

Abstract
The mechanism whereby RNA is translocated by the single subunit viral RNA-dependent RNA polymerases is not yet understood. These enzymes lack homologs of the “O-helix” structures and associated fingers domain movements thought to be responsible for translocation in many DNA-templated polymerases. The structures of multiple picornavirus polymerase elongation complexes suggest that these enzymes use a different molecular mechanism where translocation is not strongly coupled to the opening of the active site following catalysis. Here we present the 2.0–2.6 Å resolution crystal structures and biochemical data for twelve poliovirus polymerase mutants that together show how proper enzyme function and translocation activity requires conformational flexibility of a loop sequence in the palm domain B-motif. Within the loop, the Ser288–Gly289–Cys290 sequence is shown to play a major role in the catalytic cycle based on RNA binding, processive elongation activity, and single nucleotide incorporation assays. The structures show that Ser288 forms a key hydrogen bond with Asp238, the backbone flexibility of Gly289 is require for translocation competency, and Cys290 modulates the overall elongation activity of the enzyme. Some conformations of the loop represent likely intermediates on the way to forming the catalytically competent closed active site, while others are consistent with a role in promoting translocation of the nascent base pair out of the active site. The loop structure and key residues surrounding it are highly conserved, suggesting the structural dynamics we observe in poliovirus 3Dpol are a common feature of viral RNA-dependent RNA polymerases.
Keywords: polymerase, translocation, RdRP, RNA, poliovirus
Introduction
The incorporation of nucleotides onto a nucleic acid chain by polymerase enzymes is a complex process requiring more than a simple phosphoryl transfer reaction. Kinetic and thermodynamic studies of polymerases from a wide range of species reveal a common mechanism that is broadly comprised of five steps: initial NTP selection by binding in or near the active site, a conformational change to position the NTP for catalysis, closure of the active site for the phosphodiester bond formation step, a re-opening of the active site, and finally translocation of the nucleic acid to reset the enzyme for the next catalytic cycle. The latter two steps are often coupled both thermodynamically and structurally, with pyrophosphate release often being thought to be a trigger point for the conformational changes that drive translocation1; 2.
Among DNA-templated polymerases, these steps are well illustrated by structures of the bacteriophage T7 DNA-dependent RNA polymerase (T7 RNAP)1; 3; 4; 5 and the T. aquaticus DNA-dependent DNA polymerase (Taq)6 that have been captured at various stages of the catalytic cycle. These structures show initial NTP binding in the pre-insertion site that is followed by a 20–25° rotation of the fingers domain B′-motif “O-helix” that serves to reposition the nucleotide over the active site RRM motif for catalysis4. In the process, a conserved tyrosine residue from the O-helix (Y639 in T7 RNAP and Y671 in Taq) becomes stacked on the newly formed basepair. This direct contact is then thought to mediate translocation via the tyrosine pushing the nascent basepair out of the active site when the O-helix reverses its movement and the fingers domain returns to the open conformation after catalysis.
Less is known about the structural transitions that take place within RNA-templated polymerases during the catalytic cycle. This group of polymerases includes telomerases, reverse transcriptases, and the viral RNA-dependent RNA (RdRP) family of small single subunit polymerases. The RdRPs retain the common polymerase catalytic mechanism and active site geometry, but sequence and structure comparisons show they utilize different molecular movements for active site closure and translocation. Viral RdRP structures show conservation of an encircled active site topology7 where a direct contact between the fingers and thumb domain precludes the swinging movement of the fingers domain that is associated with active site closure in other polymerases. Consistent with this, structures of the poliovirus polymerase elongation complex trapped at various points during the catalytic cycle show that the viral RdRPs close their active sites via a unique structural transition in the palm domain8. The RdRPs also differ from other polymerases in that they do not contain the B′-motif helix located above the active site and are thus missing the conserved tyrosine residue that mediates translocation in the DNA-templated enzymes. Interestingly, the poliovirus polymerase elongation complex structures also showed that the enzyme can re-open the active site after catalysis without translocation8, resulting in a unique structural state that has not been captured in other polymerases where these two events appear to be tightly coupled. A comparison of several viral RdRP structures have shown that a loop within the B-motif exhibits significant structural variability and this may expose a novel target site for the development of antiviral polymerase inhibitors9. Based on its flexibility and proximity to the template RNA strand, this loop is also postulated to play an important role in modulating polymerase activity, perhaps through effects on translocation, but direct evidence of this has not yet been obtained.
In the course of investigating low ionic strength conditions for maintaining 3Dpol crystals grown without RNA, we discovered electron density evidence for an alternate conformation of this short loop that connects the base of the middle finger to the major α-helix of the B-motif in the palm domain (Figure 1A). Comprised of residues 288–292, the loop is in direct proximity to the active site of the polymerase and lies immediately adjacent to the templating RNA strand in the poliovirus 3Dpol-RNA elongation complex. This loop is highly conserved as part of the RdRP B-motif that differs structurally from the B′-motif of the DNA-templated polymerases and we hypothesized that the loop movement could play a role in mediating RNA translocation following catalysis. To address this, we generated a set of twelve mutations in the loop itself that would modulate the interactions it was making with the rest of the polymerase, and here we report the structural and biochemical analysis of these mutants. Our crystal structures indicate that the loop can exist in two stable conformations and biochemical data show that restricting the inter-conversion between these conformations affects RNA binding and overall polymerase activity, with some mutations resulting in translocation deficient polymerases.
Figure 1.

Conformations of the translocation loop. A. Structure of poliovirus 3Dpol showing the location of the loop in cyan with spheres for Cα atoms. The individual fingers are colored as in our prior publications10 (palm in grey, thumb in blue, index finger in green, middle finger in orange, ring finger in yellow, and pinky finger in red) B. Detailed view of the hydrophobic pocket into which residue 290, shown with sphere for its Cβ atom, is inserted when the loop is in the In conformation. C. Comparison of the backbone traces of 13 loop structures showing they adopt either an in (tan) or out (cyan) conformation, except for a few whose structure is perturbed by a dimethylarsenic adduct on Cys290 (black, see text).
