

Abstract
The best-established function of the melanoma-suppressor p16 is mediation of cell senescence, a permanent arrest following cell proliferation or certain stresses. The importance of p16 in melanoma suggests indolence of the other major senescence pathway through p53. Little or no p53 is expressed in senescent normal human melanocytes, but p16-deficient melanocytes can undergo p53-mediated senescence. As p16 expression occurs in nevi but falls with progression toward melanoma, we here investigated whether p53-dependent senescence occurs at some stage and, if not, what defects were detectable in this pathway, using immunohistochemistry. Phosphorylated checkpoint kinase 2 (CHEK2) can mediate DNA-damage signaling, and under some conditions senescence, by phosphorylating and activating p53. Remarkably, we detected no prevalent p53-mediated senescence in any of six classes of lesions. Two separate defects in p53 signaling appeared common: in nevi, lack of p53 phosphorylation by activated CHEK2, and in melanomas, defective p21 upregulation by p53 even when phosphorylated.
Keywords: melanoma progression, nevus, senescence, p53, p16, DNA-damage signaling, diagnosis
Significance.
Further elucidation of relationships between cell senescence and melanoma may yield much-needed prognostic and diagnostic markers for primary pigmented lesions, as escape from senescence is linked to progression. The absence of p53–p21-mediated senescence at any stage of progression sheds light on why p16, the other best-known mediator of senescence, is so important in melanoma. There is interest in the mechanisms of dysregulation of p53 signaling in melanoma, and we elucidate two common mechanisms here. We separately report a strong association between nuclear p16 and lack of vertical growth, not seen in studies of total p16, and meriting larger-scale studies.
Introduction
Cell senescence is now recognized as a major mechanism of tumor suppression (Artandi and DePinho, 2010; Collado and Serrano, 2010; reviews: Herbig and Sedivy, 2006; Peeper, 2011), so that understanding how cancer cells evade senescence is a promising route toward new diagnostic markers and therapeutic targets. Cell senescence is a permanent proliferative arrest, after either extensive cell division (replicative senescence) or certain stresses including activation of an oncogene (oncogene-induced senescence). Its anticancer role is particularly clear in melanoma, where the commonest familial melanoma locus CDKN2A encodes two effectors of cell senescence: p16 (also called INK4A or CDKN2A, cyclin-dependent kinase inhibitor 2A) and ARF (alternative reading frame; Bennett, 2008; Peters, 2008). ARF, an activator of p53, has not been clearly linked to senescence in human cells (Peters, 2008), but is central to senescence of mouse cells, including melanocytes (Ha et al., 2007, 2008). Moreover, another familial melanoma locus encodes p16's principal target, cyclin-dependent kinase 4 (CDK4). Melanoma-associated mutations in CDK4 affect the p16-binding site (Molven et al., 2005).
Oncogene-induced senescence can block carcinogenesis in vivo (Collado and Serrano, 2010; Kang et al., 2011; Peeper, 2011). In melanocytes in skin, oncogenic mutations (usually in BRAF or NRAS) appear to be followed typically by proliferation then arrest, giving a static benign mole or nevus (Bennett, 2008). Nevi show markers of senescence (Gray-Schopfer et al., 2006; Michaloglou et al., 2005), and most explanted nevus cells fail to divide at all (Soo et al., 2011).
Two major pathways mediating human replicative senescence are known. At least one involves telomeres, the protective repetitive DNA at chromosome ends. In the germline and some cancer cells, telomere length is maintained by the enzyme telomerase. However, most somatic cells lack TERT (telomerase reverse transcriptase, the catalytic subunit), so telomeres shorten at each division (Artandi and DePinho, 2010; Baird et al., 2003; Herbig and Sedivy, 2006). When they become critically short, this activates DNA-damage signaling (DDS; Figure S1), involving activation of p53 by checkpoint kinase 2 (CHEK2, formerly CHK2; d'Adda di Fagagna, 2008; Collado and Serrano, 2010; Herbig and Sedivy, 2006). In senescence, p53 transcriptionally activates mediators of growth arrest, especially p21 (CDKN1A; d'Adda di Fagagna, 2008; Collado and Serrano, 2010; Passos et al., 2010). This pathway can also directly inactivate mitotic regulatory proteins to arrest cells in G2 independently of p53 (Figure S1; van Vugt and Medema, 2005). The other major senescence pathway, the p16 pathway, is active in stress-and oncogene-induced senescence and can also respond to telomere dysfunction, although by uncertain mechanisms (Jacobs and de Lange, 2005). p16 becomes expressed in senescence, inhibiting CDK4 which ceases to phosphorylate substrates. This gives RB-family-mediated G1 arrest (Bennett, 2008), and potentially G2 arrest also through cessation of FOXM1 activation by CDK4 (Anders et al., 2011).
