indistinguishable from that of [18F]12, and while the mean brain uptake at 5 min p.i. was lower for [18F]11 than for [18F]12, the difference was not significant (3.5% ± 0.6% ID/g vs 3.9% ± 0.5% ID/g). Extension of the fluoroalkyl side chain to give [18F]13 did not alter the initial brain uptake (3.9% ± 0.3% ID/g at 5 min p.i.), but clearance of radioactivity from the brain was significantly faster than for [18F]11 and [18F]12 (Figure 2c). A gradual increase of radioactivity levels in bone tissue (4.3% ± 1.6% ID/g in femur at 60 min p.i.) indicated cleavage of the C–F bond in vivo. Because compound [18F]12 showed a favorable biodistribution and the highest nonenzymatic GSH conjugation rate within the series, it was selected as a lead pro-drug tracer for further biological evaluation.
Figure 2.

Total uptake of radioactivity in selected tissues expressed as % ID/g ± standard deviation (n = 3) after administration of (a) [18F]12, (b) [18F]11, and (c) [18F]13 in wild-type BALB/c mice over time.
Enzymatic Kinetic Studies
The pseudo-first-order enzymatic conjugation rate (kenz) of compound 12 (50 μM) with GSH (10 mM) was measured in mouse brain homogenates. HPLC analysis was used to monitor consumption of 12 over time and revealed a clean conversion to the corresponding GSH conjugate 17 within 15 min (Figure 3). The rate of enzymatic conjugation (kenz = 39.1 ± 6.1 h–1 g–1 mL–1) was approximately 2 orders of magnitude higher than the rate for the nonenzymatic reaction (knon = 0.49 ± 0.001 h–1), demonstrating that 12 is a good substrate for glutathione S-transferases (GSTs).
Figure 3.

UV chromatograms showing the enzymatic conjugation of GSH (10 mM) with 6-bromo-7-(2-fluoroethyl)purine 12 (50 μM) over a period of 2–15 min.
Metabolite Analysis
Radio-HPLC was used to determine the metabolic profile in plasma and brain tissue after administration of [18F]12 to wild-type BALB/c. Analysis of plasma samples showed that [18F]12 was rapidly metabolized, resulting in formation of the GSH conjugate [18F]17, as well as additional metabolites. The GSH conjugate [18F]17 represented 25% ± 3% and 64% ± 5% (n = 3) of the total radioactivity at 15 and 60 min p.i., respectively, with other unknown metabolites accounting for the remaining activity (Figure 4a). In the brain, the metabolic profile remained largely unchanged over this period of time, with [18F]17 constituting 44% ± 1% (n = 3) and 48% ± 6% (n = 4) of the total radioactivity at 15 and 60 min p.i., respectively. A similar metabolic profile was observed in the brain of wild-type FVB mice (Figure 4b), whereas analysis of brain tissue from mrp1 knockout (KO) FVB mice demonstrated a remarkably clean conversion of [18F]12 to [18F]17, with no other radioactive metabolites detected (Figure 4c). Although we have not determined the structural identity of the metabolites other than [18F]17, the results are consistent with previous studies of the metabolism of 6-chloropurine, which after GST-mediated conjugation with GSH undergoes stepwise breakdown mediated by γ-glutamyltranspeptidase, dipeptidase, and cysteine conjugate β-lyase to eventually give 6-mercaptopurine.21,31,32 Because this metabolic pathway occurs in the extracellular space, it relies on MPR1 mediated efflux of the glutathione conjugate from the cytosol,33−35 which could explain why [18F]17 was the only radioactive metabolite observed in brain tissue from mrp1 KO FVB mice.
Figure 4.

HPLC profile of radioactive metabolites after administration of [18F]12. (a) Brain and plasma samples 60 min p.i. in WT BALB/c mice; (b) brain at 15 min (solid line) and 60 min (dotted line) p.i. in WT FVB mice; (c) brain at 15 min (solid line) and 60 min (dotted line) p.i. in mrp1 KO FVB mice.
Measurement of the Brain Efflux Rates in WT and mrp1 KO FVB Mice with PET
Dynamic PET was used to measure the brain radioactivity levels in WT and mrp1 KO FVB mice after administration of [18F]12. In WT mice, the brain uptake peaked within 2 min after injection (data not shown), and in accordance with the biodistribution studies, the radioactivity levels gradually decreased from 15 min onward. In mrp1 KO mice, a similar profile was observed at the early time points; however, in the period from 15 to 60 min, the radioactivity levels remained largely unchanged in the brain, as well as in peripheral tissues (Figure 5a,b). The efflux rates of radioactivity from the brain in the period from 15 to 60 min after administration of [18F]12 were 1.6 ± 0.13 and 0.17 ± 0.02 h–1 in WT and mrp1 KO mice, respectively (Figure 5c). The results are in excellent agreement with the previously reported efflux rates for [11C]7m6BP (1.4 ± 0.24 and 0.15 ± 0.01 h–1 in WT and mrp1 KO mice, respectively).25 The marked difference in brain efflux rates between the two groups of mice strongly suggests that MRP1 plays a key role in the clearance of the GSH conjugate [18F]17 from the brain. However, the presence of multiple radioactive metabolites in the brain after administration of [18F]12 in WT mice raises questions about the relative contribution of phase III metabolites to the efflux rate and to what degree the clearance can be affected by alterations to the enzymatic machinery or expression levels of other efflux pumps. While the brain clearance rate measured after administration of [18F]12 is likely to reflect the efflux rate of drugs that are susceptible to GSH conjugation, further characterization is required to determine the correlation of the efflux rate to MRP1 activity.
Figure 5.

Coronal and sagittal summed images during 32.5–57.5 min after administration of [18F]12 in (a) mrp1 KO FVB mice and (b) WT FVB mice. (c) Efflux rates in WT and in mrp1 KO FVB mice. (d) Brain radioactivity levels expressed as % ID/mL over the period from 15 to 60 min after injection of [18F]12. The results are expressed as the mean ± standard deviation (n = 4).
Conclusion
A series of 6-halopurines decorated with fluorinated side chains have been synthesized and evaluated as putative pro-drug tracers for imaging of MRP1 activity in the brain with PET. Following measurements of the conjugation rates with GSH, selected derivatives were labeled with fluorine-18, and subjected to biodistribution and metabolite studies. 6-Halopurines decorated with fluoroalkyl side chains showed high initial brain uptake, whereas introduction of triazole containing side chains proved detrimental for BBB penetration. The lead compound, 6-bromo-7-(2-[18F]fluoroethyl)purine ([18F]12), gave a peak brain uptake within 2 min p.i. and was found to undergo rapid GSH conjugation in vivo, although additional radioactive metabolites were observed in the brain as well as in plasma. In biodistribution studies in wild-type mice, a favorable clearance rate of radioactivity from the brain was observed from 15 to 90 min after administration of [18F]12. Dynamic PET imaging of wild-type versus mrp1 knockout FVB mice showed a marked difference in brain efflux rates between the two groups of mice, suggesting that [18F]12 is a promising pro-drug tracer for noninvasive measurement of MRP1 activity in the brain. However, the presence of secondary metabolites in the brain of wild-type FVB mice raises questions as to their relative contribution to the clearance rate. Further characterization is therefore required to determine to what degree the clearance of radioactivity from the brain following administration of [18F]12 correlates with MRP1 activity.
