

Summary
The regulatory (R) subunit is the cAMP receptor of protein kinase A. Following cAMP binding, the inactive PKA holoenzyme complex separates into two active catalytic (C) subunits and a cAMP-bound R dimer. Thus far, only monomeric R structures have been solved, which fell short in explaining differences of cAMP binding for the full-length protein as compared to the truncated R subunits. Here we solved a full-length R-dimer structure that reflects the biologically relevant conformation, and this structure agrees well with small angle X-ray scattering. An isoform-specific interface is revealed between the protomers. This interface acts as an intermolecular sensor for cAMP and explains the cooperative character of cAMP binding to the RIα dimer. Mutagenesis of residues on this interface not only leads to structural and biochemical changes, but is also linked to Carney complex disease.
Introduction
Since protein kinase A (PKA) was discovered in 1968 (Walsh et al., 1968), it has emerged as the most ubiquitous cAMP-responsive enzyme in mammalian tissues (Kuo and Greengard, 1969). As such, it confers a myriad of cellular responses including cell differentiation, regulation of metabolism, and cell growth (Cho-Chung et al., 1995). In the absence of cAMP, PKA exists in an inhibited state composed of a regulatory (R) dimer bound to two catalytic (C) subunits. Compromised regulation of PKA activity has detrimental consequences. This is exemplified by RIα haploinsufficiency causing Carney complex disease (CNC), which is a familial multiple neoplasia syndrome characterized by cardiac and extracardiac myxomas (Veugelers et al., 2004). In mice, RIα knockout results in embryonic lethality (Amieux and McKnight, 2002), and it was identified as the tissue-specific extinguisher of cAMP-mediated gene expression (Boshart et al., 1991). Although RIα is ubiquitously expressed in every cell, it is especially abundant in the central nervous system and the heart (Cadd and McKnight, 1989). Not only is expression of the four R isoforms RIα, RIβ, RIIα, and RIIβ tissue specific, but their functional roles are nonredundant.
Elucidating monomeric regulatory subunit structures has provided basic understanding for PKA-mediated signaling, yet each full-length R subunit exists in cells as a stable dimer (Potter and Taylor, 1979; Rubin, 1979). The full-length R subunits are composed of an N-terminal dimerization/docking (D/D) domain, a flexible linker, and the C-terminally positioned tandem cyclic nucleotide binding (CNB) A and B domains, which are connected to each other by the αB/C helix (Figure 1A). Each CNB has a β-subdomain that forms a beta sandwich, which is flanked by a helical α-subdomain. Embedded within the beta sandwich is the phosphate-binding cassette (PBC), which serves as the docking site for cAMP in all cAMP-binding proteins (Su et al., 1995). The α-subdomains are in turn composed of an αN helix, a short 310 -loop, and an αA helix, together termed the N3A-motif (Figure 1A), which is conserved in other cAMP-binding proteins such as exchange protein directly activated by cAMP (EPAC) and potassium/sodium hyperpolarization-activated cyclic nucleotide-gated channel (Kornev et al., 2008).
Figure 1. CNB-A Structural Motif Organization.
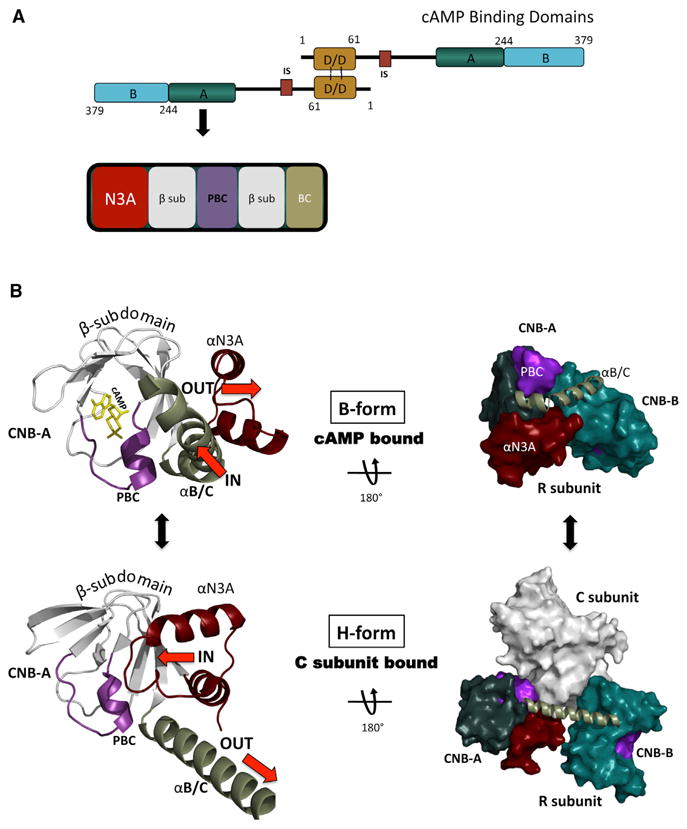
(A) Shown is the overall functional domain organization of the full-length RIα construct used in this study. This included the D/D domain, the linker with the inhibitor site that can bind to the C subunit in the holoenzyme, and the two tandem CNB domains. The bottom figure highlights the CNB-A structural motif makeup containing the N3A motif, the β sandwich, the PBC, and the αB/C helix.
(B) Current understanding of the structural motif conformational changes within the CNB-A are demonstrated as they differ in the cAMP bound (B-Form, top) and the C subunit bound form (H-Form, bottom). The N3A is colored red, the αB/C helix green, the PBC purple, the β sandwich white, and cAMP yellow. Top left figure is the CNB-A in the cAMP bound (B-Form) and bottom left is the CNB-A in the C subunit bound (H-Form). Top right shows the overall R monomer conformation in the B-Form. Bottom right shows the overall R conformation of the heterodimer R1:C1 (R is in the H-Form).
Structural studies of R monomers have uncovered some of the conformational changes as it toggles between its cAMP bound B-form and its C subunit bound H-form (Figure 1B; Kim et al., 2005, 2007). The N3A-motif and αB/C helix were revealed to be pivotal and correlated; while one moves in toward the PBC, the other moves out. In the cAMP-bound form, the kinked conformation of the αB/C helix positions it close to the PBC. In the C-subunit bound state, the αB/C helix is contiguous and positioned “out” from the PBC. Conversely, the N3A motif slides to the “in” position when C is bound to R, which allows the N3A-motif to interact with the PBC. In the cAMP-bound state, on the other hand, the N3A is in the “out” position where it is moved up and away from the PBC, which as this study reveals is crucial for the cAMP-bound RIα dimer conformation.
While truncated structures of the R subunits provided fundamental knowledge for our understanding of PKA, a full-length dimer structure has not been elucidated even though there are significant biochemical differences compared to the monomeric form. A deletion mutant of RIα that is missing most of the linker and the D/D domain and has typically been utilized for crystallography studies, displays equal cAMP binding affinities of 50 nM for the two tandem CNB domains. This monomeric protein also lacks cooperative binding (Herberg et al., 1994). In contrast, the full-length dimer binds cAMP with a cooperative profile with a Hill Slope of 1.62. Additionally, differential affinities for cAMP of the tandem CNB sites were noted: CNB-A has a Ka of 60 nM and CNB-B has a higher affinity of 15 nM (Herberg et al., 1994).
