

Abstract
Previously reported pyrrolones, such as TDR32570, exhibited potential as antimalarial agents; however, while these compounds have potent antimalarial activity, they suffer from poor aqueous solubility and metabolic instability. Here, further structure–activity relationship studies are described that aimed to solve the developability issues associated with this series of compounds. In particular, further modifications to the lead pyrrolone, involving replacement of a phenyl ring with a piperidine and removal of a potentially metabolically labile ester by a scaffold hop, gave rise to derivatives with improved in vitro antimalarial activities against Plasmodium falciparum K1, a chloroquine-and pyrimethamine-resistant parasite strain, with some derivatives exhibiting good selectivity for parasite over mammalian (L6) cells. Three representative compounds were selected for evaluation in a rodent model of malaria infection, and the best compound showed improved ability to decrease parasitaemia and a slight increase in survival.
Keywords: antiprotozoal agents, malaria, Plasmodium falciparum, pyrrolones, structure–activity relationships
Introduction
Malaria is a serious disease endemic in tropical and subtropical regions of the world. It is a major threat to public health in more than 100 countries and is responsible for >200 million clinical cases each year and probably about 1 million deaths, the majority of whom are young children and pregnant women.[1–4] In addition, malaria is responsible for a huge economic impact in endemic countries.[2] With increasing globalisation and global warming, there is a risk of malaria spreading to new areas.[3] No vaccine is currently available for malaria, and the resistance of the protozoa to clinically used chemotherapeutic agents is increasingly common. Therefore, an urgent need exists to develop new classes of antimalarial drugs that operate by novel mechanisms of action.
We have previously reported on the discovery and structure–activity relationship (SAR) of a series of pyrrolones, which have potent antimalarial activity;[5] these were initially discovered through a phenotypic screening process conducted by the World Health Organisation (WHO) Programme for Research and Training in Tropical Diseases. The prototypical compound TDR32750 (Figure 1 a) showed potent antimalarial activity against a panel of strains of Plasmodium falciparum and significant depression of parasitaemia (>99 %) in the Plasmodium berghei mouse model of malaria when dosed intraperitoneally. Unfortunately, the compound has low oral bioavailability; good oral bioavailability is a requirement for most target product profiles for malaria. While the precise reason for the low bioavailability is unknown, it is most likely due to a combination of metabolic instability (as evidenced by studies in hepatic microsomes) and low aqueous solubility.[5] The ester linkage is also susceptible to hydrolysis in blood, which contributes to the overall systemic clearance, as previously reported. Our previous SAR studies around TDR32750[5] encompassed changes to the A-, B-and C-rings (Figure 1 a), which led to the conclusion that the A-ring was reasonably tolerant of changes, unlike the B-ring. Modifications to the C-ring were not investigated apart from replacement of the ester with a variety of amides, which led to compounds with decreased activity.
Figure 1.
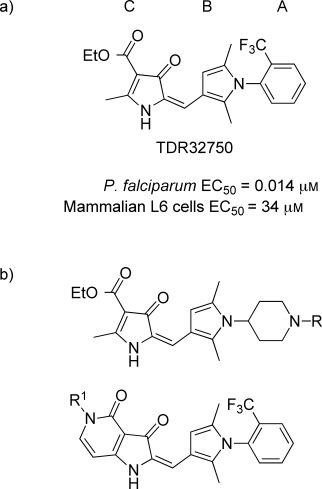
a) Prototypical compound TDR32750, structure and in vitro activities.[5] b) Generic structure of compounds prepared in this study.
In order to improve the physicochemical properties of the molecules and the prospects for further development of the compound series, our aim was to decrease the clog D value to less than three. Here, we report on changes to the A-ring and C-ring, which have led to compounds with enhanced antimalarial activity and improved physicochemical properties. In particular, our work focused around some piperidine analogues of the A-ring in our lead molecule (Figure 1 b). The piperidine moiety has an amino group, which when unconjugated is basic and should improve the solubility properties.
The lead compound, TDR32750, contains an ester in the C-ring; we were concerned that this might be a metabolic liability due to hydrolysis by esterases, since previous work has confirmed that there is gradual degradation of the ester in vivo.[5] However, this is probably not the only mode of metabolism, as we have also shown that the compound undergoes cytochrome P450 (CYP450)-mediated degradation in hepatic microsomes.[5] Nevertheless, we thought it prudent to try and replace the ester to improve the clearance properties. Previously, we reported efforts at replacing the ester with an amide, but this led to loss of activity; possibly due to the amide causing a conformational change compared to the ester.[5] In this paper, we report the effect of cyclising the ester to form a fused pyridone, which might lock the compound into a more favourable conformation (Figure 1 b).
Results and Discussion
Chemistry
The starting point for the synthesis of the A-ring variants incorporating a piperidine substituent was pyrrolone 3, which was prepared as reported previously (Scheme 1).[5] Pyrrolone 3 (which was not stored but used immediately) was then condensed with the required 3-formylpyrrole 4-aminopiperidine (7, 16 a–z, 16 aa–aj) in the presence of potassium hydrogen sulfate,[6], [7] to give predominantly the (E)-isomer of the requisite pyrrolone derivative (17–54).1
Scheme 1.