Both p16 and p53 pathways can contribute to both replicative and oncogene-induced senescence; their relative contributions differ between cell types and conditions (Collado and Serrano, 2010; Kiyono et al., 1998; Sviderskaya et al., 2003). The p16 pathway appears predominant in human melanocyte replicative senescence, as senescence is delayed without it (Sviderskaya et al., 2003), or absent in mouse melanocytes (Ha et al., 2007; Sviderskaya et al., 2002). p53 and p21 appear absent from senescent normal human melanocytes (Bandyopadhyay et al., 2001; Sviderskaya et al., 2003), but were implicated in the delayed arrest of p16-null melanocytes (Sviderskaya et al., 2003). Similarly, various human epithelial cell types underwent p16-dependent senescence, but on escaping from this could enter p53-dependent senescence (Kiyono et al., 1998).
If the p16 and p53 pathways are both disrupted, this can lead to telomeric crisis, a third net growth arrest where continuing proliferation is balanced by cell death (Artandi and DePinho, 2010; Bond et al., 1999; Gisselsson and Höglund, 2005). Here, telomeres have shortened further until some are lost or dysfunctional. Such chromosome ends can be ligated together by DNA repair enzymes, leading to anaphase bridging, chromosome breakages, and mitotic aberrations, often lethal (Artandi and DePinho, 2010; Bond et al., 1999; Gisselsson and Höglund, 2005). Cells can escape from telomeric crisis only by restoration of telomeres, usually by expression of TERT (Artandi and DePinho, 2010; Bennett, 2008; Bond et al., 1999; Gray-Schopfer et al., 2006). This overcomes the third hurdle and confers immortality, the ability to proliferate without limit.
Explanted human melanoma metastases have frequently yielded immortal cell lines (Hsu et al., 2000), while cultures from primary melanomas often appear not to be immortal (Soo et al., 2011), as proposed for breast cancer and other malignancies (Chin et al., 2004; reviews by: Artandi and DePinho, 2010; Soo et al., 2011). It seems plausible that melanocytes during melanoma progression may pass through the same three proliferative barriers as cultured cells (mediated by p16, p53 and telomeric crisis), as proposed in an updated genetic model for melanoma (Soo et al., 2011).
There is good evidence for p16-associated senescence in nevi (Gray-Schopfer and Bennett, 2006; Gray-Schopfer et al., 2006; Michaloglou et al., 2005; Zhu et al., 2007), and for escape from this arrest in melanomas, with p16 attenuation or loss (Alonso et al., 2004; Bartkova et al., 1996; Gray-Schopfer et al., 2006; Keller-Melchior et al., 1998; Talve et al., 1997), detectable also in dysplastic nevi (Gray-Schopfer et al., 2006). However, it is unclear whether p53-p21 arrest occurs subsequent to p16 loss, as it does in culture. Most uncultured advanced human melanomas apparently express wild-type p53 (IARC TP53 database: http://www-p53.iarc.fr/; Gray-Schopfer et al., 2006), despite still growing. But p53-mediated senescence might occur earlier in progression.