Results
Crystallization and Structure Determination
The twelve separate 3Dpol proteins with mutations in the 288–292 loop were generated, expressed, and purified as previously described10. Eleven of these crystallized to provide structures in both the presence and absence of bound GTP and their structures were refined to typical resolution limits of 2.2–2.3 Å with Rfree values of 23–25% (Table I). The overall structure of each loop mutant 3Dpol does not change significantly as compared to the wildtype polymerase and a maximum likelihood multiple structure alignment performed by the program Theseus11; 12 indicates that all changes resulting from each mutation are isolated to movements within and around residues 288–292 (Figure 1C). The binding of GTP does not significantly change the structure of the mutants as both the apo-structures and NTP complexes have essentially identical backbone traces with the GTP binding to the same site as in the wildtype enzyme (PDB entry 1RA710; 13). The exception to this is the weakly ordered and chemically modified S288A loop that moves to the In conformation due to a steric clash with the bound GTP. Composite simulated annealing omit maps of all the structures are shown in Figure 2.
Table I.
Summary of Structural and Biochemical Data
| Mutant | Class | Loop Conformation | Nucleotide Additions | Activity (%) | RNA Affinity (Kd) | Structure R/Rfree (%) | Structure Resolution | PDB Code | Affected Step |
|---|---|---|---|---|---|---|---|---|---|
| 3Dpol | - | In/up | All Three | 100 ± 30 | 1.71 ± 0.06 | 24.3/25.6 | 2.0 Å | 1RA6 | - |
| C290I | I+ | In/down | All Three | 170 ± 30 | 1.80 ± 0.10 | 22.6/25.1 | 2.2 Å | 4NLO | Increased rate of reset step |
| C290V | I+ | In/down | All Three | 320 ± 60 | 1.56 ± 0.05 | 22.2/24.8 | 2.2 Å | 4NLP | Increased rate of reset step |
| C290F | I− | Out/down | All Three | 39 ± 4 | 1.42 ± 0.09 | 22.2/24.5 | 2.3 Å | 4NLQ | Decreased rate of reset step |
| C290S | I− | In/up | All Three | 20 ± 4 | 1.21 ± 0.04 | 22.7/24.0 | 2.0 Å | 4NLR | Decreased rate of reset step |
| S288A | I− | Distorted | All Three | 13 ± 5 | 4.0 ± 0.3 | 23.4/25.6 | 2.0 Å | 4NLS | Decreased rate of NTP positioning |
| S291P | I− | Distorted | All Three | 2 ± 1 | 2.39 ± 0.05 | 22.2/25.5 | 2.5 Å | 4NLT | Decreased rate of translocation Decreased rate of reset |
| G289A | II | Distorted | Only One | −0.5 ± 2 | 2.80 ± 0.10 | 22.5/24.6 | 2.1 Å | 4NLU | Prevents Translocation step Decreased rate of NTP positioning |
| G289A/C290F | II | In/up | Only One | 0.4 ± 0.1 | 3.25 ± 0.08 | 21.7/24.0 | 2.3 Å | 4NLV | Prevents Translocation Decreased rate of NTP positioning |
| G289A/C290I | II | In/up | Only One | 0.12 ± .05 | 2.40 ± 0.10 | 22.2/24.2 | 2.1 Å | 4NLW | Prevents Translocation Decreased rate of NTP positioning |
| G289A/C290V | II | In/up | Only One | 1.8 ± 0.7 | 3.15 ± 0.08 | 20.8/23.6 | 2.6 Å | 4NLX | Prevents Translocation Decreased rate of NTP positioning |
| C290E | III | Out/down | None | 0.9 ± 0.7 | 8 ± 1 | 21.5/23.9 | 2.3 Å | 4NLY | Prevents RNA binding |
| C290D | III | NA | None | 1.0 ± 0.9 | 4.2 ± 0.6 | NA | NA | NA | Prevents RNA binding |
NA – Not available because C290D mutant did not crystallize
Figure 2.

Composite simulated-anneal omit electron density maps (1000K) contoured at 1.6σ showing the quality of the crystallographic data in the loop region. The structures were initially solved using molecular replacement with a search model missing the entire loop (residues 287–293), and the resolution limits for each structure are listed in Table I. DMA refers to the dimethylarsenic adduct that is sometimes found on Cys290 due to the crystallization conditions 10.
The Loop Conformations
An overview of all eleven polymerase structures shows that the 288–292 loop adopts three distinct conformations that have been designated in/up, in/down and out/down (Figure 3). The separate conformations are distinguished first by the location of residue 290 that can either be buried in a hydrophobic pocket located directly behind the loop or come out of the pocket to be solvent exposed above the palm. Second, the conformations can also be distinguished by the orientation of the Ser288 side chain, which can either point up toward the ring finger or down toward the active site.
Figure 3.
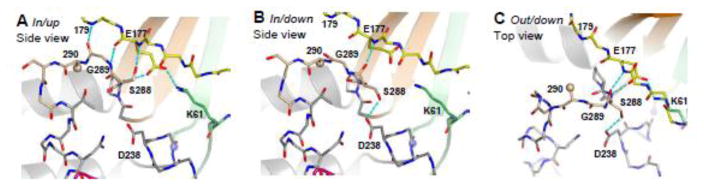
Detailed views of the three stable conformations observed for the 288–292 loop. The loop is located at the base of the middle finger motif (orange β-strands) and forms several hydrogen bonds (cyan) with the base of the ring finger (yellow). A. The In/up conformation involves four hydrogen bonds to the ring finger and an Asp177–Lys61 salt bridge between the ring and index finger. B. The in/down conformation is characterized by a Gly289 dependent rotation of Ser288 whose sidechain now interacts with the NTP-binding Asp238 residue, resulting in the loss of three hydrogen bonds to the ring finger and disruption of the Asp177–Lys61 salt bridge. C. The out/down conformation has Cys290 flipped out of its hydrophobic pocket and as a result the loop protrudes out into the template RNA channel.