Materials and Methods
General
All reagents were purchased from Sigma-Aldrich and used without further purification. NMR spectra were recorded on Bruker Avance spectrometers operating at a frequency of 500 or 600 MHz for 1H and 125 or 150 MHz for 13C. Chemical shifts (δ) are reported in ppm downfield from the internal standard tetramethylsilane. High resolution mass spectra were recorded on a thermo Finnigan MAT900xp (CI/EI) or a Waters LCT Premier XE (ES) mass spectrometers. Melting points were determined using a Gallenkamp Sanyo melting point apparatus. No-carrier-added aqueous [18F]fluoride was provided by St Thomas’ Hospital, King’s College London, U.K. HPLC analyses were performed with an Agilent 1200 series system equipped with a diode array UV detector (results described are for UV at λ = 265 nm), and a Raytest GABI star NaI detector. Automated synthesis was carried out on a HBIII module (Scintomics, Germany). Isolated radiochemical yields (RCY) were measured using a Curiementor 4 ion chamber (PTW, Germany). IR spectra were obtained using a PerkinElmer Spectrum 100 FT-IR spectrometer. All animal experiments conducted at UCL (London, U.K.) were in compliance with the United Kingdom Home Office Animal Procedures Act of 1986 with approval of the University College London Animal Ethics Committee. Female BALB/c WT mice were purchased from Charles River, U.K. All animal experiments conducted at the Molecular Imaging Center (Chiba, Japan) were in compliance with the “Recommendations for Handling of Laboratory Animals for Biomedical Research” compiled by the Committee on Safety and Ethical Handling Regulations for Laboratory Animal Experiments at the National Institute of Radiological Sciences. Male FVB WT mice and mrp1 KO mice were purchased from Taconic Farms, Inc. (Hudson, NY). Mice weighing 27–33 g (aged 17–22 wk) were used throughout this study and were allowed free access to food and water. The nonradioactive reference compounds (7m6BP, 7–13) were obtained with a purity >95% as determined by analytical HPLC (λ = 265 nm). The radioactive tracers ([18F]10–[18F]13) were obtained with radiochemical purity ≥97% as determined by analytical radio-HPLC.
General Procedure for Preparation of the Alkylated 6-Halopurines
To a solution of 6-halopurine (1 equiv) and NaH (60% dispersed in oil, 1.1 equiv) in anhydrous DMF was added the alkylating agent (1.3 equiv) under inert atmosphere. After 24 h at room temperature, the reaction mixture was diluted with dichloromethane (DCM) and washed three times with water. The organic layer was dried with MgSO4, and the solvent was removed in vacuo. The residue was purified by flash chromatography (silica gel as solid phase) using a gradient of DCM and ethyl acetate (from 0% to 60% ethyl acetate) to give the N7- and the N9-alkynyl analogues.
6-Chloro-7-(2-fluoroethyl)purine (11)
A mixture of 6-chloropurine (1) (500 mg, 3.23 mmol), (2-fluoroethyl)tosylate (910 mg, 4.21 mmol), and NaH (60% dispersed in oil, 142 mg, 3.56 mmol) in anhydrous DMF (15 mL) was reacted according to the general alkylation procedure to give 11 (43 mg, 7%) and the corresponding N9-isomer 11a (246 mg, 38%) as white solids. Compound 11. 1H NMR (CDCl3, 500 MHz): δ 8.86 (s, 1 H, H-2), 8.30 (s, 1 H, H-8), 4.86 (s, 2 H, CH2cCH2aF), 4.80 (m, 1 H, CH2dCH2F), 4.76 (m, 1 H, CH2CH2bF). 13C NMR (CDCl3): δ 162.2, 152.7, 149.8, 142.8, 122.2, 82.2, 80.9, 47.6, 47.4. Accurate mass (EI) m/z calcd for C7H6ClFN4 (M)+ 200.02595, found 200.02670. IR (cm–1): υmax 3096, 3068, 2924, 1845, 1692. Mp: 110 ± 2 °C. Compound 11a. 1H NMR (CDCl3, 500 MHz): δ 8.69 (s, 1 H, H-2), 8.18 (s, 1 H, H-8), 4.77 (dt, JH,F = 46.78 Hz, J1,2 = 4.62 Hz, 2 H, CH2F), 4.59 (t, JH,F = 27.16 Hz, J1,2 = 4.48 Hz, 2 H, CH2CH2F). 13C NMR (CDCl3): δ 152.1, 151.7, 151.2, 145.6, 131.5, 81.7, 80.3, 44.7, 44.5. Accurate mass (EI) m/z calcd for C7H6ClFN4 (M)+ 200.02595, found 200.02541. IR (cm–1): υmax 3097, 3076, 2992, 2969, 1876, 1811. Mp: 90 ± 1 °C.
6-Bromo-7-(2-fluoroethyl)purine (12)
A mixture of 6-bromopurine (2) (500 mg, 2.51 mmol), (2-fluoroethyl)tosylate (780 mg, 3.27 mmol), and NaH (60% dispersed in oil, 111 mg, 2.76 mmol) in anhydrous DMF (15 mL) was reacted according to the general alkylation procedure to give 12 (39 mg, 6%) and the corresponding N9-isomer 12a (266 mg, 43%) as white solids. Compound 12. 1H NMR (CDCl3, 600 MHz): δ 8.86 (s, 1 H, H-2), 8.31 (s, 1 H, H-8), 4.9 (bs, 2 H, CH2cCH2aF), 4.85 (t, J1,2 = 4.32 Hz, 1 H, CH2dCH2F), 4.80 (t, J1,2 = 4.20 Hz, 1 H, CH2CH2bF). 13C NMR (CDCl3): δ 161.4, 152.7, 150.1, 133.0, 124.4, 82.1, 80.0, 47.2, 47.1. Accurate mass (CI) m/z calcd for C7H7BrFN4 (M + H)+ 245.98381, found 245.98282. IR (cm–1): υmax 3087, 3051, 2973, 2913, 1873. Mp: 123 ± 2 °C. Compound 12a. 1H NMR (CDCl3, 600 MHz): δ 8.70 (s, 1 H, H-2), 8.22 (s, 1 H, H-8), 4.80 (dt, JH,F = 46.8 Hz, J1,2 = 4.44 Hz, 2 H, CH2F), 4.60 (dt, JH,F = 27.1 Hz, J1,2 = 4.40 Hz, 2 H, CH2CH2F). 13C NMR (CDCl3): δ 152.1, 150.4, 145.5, 143.4, 134.2, 81.7, 80.3, 44.7, 44.5. Accurate mass (CI) m/z calcd for C7H7BrFN4 (M + H)+ 245.98381, found 245.98291. IR (cm–1): υmax 3127, 3089, 3065, 2976, 2957, 1851, 1776. Mp: 99 ± 1 °C.