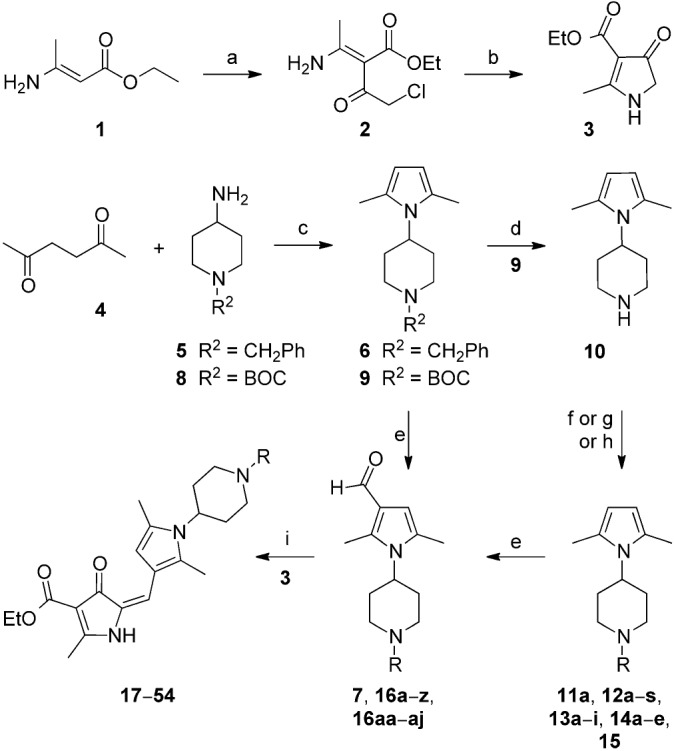
General strategy for the synthesis of piperidine derivatives. Regents and conditions: a) chloroacetyl chloride, pyridine, 0 °C, 30 min, 75 % yield; b) KOH, EtOH, 3 h, 0 °C, 95 % yield; c) p-TsOH bound on silica gel, microwave (0–400 W at 2.45 GHz), 180 °C, 15–20 min, 80–90 % yield; or p-TsOH, toluene, Dean–Stark apparatus, 90 °C, 3 h, 80–90 %; d) 4 m HCl/dioxane, 0 °C, 3 h, 66 % yield; e) POCl3, DMF, 100 °C, 3 h, 80–95 % yield; f) ROSO2CH3, Et3N, NaHCO3, CH3CN, 85 °C, 12 h, 50–70 %; g) RBr, DIPEA, DMF, 85 °C, 3 h, 40–70 % yield; h) alkyl/aryl/heterocyclic aldehyde, sodium triacetoxyborohydride, MeCN, RT, 8 h, 60–80 % yield; i) KHSO4, EtOH, 3 h, reflux, 80–95 % yield. R groups are given in Table 1.
3-Formylpyrrole intermediates 16 a–z and 16 aa–aj were prepared in a four-step sequence: 1) condensation of tert-butyloxycarbonyl (BOC)-protected 4-aminopiperidine (8) with 2,5-hexandione 4 (Paal–Knorr pyrrole synthesis); 2) deprotection of the BOC group; 3) coupling either by alkylation or reductive amination to give a range of piperidine derivatives (11 a–15); 4) carbonylation using a Vilsmeier–Haack reaction (85–95 % yield).[8], [9] The complete range of compounds prepared is shown in Table 1.
Table 1.
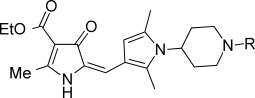
Activity data for A-ring variants against Plasmodium falciparum.
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Compd[a] | R | EC50[b] [μm] | cLog D[c] | Solubility[d] [μm] | CLint[e] | |||
| P. falc. | L6 cells | (pH 7.4) | pH 2.0 | pH 6.5 | water | [μL min−1 mg−1] | ||
| TDR32750 | 0.014 | 34 | 5.0 | 15–30 | 7–15 | >125 | 50# | |
| 17 |  |
0.035 | 20 | 2.1 | 56–110 | 14–28 | >125 | 60# |
| 18 | -H | 12 | >250 | 0.7 | n.d. | n.d. | n.d. | n.d. |
| 19 | -CH3 | 1.4 | 7.8 | 0.9 | n.d. | n.d. | n.d. | n.d. |
| 20 | -C2H5 | 2.4 | 7.6 | 1.2 | >250 | >250 | n.d. | 11# |
| 21 | -(CH2)2C(CH3)2 | 0.19 | 22 | 2.0 | n.d. | n.d. | n.d. | n.d. |
| 22 | -(CH2)2CH3 | 1.1 | 6.6 | 1.4 | n.d. | n.d. | n.d. | n.d. |
| 23 | -CH2C(CH3)3 | 0.058 | 88 | 2.0 | n.d. | n.d. | n.d. | n.d. |
| 24 | 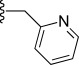 |
0.38 | 5.9 | 1.6 | n.d. | n.d. | n.d. | n.d. |
| 25 | 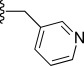 |
0.46 | 27 | 1.6 | n.d. | n.d. | n.d. | n.d. |
| 26 | 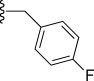 |
0.024 | 51 | 2.2 | n.d. | n.d. | >125 | 117# |
| 27 | 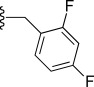 |
0.023 | 23 | 2.2 | n.d. | n.d. | >125 | 149# |
| 28 | 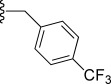 |
0.002 | 5 | 2.7 | 48-97 | 6–12 | >125 | 217§ |
| 29 |  |
0.0005 | 1.7 | 2.6 | n.d. | n.d. | >125 | 952§ |
| 30 | 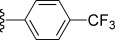 |
0.024 | 10 | 2.7 | n.d. | n.d. | >125 | n.d. |
| 31 | 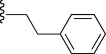 |
0.32 | 107 | 2.2 | n.d. | n.d. | n.d. | 76# |
| 32 |  |
0.083 | 72 | 2.2 | 100–200 | 25–51 | n.d. | 120# |
| 33 | 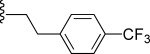 |
0.13 | 19 | 2.6 | n.d. | n.d. | n.d. | 215# |