In/up
The in/up conformation is identical to that found in the native 3Dpol structure in the absence of bound RNA10 where residue 290 is buried in the hydrophobic pocket made up of Val154, Met187, Tyr267, Val268 and Pro287 from the palm and fingers domains (Figure 1B). This conformation is further characterized by four hydrogen bonds between the 288–292 loop and the base of the ring finger; two between the Gly289 backbone and residues 177 and 179 and two between Ser288 and residue 177 (Figure 3A). The side chain of Ser288 points up and away from the active site to form a hydrogen bond with the amide nitrogen of Asp177. It is also important to note that Asp177 forms a total of three hydrogen bonds with the B-motif loop and a well-ordered salt bridge with Lys61. Substitution of Lys61 with leucine results in a catalytically inactive polymerase14, possibly because it removes this salt bridge.
In/down
The second conformation of the loop, in/down, has the Ser288 hydroxyl flipped so that it is now pointing down toward the active site to form a hydrogen bond with Asp238, while residue 290 remains buried in the hydrophobic pocket (Figure 3B). Comparisons of the in/up and in/down conformations show that the backbone torsion angles of residues 288 and 289, in particular the ϕ angle of glycine 289, are significantly different. One interesting observation is that when the loop is in the in/down conformation, the carbonyl of Ser288 replaces the side chain’s hydrogen bond with the Asp177 backbone, which then allows the serine hydroxyl to point down toward the active site and interact with Asp238. Consequently, the loop now forms only a single hydrogen bond with the base of the ring finger, as compared to the four hydrogen bonds seen in the in/up conformation. In addition, the Asp177 side chain moves such that the salt bridge between it and Lys61 on the index finger is broken.
Out/down
The final conformation of the loop is designated out/down and is characterized by a major ~5 Å displacement of residue 290 that moves it out of the hydrophobic pocket (Figure 3C). Like in/down, the Ser288 side chain forms a hydrogen bond with Asp238, but unlike the in/down conformation, the salt bridge between Asp177 and Lys61 is restored. Due to the displacement of the loop, Gly289 no longer forms hydrogen bonds with the ring finger and only two hydrogen bonds between the loop and the ring finger are retained. Similar to the transitions to the in/down conformation, the backbone torsion angles of 288 and 289 have been significantly perturbed in out/down as compared to the in/up conformation.
Distorted loop structures
The above observations hold true for the majority of the structures solved, but the presence of a cysteine at residue 290 sometimes leads to a crystallization artifact when the free sulfhydryl is covalently modified by a dimethy-arsenic adduct. This was previously observed at multiple cysteine residues in our initial 3Dpol structure and is the result of the cacodylic acid used in the crystallization buffer10; 15. Formation of the adduct results in a distortion of the structure in three of the 11 mutant structures presented here (S288A, G289A and S291P; Figure 2F–H). The distortion itself is probably not biologically relevant, but the fact that the adduct is formed suggests the loop is flexible enough to allow for the transient exposure of the cysteine 290 residue during crystallization.
Three biochemical categories of Loop mutants
To analyze the functional significance of the loop, each mutant polymerase was subjected to three separate biochemical assays (Table I). The first was a fluorescence polarization based RNA-binding assay to determine the affinity of each mutant for a small RNA-hairpin 16. The majority of mutants did not significantly affect RNA binding, but C290D and C290E did result in weakened affinities with 4-fold and 8-fold increases in Kd, respectively. The second assay was a classical oligo-dT/polyA extension assay that measures the bulk incorporation of α-32P-UMP nucleotides upon primer extensions with 50–300 nucleotide long templates. Activities are reported as a percentage of the wildtype control activity that has been set to 100%. Due to the nature of this extension assay, higher activity is generally associated with polymerases that can processively elongate long stretches of RNA in a short amount of time, which is due to minimizing the number of slow initiation events. Last, we investigated the ability of each mutant to undergo three successive rounds of nucleotide addition in a stepwise nucleotide incorporation assay using a RNA-hairpin substrate like that used in the RNA binding assay.
The resulting data have been used to divide the mutant polymerases into three biochemical activity classes (Table I). Class I mutants retain the ability to undergo three rounds of nucleotide addition and have activities in the polyA elongation assay that differ significantly from that of the wildtype enzyme. This class has been further subdivided into hyperactive Class I+ (>100%) and hypoactive Class I− (<100%) mutants. Class II mutants are able to undergo only a single round of nucleotide addition, demonstrating that these polymerases retain the ability to bind and position an NTP over the active site, but have lost the ability to rapidly carry out subsequent rounds of processive nucleotide incorporation. Last, Class III mutants have lost the ability to add even a single nucleotide, suggesting that these mutations have significantly disrupted the structure and function of the active site.
Ser288 Mutations
We first examined the effects of an alanine mutation at Ser288, a residue that adopts two different conformations so that its side chain can form hydrogen bonds with either the backbone amide nitrogen of Asp177 (/up) or with the side chain of Asp238 (/down) (Figure 3). The S288A mutation retained the ability to incorporate multiple nucleotides (Figure 4B), but the 32P-UMP incorporation activity was reduced 15-fold, indicating that the hydrogen bonding to the ring finger and Asp238 is important but not essential for proper polymerase function. The crystal structure of the S288A loop mutant is complicated by the covalent modification of Cys290 with a dimethyl-arsenic adduct and the remaining electron density of the loop is very weak, precluding identification of a preferred loop conformation and suggesting it may be more flexible than in the wildtype and the other mutant polymerases (Figure 2F).
Figure 4.

Single nucleotide RNA elongation by the various loop mutant 3Dpol enzymes after a 30 minute incubation. The polymerases were incubated with a short 8-bp hairpin RNA that included a 6-nt templating region coding for the sequential addition of G, U, and C, as shown by the first three lanes for each mutant. The last lane shows that elongation does not occur if the first nucleotide (G) is omitted from the reaction. The mutants are grouped into four classes based on these data and processive elongation data from the polyA templated reactions (see Table I). Class I+ mutants are hyperactive, Class I− mutants are hypoactive, Class II mutants bind RNA normally but only add a single nucleotide, and Class III mutants show no activity and exhibit reduced RNA binding affinity.