Primary pigmented proliferative lesions can be classified into benign nevi, dysplastic (usually atypical, large) nevi (DN), radial growth-phase (RGP) melanomas, which grow only in or near the epidermis, and vertical growth-phase (VGP) melanomas, which invade more deeply and are believed competent for metastasis (Clark et al., 1989). A good many groups including ours have shown that there is little p53 in benign or dysplastic nevi, and little p21 (from a smaller number of studies); and that p53 abundance seemed to increase only in advanced melanoma (reports including Gray-Schopfer et al., 2006; Lassam et al., 1993; Stefanaki et al., 2008). Here, we have tested for specific defects in mediators of the telomere-p53 pathway, in a series of 75 primary pigmented lesions in 6 subclasses, by immunostaining. Mediators studied included activated CHEK2 phosphorylated by ATM on Thr68 (pCHEK2), p53 phosphorylated on Ser20 (the CHEK2 substrate site; p-p53; Chehab et al., 2000; Lukas et al., 2003), and p21 (Figure S1). p16 was included to test for a relation between its loss and p53 pathway activation, and to examine whether nuclear location affected the observed trend; and nucleolin, a p53-interacting protein that can suppress activation of p53 by ATM (Nalabothula et al., 2010; Yang et al., 2011), was also evaluated, for any correlation with diagnosis or p53 status. We report evidence for two separate common defects in p53 activation during melanoma development, helping to explain the importance of p16 in melanoma.
Results
A detailed spectrum of six types of pigmented lesions was studied, by subdividing the above-mentioned categories. Benign compound nevi (BCN, containing both epidermal and dermal components) were considered separately from benign intradermal nevi (BIN, with no epidermal component; Mooi and Krausz, 2007). RGP melanomas limited to the epidermis were classed as melanoma in situ (MIS), while those with microinvasion in the papillary dermis (Gershenwald et al., 2010) were given the abbreviation MIV (microinvasion). All specimens were scored by two consultant pathologists independently, including MGC, a leading European expert on melanoma diagnosis. Diagnosis and the location of the diagnostic area were reconfirmed by both consultants from an adjacent section stained with hematoxylin and eosin (not shown). Numbers of samples scored per marker are given in Table S1.
Nuclear p16 in melanoma progression
p16 loss in melanoma is well known (citations in Introduction). p16 reanalysis here was for correlation with other markers and to characterize the additional lesional categories. We also evaluated two refinements. Firstly, only the area corresponding to the diagnosis was assessed (as with all markers here). Thus, if an RGP section also contained nevus, only the RGP area was scored. Secondly, only nuclear p16 immunoreactivity was scored, with or without cytoplasmic reaction, as cytoplasmic-only p16 can be mutated (Gray-Schopfer et al., 2006).
With these refinements, p16 expression showed a markedly stronger negative correlation with advancing lesion category than previously reported, as shown in Figure 1(A) and Table 1 (r = −0.48, P = 1.8 × 10−5). Only 3/17 VGP melanomas showed any detectable nuclear p16, and only one had more than 50% of cells positive. BINs showed the highest p16 expression (Figures 1A, 2A,B), with significantly more than DN, MIS, or VGP, while VGPs showed significantly less p16 than all other categories (Table 1).
Figure 1.