Hence, we hypothesized that there may be communication as a result of additional interaction sites between the dimer protomers in the full-length protein that could explain the cAMP binding differences as compared to the monomeric deletion mutant protein. This crystallographic study of the RIα homodimer uncovered an interaction surface between the protomers that allows communication in the dimer protein. The docking of two CNB-A domains within the dimer creates a helical-helical bundle by utilizing their N3A structural motifs. The structure is consistent with small-angle scattering X-ray (SAXS) data and reveals a biologically relevant conformation. In addition, mutations within this interface result in structural and biochemical changes. Strong evidence for the biological relevance of the discovered interface is further established by the presence of Carney complex disease mutations within this region, which thus far lacked a structural rationale for altered RIα function. While present in the crystals, the D/D domain and most of the linkers do not give rise to electron density, which is due to the linker region's intrinsic flexibility. However, our structure reveals that, in addition to the D/D domain preventing long-distance dissociation by functioning as an N-terminal tether for the two protomer chains, it also enables formation of an intermolecular interface by the protomers' CNB domains. This interface, in turn, allows cross-communication between the two CNB-A domains, which results in cooperative cAMP binding across the dimer molecule. Thus, this study presents an R homodimer structure that can explain the cooperative cAMP binding by RIα and reveals an interface with implications for Carney complex mutations.
Results
The Dimer Structure Reflects the Biologically Relevant Conformation
To crystallize the full-length RIα homodimer, excess cAMP was added to the purified protein before setting up crystallization screens. Excess cAMP improves the stability of the protein in solution while preventing early degradation. Crystals were obtained in this way and resulted in a distinct morphology and space group (P41212) compared to previous structures of the deletion mutant monomers, which had hexagonal space groups of P6 (Badireddy et al., 2011; Su et al., 1995; Wu et al., 2004a, 2004b). While in the previous structures each asymmetric unit contained one monomer, the present structure now contains the homodimer in the asymmetric unit with an inverted U-shaped architecture and a rotational NCS 2-fold symmetry (Figure 2A; Table 1). While the tandem CNB-A and CNB-B domains for each of the two chains (denoted R and R′) are resolved with cAMP bound, the D/D domain and N-terminal part of the linkers are not visible in the electron density maps. However, the crystal arrangement contains enough space above the CNB-A domains in the crystal lattice for the D/D domain. To establish if the D/D domain is indeed present in our crystals and verify that the crystals are not composed of degraded protein, we washed and ran the crystals on a SDS gel. According to the SDS gel analysis, the protein runs at the full-length molecular weight and shows that the crystals are composed of full-length RIα (Figure S1 available online). The lack of electron density can thus be explained by the linker region's flexibility that may prevent the D/D domain from being captured in a consistent position. Previously, site-directed labeling experiments showed that these linkers are highly flexible (Li et al., 2000). In fact, the flexibility of the linker region and its resulting inherent instability was observed already at the early stages of studying PKA (Erlichman et al., 1973; Rannels and Corbin, 1980). Moreover, the linkers are predicted to be intrinsically disordered regions (Figure S2; Romero et al., 2001) and together with the above-mentioned studies can explain the absence of electron density for the D/D domain in our structure.
Figure 2. Overview of the RIα Dimer Molecular Architecture.
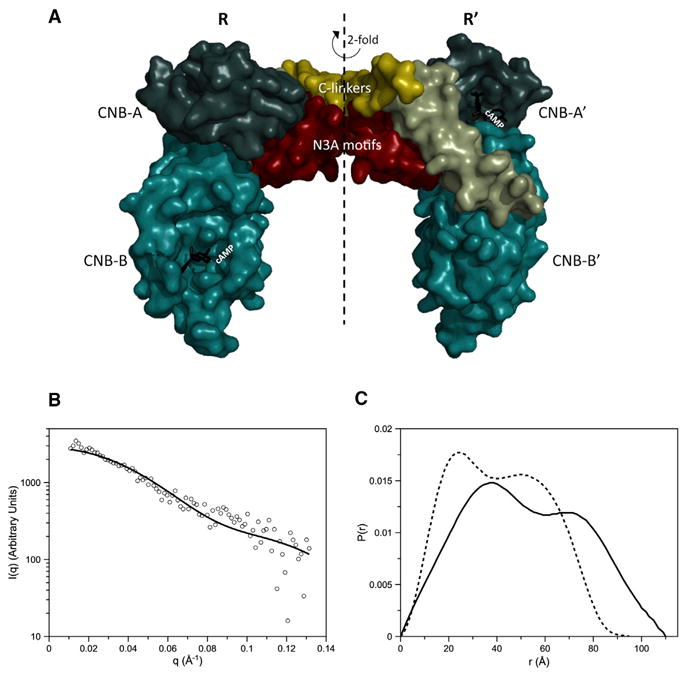
(A) A space-filling depiction of the RIα homodimer crystal structure is shown. The CNB-A domains are in deep teal, the CNB-B domains in light teal. cAMP is colored black, the linker gold, and the αB/C helix green.
(B) Plot of the theoretical scattering profile of the RIα homodimer crystal structure as calculated with FoXS is shown as a solid black line in comparison to the experimental scattering profile (Vigil et al., 2004) shown as open black circles.
(C) P(r) curve computed from the RIα homodimer crystal structure (dashed line) compared to the P(r) of RIα homodimer from SAXS (black line) (Vigil et al., 2004).
See also Figures S1 and S2.
Table 1. Data Collection and Refinement Statistics.
| RIα2 | |
|---|---|
| Data collection | |
| Space group | P41212 |
| Cell dimensions (Å) | |
| a = b | 104.7 |
| c | 218.3 |
| No. of molecules per asymmetric unit | 2 |
| Resolution (Å) | 3.88 |
| Rmerge | 0.088 (0.575)a |
| Completeness (%) | 99.9 (100.0) |
| I/σ | 35.9 (6.2) |
| No. of reflections | 11,860 |
| Multiplicity | 9 (9.4) |
| Refinement | |
| Resolution (Å) | 50–3.88 |
| Rwork/Rfree (%) | 26.0/28.7 |
| Rmsds | |
| Bond lengths (Å) | 0.017 |
| Bond angles (°) | 2.0 |
| Ramachandran angles (%) | |
| Most favored | 71.5 |
| Disallowed | none |
Values in parentheses are for highest-resolution shell (3.88–4.02 Å).
The overall shape, as revealed by our crystal structure, also agrees with early studies investigating the structural characteristics of RIα (Zoller et al., 1979). We thus compared our crystal structure to the solution structure of the protein, previously determined by SAXS analysis (Vigil et al., 2004). The small-angle scattering function was calculated from the crystal structure using FoXS (Schneidman-Duhovny et al., 2010, 2013) and the resulting pair-distance distribution function (P(r); Figure 2C) was compared to the previously published P(r) determined from SAXS of the RIα homodimer (Vigil et al., 2004). The calculated data from the crystal structure compare to the in-solution data with a χ2 = 1.1 (Figure 2B) and despite the lack of electron density for the D/D domain, the P(r) from the crystal structure has a similar double-peak shape as obtained experimentally with SAXS for the full-length protein. The Dmax value of the crystal structure is 95 Å rather than 110 Å as determined from SAXS experiments, and the Rg value of the crystal structure is also less than the SAXS of the full-length homodimer (Table 2). However, the smaller dimensions of the predicted SAXS of the crystal structure are consistent with the D/D domain and most of the linker not being represented in the dimer crystal structure. The double-peak shape of the P(r) curves of both the crystal structure and the experimental SAXS are indicative of a bilobal structure (Figure 2C) where the first peak corresponds to interatomic distances within each lobe and the second peak corresponds to interatomic distances between the two lobes. The greater Dmax seen in the P(r) of the full-length RIα SAXS is consistent with a Y-shaped structure where the D/D domain and linker form the stem of the Y and the CNB domains form the arms or lobes of the Y (see Figure 4A in Vigil et al., 2004). Indeed, the relative positions of the CNB domains in the Y-shaped model presented earlier (Vigil et al., 2004) are in very good agreement with the crystal structure. Considering the inherent resolution limitations of SAXS, combined with the fact that experimental SAXS is the result of the time- and ensemble-average of all conformations of the protein, the published SAXS-based model nevertheless does validates the crystal structure and supports the idea that these contacts could readily form.