Gly289 Mutations
Comparison of all the 3Dpol structures indicates that both Ser288 and Gly289 adopt significantly different backbone torsion angles in the separate loop conformations. We hypothesized that the ability of the loop to adopt both the in/down and out/down conformation was dependent upon the flexibility a glycine at residue 289. To further examine this we mutated Gly289 to alanine, a residue that limits backbone flexibility while minimizing the potential for adverse side chain interactions. The G289A mutation was also generated in combination with the Class I+ and I− mutants at residue 290. In all cases, the G289A mutation completely abolishes processive elongation activity and not even the hyperactive isoleucine or valine substitutions at residue 290 could compensate for the effects of the alanine (Table I). Interestingly, the single nucleotide addition assay results indicate that all G289A mutants can incorporate the first nucleotide, albeit at reduced efficiency, but then fail to efficiently carry out additional rounds of catalysis (Figure 4C). The structures show that the G289A mutation locks the polymerase in the in/up conformation, even when combined with mutations that by themselves have either the in/down (C290I or C290V) or out/down (C290F) conformation (Figure 2I–K). Like S288A and S291P, the G289A alone mutant is modified by dimethyl-arsenic adduct and the conformation of the loop has been significantly distorted as a result (Figure 2H).
Cys290 Mutations
The Cys290 residue is buried in a hydrophobic pocket when the loop adopts the in conformation, but is largely solvent exposed when the loop has the out conformation. We therefore probed its function with mutations that would energetically stabilize either state. Mutations to small hydrophobic side chains such as isoleucine and valine resulted in hyperactive polymerases with ~2- and ~3-fold increases in product formation in the poly-A template assay. This suggests that the mutant polymerases may be faster and/or that they form more stable or processive elongation complexes than the wildtype enzyme (Table I). These non-polar mutants also show efficient stepwise elongation of the RNA hairpin substrate (Figure 4A) and their structures consistently show the in/down conformation where residue 290 is buried in the hydrophobic pocket, Ser288 is down, and the Asp177 – Lys61 salt bridge remains intact (Figure 2B&C). In contrast, introducing a small polar residue with a C290S mutation results in a structure that is essentially identical to native 3Dpol with an in/up loop conformation (Figure 2E), but whose elongation activity is reduced 5-fold (Table I). Increasing the polarity of residue 290 thus decreases activity while more hydrophobic residues increase activity.
Three mutations that were not expected to be structurally compatible with the loop in conformation severely impaired 3Dpol function. First, the large hydrophobic substitution to phenylalanine at residue 290 leads to a ~2.5-fold decrease in product formation activity that is likely due to a kinetically slow step of burying the large and conformationally restricted phenylalanine residue into the hydrophobic pocket. Consistent with this, the structure of C290F shows an out/down conformation for the loop. Second, negatively charged side chains are unlikely to be buried in the hydrophobic pocket and would thus prevent the formation of either the in/up or in/down loop conformations, and the structure of C290E reveals an out/down conformation that places the negatively charged side chain in the polar environment above the palm (Figure 2L). Several attempts to crystallize C290D failed and thus the structure of this mutant is unknown. Both C290E and C290D mutations also completely abolish both the processive elongation activity and the ability of the enzyme to add even a single nucleotide (Figure 4D). These mutants also have reduced RNA affinity, suggesting there is likely a clash between a loop that is in the out conformation and the bound RNA.
Ser291 Mutations
At the final residue of the loop, Ser291, a proline mutation has previously been shown to significantly reduce viral growth via a dominant negative mutation that interfered with wildtype virus replication17. In our in vitro assays this mutant resulted in a 50-fold reduction in elongation activity. The mutant polymerase does incorporate multiple nucleotides in the stepwise assay, but it does so slowly and inefficiently, leaving behind a significant amount of starting material (Figure 4B). Structurally, this loop mutant appears to adopt the out/down conformation, but the loop has been covalently modified by dimethyl-arsenic during crystallization and its true conformation is unknown (Figure 2G).
Discussion
Based on an initial crystallographic indication of structural flexibility in a protein loop that lies adjacent to the poliovirus polymerase active site and contacts the RNA template strand in the elongation complex structure, we hypothesized that this structural element may play a key role in the polymerase catalytic cycle. We designed a dozen mutations to probe the function of specific residues within the loop, solved their structures, and examined their biochemical activities. The results indicate that this loop can exist in two dominant orientations and that mutations which restrict loop flexibility result in translocation deficient polymerases. Together, the results lead us to propose that conformational changes within this loop play a role in mediating RNA translocation after catalysis in picornaviral RNA-dependent RNA polymerases.
The structures of eleven different mutants within residues 288–292 suggests that this loop can exist in two stable conformations where it is either tucked into a pocket at the junction of the palm and fingers domains (in) or flipped out toward the active site in the palm domain (out). Closer inspection of the in conformations reveals two orientations of Ser288 whose side chain can flip ≈150° through a combination of a rotamer change and backbone movement. This allows the serine hydroxyl group to point up and away from the active site to interact with the ring finger motif (in/up) or down toward the active site to form a hydrogen bond with Asp238 (in/down). The in/down conformation is homologous to that observed in the apo 3Dpol structures from rhinovirus18 and coxsackievirus19; 20 and in the catalytically active closed conformation of the poliovirus elongation complex8; 21. The out conformation is the result of a ~5 Å movement of residue 290 out of the hydrophobic pocket and towards the active site that is always accompanied by Ser288 in a down conformation and has therefore been designated as out/down. When the conformational change to out/down is modeled into the poliovirus 3Dpol elongation complex structure8 there is a clear steric clash between the loop and the backbone of the RNA template strand (Figure 5A). This suggests that the out conformation cannot coexist with RNA in a catalytically competent register and that the observed structural transitions within the loop may be involved in facilitating the movement of RNA through the active site.
Figure 5.

A. Structure of the RNA from the poliovirus elongation complex8; 21 superimposed on the structures of several mutants showing the in and out conformations of the residue 288–292 loop. The side view shows how the ribose of the −1 position template strand nucleotide (magenta) makes direct contact with the loop in the In conformation, but has a clear steric clash when the loop is in the Out conformation. The top view shows the direction and magnitude of the loop movement relative to the active site, nascent base pair, and ddCTP base paired to the templating +1 site nucleotide. B. Kinetic scheme of the polymerase elongation cycle illustrating the model for the different steps involving conformational changes in the loop. The EC states reflect the six state cycle based on the picornaviral polymerase elongation complex structures8; 21. The flip of Ser288 from up to down is a pre-catalytic transition involved in NTP repositioning while the movement of the loop from In to Out is associated with the post-catalysis translocation step. Our finding that hydrophobic residues at position 290 increase polymerase activity while polar residues reduce activity further suggests that the viral RdRPs may have a distinct 6th step following pyrophosphate release whereby residue 290 is reinserted into its pocket, returning the loop to the in/up conformation and resetting the active site for the next round of catalysis.