| 34 |  |
0.096 | 22 | 2.3 | 52–100 | 13–26 | n.d. | 114# |
| 35 | 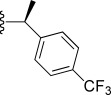 |
0.007 | 4.26 | 2.9 | n.d. | n.d. | n.d. | 88§ |
| 36 | 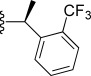 |
0.001 | 3 | 2.9 | n.d. | n.d. | >125 | 190§ |
| 37 | 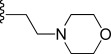 |
5.3 | 89 | 1.3 | n.d. | n.d. | n.d. | n.d. |
| 38 | 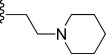 |
2.4 | 97 | 1.5 | n.d. | n.d. | n.d. | n.d. |
| 39 | 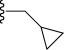 |
3.5 | 14 | 1.7 | n.d. | n.d. | n.d. | n.d. |
| 40 | 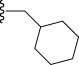 |
0.016 | 13 | 2.1 | n.d. | n.d. | >125 | 78§ |
| 41 | 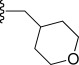 |
0.11 | 43 | 1.6 | n.d. | n.d. | n.d. | n.d. |
| 42 | 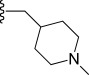 |
1.4 | 81 | 1.4 | n.d. | n.d. | n.d. | n.d. |
| 43 | 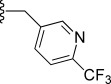 |
0.004 | 49 | 2.2 | n.d. | n.d. | >125 | 177§ |
| 44 |  |
0.0004 | 1.2 | 2.6 | n.d. | n.d. | >125 | 248§ |
| 45 | 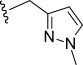 |
0.15 | 36 | 1.2 | n.d. | n.d. | n.d. | n.d. |
| 46 |  |
1.2 | 28 | 1.1 | n.d. | n.d. | n.d. | n.d. |
| 47 | 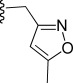 |
0.10 | 42 | 1.5 | n.d. | n.d. | n.d. | n.d. |
| 48 | 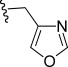 |
0.018 | 0.16 | 1.3 | n.d. | n.d. | >125 | <10§ |
| 49 | 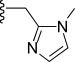 |
1.0 | 16 | 1.0 | n.d. | n.d. | n.d. | n.d. |
| 50 | 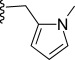 |
0.012 | 3 | 1.4 | n.d. | n.d. | n.d. | 55§ |
| 51 | 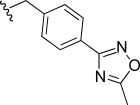 |
0.001 | 11 | 2.4 | n.d. | n.d. | >125 | n.d. |
| 52 | 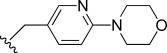 |
1.5 | 50 | 2.2 | n.d. | n.d. | n.d. | n.d. |
| 53 | 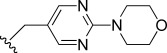 |
0.075 | 28 | 1.8 | n.d. | n.d. | n.d. | n.d. |
| 54 |  |
2.6 | 53 | 2.5 | n.d. | n.d. | n.d. | n.d. |
Overall yields: 40–85 %; reference compounds: chloroquine, EC50=0.095–0.172 μm (P. falciparum K1); podophyllotoxin, EC50=0.009–0.022 μm (L6 cells).
The EC50 values are the mean of two independent assays, which varied less than ±50 %; n.d=not determined.
Calculated using StarDrop (http://www.optibrium.com).
Measured using nephelometry.
Intrinsic clearance determined in vitro using mouse liver microsomes at Monash University (#) or at the University of Dundee (UK) (§).
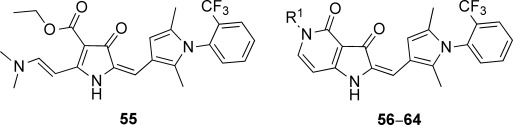
The pyrrolo [3,2-c] pyridine-3, 4-dione series, that is the C-ring analogues of TDR32750 incorporating a fused pyridone ring, were prepared according to Scheme 2. Reaction of the pyrrolone ring of TDR32750 with the diethyl acetal of N,N-dimethylformamide (DMF) gave key intermediate 55.[10] Reaction with an amine caused displacement of the dimethylamine and cyclisation to the required pyrrolo[3,2-c] derivatives (56–64).
Scheme 2.

a) DMF–diethyl acetal, 85 °C, 15 min, 100 % yield; b) R1NH2, iso-propyl alcohol, microwave (0–400 W at 2.45 GHz), 110 °C, 15–30 min, 30–70 % yield.