6-Chloro-7-(5-fluoropentyl)purine (13)
A mixture of 6-chloropurine (1) (450 mg, 2.91 mmol), (5-fluoropentyl)tosylate (1.3 g, 3.78 mmol), and NaH (60% dispersed in oil, 128 mg, 3.2 mmol) in anhydrous DMF (15 mL) was reacted according to the general alkylation procedure to give 13 (90 mg, 13%, white solid) and the corresponding N9-isomer 13a (327 mg, 46%, colorless oil). Compound 13. 1H NMR (CDCl3, 600 MHz): δ 8.88 (s, 1 H, H-2), 8.22 (s, 1 H, H-8), 4.49 (t, J = 7.32 Hz, 2 H, CH2(CH2)4F), 4.45 (dt, JF,H = 47.29 Hz, J1,2 = 5.76 Hz, 2 H, CH2F), 1.99 (m, 2 H, CH2(CH2)3F), 1.80–1.71 (m, 2 H, CH2CH2F), 1.52 (m, 2 H, CH2(CH2)2F). 13C NMR (CDCl3): δ 162.2, 152.6, 149.0, 143.1, 122.4, 84.1, 83.0, 47.5, 31.5, 29.9, 29.8, 22.6. Accurate mass (EI) m/z calcd for C10H12ClFN4 (M)+ 242.07345, found 242.07359. IR (cm–1): υmax 3105, 3076, 2932, 2865, 1909, 1811. Mp: 97 ± 1 °C. Compound 13a. 1H NMR (CDCl3, 600 MHz): δ 8.73 (s, 1 H, H-2), 8.12 (s, 1 H, H-8), 4.42 (dt, JF,H = 47.29 Hz, J1,2 = 5.82 Hz, 2 H, CH2F),4.31 (t, J = 5.82 Hz 2 H, CH2(CH2)4F), 1.98 (m, 2 H, CH2(CH2)3F), 1.78–1.69 (m, 2 H, CH2CH2F), 1.47 (m, 2 H, CH2(CH2)2F). 13C NMR (CDCl3): δ 152.0, 151.9, 151.9, 415.2, 131.7, 84.2, 83.1, 44.5, 29.8, 29.7, 29.5, 22.5. Accurate mass (EI) m/z calcd for C10H12ClFN4 (M)+ 242.07345, found 242.07342. IR (cm–1): υmax 3074, 2943, 2867. Analytical HPLC: Luna C18(2) column (3 μm, 50 mm × 4.6 mm, Phenomenex) using water and methanol containing 0.1% of formic acid as eluents (methanol content from 15% to 50% over 20 min, flow 1 mL/min, λ = 265 nm). Retention time: 12.9 min.
6-Chloro-7-(2-tosyloxyethyl)purine (14)
A mixture of 6-chloropurine (1) (500 mg, 3.24 mmol), ethylene di(p-toluenesulfonate) (1.6 g, 4.21 mmol), and NaH (60% dispersed in oil, 142 mg, 3.56 mmol) in anhydrous DMF (15 mL) was reacted according to the general alkylation procedure to give 14 (109 mg, 19%) and the corresponding N9-isomer 14a (466 mg, 41%) as white solids. Compound 14. 1H NMR (CDCl3, 600 MHz): δ 8.83 (s, 1 H, H-2), 8.19 (s, 1 H, H-8), 7.46 (d, J = 7.8 Hz, 2 H, ArH), 7.07 (d, J = 8.4 Hz, 2 H, ArH), 4.69 (t, J = 4.8 Hz, 2 H, CH2CH2OTs), 4.45 (t, J = 4.8 Hz, 2 H, CH2OTs), 2.33 (s, 3 H, CH3). 13C NMR (CDCl3): δ 162.2, 152.5, 149.9, 145.6, 142.3, 131.1, 130.0, 127.5, 121.6, 67.3, 46.2, 29.8, 21.7. Accurate mass (EI) m/z calcd for C14H13N4O3SCl (M)+ 352.03914, found 352.03958. IR (cm–1): υmax 3110, 3082, 3016, 2970, 1831, 1718. Mp: 166 ± 1 °C. Compound 14a. 1H NMR (CDCl3, 600 MHz): δ 8.56 (s, 1 H, H-2), 8.06 (s, 1 H, H-8), 7.45 (d, J = 8.4 Hz, 2 H, ArH), 7.09 (d, J = 7.8 Hz, 2 H, ArH), 4.51 (t, J = 4.8 Hz, 2 H, CH2CH2OTs), 4.45 (t, J = 4.8 Hz, 2 H, CH2OTs), 2.37 (s, 3 H, CH3). 13C NMR (CDCl3): δ 152.2, 151.15, 151.12, 145.6, 145.59, 131.6, 131.4, 129.8, 127.6, 66.2, 43.7, 21.7. Accurate mass (EI) m/z calcd for C14H13N4O3SBr (M)+ 352.03914, found 352.03974. IR (cm–1): υmax 3112, 3083, 2960, 1765. Mp: 175 ± 1 °C.