Kinetic studies of 3Dpol have shown that, like in other polymerases, the catalytic cycle can be broadly divided into five separate steps22. Within these steps, there are typically two conformational changes that flank phosphoryl transfer; the first positions the NTP over the active site, and the second translocates the nucleic acid following catalysis. The viral RNA-dependent RNA polymerases have fingers domains that reach across the active site and are tethered to their thumb domains, precluding large-scale fingers domain movements that could act to reposition NTPs for catalysis. These enzymes instead use more subtle conformational changes to close their active sites via a novel structural rearrangement of Motifs A and D that are part of the palm domain8; 23. The viral RdRPs lack a structural analog of Tyr639 and the fingers domain O-helix motif, making it likely that the molecular details of the translocation mechanism differs considerably from that of DNA templated polymerases.
Based on our data, we present a model for the structural changes during the poliovirus 3Dpol catalytic cycle wherein the alternate conformations of the 288–292 loop are important for NTP positioning and translocation (Figure 5B). These conformations also represent plausible intermediates along the six state catalytic cycle observed in the poliovirus elongation complex (EC) structures8. The catalytic cycle starts with a loop in the in/up conformation that is found in the original poliovirus 3Dpol apo structure10, those of all the 3Dpol-NTP complexes13, and the EC state 1 structures of the elongation complexes8; 21. Next, Ser288 flips down towards the active site, resulting in the in/down conformation seen in our C290I and C290V single mutants. The Ser 288 flip from up to down results in the formation of a hydrogen bond between Ser288 and Asp238 that competes with and weakens the existing hydrogen bond between Asp238 and Asn297, which must be broken to accommodate the NTP in the active site of the EC structure. The transition from in/up to in/down can be interpreted as an intermediate step between the initial NTP binding event (EC state 2) and the NTP dropping into the catalytically competent closed active site (EC state 3) where the newly formed Ser288–Asp238 hydrogen bond plays a key role in rearranging Motif A for catalysis. The Ser288 flip to down is accompanied by the loss of four hydrogen bonds between the base of the ring finger and the loop itself as well as the disruption of the salt bridge between Asp177 and Lys61 (Figure 3A&B). The release of these energetic constraints reflects key changes in the active site hydrogen bonding geometry that set the stage of catalysis in the EC state 3 structure8. After catalysis, the RNA must be translocated in order to reset the active site for the next round of nucleotide addition. We propose that the transition from the in to the out loop conformation is required for this step, and that this transition requires the backbone flexibility of the Gly289 residue that is absolutely conserved in RdRPs. The G289A mutation limits backbone flexibility and structurally locks the loop in the in conformation, resulting in translocation deficient polymerases. Following translocation, the loop must then move from the out/down conformation back to the in/up to reset the enzyme active site for the next round of catalysis.
The data reported herein supports this model in that each class of mutants differentially compromises the rate at separate molecular events, leading to the observed effects on single nucleotide incorporation and overall activity. Class I mutants have native like properties in that they incorporate all three nucleotides in the single nucleotide incorporation assay, yet they display a wide range of activities in the poly-A templated elongation assay. Class II mutants are translocation deficient because Gly289 is replaced with alanine, limiting backbone flexibility within the loop and resulting in unambiguous electron density showing loops structurally locked in the in/up conformation. This locking of the loop is a dominant effect in that the hyperactive C290I and C290V mutants lose processive elongation activity when combined with G289A. Class III mutants replace Cys290 with negatively charged amino acids that cannot be buried in the hydrophobic pocket and are characterized by their inability to undergo even a single round of nucleotide addition.
There is also evidence for dynamics of the poliovirus polymerase loop from NMR studies of 3Dpol-RNA complexes in the presence and absence of added NTPs. In a 13C study of methionines, Yang et al.24 observed that the chemical shift of the Met187 methyl resonance changes significantly upon RNA binding and then more subtly upon nucleotide addition to form a non-productive catalytic complex. Met187 forms one side of the binding pocket for Cys290 and in light of our structural data the major change in chemical shift upon RNA binding probably reflects a burial of the loop into the pocket. The more subtle change upon NTP addition may be due to closure of the active site in which Ser288 flips from up to down with only minor changes in the positioning of Cys290. It is also noteworthy that Met286, which is diagonally opposite of the pocket from Met187, does not produce an observable NMR resonance, perhaps because of protein dynamics that result in Met286 interconverting between multiple structural states in an intermediate chemical shift exchange regime.
Conservation Among RdRPs
The 288–292 loop is also highly conserved among viral RdRPs at both the sequence and structural level (Table II) and we anticipate that our findings pertain to a wide range of positive strand RNA virus polymerases. As was well described in a recent Perspective article9, the structures of RdRPs from picornaviridae18; 19; 20; 25; 26; 27; 28, caliciviridae29; 30, flaviviridae31; 32; 33; 34; 35; 36 and double-stranded RNA viruses37; 38 clearly show that the loop is flexible and that the central residue of the loop is positioned for burial in a pocket between the finger and palm domains. In the picornaviral polymerases this is generally a cysteine that is buried in a hydrophobic pocket, while other RdRPs tend to have a small hydrophobic residue that is buried in a hydrophobic pocket. Interestingly, the bovine viral diarrhea virus polymerase has a polar glutamine at the central position, and its pocket contains complementary polar glutamate and arginine residues. Among the various polymerase structures without bound RNA, the loops have been seen in all three distinct conformations (in/up, in/down, and out/down), indicating that the structural variability observed in our engineered poliovirus 3Dpol mutants is an inherent feature of this loop in viral polymerases. For example, the structure of Norwalk virus polymerase (PDB code 1SH3) was solved with two independent molecules in the asymmetric unit, one of which is in the in conformation while the other is out30, and the structures of enterovirus 71 polymerase show one instance of the loop in and two instances of the loop out26. As in the poliovirus EC structures, the loop is also in direct contact with the template strand in the FMDV 3Dpol-RNA complexes25; 27. Additionally, the insertion of an extra serine after Ser29139 and a Ser291Pro17 mutation both result in loss of poliovirus infectivity, demonstrating the importance of the loop for viral replication. Finally, while the loop appears to be highly flexible based on the heterogeneity seen in a comparison of multiple polymerase structures9, direct comparisons of the conformations observed in multiple structures of any one polymerase shows that the loop has two predominant conformations that largely correspond to the in and out forms described in this work.