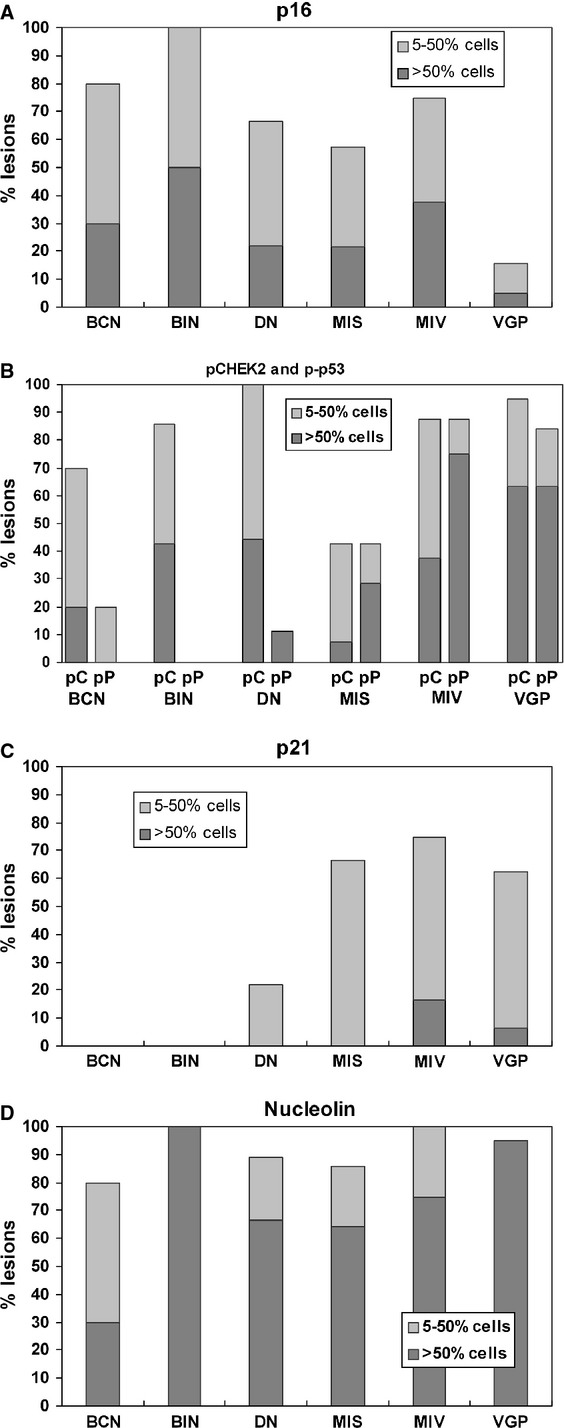
Prevalence of five regulatory proteins in pigmented lesions by immunostaining, and changes with progression. The numbers of lesions studied per category per marker are detailed in Table S1; the average was 12 (range 8–17). Key: BCN, benign compound nevus; BIN, benign intradermal nevus; DN, dysplastic nevus; MIS, melanoma in situ (RGP limited to epidermis); MIV, RGP with microinvasion; VGP, vertical growth phase; pC, pCHEK2; pP, p-p53. Lesions were classified as showing >50% of lesion (diagnostic area only) positive, 5–50, or <5%. The bars show the upper two categories, as indicated by shading. Significance of individual differences in markers between lesion categories are listed in Table 1, as are trends in prevalence of markers with advancing lesion category.
Table 1.
Differences in immunostaining prevalence between pigmented lesion categories: significance testing
| Diagnostic categorya | Diagnostic category | Significance level (P)b | ||||||
|---|---|---|---|---|---|---|---|---|
| p16 | pCHEK2 | p-p53 | p21 | Nucleolin | ||||
| BCN | BIN | NS | NS | NS | NS | <0.01 | ||
| BCN | DN | NS | NS | NS | NS | NS | ||
| BCN | MIS | NS | NS | NS | <0.05 | NS | ||
| BCN | MIV | NS | NS | <0.01 | <0.01 | <0.05 | ||
| BCN | VGP | <0.01 | <0.05 | <0.001 | <0.01 | <0.01 | ||
| BIN | DN | <0.05 | NS | NS | NS | NS | ||
| BIN | MIS | <0.05 | <0.01 | <0.05 | <0.01 | NS | ||
| BIN | MIV | NS | NS | <0.001 | <0.001 | NS | ||
| BIN | VGP | <0.001 | NS | <0.001 | <0.01 | NS | ||
| DN | MIS | NS | <0.01 | NS | NS | NS | ||
| DN | MIV | NS | NS | <0.01 | <0.05 | NS | ||
| DN | VGP | <0.05 | NS | <0.01 | <0.05 | NS | ||
| MIS | MIV | NS | <0.05 | <0.05 | NS | NS | ||
| MIS | VGP | <0.05 | <0.001 | <0.05 | NS | NS | ||
| MIV | VGP | <0.01 | NS | NS | NS | NS | ||
| Overall Kruskal–Wallis significancec | 0.0002 | 0.0018 | <0.0001 | <0.0001 | 0.0018 | |||
| Correlation coefficient (r)d | −0.48 | 0.196 | 0.647 | 0.528 | 0.268 | |||
| Significance of r | 1.8 × 10−5 | NS | <10−6 | 3 × 10−6 | 0.022 | |||
Categories are as in the text: BCN, benign cellular nevus; BIN, benign intradermal nevus; DN, dysplastic nevus; MIS, melanoma in situ, epidermal-only RGP; MIV, RGP melanoma with microinvasion; VGP, vertical growth phase.