6-Bromo-7-(2-tosyloxyethyl)purine (15)
A mixture of 6-bromopurine (2) (500 mg, 2.51 mmol), ethylene di(p-toluenesulfonate) (1.2 g, 3.27 mmol), and NaH (60% dispersed in oil, 111 mg, 2.76 mmol) in anhydrous DMF (15 mL) was reacted according to the general alkylation procedure to give 15 (40 mg, 4%) and the corresponding N9-isomer 15a (399 mg, 40%) as white solids. Compound 15. 1H NMR (CDCl3, 600 MHz): δ 8.78 (s, 1 H, H-2), 8.19 (s, 1 H, H-8), 7.46 (d, J = 8.22 Hz, 2 H, ArH), 7.07 (d, J = 8.10 Hz, 2 H, ArH), 4.72 (t, J = 4.86 Hz, 2 H, CH2CH2OTs), 4.48 (t, J = 5.04 Hz, 2 H, CH2OTs), 2.33 (s, 3 H, CH3). 13C NMR (CDCl3): δ 161.3, 152.4, 150.1, 145.6, 1325.5, 131.1, 129.9, 127.5, 123.8, 67.3, 45.7, 21.7. Accurate mass (CI) m/z calcd for C14H14N4O3SBr (M + H)+ 396.99700, found 396.99825. IR (cm–1): υmax 2996, 2965, 2920, 1917, 1772. Mp: 165 ± 1 °C. Compound 15a. 1H NMR (CDCl3, 600 MHz): δ 8.53 (s, 1 H, H-2), 8.07 (s, 1 H, H-8), 7.45 (d, J = 8.28 Hz, 2 H, ArH), 7.09 (d, J = 7.98 Hz, 2 H, ArH), 4.50 (t, J = 4.44 Hz, 2 H, CH2CH2OTs), 4.45 (t, J = 4.98 Hz, 2 H, CH2OTs), 2.37 (s, 3 H, CH3). 13C NMR (CDCl3): δ 151.8, 149.8, 145.6, 145.5, 143.3, 134.2, 131.3, 129.8, 127.5, 66.2, 43.7, 21.7. Accurate mass (CI) m/z calcd for C14H14N4O3SBr (M + H)+ 396.99700, found 396.99792. IR (cm–1): υmax 3113, 2989, 2959, 2914, 1921. Mp: 175 ± 1 °C.
6-Chloro-7-(5-tosyloxypentyl)purine (16)
A mixture of 6-chloropurine (1) (500 mg, 3.24 mmol), pentane di(p-toluenesulfonate) (1.7 g, 4.21 mmol), and NaH (60% dispersed in oil, 142 mg, 3.56 mmol) in anhydrous DMF (15 mL) was reacted according to the general alkylation procedure to give 16 (115 mg, 9%) and the corresponding N9-isomer 16a (810 mg, 64%) as yellow oils. Compound 16. 1H NMR (CDCl3, 600 MHz): δ 8.86 (s, 1 H, H-2), 8.22 (s, 1 H, H-8), 7.74 (d, J = 7.80 Hz, 2 H, ArH), 7.33 (d, J = 7.80 Hz, 2 H, ArH), 4.44 (t, J = 7.20 Hz, 2 H, CH2(CH2)4OTs), 4.00 (t, J = 6.0 Hz, 2 H, CH2OTs), 2.43 (s, 3 H, CH3), 1.91 (m, 2 H, CH2(CH2)3OTs), 1.71 (m, 2 H, CH2CH2OTs), 1.41 (m, 2 H, CH2(CH2)2OTs). 13C NMR (CDCl3): δ 162.1, 152.6, 149.1, 145.1, 143.0, 132.9, 130.1, 127.9, 122.4, 69.8, 47.3, 44.5, 31.2, 29.6, 22.6, 21.8. Accurate mass (ES+) m/z calcd for C17H20ClN4O3S (M + H)+ 395.0945, found 395.0944. IR (cm–1): υmax 3059, 2937, 2865. Analytical HPLC: Chromolith performance column, RP18-e (100 mm × 4.6 mm, Merck), using water and methanol containing 0.1% of formic acid as eluents (methanol content from 15% to 70% over 9 min, flow 3 mL/min, λ = 265 nm). Retention time: 6.8 min. Compound 16a. 1H NMR (CDCl3, 600 MHz): δ 8.73 (s, 1 H, H-2), 8.06 (s, 1 H, H-8), 7.40 (d, J = 7.80 Hz, 2 H, ArH), 7.32 (d, J = 8.40 Hz, 2 H, ArH), 4.27 (t, J = 7.20 Hz, 2 H, CH2(CH2)4OTs), 4.00 (m, 2 H, CH2OTs), 2.43 (s, 3 H, CH3), 1.91 (m, 2 H, CH2(CH2)3OTs), 1.70 (m, 2 H, CH2CH2OTs), 1.39 (m, 2 H, CH2(CH2)2OTs). 13C NMR (CDCl3): δ 152.0, 151.9, 151.2, 145.2, 145.0, 132.9, 131.7, 130.0, 127.9, 69.9, 44.3, 29.4, 29.3, 22.8, 21.8. Accurate mass (ES+) m/z calcd for C17H20ClN4O3S (M + H)+ 395.0945, found 395.0941. IR (cm–1): υmax 3070, 2942, 2867, 1737. Analytical HPLC: Luna C18(2) column (3 μm, 50 mm × 4.6 mm, Phenomenex) using water and methanol containing 0.1% of formic acid as eluents (methanol content from 25% to 65% over 20 min, flow 1 mL/min, λ = 265 nm). Retention time: 15.6 min.
5-Tosyloxy-1-pentanol
1,5-Pentanediol (4.0 g, 40 mmol), p-toluenesulfonyl chloride (7.3 g, 42 mmol), and pyridine (3.9 mL, 50 mmol) were dissolved in DCM (20 mL), and the resulting solution was stirred overnight at room temperature. The reaction mixture was diluted with DCM and washed three times with water. The organic layer was dried with MgSO4, and the solvent was removed in vacuo. The crude mixture was suspended in methanol to precipitate pentane di(p-toluenesulfonate). The white solid was removed by filtration, and the filtrate was concentrated in vacuo. The resulting crude product was purified by liquid chromatography on silica gel using a gradient of petroleum ether and ethyl acetate (from 0% to 60% of ethyl acetate). 5-Tosyloxy-1-pentanol: 53% yield (3.8 g). 1H NMR (CDCl3, 600 MHz): δ 7.78 (d, J = 8.40 Hz, 2 H, ArH), 7.34 (d, J = 7.80 Hz, 2 H, ArH), 4.02 (t, J = 6.60 Hz, 2 H, CH2OH), 4.59 (t, J = 6.60 Hz, 2 H, CH2OTs), 2.44 (s, 3 H, CH3), 1.68–1.53 (m, 2 H, CH2CH2OH), 1.52–1.49 (m, 2 H, CH2CH2OTs), 1.41–1.36 (m, 2 H, CH2CH2CH2OH). 13C NMR (CDCl3): δ 144.9, 133.1, 129.8, 128.0, 70.6, 62.6, 60.6, 53.6, 31.9, 28.7, 21.6. Accurate mass (EI) m/z calcd for C12H18O4S (M+) 258.09203, found 258.09246. Pentane di(p-toluenesulfonate): 18% yield (2.8 g). 1H NMR (CDCl3, 600 MHz): δ 7.76 (d, J = 8.40 Hz, 4 H, ArH), 7.34 (d, J = 7.80 Hz, 4 H, ArH), 3.96 (t, J = 6.00 Hz, 4 H, CH2OTs), 2.45 (s, 6 H, CH3), 1.60 (m, 4 H, TsOCH2CH2), 1.34 (m, 2 H, TsOCH2CH2CH2). 13C NMR (CDCl3): δ 145.0, 133.0, 129.8, 128.2, 70.1, 28.3, 21.6, 21.5. Accurate mass (CI) m/z calcd for C19H25O6S2 (M + H)+ 413.10925, found 413.10940.