Table II.
Comparison of loop sequences in viral RdRPs
| Virus | PDB | Structural motifa | ||
|---|---|---|---|---|
| mid | | loop | | Bhelix | ||
| Poliovirus | 1RA6 | 285- GMP | SGCSGT | SIFNSM -299 |
| Coxsackievirus | 3DDK | 286- GMP | SGCSGT | SIFNSM -300 |
| Enterovirus 71 | 3N6L | 286- GMP | SGCSGT | SIFNSM -300 |
| HRV14 | 1XR5 | 284- GMP | SGCSGT | SIFNSM -298 |
| HRV16 | 1XR7 | 284- GVP | SGCSGT | SIFNTM -298 |
| FMDV | 1U09 | 295- GMP | SGCSAT | SIINTI -309 |
| RHDV | 1KHV | 305- GLP | SGMPFT | SVINSI -319 |
| Norwalk | 1SH0 | 297- GLP | SGVPCT | SQWNSI -311 |
| Dengue | 2J7U | 597- QRG | SGQVGT | YGLNTF -611 |
| JEV NS5 | 4K6M | 601- QRG | SGQVVT | YALNTF -615 |
| BVDV | 1S48 | 402- QRG | SGQPDT | SAGNSM -416 |
| Hepatitis C | 1C2P | 279- CRA | SGVLTT | SCGNTL -293 |
mid=Middle finger, loop=SGXXXT, helix=Motif B helix that is a core feature of the of palm domain.
Conclusions
Our results demonstrate that flexibility within the residue 288–292 loop of poliovirus 3Dpol is needed for completion of the enzyme’s catalytic cycle, in particular the post-catalysis translocation step. Mutations within the loop have drastic effects on the activity of the polymerase and a glycine is required at position 289. These effects are likely due to the disruption of a delicate balance of loop conformations that must exist for proper function. This loop exhibits high sequence and structural conservation among viral polymerases, suggesting that its conformational dynamics are a common feature of the RNA-dependent RNA polymerases from positive strand RNA viruses.
Materials and Methods
Protein Purification and Crystallization
The 3Dpol proteins all contained the solubility enhancing L446D/R455D mutations on the thumb domain and were expressed, purified, and crystallized as previously described10; 13. Crystals were transferred to 4°C in Hampton micro-bridges and slowly equilibrated into a final solution containing 250 mM sodium acetate, 30% (w/v) PEG-400, 0.1 M cacodylic acid (pH 7.0) and 2 mM DTT. NTP soaks were carried out using this same solution supplemented with 10 mM NTP and 10 mM MgCl2 for ~1 hour, and crystals were flash frozen with liquid nitrogen.
Structure Determination
Diffraction data were collected on the MBC beamline 4.2.2 at the Advanced Light Source (Berkeley, CA) and on a home-source R-AXIS IV++ detector with CuKα radiation. Reflections were integrated, scaled, and merged using the d*TREK suite of programs and the reciprocal space lattice was reindexed to the orientation observed for the wild type protein (PDB code 1RA6) using the program dtcell40. The mutant structures were solved by molecular replacement with CNS41 using PDB entry 1RA6 with the loop deleted to minimize any model bias and the new Rfree data selections were uncoupled from the remainder of the data using a 1500K simulated annealing step. The structures were refined using CNS with the MLI target, manual model building was performed using O42, and figures were generated with the PyMol Molecular Graphics System (www.pymol.org)43.
Biochemical Assays
Polymerase elongation activity assays on oligo-dT/poly(A) substrates were carried out by measuring 32P-uracil incorporation in quenched reaction time points from 10–30 minutes, as previously described13. Data from the mutants were always normalized to a wild type control reaction done in parallel. RNA binding affinity was assessed with a fluorescence polarization assay utilizing a fluorescein 5′ end-labeled self-priming RNA hairpin substrate with a 6 nucleotide single stranded templating sequence; 5′-UUUGACGGCCGGCCGAAAGGCCGGCC-3′ (stem sequence underlined)16. Final reaction conditions for the RNA binding experiments were 10 nM RNA with 3Dpol concentrations ranging from 200 nM to 30 uM in 50 mM NaCl, 50 mM Hepes (pH 7.0), 4 mM DTT, 1.5 mM magnesium acetate, 60 uM ZnCl2, 0.1% NP40.
The single nucleotide incorporation assays were carried out using an identical RNA hairpin sequence that was 5′ 32P end labeled instead of fluorescein labeled. Reaction samples contained 2.5 uM 3Dpol, 150 nM RNA, 5.5 mM NaCl, 50 mM Hepes (pH 7.5), 1.5 mM MgCl2, 60 uM ZnCl2, 0.1% NP40, and 4 mM DTT, and the appropriate NTPs at a concentration of 25 uM each. The samples were assembled and pre-incubated on ice for 30 minutes and then transferred to a room temperature water bath for 30 minutes prior to quenching the reactions with EDTA and running products on a denaturing 20% acrylamide gel containing 7M urea and 1x TBE buffer. An analysis of the single nucleotide incorporation assays as a function of reaction time showed essentially identical results for incubation times from 20 to 45 minutes.
Highlights.
Two distinct conformations of a conserved loop in viral RNA-dependent RNA polymerases.
Interconversion between conformational states needed for efficient catalysis.
Loop in Out conformation contacts RNA template strand and may facilitate translocation.