Table 2. Effect of RIα Mutations on SAXS.
| RIα Homodimer | Rg (Å) | Dmax (Å)a |
|---|---|---|
| Wild-type RIα (experimental)b | 39.8 ± 1.2b | 110b |
| Wild-type RIα (calculated from crystal structure) | 32.1 ± 0.001a | 95 |
| RIα Y120A | 48.8 ± 0.3a | 150 |
| RIα K121A | 43.7 ± 0.3a | 130 |
Calculated using GNOM.
Data from Vigil et al. (2004).
Conserved Features of the Dimer Architecture
At the vertex of the dimer, the two R subunits converge. Electron density is visible for the two RIα chains beginning with residue E105 (R) and A108 (R′) respectively (Figure 3), which are extended compared to the monomer cAMP bound structure (1rgs) that starts with residue 113 (Wu et al., 2004a). At the C-terminal ends, residues are visible up to S376 and each chain includes two bound cAMP molecules according to the Fo-Fc electron density maps (Figure S3). The C-terminal portions of the linkers (C-linkers) that are resolved run antiparallel and are positioned on top of their respective helical α-subdomains (Figure 3). This C-linker interaction between the two monomers seals an intermolecular interface below it. Docking of CNB-A and CNB-A′ results in an interface, which will be described in detail later. The CNB-B domains are extended down and away from their CNB-A domains (Figure 3). Structural features of previous cAMP-bound truncation monomers are conserved in the dimer structure as well. The CNB-A domains of the dimer compare to the 1rgs structure with an rmsd of 0.51 Å2, whereas the CNB-B domains have an rmsd of 0.98 Å due to its highly dynamic nature (Cheng et al., 2009).
Figure 3. Crystal Structure of the RIα Homodimer Complex.
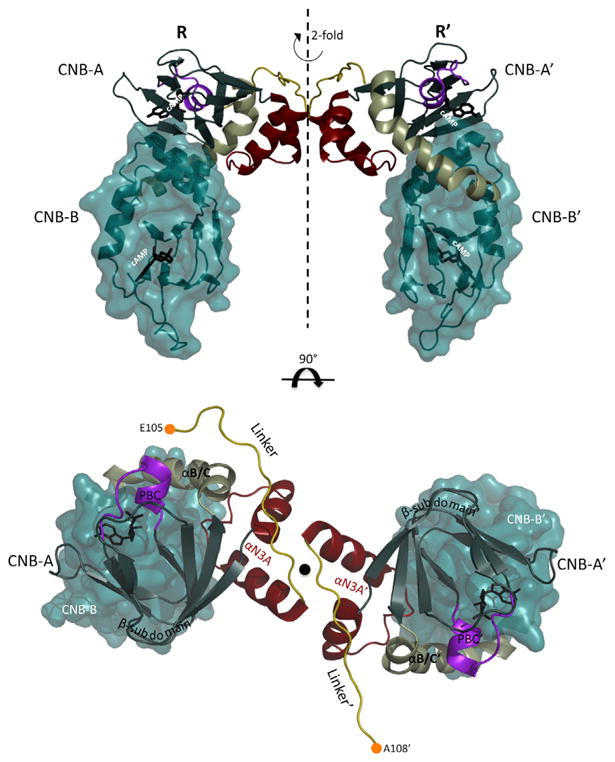
The top shows the CNB-A domains of R and R′ dock into each other creating an upside down U-shape. The CNB-B domains are extended from there. The previously seen architecture within each R and R′ monomer is conserved. The bottom shows a view from the top down: the symmetry axis and thus the CNB-B domains are positioned underneath the CNB-A domains. The N-terminal Cα of the linkers are shown as orange circles.
See also Figure S3.
Furthermore, the interface created by R and R′ in our dimer structure is also present in previous high-resolution RIα structures. All previous structures are of deletion mutant monomer constructs that are missing at least the D/D and N-linker of RIα and include but are not limited to Protein Data Bank (PDB) accession codes 1RGS, 3IIA, 1NE4, 1RL3, and 1NE6 (Badireddy et al., 2011; Su et al., 1995; Wu et al., 2004a, 2004b). Even in these previous crystal structures, the monomers utilize the same interface as an interaction to pack in the crystals. Because the monomeric structures are missing the N-linker and D/D domain, the observed monomer contacts were interpreted as crystal packing only and thus not appreciated or analyzed as a functionally relevant interaction.
An RIα-Specific Homodimer Interface Is Revealed
Each R monomer contains a previously described molecular architecture (Su et al., 1995), and we will use the same nomenclature here. The two linker segments from each monomer that immediately flank the N-terminal N3A motifs run antiparallel and are positioned just above their helical α-subdomains (Figure 4, top). The N3A and N3A′ motifs of each protomer in CNB-A and CNB-A′ are sandwiched between the two cAMP--binding β-subdomains (Figure 4, middle). The adjacent N3A-motifs are thus wedged against each other, creating hydrophobic interactions along the center of a helical-helical bundle (Figure 4, bottom). The interface is symmetrical with each αA helix oriented nearly perpendicular against the opposing monomers' αN′ helix. Clusters of residue interactions enable this docking of the αA helix against the αN′ helix and αA′ helix against the αN helix at opposite ends of the interface. Electron density for residue side chains positioned within the interface is visible in this homodimer (Figure S4B). The orientation of the interface residues also compares well to that of the higher resolution monomeric structures (Figure S4A). Each residue cluster is positioned so that they seal the hydrophobic interface resulting in a buried area of 485 Å2. The energy of desolvation is −6.2 kcal/mol, making it an energetically favorable interaction (Krissinel and Henrick, 2007).
Figure 4. CNB-A Domain Docking Creates a Hydrophobic Interface.
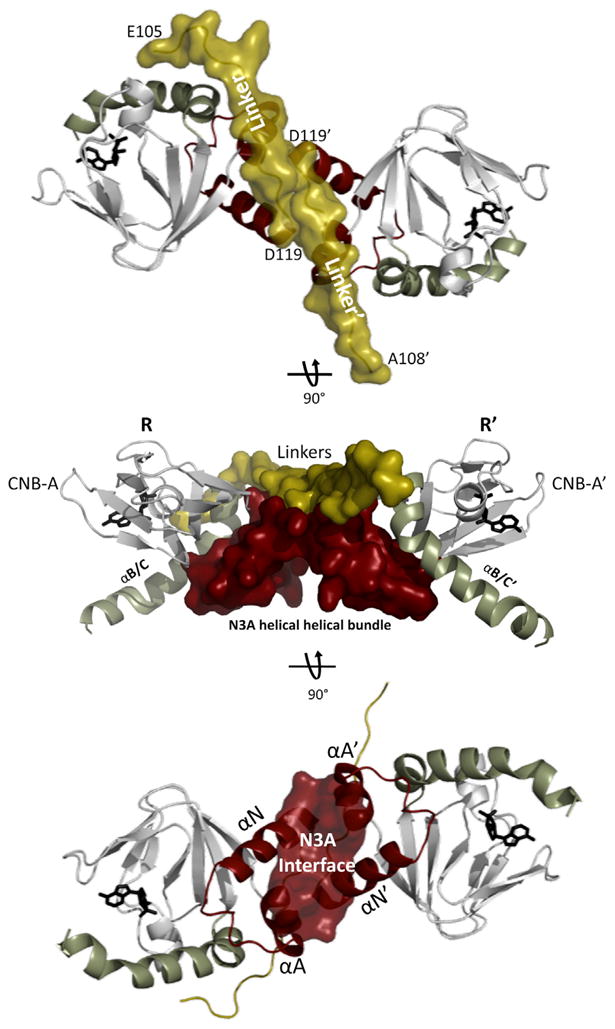
The CNB-A domains only of each protomer are shown with the N3A motifs in red and the linkers in gold.
Top: view down the symmetry axis to illustrate how the linkers are running antiparallel and sealing the hydrophobic interface below. Middle: shows docking of the CNB-A domains with their respective N3A motifs creating a helical-helical bundle.