In vitro activity
Compounds were evaluated for activity against P. falciparum K1, a chloroquine-and pyrimethamine-resistant parasite strain, and counter-screened against mammalian L6 cells (Table 1). Many of the compounds showed potent antimalarial activity, with eight compounds showing EC50 values of less than 10 nm and two compounds (29 and 44) showing sub-nanomolar potencies. Most of the compounds showed good selectivity for parasite over mammalian cells.
A-Ring modifications
The following observations were made regarding the in vitro antimalarial activity for compounds with variations around the A-ring:
In general, simple alkyl substituents (18–23) led to compounds that exhibit a large decrease in activity compared with TDR32750. This also applied to a methylene cyclopropyl derivative (39). Interestingly, methylene cyclohexyl derivative 40 showed only a slight drop in activity compared with TDR32750, although adding heteroatoms to the ring to improve the physicochemical properties of the compounds was counter-productive for activity (41 and 42). Similarly, cyclohexyl analogues linked via an ethylene linker (37 and 38) showed decreased activity.
Where the substituent was an aromatic ring directly attached to the piperidine ring (30), the compound retained reasonably potent activity (EC50=0.024 μm); however, this compound would have decreased basicity at the piperidine ring.
Compounds with a phenyl ring separated from the piperidine by a methylene linker in general showed good potency (17, 26–29, 35, 36, 43, 44, 51).
Replacement of the phenyl ring by a heteroaromatic ring (to improve physicochemical properties) was generally detrimental to activity (24, 25, 45–47, 49, 52, 53). The marked exception to this was compound 43, a pyridine, albeit with an electron-withdrawing trifluoromethyl group, which is likely to significantly decrease the basicity of the pyridine nitrogen. The oxazole (48) and N-methylpyrrole (50) derivatives also retained activity.
Increasing the spacer length between the piperidine and the aromatic ring also led to a drop in activity (31–34).
The phenyl ring was tolerant to different substitutions, although the morpholine analogue (54) had greatly decreased activity.
C-Ring modifications
Modifications to the C-ring showed interesting activities in vitro against P. falciparum K1 (Table 2). The most active compounds displayed sub-micromolar activities; compounds 56, 57, 58 and 63 were found to be the most active. The following observations were made regarding the SAR of the C-ring-modified analogues:
Table 2.
Activity data for C-ring variants against Plasmodium falciparum.
 | ||||||
|---|---|---|---|---|---|---|
| Compd[a] | R1 | EC50[b] [μm] | cLog D[c] | Solubility [μm] | CLint[d] | |
| P. falc. | L6 cells | (pH 7.4) | water | [μL min mg−1 protein] | ||
| 55 | – | 0.061 | 56 | 2.8 | >125 | n.d. |
| 56 | H | 0.047 | 54 | 4.0 | >125 | 69 |
| 57 | CH3- | 0.014 | 29 | 4.2 | >125 | 286 |
| 58 | CH3CH2 | 0.021 | 36 | 4.5 | >125 | 381 |
| 59 | CH3CH2CH2 | 0.15 | 34 | 4.7 | n.d. | n.d. |
| 60 | 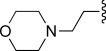 |
0.23 | 65 | 2.8 | n.d. | n.d. |
| 61 | (CH3)2NCH2CH2- | 0.25 | 35 | 2.4 | n.d. | n.d. |
| 62 | 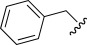 |
0.23 | 43 | 5.5 | n.d. | n.d. |
| 63 |  |
0.046 | 54 | 5.9 | 89 | n.d. |
| 64 | 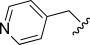 |
0.077 | 38 | 4.5 | >125 | n.d. |
Overall yields: 50–90 %; reference compounds: chloroquine, EC50=0.019–0.066 μm (P. falciparum K1); podophyllotoxin, EC50=0.012 μm (L6 cells).
The EC50 values are the mean of two independent assays, which varied less than ±50 %; n.d=not determined.
Calculated using StarDrop (http://www.optibrium.com).
Intrinsic clearance determined in vitro using mouse liver microsomes at the University of Dundee (UK).
Compound 56 with a free NH group on the pyrrolo[3,2-c]pyridine ring showed an EC50 value of 0.047 μm. Small alkyl substituents on the “pyridone” nitrogen were tolerated (57, R=Me: EC50=14 nm; 58, R=Et: EC50=21 nm), whilst a propyl substituent (59) led to a tenfold drop in activity.
Basic substituents did not appear to be tolerated, with compounds (60 and 61) exhibiting approximately a fivefold decrease in EC50 value relative to the unsubstituted analogue 56.
The para-trifluoromethylbenzyl (63) and para-pyridyl (64) analogues appeared to substantially retain potency compared with TDR32750, although the straight benzyl derivative (62) showed around a fivefold loss in activity.
In vitro drug metabolism and pharmacokinetics (DMPK)
For key compounds, metabolic stability when incubated with mouse liver microsomes was measured, and for some compounds solubility was also assessed. For the A-ring variants, the solubility increased, particularly at low pH, probably due to the basic nature of the piperidine ring. In those derivatives with an aromatic or benzyl substituent, the basicity of the amine is decreased, which probably explains the lower solubility at pH 6.5.