Acknowledgments
We would like to thank Aaron Thompson and Grace Campagnola for their assistance with experiments. This work was supported by National Institutes of Health grant R01-AI059130 to O.B.P.
Footnotes
Accession Numbers
The coordinates and structure factors have been deposited in the Protein Data Bank with accession numbers as listed in Table I (4NLO, 4NLP, 4NLQ, 4NLR, 4NLS, 4NLT, 4NLU, 4NLV, 4NLW, 4NLX, and 4NLY).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Steitz TA. Visualizing polynucleotide polymerase machines at work. Embo J. 2006;25:3458–68. doi: 10.1038/sj.emboj.7601211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Svetlov V, Nudler E. Macromolecular micromovements: how RNA polymerase translocates. Curr Opin Struct Biol. 2009;19:701–7. doi: 10.1016/j.sbi.2009.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cheetham GM, Jeruzalmi D, Steitz TA. Structural basis for initiation of transcription from an RNA polymerase-promoter complex. Nature. 1999;399:80–3. doi: 10.1038/19999. [DOI] [PubMed] [Google Scholar]
- 4.Yin YW, Steitz TA. The structural mechanism of translocation and helicase activity in T7 RNA polymerase. Cell. 2004;116:393–404. doi: 10.1016/s0092-8674(04)00120-5. [DOI] [PubMed] [Google Scholar]
- 5.Yin YW, Steitz TA. Structural basis for the transition from initiation to elongation transcription in T7 RNA polymerase. Science. 2002;298:1387–95. doi: 10.1126/science.1077464. [DOI] [PubMed] [Google Scholar]
- 6.Li Y, Korolev S, Waksman G. Crystal structures of open and closed forms of binary and ternary complexes of the large fragment of Thermus aquaticus DNA polymerase I: structural basis for nucleotide incorporation. Embo J. 1998;17:7514–25. doi: 10.1093/emboj/17.24.7514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ferrer-Orta C, Arias A, Escarmis C, Verdaguer N. A comparison of viral RNA-dependent RNA polymerases. Curr Opin Struct Biol. 2006;16:27–34. doi: 10.1016/j.sbi.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 8.Gong P, Peersen OB. Structural basis for active site closure by the poliovirus RNA-dependent RNA polymerase. Proc Natl Acad Sci U S A. 2010;107:22505–10. doi: 10.1073/pnas.1007626107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Garriga D, Ferrer-Orta C, Querol-Audi J, Oliva B, Verdaguer N. Role of Motif B Loop in Allosteric Regulation of RNA-Dependent RNA Polymerization Activity. J Mol Biol. 2013;425:2279–87. doi: 10.1016/j.jmb.2013.03.034. [DOI] [PubMed] [Google Scholar]
- 10.Thompson AA, Peersen OB. Structural basis for proteolysis-dependent activation of the poliovirus RNA-dependent RNA polymerase. Embo J. 2004;23:3462–71. doi: 10.1038/sj.emboj.7600357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Theobald DL, Wuttke DS. Empirical Bayes hierarchical models for regularizing maximum likelihood estimation in the matrix Gaussian Procrustes problem. Proc Natl Acad Sci U S A. 2006;103:18521–7. doi: 10.1073/pnas.0508445103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Theobald DL, Wuttke DS. THESEUS: maximum likelihood superpositioning and analysis of macromolecular structures. Bioinformatics. 2006;22:2171–2. doi: 10.1093/bioinformatics/btl332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thompson AA, Albertini RA, Peersen OB. Stabilization of poliovirus polymerase by NTP binding and fingers-thumb interactions. J Mol Biol. 2007;366:1459–74. doi: 10.1016/j.jmb.2006.11.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Richards OC, Baker S, Ehrenfeld E. Mutation of lysine residues in the nucleotide binding segments of the poliovirus RNA-dependent RNA polymerase. J Virol. 1996;70:8564–70. doi: 10.1128/jvi.70.12.8564-8570.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tsao DH, Maki AH. Optically detected magnetic resonance study of the interaction of an arsenic(III) derivative of cacodylic acid with EcoRI methyl transferase. Biochemistry. 1991;30:4565–72. doi: 10.1021/bi00232a029. [DOI] [PubMed] [Google Scholar]
- 16.Mestas SP, Sholders AJ, Peersen OB. A fluorescence polarization-based screening assay for nucleic acid polymerase elongation activity. Anal Biochem. 2007;365:194–200. doi: 10.1016/j.ab.2007.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Crowder S, Kirkegaard K. Trans-dominant inhibition of RNA viral replication can slow growth of drug-resistant viruses. Nat Genet. 2005;37:701–9. doi: 10.1038/ng1583. [DOI] [PubMed] [Google Scholar]
- 18.Love RA, Maegley KA, Yu X, Ferre RA, Lingardo LK, Diehl W, Parge HE, Dragovich PS, Fuhrman SA. The crystal structure of the RNA-dependent RNA polymerase from human rhinovirus: a dual function target for common cold antiviral therapy. Structure. 2004;12:1533–44. doi: 10.1016/j.str.2004.05.024. [DOI] [PubMed] [Google Scholar]
- 19.Campagnola G, Weygandt M, Scoggin K, Peersen O. Crystal structure of coxsackievirus B3 3Dpol highlights the functional importance of residue 5 in picornavirus polymerases. J Virol. 2008;82:9458–64. doi: 10.1128/JVI.00647-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gruez A, Selisko B, Roberts M, Bricogne G, Bussetta C, Jabafi I, Coutard B, De Palma AM, Neyts J, Canard B. The crystal structure of coxsackievirus B3 RNA-dependent RNA polymerase in complex with its protein primer VPg confirms the existence of a second VPg binding site on Picornaviridae polymerases. J Virol. 2008;82:9577–90. doi: 10.1128/JVI.00631-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gong P, Kortus MG, Nix JC, Davis RE, Peersen OB. Structures of coxsackievirus, rhinovirus, and poliovirus polymerase elongation complexes solved by engineering RNA mediated crystal contacts. PLoS One. 2013;8:e60272. doi: 10.1371/journal.pone.0060272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arnold JJ, Cameron CE. Poliovirus RNA-dependent RNA polymerase (3Dpol): pre-steady-state kinetic analysis of ribonucleotide incorporation in the presence of Mg2+ Biochemistry. 2004;43:5126–37. doi: 10.1021/bi035212y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zamyatkin DF, Parra F, Alonso JM, Harki DA, Peterson BR, Grochulski P, Ng KK. Structural insights into mechanisms of catalysis and inhibition in Norwalk virus polymerase. The Journal of biological chemistry. 2008;283:7705–12. doi: 10.1074/jbc.M709563200. [DOI] [PubMed] [Google Scholar]