(5-Fluoropentyl)tosylate
5-Tosyloxy-1-pentanol (1 g, 3.87 mmol) and diethylaminosulfur trifluoride (DAST) (2.6 mL, 19.4 mmol) were dissolved in DCM (20 mL) at 0 °C, and the resulting solution was allowed to gradually reach room temperature with stirring over 4 h. The reaction mixture was diluted with DCM (100 mL) and washed with brine (100 mL × 1) and twice with water (100 mL × 2). The organic layer was dried with MgSO4, and the solvent was removed in vacuo. The product was purified by liquid chromatography on silica gel using petrol ether and ethyl acetate (90:10), to give the title compound as colorless oil in 70% yield (700 mg). 1H NMR (CDCl3, 600 MHz): δ 7.78 (d, J = 8.28 Hz, 2 H, ArH), 7.34 (d, J = 8.11 Hz, 2 H, ArH), 4.39 (dt, JF,H = 47.23 Hz, J1,2 = 6.00 Hz, 2 H, CH2F), 4.03 (t, J = 6.36 Hz, 2 H, TsOCH2), 2.44 (s, 3 H, CH3), 1.71–1.65 (m, 2 H, TsOCH2CH2), 1.64–1.59 (m, 2 H, FCH2CH2), (m, 2 H, FCH2CH2CH2). 13C NMR (CDCl3): δ 144.9, 133.1, 130.0, 127.8, 84.3, 83.2, 70.4, 29.9, 29.7, 28.6, 27.8, 21.5, 21.4. Accurate mass (EI) m/z calcd for C12H17FO3S (M)+ 260.08769, found 260.08743.
S-[6-(7-(2-Fluoroethyl)purinyl)]glutathione (17)
Compound 17 was prepared following a modified literature procedure.24 Briefly, 11 (10 mg, 0.05 mmol) and GSH (33.7 mg, 0.11 mmol) dissolved in 300 μL of a solution of NaOH (1 M) and ethanol (1:1) were stirred for 4 h at 50 °C. The crude mixture was filtered over a plug of silica, and the silica plug was washed with a solution of ethyl acetate/methanol (1:1), and the product was eluted with methanol. The fraction containing the product was concentrated under reduced pressure, and the crude product was further purified by semipreperative HPLC using a Zorbax ODS column (C18, 5 μm, 9.4 mm × 250 mm, Agilent). The flow rate was 2 mL/min with a mobile phase of water and methanol containing 0.1% trifluoroacetic acid (TFA). The methanol content was increased from 10% to 30% over 20 min and then kept constant at 30% for 7 min. The UV absorbance detector was set at 288 nm. Retention time of 17: 23.4 min. Compound 17 was isolated in 84% yield (25 mg). 1H NMR (D2O, 500 MHz): δ 8.72 (s, 1 H, H-2), 8.49 (s, 1 H, H-8), 4.83 (s, 2 H, CH2aCH2cF), 4.78 (m, 2 H, CH-Cys, CH2bCH2F), 4.75 (t, J = 1.86 Hz, 1 H, CH2CH2dF), 3.95 (dd, J = 14.43 Hz, J = 5.12 Hz, 1 H, CH2a-Cys), 3.93 (s, 2 H, CH2–Gly), 3.81 (t, J = 6.56 Hz, 1 H, CH-Glu), 3.66 (dd, J = 14.46 Hz, J = 8.09 Hz, 1 H, CH2b-Cys), 2.39–2.33 (m, 2 H, CH2α-Glu), 2.01–1.98 (m, 2 H, CH2β-Glu). 13C NMR (D2O): δ 175.0, 173.5, 173.0, 172.6, 156.7, 154.9, 152.1, 150.1, 124.0, 83.6, 82.5, 53.7, 53.5, 48.9, 48.7, 41.8, 31.7, 31.2, 26.3. Accurate mass (ES–) m/z calcd for C17H21FN7O6S (M – H)− 470.1258, found 470.1243. IR (cm–1): υmax 3286, 2932, 2595. Mp: 191 ± 2 °C.
General Procedure for the Radiosynthesis of [18F]11–13
[18F]fluoride in water (∼1–5 GBq) was trapped on a QMA cartridge (Waters Sep-Pak light) and released with 0.5 mL of Kryptofix 222 and potassium carbonate mixture (30:15 mM) dissolved in acetonitrile/water (85:15). The solvent was removed by heating at 90 °C under a stream of nitrogen. Acetonitrile (0.5 mL) was added, and the distillation was continued. This procedure was repeated twice. The dried mixture was cooled to room temperature by a stream of nitrogen, and a solution of precursor (14–16, 7 μmol in 0.4 mL anhydrous acetonitrile) was added. The resulting mixture was stirred at 80 °C for 15 min. The solvent volume was reduced to 0.2 mL under a stream of nitrogen, then diluted with 2 mL water and passed through a C18 cartridge (Waters Sep-Pak light). After elution, the fraction containing the radioactive product was subsequently purified by semipreparative HPLC. C18 cartridge formulation: the fraction containing the radioactive product ([18F]11–[18F]13) was diluted with water to reduce the organic content to 5–10% depending on the lipophilicity of the radiolabeled product and loaded onto a C18 cartridge. The cartridge was washed with 2 mL of water, and the product was then released with 1 mL of pure ethanol. The volume fraction was reduced to ∼100 μL and diluted with saline to give an ethanol concentration of 5% before sterile filtration.
6-Chloro-7-(2-[18F]fluoroethyl)purine ([18F]11)
Hands-on reaction: analytical RCY, 27% ± 6% (n = 3). Automated synthesis: isolated RCY, 10% ± 2% (n = 7). [18F]11 was obtained with a radiochemical purity of 99% and SA of 6 GBq/μmol after sterile filtration. Precartridge purification with a C18 cartridge (Waters Sep-Pak light): the product [18F]11 was released with 1 mL of 20% ethanol in water, and the resulting fraction was further diluted with 1 mL of water. HPLC purification was performed on a Chromolith performance column, RP18-e (100 mm × 10 mm, Merck) using water and methanol containing 0.1% of formic acid as mobile phase (water/methanol 95:5 v/v, flow rate 5 mL/min, λ = 254 nm). Retention time: 10 min.