Disappointingly, the in vitro intrinsic clearance (CLint) values were all rather high—the preferred value is <50 μL min−1 mg−1 protein, although the aim is for a value of less than 20 μL min−1 mg−1. The most stable compounds were 17, 20, 31, 35, 40, 48, and 50, with derivatives 20 and 31 being relatively inactive. Compound 48 is particularly stable, presumably due to the relatively low clog D value and the metabolic stability of the isoxazole ring.
For C-ring variants, the most active compounds (56–58) were investigated for microsomal stability. Compound 56 showed moderate stability, whilst the N-alkyl congeners were less stable, possibly due to CYP450-mediated metabolism of the alkyl groups.
In vivo studies
Three compounds were selected for further study in vivo. Compound 43 was selected from the A-ring-modified series, as it represents a compound with good potency against the malaria parasite and very good selectivity compared with mammalian cells, albeit with a slightly increased microsomal instability. Compounds 56 and 57 were chosen as C-ring-modified derivatives, and also for their potency, selectivity and clog D value. Compounds 57 and 58 have very similar properties; compound 57 was chosen as it has marginally better properties. Multidose oral efficacy studies of compounds 43, 56, and 57 against green fluorescent protein (GFP)-transfected P. berghei ANKA-infected mice were conducted according to the standard protocol (Peters), with test compounds administered either per oral (po) or via intraperitoneal (ip) injection (Table 3).
Table 3.
In vivo antimalarial activity against green fluorescent protein (GFP)-transfected Plasmodium berghei ANKA.[a]
| Compd | Dose [mg kg−1 day−1] | Route | [%] Reduction parasitaemia | Survival [days] |
|---|---|---|---|---|
| 43 | 50 | ip | 10.0 | 4 |
| 56 | 50 | ip | 21.8 | 4 |
| 57 | 50 | ip | 99.9 | 13.7 |
| 43 | 100 | po | 0.0 | 4 |
| 56 | 100 | po | 13.6 | 4 |
| 57 | 100 | po | 50.5 | 8 |
| Chloroquine | 10 | ip | 99.97 | 20 |
| TDR32570 | 100 | po | 25.5 | 7.7 |
| Untreated control | – | – | – | 7 |
Animals were dosed four times a day at the stated dose. Route of administration: per oral (po); intraperitoneal (ip). Formulation: 10 % DMSO in water.
Both via po and ip administration, compound 57 was the most efficacious in terms of decrease of parasitaemia. At a dose of 50 mg kg−1 ip, compound 57 decreased the level of parasitaemia by 99.9 %, and there was a significant increase in survival time. When dosed orally, the decrease in parasitaemia was less at 50.5 %. Compound 57 shows limited microsomal stability, so the decrease in oral activity of the compound is likely to be due to poorer exposure as a consequence of first-pass metabolism, but further work is required to confirm this hypothesis. Compound 43 failed to show significant activity in vivo, despite showing good potency in vitro. This might be due to poor pharmacokinetics, but this needs to be further investigated.
Conclusions
Modifications to our prototypical lead compound TDR32750 have been shown to yield compounds that retain in vitro activity. These changes include removal of the ester functionality that might be a point of metabolism and addition of a basic centre that should be helpful in increasing the solubility. Although these changes have not yet provided compounds with good oral in vivo activity, the results do suggest that there is further scope for the optimisation of the metabolic and physicochemical properties of this series, which could potentially lead to orally active antimalarial compounds.
Experimental Section
Profiling software: StarDrop version 5.3 with the P450 metabolism plug-in module (http://www.optibrium.com) was used to predict the sites of metabolism of the compounds. The same software was used to calculate the cLog D values for all compounds.
Chemistry: Chemicals and solvents were purchased from Sigma–Aldrich or Fluka and were used as received unless otherwise stated. Air-and moisture-sensitive reactions were carried out under an inert atmosphere of argon in oven-dried glassware. Analytical thin-layer chromatography (TLC) was performed on precoated TLC plates (layer 0.20 mm silica gel 60 with fluorescent indicator UV254; Merck). Developed plates were air-dried and analysed under a UV lamp (254/365 nm). Flash column chromatography was performed using prepacked silica gel cartridges (230–400 mesh, 40–63 μm; SiliCycle) using a Teledyne ISCO Combiflash Companion or Combiflash Retrieve. Microwave irradiation was conducted using a Biotage Initiator unit. The machine consists of a continuous focused microwave power delivery system with operator-selectable power output (0–400 W at 2.45 GHz). 1H NMR and 13C NMR spectra were recorded on a Bruker Avance II 500 spectrometer (1H at 500.1 MHz; 13C at 125.8 MHz) or a Bruker DPX300 spectrometer (1H at 300.1 MHz). Chemical shifts (δ) are expressed in ppm recorded using the residual solvent as the internal reference in all cases. Signal splitting patterns are described as singlet (s), doublet (d), triplet (t), quartet (q), pentet (p), multiplet (m), broad (br), or a combination thereof. Coupling constants (J) are quoted to the nearest 0.1 Hz. LCMS analyses were performed with either an Agilent HPLC 1100 series connected to a Bruker Daltonics MicroTOF or an Agilent Technologies 1200 series HPLC connected to an Agilent Technologies 6130 quadrupole spectrometer, where both instruments were connected to an Agilent diode array detector. Liquid chromatography–mass spectrometry (LCMS chromatographic separations were conducted with a Waters X bridge C18 column (50 mm×2.1 mm, 3.5 μm particle size), with a mobile phase of water/acetonitrile+0.1 % HCOOH, or water/acetonitrile+0.1 % NH3, using a linear gradient from 80:20 to 5:95 over 3.5 min and then held for 1.5 min, at a flow rate of 0.5 mL min−1. All compound samples evaluated in biological assays had a measured purity of ≥95 % (by total ion current (TIC) and UV) as determined using this analytical LCMS system. High-resolution mass spectrometry (HRMS) using electrospray ionisation was performed on a Bruker Daltonics MicrOTOF mass spectrometer.