Significance levels for all pairwise comparisons between lesion categories in prevalence of immunostaining for each marker (Figure 1), by the Kruskal–Wallis test (Siegel and Castellan, 1998). Significant differences are shown in boldface. NS, not significant.
Overall Kruskal–Wallis significance per marker is the probability of no difference in prevalence between any categories.
Spearman's correlation coefficient (r) between prevalence and progression, and its significance (Siegel and Castellan, 1998) are shown per marker.
Figure 2.
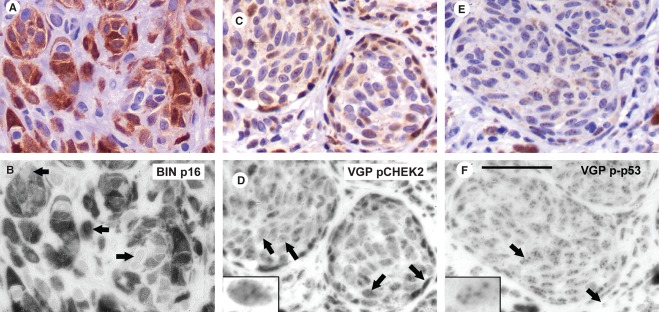
Expression patterns for p16, pCHEK2, and p-p53. Positive reaction is brown, with purple hematoxylin counterstain. Upper panels show normal color and lower panels the blue RGB channel, emphasizing the immunostain as dark (absorbing blue light). Control stains of the same lesions with no or nonspecific first antibody (results indistinguishable) are in Figure S2. Scale bar for all panels: 50 μm. (A,B) p16 in a BIN, illustrating widespread, strong expression, usually nuclear and cytoplasmic (middle arrow), although some cells had cytoplasmic-only expression (upper arrow) or none (lower arrow). (C,D) pCHEK2 in a VGP melanoma. Reaction was nuclear, with varied levels. Arrows show examples respectively of (left to right): no reaction, one subnuclear focus, several subnuclear foci (commonest; enlarged in inset), or pan-nuclear. (Note: contrast slightly enhanced in this inset.) Cytoplasmic reaction sometimes accompanied strong nuclear expression; however, cytoplasmic brown material in this melanoma was melanin (Figure S2c,d). (E,F) p-p53 in a VGP melanoma: typically in several subnuclear foci (left arrow and enlarged in inset). Unstained nuclei outside the melanoma nest (right arrow) may belong to fibroblasts. Two melanophages (dark brown) are seen at the bottom center and lower right.
pCHEK2 and p-p53 in melanoma progression
Expression of phospho-CHEK2 (Thr68; pCHEK2) was nuclear, in one or several subnuclear foci, as expected for DDS foci such as short-telomere-induced foci (TIFs; Herbig and Sedivy, 2006; Figure 2C,D). Sometimes, pCHEK2 was more highly expressed and pan-nuclear. This pattern was not systematically related to lesion type, but appeared most common in VGP specimens where high expression levels were typical (see below). pCHEK2 was readily detected in most lesions of all categories except MIS (only around 40% of which had >5% positive cells; Figure 1B). There was no sustained trend in expression levels with progression, but prevalence appeared to fall between BIN/DN and MIS, and then rise, as both MIV and VGP lesions showed significantly more than MIS (Table 1). In some samples, pCHEK2 nuclear foci were seen in some areas of epidermis and other normal tissues such as sebaceous and sweat glands, for unknown reasons (Figure S2e,f; some possibilities are age-related cell senescence, or localized oxidative or sun damage).