Bottom: the A helix of each N3A stacks against the N helix of the opposite N3A motif creating a hydrophobic interface between them.
While the CNB-B domains are less well resolved, the CNB-A domains and the interface show electron density for several side chains (Figure S4B). A detailed look at the interface reveals that Phe148 of the αA helix pi stacks against Tyr120′ in the opposing αN′ helix. Tyr120′ also hydrogen bonds to Arg144 of the aA helix (Figure 5B). Next to this Arg on the A-helix is Ser145 which, in turn, hydrogen bonds to Lys121′ in the αN′ helix of R′. This H-bonding cluster is present twice, at opposite ends of the interface, due to the 2-fold symmetry within the dimer. In the center of this elongated interface are two opposing methionines, Met123 from R and Met123′ from R′ (Figure 5A), which serve as a strong hydrophobic core by being 90% buried within the interface (Krissinel and Henrick, 2007). Further stabilizing this conformation is Lys118 from each linker, which caps the corresponding A-helix by hydrogen bonding with the backbone oxygens of F148, D149, and M151 (Figure 5C). Lys118 and Lys118′ are hydrogen bonding with each other's backbone oxygen and nitrogen atoms, thus stabilizing the protomers' linker interaction (Figure 5A).
Figure 5. Isoform-Specific N3A Motif Residues Support the RIα Dimer Interface.
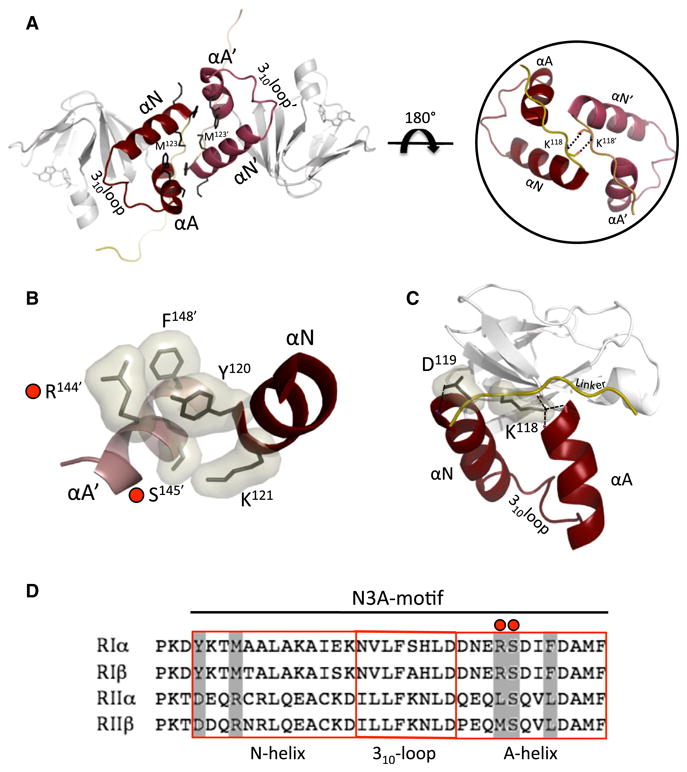
(A) Left: N3A helical-helical bundle creates H-bonding network on the interface. At the center the two opposing M123 from each protomer constitute a strong hydrophobic core. Right: a zoomed-in view of the linker interactions between the protomers' K118 backbone nitrogens and oxygens.
(B) Specific residue interaction present at opposite ends of the interface. Red circles denote CNC mutations.
(C) The linkers are antiparallel to each other and crossover their respective N3A motif. K118 H-bonds to backbone residues of the A helix thereby capping the A helix.
(D) R subunit isoform sequence alignment of the N3A-motif. Boxed in gray are residues interacting on the interface of RIα. CNC mutations in RIα are marked with a red circle.
See also Figure S4.
Previously published SAXS comparisons of the RI and RII subunits have demonstrated they have very different molecular shapes even though their sequences and domain organization are similar (Vigil et al., 2004). The published P(r) functions for RIα and RIβ homodimers have a similar Dmax of ∼100 Å and are also predicted to have a similar bilobal shape (Ilouz et al., 2012). This prediction is supported by a comparison of their CNB-A N3A sequence, which shows high sequence similarity (Figure 5D). In contrast, the SAXS P(r) functions for the RII homodimers are much more extended and suggest a different shape compared to the RI subunits (Vigil et al., 2004). This could, in part, be due to the longer linker regions of the RII subunits. However, alignment of the CNB-A N3A-motif sequences of RIα, RIIα, and RIIβ reveal very different side chains at the key residues that form the intermolecular contacts that would presumably rule out a similar interface for the RII subunits. While the crucial Tyr120 of RIα is conserved in RIβ, in the corresponding position in the N-helix, RIIα and β have an aspartate at this position. We predict that this aspartate (Asp137) of the N-helix in RIIβ would interfere with the interface formation because it is opposite Asp166 in the A-helix of the other RIIβ protomer. Lysine 121 of RIα, which also appears to be important, is replaced with either a Glu in RIIα or an Asp in RIIβ. Met123 in RIα forms the hydrophobic core of the inter-face and is an Arginine in both RII isoforms. These differences in sequence at key positions might largely explain the extended shape of the RII homodimers in solution because the crucial interface residues seen in the RIα homodimer structure are not conserved and would presumably prevent this same dimer interaction.
Mutational Analysis of the Interface Uncovers Its Functional Significance
To study the homodimer interface in more detail, we mutated interface residues, namely Lys121 and Tyr120 to alanine. We characterized and evaluated changes in the mutant proteins by SAXS, catalytic subunit inhibition, and holoenzyme activation assays. Because neither residue is part of the heterodimer R:C interface (Kim et al., 2007) or the R:cAMP binding interactions, changes in these residues would likely cause the R:R′ interface to be perturbed. Comparison of the SAXS P(r) curves of the mutant proteins to the wild-type protein indicate the mutations cause significant structural changes in the RIα homodimer. While the predicted scattering of the crystal structure and the experimental SAXS data of the wild-type homodimer are indicative of compact structures (Table 2), the mutant homodimers are much more extended. The maximal distance (Dmax) shifts from 110 Å for the wild-type R-subunit to 150 Å and 130 Å for the Tyr120 and Lys121 to Ala mutants, respectively (Figure 6; Table 2). Moreover, the distinct double-peak shape of the P(r) becomes blurred, which suggests that the lobes of the Y-shaped structure are more dynamic. These results indicate that the compact relatively rigid structural integrity of the RIα homodimer is lost when the interface is disrupted.
Figure 6. SAXS Profiles of RIα Y120A and K121A Mutants.
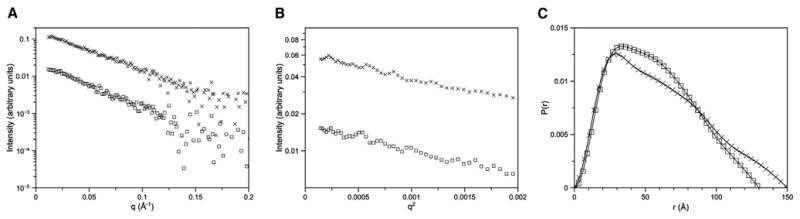
I(q) versus q (A), Guinier plots (B), and P(r) curves (C) calculated from solution small-angle X-ray scattering data for RIα Y120A mutant (crosses) and K121A mutant (squares). Wild-type P(r) curve is from Vigil et al. (2004). P(r) curves have been normalized to the area under the curve.