1-Benzyl-4-(2,5-dimethyl-1H-pyrrol-1-yl)piperidine (6): 2,5-Hexandione (4) (2.0 g, 17.5 mmol), 4-amino-1-benzylpiperidine (5) (4.0 g, 21.0 mmol) and para-toluenesulfonic acid (p-TsOH) bound to silica gel (0.4 equiv mol−1) were mixed in an oven-dried pressure vials with magnetic stir bars, and heated twice (180 °C, 15 min) under microwave irradiation (0–400 W at 2.45 GHz) and then stirred for 15 min at RT. The reaction mixture was filtered, and the silica gel residue was washed with CH2Cl2 (10 mL). The solvent was removed in vacuo to give compound 6 as a light brown oil (3.8 g, 90 %): 1H NMR (500 MHz, CDCl3): δ=7.43–7.33 (m, 5 H), 5.82 (s, 2 H), 4.01–3.94 (m, 1 H), 3.64 (s, 2 H), 3.11–3.09 (m, 2 H), 2.39 (br s, 6 H), 2.38–2.32 (m, 2 H), 2.19–2.12 (dt, 2 H, J=11.9 Hz), 1.89–1.85 ppm (m, 2 H); MS (ESI+): m/z (%): 269.4 [M+H]+ (100).
1-(1-Benzylpiperidin-4-yl)-2,5-dimethyl-1H-pyrrole-3-carbaldehyde (7): General procedure A (below) was used to give desired product 7 as a dark brown oil (3.5 g, 92 %): 1H NMR (500 MHz, CDCl3): δ=9.78 (s, 1 H, CHO), 7.36–7.25 (m, 5 H), 6.25 (s, 1 H), 4.00–3.93 (m, 1 H), 3.56 (m, 2 H), 3.08–3.01 (m, 2 H), 2.58 (s, 3 H), 2.39–2.33 (m, 2 H), 2.31 (s, 3 H), 2.13–2.08 (dt, 2 H, J=11.8 Hz), 1.80–1.77 ppm (m, 2 H); MS (ESI+): m/z (%): 297.9 [M+H]+ (100).
tert-Butyl 4-(2,5-dimethyl-1H-pyrrol-1-yl)piperidine-1-carboxylate (9): The procedure for 6 (see above) was used to give compound 9 as a brown oil (3.0 g, 62 %): 1H NMR (500 MHz, CDCl3): δ=5.68 (s, 2 H), 4.20 (br s, 2 H), 3.96 (tt, 1 H, J=7.5, 4.5 Hz), 2.67 (t, 2 H, J=7.5 Hz), 2.21 (s, 6 H), 2.04 (td, 2 H, J=12.4 Hz), 1.74 (d, 2 H, J=13.5 Hz), 1.41 ppm (br s, 9 H); MS (ESI+): m/z (%): 279.4 [M+H]+ (100).
4-(2,5-Dimethylpyrrol-1-yl)piperidine (10): A solution of 9 (3.0 g, 0.010 mol) in 4 m HCl/dioxane (30 mL) was stirred at 0 °C for 3 h. Once the reaction was complete, the solvent was removed in vacuo. The residue was redissolved in CH2Cl2 (30 mL), washed with 10 % aq NaOH (30 mL), dried over MgSO4, filtered and concentrated in vacuo to afford product 10 as a colourless oil (1.8 g, 94 %): 1H NMR (500 MHz, CD3OD): δ=5.79 (s, 2 H), 3.94–3.88 (m, 1 H), 3.17–3.13 (m, 2 H), 2.66–2.60 (m, 2 H), 2.23 (s, 6 H), 2.10–2.02 (m, 2 H), 1.77–1.74 ppm (m, 2 H); MS (ESI+): m/z (%): 179.5 [M+H]+ (100).
Alternate method for the synthesis of 4-(2,5-Dimethylpyrrol-1-yl)piperidine (10): A solution of 1-benzyl-4-(2,5-dimethyl-1H-pyrrol-1-yl)piperidine (6) (3.8 g, 0.014 mol) in MeOH (10 mL) was treated with a catalytic amount of Pd(OH)2. The mixture was stirred under a hydrogen atmosphere at RT overnight. Upon completion, the reaction mixture was filtered through Celite, and the filtrate was concentrated in vacuo to afford product 10 as a colourless oil, which was used without further purification (2.4 g, 96 %); 1H NMR (500 MHz, CD3OD): δ=5.79 (s, 2 H), 3.94–3.88 (m, 1 H), 3.17–3.13 (m, 2 H), 2.66–2.60 (m, 2 H), 2.23 (s, 6 H), 2.10–2.02 (m, 2 H), 1.77–1.74 ppm (m, 2 H); MS (ESI+): m/z (%): 179.5 [M+H]+ (100).