p-p53 (Ser20) was examined in conjunction with pCHEK2, being a pCHEK2 substrate site. p-p53 was detected typically in subnuclear foci, again consistent with TIFs or other DDS foci (Figure 2E,F). There was a strong trend of rising expression with progression (Figure 1B; r = 0.647; P < 0.0001). Some VGP areas had very numerous p-p53 foci (Figure S2i). Somewhat surprisingly, pCHEK2 and p-p53 showed little quantitative association, either in overall trends or lesion by lesion. Most benign and dysplastic nevi had some or much pCHEK2, but little or no p-p53 (in line with previous reports of very little total p53 in nevi). Among the three categories of melanomas, pCHEK2 and p-p53 seemed well correlated numerically (Figure 1B); however, areas of lesions could be found showing each marker without the other, in adjacent or nearby sections, and a bubble plot and statistical analysis (Figure 3A) showed no significant correlation between the two among lesions. Thus, p-p53 is not detectably associated with active CHEK2 in these lesions, suggesting possible involvement of another kinase(s). Of note, both pCHEK2 and p-p53 foci could be found in some mitotic cells in VGP melanomas, showing that both were failing to signal growth arrest in these cells (Figure S2j,k).
Figure 3.
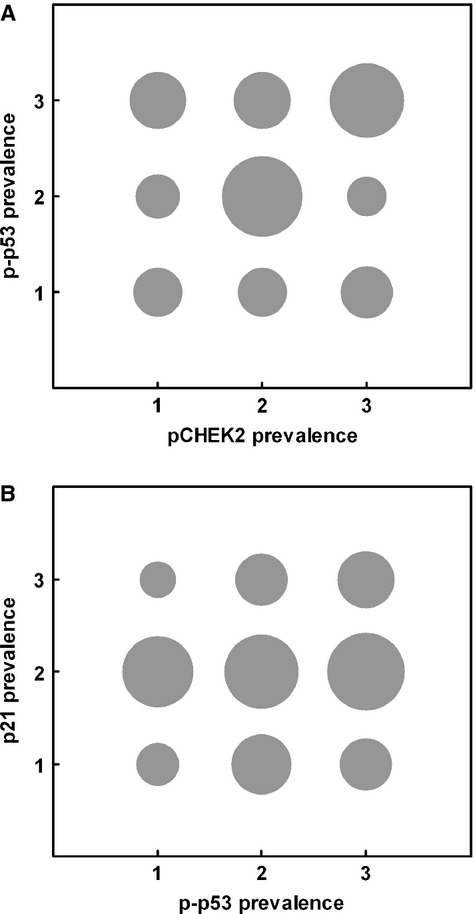
Lack of relation between prevalence of p-p53 and those of pCHEK2 and p21. Grouped scatter plots, all categories of lesions pooled. The area of each circle is proportional to the number of lesions in that class for each marker. (A) pCHEK2 and p-p53; (B) p-p53 and p21. For pCHEK2 and p-p53, prevalence classes 1, 2, and 3 here are as in Figure 1 (<5, 5–50, and >50% positive cells), while classes 1, 2, and 3 for p21 were chosen to span the lower levels of expression seen, namely 0, >0–10, and >10% positive cells. No correlation was found in either case: Spearman rank correlation coefficient r = −0.19 for (a) and 0.06 for (B) (NS).
p21 in melanoma progression
Protein p21 (CDKN1A) is a principal effector of p53-mediated cell senescence, arresting cells by inhibiting several CDKs including CDK4, 1, and 2. We reanalyzed p21 expression in this more detailed series of lesions and to test relatedness to DDS. p21 localization was nuclear, as previously reported (Figure 4A). All benign and most dysplastic nevi showed little or no p21 expression (<5% of cells; Figure 1C). p21 prevalence differed significantly between most nevus and melanoma categories, and the overall trend with progression was highly significant (r = 0.528, P = 3 × 10−6; Table 1). Thus, most melanomas contained some p21-positive cells, but the prevalence was consistently lower than that of p-p53 (Figure 1B,C), indicating failure of p-p53 to activate p21 transcription substantially.
Figure 4.