To establish if the interface plays a role in the activation of PKA, the mutant proteins Tyr120A and Lys121A were tested for their ability to regulate the catalytic subunit. Both mutants are able to form holoenzymes with the C subunit; however, they show significant differences in their cAMP activation profiles. While the wild-type complex has a half-maximal effective concentration (EC50) for cAMP of about 54 nM, the mutant complexes require less cAMP to release the catalytic subunit and have EC50 values of around 35 nM (Figure 7A). The mutants are thus more sensitive to cAMP and are more readily activated. In addition, the mutant holoenzymes show almost complete loss of cooperativity for cAMP activation with a shift in the Hill coefficient from 1.73 for wild-type to ∼1.0 for the mutants (Figure 7A). These results suggest that the N3A interface is of functional as well as structural significance.
Figure 7. cAMP-Induced Activation of Holoenzymes by Fluorescence Polarization.

(A) Alanine mutations of interface residues were analyzed with a fluorescent polarization assay to monitor changes in the mutant compared to wild-type holoenzymes. Left: Activation profiles of holoenzymes formed with RIα wild-type (squares), RIα K121A (triangles), RIα Y120A (circles). The curves for RIα K121A and Y120A are similar and lay on top of each other. Right: GraphPad Prism was used to fit binding curves and the SEM is shown with error bars.
(B) CNC interface mutants were analyzed for their holoenzyme activation properties. Left: Activation profiles of holoenzymes formed with RIα wild-type (squares), RIα S145G (triangles), and RIα R144S (circles). The curves for RIα S145G and R144S are similar and lay on top of each other. Right: GraphPad Prism was used to fit binding curves and the SEM is shown with error bars.
Carney Complex Disease Mutations Are Localized at the Dimer Interface
Because our mutational analysis had a significant impact on RIα, we searched for known disease mutations of RIα that could be linked to the interface. RIα is the only isoform that behaves as the tissue-specific extinguisher of cAMP-mediated gene expression (Boshart et al., 1991; Jones et al., 1991; Yin and Kirschner, 2009) and is linked to diseases such as systemic lupus erythematosus (Kammer, 2002; Kammer et al., 1996; Laxminarayana et al., 1999), Carney complex disease (CNC), and breast cancer (Miller, 2002). Although most CNC mutations that escape nonsense-mediated mRNA decay (NMD) cluster around the cAMP binding site, we found three CNC mutations in the published literature that could be mapped to the interface of the homodimer structure including Arg144 to Ser, Ser145 to Gly, as well as Ser145 to Asp (Horvath et al., 2010). Whereas more than 100 mutations of the RIα gene in humans have been identified, the majority are subject to NMD (Horvath et al., 2010; Kirschner et al., 2000). The CNC mutations located on the homodimer interface are residues that hydrogen bond to Tyr120 and Lys121, mutations of which were analyzed structurally and functionally in the previous section (Figure 5B). Also of great interest is that these CNC mutations result in expressed altered protein and cause disease as a result of dysfunctional RIα. A publication by Greene and colleagues in 2008 analyzed the Arg144 to Ser CNC mutation and found that it exhibited an increase in PKA-specific activation compared to wild-type (Greene et al., 2008). These data are consistent with the functional data for Tyr120 and Lys121 reported here.
To compare the effects of these CNC mutations with our study, we introduced the Carney complex mutations Arg144 to Ser and Ser145 to Gly and Ser145 to Asp in the full-length RIα protein. While we saw changes in the SAXS scattering behavior and cAMP activation profile for Arg144 to Ser and Ser145 to Gly, the Ser145 to Asp mutation caused the protein to be insoluble during expression and we were thus unable to characterize this mutant. Preliminary results of the scattering of the Arg144 to Ser protein compared to the wild-type are indicative of structural changes and we are now investigating the implications of these mutants further by SAXS and crystallography. The Arg144 to Ser and Ser145 to Gly mutants also displayed an increase in sensitivity for cAMP in their activation of the corresponding holoenzymes (Figure 7B), which is consistent with the analysis of their hydrogen bonding partners on the interface. Our previous structural knowledge could not explain the reason for the dysfunction of these RIα mutations, but this study can now link these Carney complex mutations to a specific structural feature. In addition, the Hill coefficient of the mutants is lowered to about 1.4, comparable to the Hill coefficient obtained for the hetero-dimer R:C complex (Figure 7B).
Discussion
The many previous structures of PKA served as a foundation for functional studies. Yet, the importance of integrating this structural information with PKA's biological functions has not subsided. On the contrary, the challenges have increased while attempting to solve the larger PKA macromolecular complexes. While the recently solved RIβ and RIIβ holoenzyme crystal structures represent a major milestone for PKA research, obtaining a regulatory subunit homodimer structure that lies at a lower level of complexity has been elusive (Ilouz et al., 2012; Zhang et al., 2012). We finally report here the structure of the PKA RIα homodimer and the detection of a functionally significant interface.
The compact homodimer structure of RIα is consistent with previous SAXS observations and the basis for this compact structure, an isoform-specific intermolecular interface, is revealed at the vertex of the two interacting monomers. This interface is conserved crystallographically, and is independent of the D/D domain as it is seen in many monomeric RIα structures, which further implies its significance. The interface is energetically favorable and interaction specific, and not a trivial artifact of crystal packing. Furthermore, we propose that the interface is isoform-specific and can account for the large differences in the solution structures of the type RI and RII homodimers observed by SAXS, where the RI homodimers are compact and the RII homodimers are extended (Vigil et al., 2004). Although the RII subunits contain longer linker regions, the large differences in the solution structures are most likely the result of interactions involving the R subunits' N3A-motif. Mutations on the interface of RIα result in extended structures as analyzed by SAXS and now look more like the RII subunits in solution. Moreover, and in contrast to RI structures, monomeric deletion mutant structures of RIIα and RIIβ, that lack the D/D domain and the N-linker, do not have the N3A interface (Brown et al., 2009; Diller et al., 2001; Wu et al., 2007). The RIIβ holoenzyme likewise does not use this interface (Zhang et al., 2012). Thus, the RII subunits do not utilize the N3A motif to create an interaction surface.
According to our results, the D/D domain is structurally and spatially independent from the rest of the homodimer complex. Unfortunately, the flexibility of the linker segment apparently prevents the D/D domain from assuming a stable docked conformation in the crystal lattice, which results in the lack of electron density for this region. This may account in part for the low diffracting crystals. Yet, even at higher resolution, the RIIβ holoenzyme crystal structure published recently contains no electron density for the D/D domain nor the N-linker (Zhang et al., 2012). A requirement for the regulatory subunits to inhibit the catalytic subunits is that part of the linker and the inhibitor site (IS) must bind to and become immobilized within the active site cleft of the catalytic subunit (Kim et al., 2005, 2007). Thus, while limiting their crystallographic visualization, the flexibility of the linker region is intrinsic and necessary to allow the IS to search for the catalytic subunit to then become bound and finally form the holoenzyme complex.
While the N3A interface is the structural highlight in our dimer structure, it was not previously recognized as an integral or functional part of the RIα homodimer. The D/D domain stably links the monomers at the N terminus constraining the rest of the molecule into a spatial proximity and thereby enhancing the dimer interface interaction. Yet, during crystallization, the interface is D/D domain-independent as also seen in the monomeric structures that lack the D/D domain sequence. In addition to its role as a docking site for A-kinase anchoring proteins, the D/D domain functions as a covalently linked tether that increases the interaction of its CNB-A domains by increasing their effective concentration, independent of the local concentration of RIα. Furthermore, this allows the dimer interface to form and dissociate without compromising the global dynamics of the regulatory subunit, as would potentially be the case with a high-affinity interaction. The D/D domain thus limits significant dissociation of the monomers and allows the regulatory subunit to sense its environment for binding partners such as the catalytic subunit and cAMP effectively.