General procedure A for the preparation of 2,5-dimethyl-1-(1-substituted-piperidine)-3-formylpyrroles (16 a–z, 16 aa–aj): POCl3 (6 mmol, 6 equiv) was added dropwise to ice-cooled DMF (12 mL) under a N2 atmosphere. The mixture was allowed to warm to RT over 15 min, then a solution of the appropriate 2,5-dimethyl-1-(1-substituted-piperidine)-1H-pyrrole (11 a, 12 a–s, 13 a-i, 14 a–e, 15) (1 mmol, 1 equiv) in DMF (5 mL) was added, and the mixture was heated at 100 °C for 3 h. After cooling, 30 % aq NaOH was added dropwise to adjust the solution to approximately pH 10. The resulting precipitate was isolated by filtration, and washed with water, and dried in vacuo to afford the desired 2,5-dimethyl-1-aryl/substituted-aryl-3-formylpyrrole (16 a–z, 16 aa–aj) (80–95 % yield).
General procedure B for the preparation of (E)-ethyl 5-((1-substituted-piperidin-4-yl)-2,5-dimethyl-1H-pyrrol-3-yl)methylene)-2-methyl-4-oxo-4,5-dihydro-1H-pyrrole-3-carboxylates (17–54): A solution of 3 (1.0 molar equiv) in abs EtOH (3 mL) was treated with the appropriate 2,5-dimethyl-1-(1-substituted-piperidine)-3-formylpyrrole (16 a–z, 16 aa–aj) (1.0 molar equiv) and KHSO4 (0.2 molar equiv). The reaction mixture was heated at 70–80 °C for 3 h and then poured onto crushed ice and filtered to afford the desired product (17–54) as a yellow powder (80–95 % yield).
(E)-Ethyl 5-((1-(1-benzylpiperidin-4-yl)-2,5-dimethyl-1H-pyrrol-3-yl)methylene)-2-methyl-4-oxo-4,5-dihydro-1H-pyrrole-3-carboxylate (17): Yellow powder (0.100 g, 40 %): mp: 150–155 °C; 1H NMR (500 MHz, CD3OD): δ=10.13 (s, 1 H, NH), 7.36–7.30 (m, 5 H), 6.65 (s, 1 H), 6.51 (s, 1 H), 4.12–4.07 (m, 3 H), 3.55 (br s, 2 H), 2.95 (d, 2 H, J=7.0 Hz), 2.53 (s, 3 H), 2.43 (t, 2 H, J=5.7 Hz), 2.36 (s, 3 H), 2.32 (s, 3 H), 2.16 (d, 2 H, J=9.1 Hz), 1.75 (d, 2 H, J=10.4, Hz), 1.21 ppm (t, 3 H, J=7.1 Hz); 13C NMR (125 MHz, [D6]DMSO): δ=180.3, 178.3, 169.2, 164.2, 163.7, 163.3, 134.6, 130.1, 128.8, 128.5, 128.1, 126.9, 117.4, 113.0, 110.1, 102.4, 64.0, 61.7, 58.1, 57.8, 55.1, 54.6, 52.6, 30.5, 15.7, 14.4, 11.0 ppm; MS (ESI+): m/z (%): 448.3 [M+H]+ (100); HRMS–ESI: m/z [M+H]+ calcd for C28H34N3O3: 448.2469, found: 448.2468.
(E)-Ethyl 5-((2,5-dimethyl-1-(2-(trifluoromethyl)phenyl)-1H-pyrrol-3-yl)methylene)-2-((E)-2-(dimethylamino)vinyl)-4-oxo-4,5-dihydro-1H-pyrrole-3-carboxylate (55): A mixture of TDR32750 (0.02 g, 0.004 mmol) and DMF–diethyl acetal (3 mL) was heated at reflux for 15 min. After cooling, the precipitate was isolated by filtration, washed with ice-cold water, and dried in vacuo to afford desired enamino ester 55 as a yellow powder (0.22 g, 89 %): mp: 270–275 °C; 1H NMR (500 MHz, [D6]DMSO): δ=9.05 (s, 1 H, NH), 8.14 (d, 1 H, J=13.3 Hz), 8.00 (d, 1 H, J=7.4 Hz), 7.91 (t, 1 H, J=7.4 Hz), 7.81 (t, 1 H, J=7.7 Hz), 7.47 (d, 1 H, J=7.8 Hz), 6.64 (s, 1 H), 6.32 (s, 1 H), 6.09 (d, 1 H, J=13.3 Hz), 4.13–4.07 (m, 2 H), 2.51 (q, 6 H, J=7.1 Hz), 1.95 (s, 3 H), 1.91 (s, 3 H), 1.23–121 ppm (m, 3 H, J=2.4, 7.05 Hz); 13C NMR (125 MHz, [D6]DMSO): δ=179.9, 170.2, 165.7, 164.2, 163.2, 151.7, 134.2, 132.4, 131.8, 130.2, 114.2, 113.5, 109.3, 106.4, 105.9, 102.4, 102.3, 96.8, 84.8, 57.6, 15.8, 14.5, 14.4, 12.0, 10.2 ppm; HRMS–ESI: m/z [M+H]+ calcd for C25H27F3N3O3: 474.1999, found: 474.2002.