Radiochemical purity and SA of [18F]11 were determined by analytical HPLC, using a Chromolith performance column, RP18-e (100 mm × 4.6 mm, Merck), and water and methanol containing 0.1% of formic acid as mobile phase (methanol content from 5% to 20% over 5 min, at 20% for 2 min, and up to 70% over 2 min, flow 3 mL/min, λ = 265 nm). Retention time: 4.0 min.
6-Bromo-7-(2-[18F]fluoroethyl)purine ([18F]12)
Hands-on reaction: analytical RCY, 17.2% ± 1.8% (n = 3). Automated synthesis: isolated RCY, 11% ± 3% (n = 7). [18F]12 was obtained with a radiochemical purity of 97% and SA of 180 GBq/μmol at end of synthesis. Precartridge purification with a C18 cartridge (Waters Sep-Pak classical): the product [18F]12 was released with 1 mL of 30% ethanol in water, and the resulting fraction was further diluted with 1 mL of water. HPLC purification was performed on a Zorbax ODS (C18, 5 μm, 250 mm × 9.4 mm, Agilent) at room temperature, using water and methanol containing 0.1% of formic acid as mobile phase (15% methanol for the first 5 min, and from 15% to 30% in 25 min, flow rate of 3 mL/min, λ = 254 nm). Retention time: 22.5 min.
Radiochemical purity and SA of [18F]12 were determined by analytical HPLC, using a Luna C18(2) column (3 μm, 50 mm × 4.6 mm, Phenomenex) and water and methanol containing 0.1% formic acid as mobile phase (methanol content 5% for the first 5 min, and from 5% to 50% over 25 min, flow 1 mL/min, λ = 265 nm). Retention time: 12.6 min.
6-Chloro-7-(5-[18F]fluoropentyl)purine ([18F]13)
Hands-on reaction: analytical RCY 19% ± 6% (n = 3). Automated synthesis: isolated RCY, 6% ± 1.5% (n = 3). [18F]13 was obtained with a radiochemical purity of 99% and SA of 2 GBq/μmol after sterile filtration. Precartridge purification with a C18 cartridge (Waters Sep-Pak light): the product [18F]13 was then released with 0.7 mL of pure ethanol, and the resulting fraction was further diluted with 1.3 mL of water. HPLC purification was performed on a Chromolith performance column, RP18-e (100 mm × 10 mm, Merck), using water and methanol containing 0.1% of formic acid as mobile phase (water/methanol 70:30 v/v, flow rate 5 mL/min, λ = 254 nm). Retention time: 7.1 min.
Radiochemical purity and SA of [18F]13 were determined by analytical HPLC, using a Chromolith performance column, RP18-e (100 mm × 4.6 mm, Merck), and water and methanol containing 0.1% of formic acid as mobile phase (methanol content from 20% to 36% over 6 min, and up to 60% over 3 min, flow 3 mL/min, λ = 265 nm). Retention time: 5.2 min.
Rate of GSH Conjugation under Nonenzymatic Conditions
Solutions of GSH (10 mM in 0.1 M phosphate buffer, pH = 7.4, 1 mL) were preincubated at 37 °C for 15 min. The reaction was initiated by adding a standard solution of 6-halopurine analogue (7–13 and 7m6BP, ∼1 mM in 0.1 M phosphate buffer, pH = 7.4) to each tube to achieve 50 μM as the final concentration. At designated intervals (from 5 min to 1–5 h depending on the reactivity of the purine analogues), aliquots from the resulting reaction mixtures were analyzed by HPLC, using a Chromolith performance column, RP18-e (100 mm × 4.6 mm, Merck), and water and methanol containing 0.1% trifluoroacetic acid (TFA) as mobile phase. The flow rate was 3 mL/min, and the UV absorbance detector was set at 265 nm. The pseudo-first-order rate constants were calculated by plotting the log of the concentration (mg/L) of the parent 6-halopurine against the time (min). Measurements were performed in duplicate. The results are expressed as the mean (h–1) ± standard deviation.
Kinetic Studies of GSH Conjugation in Brain Homogenates
Wild-type BALB/c mice were decapitated, and the brain was quickly removed and homogenized in 1 mL of cold phosphate buffer (0.1 M, pH 7.4). A solution of GSH (10 mM in 0.1 M phosphate buffer, pH = 7.4, 1 mL) was added. Aliquots of the resulting mixture (1 mL each) were preincubated at 37 °C for 15 min. The enzymatic GSH conjugation reaction was initiated by addition of a standard solution of 12 to each tube (∼1 mM in 0.1 M phosphate buffer, pH = 7.4) to achieve 50 μM as the final concentration. At predetermined intervals (0–15 min), the enzymatic reaction was stopped by addition of TFA (50 μL), and the resulting mixture was centrifuged (5 min, 13000 rpm). The supernatant was collected and analyzed by HPLC (Chromolith performance column, RP18-e, 100 mm × 4.6 mm, Merck), using water and methanol containing 0.1% TFA as mobile phase. The flow rate was 3 mL/min, and the UV absorbance detector was set at 265 nm. The pseudo-first-order constant was calculated by plotting the log of the concentration (mg/L) of 12 against time (min) and adjusting for brain homogenate concentration after subtracting knon. Determinations were made in triplicate. The results are expressed as the mean (h–1 g–1 mL–1) ± standard deviation.
Biodistribution Studies
Female wild-type BALB/c mice (20 g) received 1–2 MBq of the radiotracer as an intravenous injection. At designated intervals (5–90 min p.i.), the mice were anesthetized with isoflurane (5% mixed with medical air at a flow of 2 mL/min) and sacrificed. The organs of interest were removed and weighed, and the radioactivity was counted with a γ counter. The results were reported as the mean of percentage injected dose per gram (% ID/g) ± standard deviation (n = 3).
Plasma Metabolite Analysis in WT Mice
Female wild-type BALB/c mice (20 g) received 6–8 MBq of the tracer as intravenous injection. At designated intervals (15 and 60 min p.i.), the mice were anesthetized with isoflurane (5% mixed with medical air at a flow of 2 mL/min), and the blood was collected by cardiac puncture. The blood was centrifuged for 2 min at 13000 rpm. The plasma was separated, and the proteins were precipitated with TFA (25% v/v). The sample was diluted with water (1:1), and the proteins were removed by centrifugation (2 min at 13000 rpm). The supernatant was collected, a standard solution containing the reference compounds 12 and 17 (20 μL, 2.5 mM in 0.1 M phosphate buffer, pH = 7.4) was added, and the resulting mixture was analyzed by HPLC (radioactivity detector and UV detector 265 nm). A Chromolith performance RP18-e (100 mm × 4.6 mm, Merck) was used as the column, with a mobile phase consisting of water and methanol with 0.1% TFA (from 2% of methanol to 5% over 15 min), with a flow rate of 3 mL/min. Experiments were perfomed in triplicate. The recovery of radioactivity from the extraction of plasma was >95%.