Our dimer structure reveals an isoform-specific interaction site between the RIα monomers that allows the interpretation of previous cAMP binding studies. We show that there is long-range communication realized through the interface. Specifically, the interface limits access and dynamics of the CNB-A compared to CNB-B. An earlier study reported that when the cAMP-binding pocket of CNB-A is manipulated, solution structural changes are seen as reflected by the Stokes radius. Corresponding mutations in the CNB-B binding pocket on the other hand does not change the Stokes radius of RIα (Herberg et al., 1996). Thus, binding cAMP to the accessible CNB-B first is favorable over the more structurally constrained CNB-A and results in a higher affinity site for CNB-B compared to CNB-A. Urea unfolding studies also support our structure in that the CNB-B unfolds first (Cànaves et al., 2000; Leon et al., 2000). The CNB-A domain however, is much more stable and resistant to unfolding with urea. Contributing to this behavior could be that the CNB-A is positioned in the dimer so that it is protected and stably docked via the N3A interface. Finally, cooperative cAMP binding to the dimer is accomplished through the interface, where communication and sensing of the two monomers can be transmitted.
The realization that Carney complex mutations are found on the dimer interface points to its functional importance for RIα. The in vitro studies presented in this work correlate well with previous analysis of CNC mutant proteins. It was shown that CNC mutant PKA is easier to activate and results in overactive catalytic subunits (Horvath et al., 2010). In patients with CNC where haploinsufficiency of RIα is not the culprit, the increase in PKA activity is likely the result of disrupted C subunit inhibition by the mutant RIα. The holoenzyme activation of the CNC mutant proteins with interface residue changes shows this is due to a lower EC50 compared to wild-type. These mutant holoenzymes display a lowered cooperativity with a Hill-Slope of 1.4. This is similar to the cooperativity seen with the heterodimeric holoenzyme, which is composed of just one RIα monomer and one catalytic subunit and suggests that disrupting the interface essentially uncouples the established communication between CNB domains within the holoenzyme as well. A significant implication of our work is that cooperativity, while fine-tuning the response of PKA in the highly variable cellular environment, is also a crucial component to PKA function that can lead to disease when disrupted.
In summary, this study reveals an isoform-specific RIα homodimer interface that contributes to understanding the in vivo function of RIα. With the knowledge gained from this structure, it would be intriguing to test the Carney complex interface mutants in vivo and explore if their cellular localization is affected. Support for a conformational equilibrium existing for PKA's R subunits is surfacing with recent studies utilizing nuclear magnetic resonance and molecular dynamics to analyze the distribution of RIα in the H and B-forms (Akimoto et al., 2013). Accordingly, in the apo state RIα is toggling between and populating both the C subunit-bound and the cAMP-bound conformations. Addition of either binding partner pushes the equilibrium further to the corresponding conformation, suggesting that specific mutations could also alter the population of states within this equilibrium. While this study has filled a gap in the PKA structural library, the full RIα holoenzyme structure remains unsolved and is now a major target of our crystallographic efforts that will further aid in understanding the role the interface plays for PKA.
Experimental Procedures
Protein Expression and Purification
Bovine full-length (1–379) wild-type and mutant RIα proteins were purified as described previously (Su et al., 1995; Wu et al., 2004b). Mutations to analyze the interface biochemically were generated by QuickChange site-directed mutagenesis. Expression was carried out at 16°C for 16–20 hr in Escherichia coli BL21 (DE3) from Novagen suspended in YT Media. In summary, a combination of ammonium sulfate precipitation, affinity chromatography with cAMP-resin, and size-exclusion chromatography were utilized to achieve > 94% pure protein. Briefly, following expression, cells were lysed with a Microfluidizer processor at 18,000 psi, centrifuged at 16,000 rpm, and the soluble fraction was then ammonium sulfate precipitated. The ammonium sulfate pellet was resuspended and batch-bound overnight to cAMP resin. After elution from the resin with 40 mM cAMP, the elution was applied to a Superdex 200 gel filtration column utilizing a buffer containing 50 mM MES (pH 5.8), 200 mM NaCl, 2 mM EGTA, 2 mM EDTA, and 5 mM dithiothreitol (DTT).
Crystallization of the RIα Homodimer
Purified RIα (1–379) was concentrated up to 12 mg/ml with a 30K MWCO concentrator and cAMP was added to 3-fold molar exess of RIα. Protein was then centrifuged at 14,000 rpm for 10 min to remove particulate contamination and was set up by hanging-drop vapor diffusion with various commercially available screens at 50% protein plus 50% screen buffer at 4°C and 22°C. Several crystal hits with a similar tetragonal bipyramid shape were obtained but the optimized condition that yielded the best diffracting crystals was composed of 0.125 M sodium acetate (pH 5) and 2 M sodium formate with the protein at a final concentration of 4 mg/ml grown in a 2 μl drop. SDS gel analysis of the crystals confirmed the full-length RIa and not a proteolyzed monomer form.
Structure Determination and Refinement
Crystals were flash-frozen in liquid nitrogen after soaking in a cryoprotectant composed of mother liquor supplemented with 20% PEG 400. X-ray diffraction data sets were collected from one crystal at the Advanced Light Source, Berkeley California beamline 8.2.1 and processed with HKL2000 (Otwinowski, 1997). This resulted in a tetragonal P41212 space group and cell dimensions of a = b = 104.7 Å and c = 218.3 (Table 1). The best data set resulted in a structure solution with final resolution of 3.88 Å. The full-length homodimer crystals contain a distinct morphology and space group as compared to previous structures of the deletion mutant monomers, which have a hexagonal space group of P6 (Badireddy et al., 2011; Su et al., 1995; Wu et al., 2004a, 2004b). While in the previous monomer structures each asymmetric unit contained one monomer, this structure now contains the dimer in the asymmetric unit. The structure was solved using the RIα monomer structure with PDB code 1RGS as a molecular replacement probe with the resulting LLG score of 1,634 and TFZ score of 37.9 for using two copies of 1RGS as the search model (Su et al., 1995). Model building was carried out in Coot (Emsley and Cowtan, 2004). Initially, CNS with DEN restraints-assisted refinement was carried out (Schröder et al., 2010). This was followed by Refmac refinement with noncrystallographic symmetry restraints applied (Murshudov et al., 1999). The positive Fo-Fc density at 3-σ level is visible for each of the four cAMP molecules in their binding pockets. The final model includes residues 105–376 for one chain (R), where the first three residues are displayed as alanines due to lack of side chain density. Residues 108–376 are included for the other monomer chain (R′). The R and R-free are 0.26 and 0.28, respectively, and the structure model has a good geometry as evaluated with PROCHECK (Laskowski et al., 1993; Table 1).
Small-Angle X-Ray Scattering
RIα full-length mutant proteins were purified as described above and the final buffer composition was 50 mM MES (pH 5.8), 200 mM NaCl, 2 mM EGTA, 2 mM EDTA, and 5 mM DTT. The protein concentrations for the SAXS experiments were in the range of 2–5 mg/ml. The SAXS data were collected for 90 min for the Y120A experiments sample and 240 min for the K121A sample. SAXS data were acquired at 12°C using the SAXSess (Anton Paar) line collimation (10 mm) instrument at the University of Utah. Data were collected using an image plate detector, and reduced to I(q) versus q (q = (4πsinθ)/λ; 2θ is the scattering angle; λ = 1.54 Å CuKα) using the program SAXSquant 2.0. X-ray scattering from the protein was obtained by subtracting the scattering of the normalized buffer blank. The theoretical scattering profile for the RIα homodimer crystal structure was calculated using the FoXS server (Schneidman-Duhovny et al., 2010, 2013). P(r) functions for the experimental and theoretical scattering were calculated using GNOM (as implemented in ATSAS 2.5.0; Svergun, 1992). The experimental scattering data were corrected for smearing effects in GNOM using beam length profile parameters.