General procedure for the preparation of pyrrolones 56–64: A mixture of enamino ester 55 (1.0 equiv) and the appropriate primary amine (4.0 equiv) in iso-propanol was heated at reflux for 6 h or irradiated by microwave (0–400 W at 2.45 GHz) at 110 °C for 15–30 min and then cooled. The resultant precipitate was isolated by filtration, washed with ice-cold MeOH or EtOH, and dried in vacuo to afford the desired product (56–64).
(E)-5-Benzyl-2-((2,5-dimethyl-1-(2-(trifluoromethyl)phenyl)-1H-pyrrol-3-yl)methylene)-1H-pyrrolo[3,2-c]pyridine-3,4(2H, 5H)dione (62): Isolated as a yellow powder (0.22 g, 89 %): mp: 195–200 °C; 1H NMR (500 MHz, [D6]DMSO): δ=9.45 (s, 1 H, NH), 8.00 (d, 1 H, J=7.8 Hz), 7.93 (t, 2 H, J=7.6 Hz), 7.83 (t, 1 H, J=7.2 Hz), 7.68 (d, 2 H, J=8.2 Hz), 7.59 (d, 2 H, J=8.2 Hz), 7.49 (d, 1 H, J=7.8 Hz), 6.83 (br s, 2 H), 6.54 (s, 1 H), 6.38 (d, 1 H, J=7.2 Hz), 5.27–5.26 (m, 2 H), 1.82 ppm (s, 6 H); 13C NMR (125 MHz, [D4]acetone): δ=181.7, 170.8, 162.3, 161.0, 158.2, 145.2, 143.6, 136.0, 134.8, 132.6, 132.3 (2C), 131.0 (2C), 130.8, 130.3, 129.3, 129.2, 128.3, 126.3, 126.2, 115.6, 108.7, 106.5, 95.6, 50.6, 12.3, 10.7 ppm; MS (ESI+): m/z (%): 490.18 [M+H]+ (100); HRMS–ESI: m/z [M+H]+ calcd for C28H23F3N3O2: 490.1737, found: 490.1752.
In vivo efficacy studies: The protocols are described in the Supporting Information. In vivo efficacy studies in mice were conducted at the Swiss Tropical & Public Health Institute (Basel, Switzerland) according to the rules and regulations for the protection of animal rights (“Tierschutzverordnung”) of the Swiss “Bundesamt für Veterinärwesen”. They were approved by the veterinary office of Canton Basel-Stadt, Switzerland.
Supporting Information
Supporting Information contains: synthetic routes for the synthesis of intermediates and characterisation data for compounds not included in the main text; methods for physicochemical evaluation of the compounds and the data generated; methods for assessing metabolic stability; in vitro and in vivo parasite testing.
This material is available free of charge on the WWW under http://dx.doi.org/10.1002/cmdc.201300177.
Acknowledgments
This investigation was done for and received support from the UNICEF/UNDP/World Bank/WHO Special Programme for Research and Training in Tropical Diseases (TDR). The University of Dundee would also like to acknowledge the Wellcome Trust (grant 083481) for support.
Notes
Some of the compounds had a trace, inseparable amount of (Z)-isomer. This could not be detected in the 13C NMR spectra and was only detected using UPLC.
References
- [1].Snow RW, Guerra CA, Noor AM, Myint HY, Hay SI. Nature. 2005;434:214–217. doi: 10.1038/nature03342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Winzeler EA. Nature. 2008;455:751–756. doi: 10.1038/nature07361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Wells TNC, Burrows JN, Baird JK. Trends Parasitol. 2010;26:145–151. doi: 10.1016/j.pt.2009.12.005. [DOI] [PubMed] [Google Scholar]
- [4].Gamo FJ, Sanz LM, Vidal J, de Cozar C, Alvarez E, Lavandera JL, Vanderwall DE, Green DVS, Kumar V, Hasan S, Brown JR, Peishoff CE, Cardon LR, Garcia-Bustos JF. Nature. 2010;465:305–310. doi: 10.1038/nature09107. [DOI] [PubMed] [Google Scholar]
- [5].Murugesan D, Mital A, Kaiser M, Shackleford DM, Morizzi J, Katneni K, Campbell M, Hudson A, Charman SA, Yeates C, Gilbert IH. J. Med. Chem. 2013;56:2975–2990. doi: 10.1021/jm400009c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Galdino SL, Pitta IdR, Luu-Duc C, Lucena B, Lima RMOliveiraCorrea. Eur. J. Med. Chem. 1985;20:439–442. [Google Scholar]
- [7].Higino JS, Lins-Galdino S, Da Rocha-Pitta I, De Lima JG, Luu-Duc C. Il Farmaco. 1990;45:1283–1287. [PubMed] [Google Scholar]
- [8].Ragan JA, Jones BP, Castaldi MJ, Hill PD, Makowaki TW. Org. Synth. 78:63. [Google Scholar]; Org. Synth. 2002–2004:418. [Google Scholar]
- [9].Vorkapić-Furač J, Mintas M, Burgemeister T, Mannschreck A. J. Chem. Soc. Perkin Trans. 2. 1989:713–717. [Google Scholar]
- [10].Trofimkin YI, Ryabova SY, Alekseeva LM, Granik VG. Pharm. Chem. J. 1997;31:40–42. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.