Brain Metabolite Analysis in WT and mrp1 KO Mice
Wild-type and mrp1 KO FVB mice were decapitated 15 and 60 min after intravenous injection of [18F]12 (15–26 MBq in 0.2–0.3 mL saline; <0.29 nmol). The head was quickly immersed in liquid nitrogen, and the brain was removed. The brain was homogenized in 1 mL of cold phosphate buffer (0.1 M, pH 7.4). TFA (50 μL) was added to the homogenate, which was centrifuged (5 min, 13000 rpm, 4 °C). The supernatant was collected, diluted 1:1 with H2O, and analyzed by HPLC consisting of a pump (PU-2089 plus, JASCO, Tokyo, Japan), a multiwavelength detector (MD-2015 plus, JASCO), a sensitive positron detector (Ohyo Koken Kogyo Co. Ltd., Tokyo, Japan), and a Chromolith performance RP-18e (100 mm × 4.6 mm, Merk) column. The compounds were eluted with a mobile phase of water and methanol containing 0.1% TFA (from 2% of methanol to 10% over 15 min) at flow rate of 3 mL/min. In this HPLC system, the parent compound [18F]12 and the GSH conjugate [18F]17 showed a retention times of 9.5 and 13.2 min, respectively. The recovery of radioactivity from the extraction from brain was >95%.
Positron Emission Tomography Scanning Procedures
Four WT mice and four mrp1 KO mice were used for PET scanning with [18F]12. Body temperature was monitored using a rectal thermometer throughout PET scanning and was maintained within the normal range using a heating pad system. Scans were performed with an Inveon Dedicated PET system (Siemens Medical Solutions, Knoxville, TN, USA), which has a transaxial field of view of 10 cm and an axial field of view of 12.7 cm. The compound [18F]12 (3.6 MBq; <0.04 nmol) was administered as a 0.2 mL intravenous bolus injection to each mouse, which was maintained under isoflurane anesthesia during the scanning periods. Data were acquired by the animal tomograph for 60 min after injection in 20 frames divided as follows: 4 × 1 min, 8 × 2 min, and 8 × 5 min frames. Without attenuation correction, the data were reconstructed using Fourier rebinning and filtered back projection with a Hanning filter cutoff at the Nyquist frequency into images with a 128 × 128 matrix size and 3 times zoom, to give a voxel size of 0.26 × 0.26 × 0.8 mm3. After image reconstruction, volumes of interest were manually placed on a summed PET image (coronal view of the brain) and transferred to all of the frames of images to generate time–radioactivity concentration (Bq/mL) curves for whole brain, using the ASIPro VM software (CTI Concorde Microsystems, Knoxville, TN, USA).
Efflux Rate Constant in WT and mrp1 KO Mice
Brain radioactivity expressed as % injected dose per mL (% ID/mL) during the period of 15 to 60 min after injection of [18F]12 versus time was fitted to the monoexponential function by linear least-squares regression to obtain the efflux rate constant. The results were expressed as the mean (h–1) ± standard deviation (n = 4).
Acknowledgments
This research was supported by the European Community’s Seventh Framework Programme (FP7/2007–2013) under Grant Agreement Number 201380 (“Euripides”) (M.K., K.S., and E.G.). This work was undertaken at UCLH/UCL, which is funded in part by the Department of Health’s NIHR Biomedical Research Centres funding scheme.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
References
- Schinkel A. H. P-Glycoprotein, a gatekeeper in the blood-brain barrier. Adv. Drug Delivery Rev. 1999, 36, 179–194. [DOI] [PubMed] [Google Scholar]
- Schinkel A. H.; Jonker J. W. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family: an overview. Adv. Drug Delivery Rev. 2003, 55, 179–194. [DOI] [PubMed] [Google Scholar]
- Leslie E. M.; Deeley L. G.; Cole S. P. C. Multidrug resistance proteins: Role of P-glycoprotein, MRP1, MRP2, and BCRP (ABCG2) in tissue defense. Toxicol. Appl. Pharm. 2005, 204, 216–237. [DOI] [PubMed] [Google Scholar]
- Löscher W.; Potschka H. Role of drug efflux transporters in the brain for drug disposition and treatment of brain diseases. Prog. Neurobiol. 2005, 76, 22–76. [DOI] [PubMed] [Google Scholar]
- Bakos É.; Homolya L. Portrait of multifaceted transporter, the multidrug resistance-associated protein 1 (MRP1/ABCC1). Eur. J. Physiol. 2007, 453, 621–641. [DOI] [PubMed] [Google Scholar]
- DeGorter M. K.; Xia C. Q.; Yang J. J.; Kim R. B. Drug transporters in drug efficacy and toxicity. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 249–73. [DOI] [PubMed] [Google Scholar]
- Dallas S.; Miller D. S.; Bendayan R. Multidrug resistance-associated proteins: Expression and fuction in the central nervous system. Phamacol. Rev. 2006, 58, 140–161. [DOI] [PubMed] [Google Scholar]
- Deeley R. G.; Westlake C.; Cole S. P. C. Transmembrane transport of endo- and xenobiotics by mammalian ATP-binding cassette multidrug resistance proteins. Physiol. Rev. 2006, 86, 849–899. [DOI] [PubMed] [Google Scholar]
- Lee G.; Dallas S.; Hong M.; Bendayan R. Drug transporters in the central nervous system: Brain barriers and brain parenchyma considerations. Phamacol. Rev. 2001, 53, 569–59. [PubMed] [Google Scholar]
- Eckford P. D. W.; Sharom F. J. ABC efflux pump-based resistance to chemotherapy drugs. Chem. Rev. 2009, 109, 2989–3011. [DOI] [PubMed] [Google Scholar]
- Declèves X. A.; Regina A.; Laplanche J.-L.; Roux F.; Boval B.; Launay J.-M.; Scherrmann J.-M. Functional expression of p-glycoprotein and multidrug resistance-associated protein (mrp1) in primary cultures of rat astrocytes. J. Neurosci. Res. 2000, 60, 594–601. [DOI] [PubMed] [Google Scholar]