PKA Activation/IP-20
The activation of wild-type and mutants of RIα holoenzymes was investigated by a fluorescence polarization assay (Saldanha et al., 2006). Holoenzyme was formed with molar ratio of 1.2 mol RIα to 1 mol C subunit and diluted in buffer containing 20 mM HEPES (pH 7), 75 mM KCI, 0.005% Triton X-100, 1 mM ATP, 1 mM DTT, and 10 mM MgCl2. A20-residue long PKA inhibitor peptide (IP-20) labeled with succinimidyl activated carboxyfluorescein (FAM-IP20) was added to RIα holoenzymes, followed by the addition of cAMP to activate PKA. The working concentration of C subunit was 12 nM, FAM-IP20 was 2 nM, and 2-fold serial dilutions from 2,000 to 0 nM cAMP were added to initiate holoenzyme dissociation and FAM-IP20 binding to the C subunit. Fluorescence polarization readings with excitation at 485 nm and emission at 535 nm were carried out with a GENios Pro micro-plate reader (Tecan) using black flat-bottom Costar assay plates. Each protein was tested in quadruplicate and the data were analyzed with Prism 4.
Supplementary Material
Acknowledgments
This work was funded in part by NIH grant GM 034921 to S.S.T. J.G.H.B. is supported by the San Diego Fellowship. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the US Department of Energy under contract no. DE-AC02-05CH11231. We thank Dr. Jill Trewhella for the generous use of small-angle scattering and laboratory facilities at the University of Utah. X-ray scattering data were collected at the University of Utah using facilities that were supported by US Department of Energy grant DE-FG02-05ER64026 (to Jill Trewhella).
Footnotes
Accession Numbers: The PDB accession number for the coordinates and structure factors reported in this paper is 4MX3.
Supplemental Information: Supplemental Information includes four figures and can be found with this article online at http://dx.doi.org/10.1016/j.str.2013.10.012.
References
- Akimoto M, Selvaratnam R, McNicholl ET, Verma G, Taylor SS, Melacini G. Signaling through dynamic linkers as revealed by PKA. Proc Natl Acad Sci USA. 2013;110:14231–14236. doi: 10.1073/pnas.1312644110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amieux PS, McKnight GS. The essential role of RI alpha in the maintenance of regulated PKA activity. Ann N Y Acad Sci. 2002;968:75–95. doi: 10.1111/j.1749-6632.2002.tb04328.x. [DOI] [PubMed] [Google Scholar]
- Badireddy S, Yunfeng G, Ritchie M, Akamine P, Wu J, Kim CW, Taylor SS, Qingsong L, Swaminathan K, Anand GS. Cyclic AMP analog blocks kinase activation by stabilizing inactive conformation: conformational selection highlights a new concept in allosteric inhibitor design. Mol Cell Proteomics. 2011;10:M110 004390. doi: 10.1074/mcp.M110.004390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boshart M, Weih F, Nichols M, Schütz G. The tissue-specific extinguisher locus TSE1 encodes a regulatory subunit of cAMP-dependent protein kinase. Cell. 1991;66:849–859. doi: 10.1016/0092-8674(91)90432-x. [DOI] [PubMed] [Google Scholar]
- Brown SH, Wu J, Kim C, Alberto K, Taylor SS. Novel isoform-specific interfaces revealed by PKA RIIbeta holoenzyme structures. J Mol Biol. 2009;393:1070–1082. doi: 10.1016/j.jmb.2009.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadd G, McKnight GS. Distinct patterns of cAMP-dependent protein kinase gene expression in mouse brain. Neuron. 1989;3:71–79. doi: 10.1016/0896-6273(89)90116-5. [DOI] [PubMed] [Google Scholar]
- Cànaves JM, Leon DA, Taylor SS. Consequences of cAMP-binding site mutations on the structural stability of the type I regulatory subunit of cAMP-dependent protein kinase. Biochemistry. 2000;39:15022–15031. doi: 10.1021/bi001563q. [DOI] [PubMed] [Google Scholar]
- Cheng CY, Yang J, Taylor SS, Blumenthal DK. Sensing domain dynamics in protein kinase A-Ialpha complexes by solution X-ray scattering. J Biol Chem. 2009;284:35916–35925. doi: 10.1074/jbc.M109.059493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho-Chung YS, Pepe S, Clair T, Budillon A, Nesterova M. cAMP-dependent protein kinase: role in normal and malignant growth. Crit Rev Oncol Hematol. 1995;21:33–61. doi: 10.1016/1040-8428(94)00166-9. [DOI] [PubMed] [Google Scholar]
- Diller TC, Madhusudan, Xuong NH, Taylor SS. Molecular basis for regulatory subunit diversity in cAMP-dependent protein kinase: crystal structure of the type II beta regulatory subunit. Structure. 2001;9:73–82. doi: 10.1016/s0969-2126(00)00556-6. [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Erlichman J, Rubin CS, Rosen OM. Physical properties of a purified cyclic adenosine 3′:5′-monophosphate-dependent protein kinase from bovine heart muscle. J Biol Chem. 1973;248:7607–7609. [PubMed] [Google Scholar]
- Greene EL, Horvath AD, Nesterova M, Giatzakis C, Bossis I, Stratakis CA. In vitro functional studies of naturally occurring pathogenic PRKAR1A mutations that are not subject to nonsense mRNA decay. Hum Mutat. 2008;29:633–639. doi: 10.1002/humu.20688. [DOI] [PubMed] [Google Scholar]
- Herberg FW, Dostmann WR, Zorn M, Davis SJ, Taylor SS. Crosstalk between domains in the regulatory subunit of cAMP-dependent protein kinase: influence of amino terminus on cAMP binding and holoenzyme formation. Biochemistry. 1994;33:7485–7494. doi: 10.1021/bi00189a057. [DOI] [PubMed] [Google Scholar]
- Herberg FW, Taylor SS, Dostmann WR. Active site mutations define the pathway for the cooperative activation of cAMP-dependent protein kinase. Biochemistry. 1996;35:2934–2942. doi: 10.1021/bi951647c. [DOI] [PubMed] [Google Scholar]
- Horvath A, Bertherat J, Groussin L, Guillaud-Bataille M, Tsang K, Cazabat L, Libé R, Remmers E, René-Corail F, Faucz FR, et al. Mutations and polymorphisms in the gene encoding regulatory subunit type 1-alpha of protein kinase A (PRKAR1A): an update. Hum Mutat. 2010;31:369–379. doi: 10.1002/humu.21178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilouz R, Bubis J, Wu J, Yim YY, Deal MS, Kornev AP, Ma Y, Blumenthal DK, Taylor SS. Localization and quaternary structure of the PKA RIβ holoenzyme. Proc Natl Acad Sci USA. 2012;109:12443–12448. doi: 10.1073/pnas.1209538109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones KW, Shapero MH, Chevrette M, Fournier RE. Subtractive hybridization cloning of a tissue-specific extinguisher: TSE1 encodes a regulatory subunit of protein kinase A. Cell. 1991;66:861–872. doi: 10.1016/0092-8674(91)90433-y. [DOI] [PubMed] [Google Scholar]
- Kammer GM. Deficient protein kinase a in systemic lupus erythematosus: a disorder of T lymphocyte signal transduction. Ann N Y Acad Sci. 2002;968:96–105. doi: 10.1111/j.1749-6632.2002.tb04329.x. [DOI] [PubMed] [Google Scholar]
- Kammer GM, Khan IU, Kammer JA, Olorenshaw I, Mathis D. Deficient type I protein kinase A isozyme activity in systemic lupus erythematosus T lymphocytes: II. Abnormal isozyme kinetics. J Immunol. 1996;157:2690–2698. [PubMed] [Google Scholar]
- Kim C, Xuong NH, Taylor SS. Crystal structure of a complex between the catalytic and regulatory (RIalpha) subunits of PKA. Science. 2005;307:690–696. doi: 10.1126/science.1104607. [DOI] [PubMed] [Google Scholar]