- 24.Yang X, Welch JL, Arnold JJ, Boehr DD. Long-range interaction networks in the function and fidelity of poliovirus RNA-dependent RNA polymerase studied by nuclear magnetic resonance. Biochemistry. 2010;49:9361–71. doi: 10.1021/bi100833r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ferrer-Orta C, Arias A, Perez-Luque R, Escarmis C, Domingo E, Verdaguer N. Structure of foot-and-mouth disease virus RNA-dependent RNA polymerase and its complex with a template-primer RNA. J Biol Chem. 2004;279:47212–21. doi: 10.1074/jbc.M405465200. [DOI] [PubMed] [Google Scholar]
- 26.Wu Y, Lou Z, Miao Y, Yu Y, Dong H, Peng W, Bartlam M, Li X, Rao Z. Structures of EV71 RNA-dependent RNA polymerase in complex with substrate and analogue provide a drug target against the hand-foot-and-mouth disease pandemic in China. Protein & cell. 2010;1:491–500. doi: 10.1007/s13238-010-0061-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ferrer-Orta C, Arias A, Perez-Luque R, Escarmis C, Domingo E, Verdaguer N. Sequential structures provide insights into the fidelity of RNA replication. Proc Natl Acad Sci U S A. 2007;104:9463–8. doi: 10.1073/pnas.0700518104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ferrer-Orta C, Arias A, Agudo R, Perez-Luque R, Escarmis C, Domingo E, Verdaguer N. The structure of a protein primer-polymerase complex in the initiation of genome replication. Embo J. 2006;25:880–8. doi: 10.1038/sj.emboj.7600971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ng KK, Cherney MM, Vazquez AL, Machin A, Alonso JM, Parra F, James MN. Crystal structures of active and inactive conformations of a caliciviral RNA-dependent RNA polymerase. J Biol Chem. 2002;277:1381–7. doi: 10.1074/jbc.M109261200. [DOI] [PubMed] [Google Scholar]
- 30.Ng KK, Pendas-Franco N, Rojo J, Boga JA, Machin A, Alonso JM, Parra F. Crystal structure of norwalk virus polymerase reveals the carboxyl terminus in the active site cleft. J Biol Chem. 2004;279:16638–45. doi: 10.1074/jbc.M400584200. [DOI] [PubMed] [Google Scholar]
- 31.Ago H, Adachi T, Yoshida A, Yamamoto M, Habuka N, Yatsunami K, Miyano M. Crystal structure of the RNA-dependent RNA polymerase of hepatitis C virus. Structure. 1999;7:1417–26. doi: 10.1016/s0969-2126(00)80031-3. [DOI] [PubMed] [Google Scholar]
- 32.Bressanelli S, Tomei L, Roussel A, Incitti I, Vitale RL, Mathieu M, De Francesco R, Rey FA. Crystal structure of the RNA-dependent RNA polymerase of hepatitis C virus. Proc Natl Acad Sci U S A. 1999;96:13034–9. doi: 10.1073/pnas.96.23.13034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lesburg CA, Cable MB, Ferrari E, Hong Z, Mannarino AF, Weber PC. Crystal structure of the RNA-dependent RNA polymerase from hepatitis C virus reveals a fully encircled active site. Nat Struct Biol. 1999;6:937–43. doi: 10.1038/13305. [DOI] [PubMed] [Google Scholar]
- 34.O’Farrell D, Trowbridge R, Rowlands D, Jager J. Substrate complexes of hepatitis C virus RNA polymerase (HC-J4): structural evidence for nucleotide import and de-novo initiation. J Mol Biol. 2003;326:1025–35. doi: 10.1016/s0022-2836(02)01439-0. [DOI] [PubMed] [Google Scholar]
- 35.Choi KH, Groarke JM, Young DC, Kuhn RJ, Smith JL, Pevear DC, Rossmann MG. The structure of the RNA-dependent RNA polymerase from bovine viral diarrhea virus establishes the role of GTP in de novo initiation. Proc Natl Acad Sci U S A. 2004;101:4425–30. doi: 10.1073/pnas.0400660101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lu G, Gong P. Crystal Structure of the Full-Length Japanese Encephalitis Virus NS5 Reveals a Conserved Methyltransferase-Polymerase Interface. PLoS Pathog. 2013;9:e1003549. doi: 10.1371/journal.ppat.1003549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Butcher SJ, Grimes JM, Makeyev EV, Bamford DH, Stuart DI. A mechanism for initiating RNA-dependent RNA polymerization. Nature. 2001;410:235–40. doi: 10.1038/35065653. [DOI] [PubMed] [Google Scholar]
- 38.Tao Y, Farsetta DL, Nibert ML, Harrison SC. RNA synthesis in a cage--structural studies of reovirus polymerase lambda3. Cell. 2002;111:733–45. doi: 10.1016/s0092-8674(02)01110-8. [DOI] [PubMed] [Google Scholar]
- 39.Burns CC, Lawson MA, Semler BL, Ehrenfeld E. Effects of mutations in poliovirus 3Dpol on RNA polymerase activity and on polyprotein cleavage. J Virol. 1989;63:4866–74. doi: 10.1128/jvi.63.11.4866-4874.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pflugrath JW. The finer things in X-ray diffraction data collection. Acta Crystallogr D Biol Crystallogr. 1999;55 (Pt 10):1718–25. doi: 10.1107/s090744499900935x. [DOI] [PubMed] [Google Scholar]
- 41.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54 (Pt 5):905–21. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 42.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Cryst. 1991;A47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 43.DeLano WL. The PYMOL Molecular Graphics System. DeLano Scientific; San Carlos, CA USA: 2002. [Google Scholar]