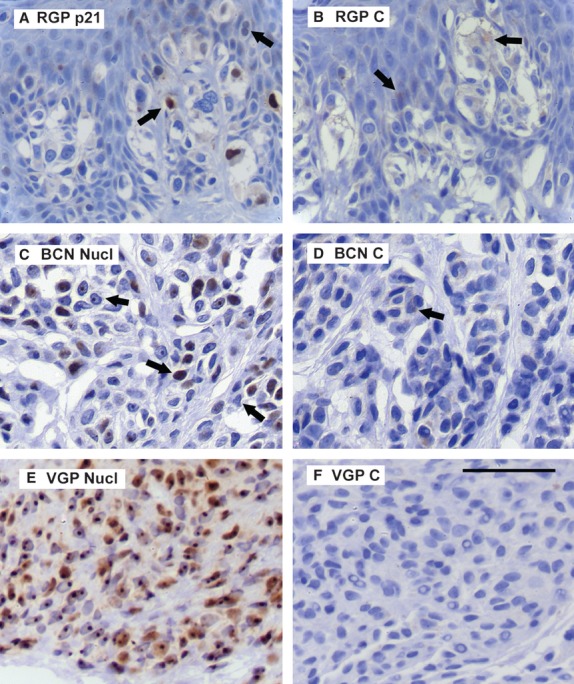
Expression patterns for p21 and nucleolin. Key: C, control section of same lesion; Nucl, nucleolin. Figure S3 shows the RGB blue channel for all of these panels. Scale bar for all panels: 50 μm. (A) Typical p21 reaction in an RGP (MIV) melanoma. Positive (nuclear) stain in a few melanoma cells, sometimes weak (right arrow, clearer in blue channel), while many cells were negative. Cytoplasmic brown here was melanin, as seen in control section (B) (compare also Figure S3). No p21 was detected in normal tissues. (C) Nucleolin in a BCN. Reaction was typically nucleolar (left arrow), while sometimes pan-nuclear or absent (middle and right arrows). Control section (D) showed traces of cytoplasmic brown melanin (arrow). (E) Nucleolin in a VGP melanoma. Nearly all cells had strong reaction in the nucleolus and weaker nucleoplasmic expression. The cytoplasmic color was not melanin, as this melanoma had little or no melanin (F).
Only in a small minority of MIV (and one VGP) melanomas were more than half the cells expressing p21 and thus possibly in p53/p21-dependent senescence. Moreover, there was no statistical support for colocalization of p21 with p-p53 (Figure 3B), suggesting an alternative pathway for p21 upregulation in these few melanomas. (Note that parallel sections were used, not necessarily immediately adjacent although within about 20 μm of each other, so microheterogeneity within lesions may possibly have masked an association.)
Nucleolin in melanoma progression
Nucleolin locates preferentially to nucleoli. It was included here as a protein that can impair the activation of p53 by ATM and downregulate p53 abundance to maintain cell ‘stemness’ (Nalabothula et al., 2010; Yang et al., 2011). Nucleolar immunoreactivity for nucleolin was detected in some normal tissues, including epidermis, hair follicles, and some endothelium, but not in nerve (Schwann cells). It was also detected in all classes of pigmented lesions studied (Figures 1D and 4C,E). There was a slight but significant trend toward increased prevalence with progression (P = 0.022). Interestingly, and contrary to this trend, expression was widespread in 100% of BIN, significantly more than in BCN (Figure 2D; Table 1). When BIN were excluded from the analysis, the trend with progression was highly significant (P = 6.0 × 10−4).
Discussion
Here, we searched for a phase of p53-mediated senescence in melanoma progression, based on the findings that cultured p16-deficient human melanocytes can still senesce belatedly through a p53-associated pathway (Sviderskaya et al., 2003) and that p16 loss is common in melanomas and DN. As normal p53-mediated senescence is triggered by telomere shortening and DDS from dysfunctional telomeres (Herbig and Sedivy, 2006), we looked for a stage of melanoma progression with these characteristics: p16 loss, DDS foci containing the signaling intermediates pCHEK2 and p-p53, and expression of the effector p21.