- Kim C, Cheng CY, Saldanha SA, Taylor SS. PKA-I holoenzyme structure reveals a mechanism for cAMP-dependent activation. Cell. 2007;130:1032–1043. doi: 10.1016/j.cell.2007.07.018. [DOI] [PubMed] [Google Scholar]
- Kirschner LS, Carney JA, Pack SD, Taymans SE, Giatzakis C, Cho YS, Cho-Chung YS, Stratakis CA. Mutations of the gene encoding the protein kinase A type I-alpha regulatory subunit in patients with the Carney complex. Nat Genet. 2000;26:89–92. doi: 10.1038/79238. [DOI] [PubMed] [Google Scholar]
- Kornev AP, Taylor SS, Ten Eyck LF. A generalized allosteric mechanism for cis-regulated cyclic nucleotide binding domains. PLoS Comput Biol. 2008;4:e1000056. doi: 10.1371/journal.pcbi.1000056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- Kuo JF, Greengard P. Cyclic nucleotide-dependent protein kinases. IV. Widespread occurrence of adenosine 3′,5′-monophosphate-dependent protein kinase in various tissues and phyla of the animal kingdom. Proc Natl Acad Sci USA. 1969;64:1349–1355. doi: 10.1073/pnas.64.4.1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski RA, Macarthur MW, Moss DS, Thornton JM. PROCHECK - a program to check the stereochemical quality of protein structures. J Appl Cryst. 1993;26:283–291. [Google Scholar]
- Laxminarayana D, Khan IU, Mishra N, Olorenshaw I, Taskén K, Kammer GM. Diminished levels of protein kinase A RI alpha and RI beta transcripts and proteins in systemic lupus erythematosus T lymphocytes. J Immunol. 1999;162:5639–5648. [PubMed] [Google Scholar]
- Leon DA, Canaves JM, Taylor SS. Probing the multidomain structure of the type I regulatory subunit of cAMP-dependent protein kinase using mutational analysis: role and environment of endogenous tryptophans. Biochemistry. 2000;39:5662–5671. doi: 10.1021/bi992819z. [DOI] [PubMed] [Google Scholar]
- Li F, Gangal M, Jones JM, Deich J, Lovett KE, Taylor SS, Johnson DA. Consequences of cAMP and catalytic-subunit binding on the flexibility of the A-kinase regulatory subunit. Biochemistry. 2000;39:15626–15632. doi: 10.1021/bi002196l. [DOI] [PubMed] [Google Scholar]
- Miller WR. Regulatory subunits of PKA and breast cancer. Ann N Y Acad Sci. 2002;968:37–48. doi: 10.1111/j.1749-6632.2002.tb04325.x. [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Lebedev A, Wilson KS, Dodson EJ. Efficient anisotropic refinement of macromolecular structures using FFT. Acta Crystallogr D Biol Crystallogr. 1999;55:247–255. doi: 10.1107/S090744499801405X. [DOI] [PubMed] [Google Scholar]
- Otwinowski ZMW. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Potter RL, Taylor SS. Relationships between structural domains and function in the regulatory subunit of cAMP-dependent protein kinases I and II from porcine skeletal muscle. J Biol Chem. 1979;254:2413–2418. [PubMed] [Google Scholar]
- Rannels SR, Corbin JD. Studies of functional domains of the regulatory subunit from cAMP-dependent protein kinase isozyme I. J Cyclic Nucleotide Res. 1980;6:201–215. [PubMed] [Google Scholar]
- Romero P, Obradovic Z, Li X, Garner EC, Brown CJ, Dunker AK. Sequence complexity of disordered protein. Proteins. 2001;42:38–48. doi: 10.1002/1097-0134(20010101)42:1<38::aid-prot50>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- Rubin CS. Characterization and comparison of membrane-associated and cytosolic cAMP-dependent protein kinases. Studies on human erythrocyte protein kinases. J Biol Chem. 1979;254:12439–12449. [PubMed] [Google Scholar]
- Saldanha SA, Kaler G, Cottam HB, Abagyan R, Taylor SS. Assay principle for modulators of protein-protein interactions and its application to non-ATP-competitive ligands targeting protein kinase A. Anal Chem. 2006;78:8265–8272. doi: 10.1021/ac061104g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneidman-Duhovny D, Hammel M, Sali A. FoXS: a web server for rapid computation and fitting of SAXS profiles. Nucleic Acids Res. 2010;38(Web Server issue):W540–W544. doi: 10.1093/nar/gkq461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneidman-Duhovny D, Hammel M, Tainer JA, Sali A. Accurate SAXS profile computation and its assessment by contrast variation experiments. Biophys J. 2013;105:962–974. doi: 10.1016/j.bpj.2013.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schröder GF, Levitt M, Brunger AT. Super-resolution biomolecular crystallography with low-resolution data. Nature. 2010;464:1218–1222. doi: 10.1038/nature08892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y, Dostmann WR, Herberg FW, Durick K, Xuong NH, Ten Eyck L, Taylor SS, Varughese KI. Regulatory subunit of protein kinase A: structure of deletion mutant with cAMP binding domains. Science. 1995;269:807–813. doi: 10.1126/science.7638597. [DOI] [PubMed] [Google Scholar]
- Svergun DI. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J Appl Cryst. 1992;25:495–503. [Google Scholar]
- Veugelers M, Wilkes D, Burton K, McDermott DA, Song Y, Goldstein MM, La Perle K, Vaughan CJ, O'Hagan A, Bennett KR, et al. Comparative PRKAR1A genotype-phenotype analyses in humans with Carney complex and prkar1a haploinsufficient mice. Proc Natl Acad Sci USA. 2004;101:14222–14227. doi: 10.1073/pnas.0405535101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigil D, Blumenthal DK, Heller WT, Brown S, Canaves JM, Taylor SS, Trewhella J. Conformational differences among solution structures of the type Ialpha, IIalpha and IIbeta protein kinase A regulatory subunit homodimers: role of the linker regions. J Mol Biol. 2004;337:1183–1194. doi: 10.1016/j.jmb.2004.02.028. [DOI] [PubMed] [Google Scholar]
- Walsh DA, Perkins JP, Krebs EG. An adenosine 3′,5′-monophosphate-dependant protein kinase from rabbit skeletal muscle. J Biol Chem. 1968;243:3763–3765. [PubMed] [Google Scholar]
- Wu J, Brown S, Xuong NH, Taylor SS. RIalpha subunit of PKA: a cAMP-free structure reveals a hydrophobic capping mechanism for docking cAMP into site B. Structure. 2004a;12:1057–1065. doi: 10.1016/j.str.2004.03.022. [DOI] [PubMed] [Google Scholar]
- Wu J, Jones JM, Nguyen-Huu X, Ten Eyck LF, Taylor SS. Crystal structures of RIalpha subunit of cyclic adenosine 5′ -monophosphate (cAMP)-dependent protein kinase complexed with (Rp)-adenosine 3′,5′-cyclic monophosphothioate and (Sp)-adenosine 3′,5′ -cyclic monophosphothioate, the phosphothioate analogues of cAMP. Biochemistry. 2004b;43:6620–6629. doi: 10.1021/bi0302503. [DOI] [PubMed] [Google Scholar]
- Wu J, Brown SH, von Daake S, Taylor SS. PKA type IIalpha holoenzyme reveals a combinatorial strategy for isoform diversity. Science. 2007;318:274–279. doi: 10.1126/science.1146447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Z, Kirschner LS. The Carney complex gene PRKAR1A plays an essential role in cardiac development and myxomagenesis. Trends Cardiovasc Med. 2009;19:44–49. doi: 10.1016/j.tcm.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Smith-Nguyen EV, Keshwani MM, Deal MS, Kornev AP, Taylor SS. Structure and allostery of the PKA RIIβ tetrameric holoenzyme. Science. 2012;335:712–716. doi: 10.1126/science.1213979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoller MJ, Kerlavage AR, Taylor SS. Structural comparisons of cAMP-dependent protein kinases I and II from porcine skeletal muscle. J Biol Chem. 1979;254:2408–2412. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.