- Minich T.; Riemer J.; Schulz J. B.; Wielinga P.; Wijnholds J.; Dringen R. The multidrug resistance protein 1 (Mrp1), but not Mrp5, mediates export of glutathione and glutathione disulfide from brain astrocytes. J. Neurochem. 2006, 97, 373–384. [DOI] [PubMed] [Google Scholar]
- Löscher W.; Potschka H. Role of multidrug transporters in pharmacoresistance to antiepileptic drugs. J. Pharmacol. Exp. Ther. 2002, 301, 7–14. [DOI] [PubMed] [Google Scholar]
- Munoz M.; Henderson M.; Haber M.; Norris M. Role of the MRP1/ABCC1 multidrug transporter protein in cancer. Life 2007, 59, 752–757. [DOI] [PubMed] [Google Scholar]
- Fletcher J. I.; Haber M.; Henderson M. J.; Norris M. D. ABC transporters in cancer: More than just drug efflux pumps. Nat. Rev. Cancer 2010, 10, 147–156. [DOI] [PubMed] [Google Scholar]
- Borst P.; Evers R.; Kool M.; Wijnholds J. A family of drug transporters: The multidrug resistance-associated proteins. J. Natl. Cancer Inst. 2000, 92, 1295–1302. [DOI] [PubMed] [Google Scholar]
- Sisodiya S. M.; Lin W.-R.; Harding B. N.; Squier M. V.; Thom M. Drug resistance in epilepsy: Expression of drug resistance proteins in common causes of refractory epilepsy. Brain 2002, 125, 22–31. [DOI] [PubMed] [Google Scholar]
- Kubota H.; Ishihara H.; Langmannd T.; Schmitz G.; Stieger B.; Wieser H.-G.; Yonekawa Y.; Frei K. Distribution and functional activity of P-glycoprotein and multidrug resistance-associated proteins in human brain microvascular endothelial cells in hippocampal sclerosis. Epilepsy Res. 2006, 68, 213–228. [DOI] [PubMed] [Google Scholar]
- Luna-Tortós C.; Fedrowitz M.; Löscher W. Evaluation of transport of common antiepileptic drugs by human multidrug resistance-associated proteins (MRP1, 2 and 5) that are overexpressed in pharmacoresistant epilepsy. Neuropharmacology 2010, 58, 1019–1032. [DOI] [PubMed] [Google Scholar]
- Chen Y.-H.; Wang C.-C.; Xiao X.; Wei L.; Xu G. Multidrug resistance-associated protein 1 decreases the concentrations of antiepileptic drugs in cortical extracellular fluid in amygdale kindling rats. Acta Pharmacol. Sin. 2013, 34, 473–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sultana R.; Butterfield D. A. Oxidatively modified GST and MRP1 in Alzheimer’s disease brain: Implications for accumulation of reactive lipid peroxidation products. Neurochem. Res. 2004, 29, 2215–2220. [DOI] [PubMed] [Google Scholar]
- Krohn M.; Lange C.; Hofrichter J.; Scheffler K.; Stenzel J.; Steffen J.; Schumacher T.; Brüning T.; Plath A.-S.; Alfen F.; Schmidt A.; Winter F.; Rateitschak K.; Wree A.; Gsponer J.; Walker L. C.; Pahnke J. Cerebral amyloid-β proteostasis is regulated by the membrane transport protein ABCC1 in mice. J. Clin. Invest. 2011, 121, 3924–3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abuznait A. L.; Kaddoumi A. Role of ABC transporters in the pathogenesis of Alzheimer’s disease. ACS Chem. Neurosci. 2012, 3, 820–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamura T.; Kikuchi T.; Fukushi K.; Arano Y.; Irie T. A novel noninvasive method for assessing glutathione-conjugate efflux systems in the brain. Bioorg. Med. Chem. 2007, 15, 3127–3133. [DOI] [PubMed] [Google Scholar]
- Okamura T.; Kikuchi T.; Okada M.; Toramatsu C.; Fukushi K.; Takei M.; Irie T. Noninvasive and quantitative assessment of the function of multidrug resistance-associated protein 1 in the living brain. J. Cereb. Blood Flow Metab. 2009, 29, 504–511. [DOI] [PubMed] [Google Scholar]
- Okamura T.; Kikuchi T.; Fukushi K.; Irie T. Reactivity of 6-halopurine analogs with glutathione as a radiotracer for assessing function of multidrug resistance-associated protein 1. J. Med. Chem. 2009, 52, 7284–7288. [DOI] [PubMed] [Google Scholar]
- Galante E.; Schoultz B. W.; Koepp M.; Årstad E. Chelator-accelerated one-pot ‘click’ labeling of small molecule tracers with 2-[18F]fluoroethyl azide. Molecules 2013, 18, 5335–5347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan R.; Sander K.; Galante E.; Rajkumar V.; Badar A.; Robson M.; El-Emir E.; Lythgoe M. F.; Pedley R. B.; Årstad E. A one-pot three-component radiochemical reaction for rapid assembly of 125I-labeled molecular probes. J. Am. Chem. Soc. 2013, 135, 703–709. [DOI] [PubMed] [Google Scholar]
- Kjellberg J.; Johansson N. G. Charaterization of N7 and N9 alkylated purine by 1H and 13C NMR. Tetrahedron 1986, 42, 6541–6544. [Google Scholar]
- Manuprasad B. K.; Murthy V. S.; Shashikanth S.; Rakshith D.; Satish S.; Raveesha K. A. Design, synthesis and antibacterial activity of novel 1,3-thiazolidine purine nucleosides. Pharma Chem. 2011, 3, 45–55. [Google Scholar]
- Hwang I. Y.; Elfarra A. A. Detection and mechanisms of formation of S-(6-purinyl)glutathione and 6-mercaptopurine in rats given 6-chloropurine. J. Pharmacol. Exp. Ther. 1993, 264, 41–46. [PubMed] [Google Scholar]
- Lash L. H.; Shivnani A.; Mai J.; Chinnaiyan P.; Krause R. J.; Elfarra A. A. Renal cellular transport, metabolism, and cytotoxicity of S-(6-purinyl)glutathione, a prodrug of 6-mercaptopurine, and analogues. Biochem. Pharmacol. 1997, 54, 1341–1349. [DOI] [PubMed] [Google Scholar]
- Backos D. S.; Franklin C. C.; Reigan P. The role of glutathione in brain tumor drug resistance. Biochem. Pharmacol. 2012, 83, 1005–1012. [DOI] [PubMed] [Google Scholar]
- Lieberman M. W.; Barrios R.; Carter B. Z.; Habib G. M.; Lebovitz R. M.; Rajagopalan S.; Sepulveda A. R.; Shi Z. Z.; Wan D. F. Am. J. Pathol. 1995, 147, 1175–1185. [PMC free article] [PubMed] [Google Scholar]
- Karp D. R.; Shimooku K.; Lipsky P. E. Expression of γ-glutamyl transpeptidase protects ramos B cells from oxidation-induced cell death. J. Biol. Chem. 2001, 276, 3798–3804. [DOI] [PubMed] [Google Scholar]