We report here, despite a detailed breakdown of primary pigmented lesions into six classes, that no such stage could be found in melanoma progression. p16 expression was numerically highest in BIN, and significant p16 loss compared with BIN was seen in DN and more advanced lesions (MIS, VGP). However, DDS foci with pCHEK2 appeared not only in DN and more advanced lesions but in all lesion classes including BCN and BIN. pCHEK2 and DDS have previously been reported in DN and melanomas, although that study compared only those two classes of lesion and did not test further correlations (Gorgoulis et al., 2005). In general, the presence of pCHEK2 yet lack of p-p53 and p21 in BCN, BIN, and most DN indicates that these have some DDS foci, but this signaling fails to activate p53. The static nature of DN is thus apparently not due to p53 and p21 and may depend instead on residual p16 and/or another mediator(s) such as p15(CDKN2B), ARF, or PTEN (Peters, 2008; Vredeveld et al., 2012).
The DDS without p53 activation parallels findings in senescent normal cultured human melanocytes, which upregulate p16 but not p53 (Gray-Schopfer et al., 2006; Sviderskaya et al., 2003) and do show DDS foci, about 1–2 per cell, immunoreactive for γH2AX and 53BP1 (R. S. Collinson and D. C. Bennett, unpublished data). We speculate that these are TIFs and that the foci in nevi may also be TIFs. As supporting evidence, dysfunctional telomeres can activate p16 (Jacobs and de Lange, 2005); DDS foci were reported in NRAS oncogene-induced, p16-associated melanocyte senescence (Haferkamp et al., 2009), and telomere length in humans is positively correlated with the number and size of their nevi (Bataille et al., 2007). This hypothesis needs further investigation.
DDS foci appear highly prevalent in VGP melanomas, with p-p53 also now typically present. This was initially surprising. If cellular immortality – requiring functional telomeres – is a hallmark of all cancers (Hanahan and Weinberg, 2011), no TIFs would be expected in advanced melanomas; and no other obvious rationale for such numerous DNA breaks suggests itself. However, the recent findings that primary melanomas are often not immortal when explanted, and often exhibit markers of telomeric crisis in vivo (Soo et al., 2011), predict that TIFs may well be present in these melanomas in vivo and that DNA breaks could also arise from the bridge-break cycles of telomeric crisis (Artandi and DePinho, 2010). The detection of pCHEK2 and p-p53 foci in mitotic as well as interphase VGP cells shows that the DDS/p53 is failing to signal arrest in at least parts of these lesions, such failure being a prerequisite for cells to approach crisis. The location of DDS foci in these mitotic figures was generally consistent with the ends of chromosomes (Figure S2j,k), that is, with their being TIFs. Cultured permanent melanoma lines represent cells that have passed through crisis and spontaneously immortalized; most of these are also reported to show defects in checkpoint responses to exogenous DNA damage, including reduced p53 transcriptional function (Kaufmann et al., 2008).
Nuclear p16, as analyzed here, provided a strong negative association with progression or VGP status, distinctly clearer than that seen using total p16 as scored in most previous reports (Alonso et al., 2004; Keller-Melchior et al., 1998; Talve et al., 1997). Only 1/17 VGP melanomas was predominantly positive for nuclear p16. This further supports the longstanding hypothesis (Bartkova et al., 1996; Bennett, 2008; Soo et al., 2011) that a defective p16-CDK4 pathway is mandatory for melanoma development. Speculatively, the one p16-positive melanoma may have had an equivalent defect such as a CDK4 or RB1 mutation, as found in p16-positive melanoma cell lines (Bartkova et al., 1996). A correlation of nuclear p16 expression in melanomas with survival was reported previously (Sirigu et al., 2006), although benign lesions were not included. The findings thus indicate potential value for a larger-scale study of nuclear p16 in melanoma diagnosis.
p53 is often both highly expressed and normal in sequence in advanced melanomas and their cell lines; p53 even rises in abundance with melanoma progression (Gray-Schopfer et al., 2006; Stefanaki et al., 2008). Ser20-phosphorylation stabilizes p53, so increasing levels of p-p53(Ser20) may contribute to the accumulation of total p53. However, no significant colocalization with pCHEK2 was observed, suggesting an alternative kinase for Ser20 here. Candidate Ser20 kinases include several other mediators of DDS or stress signaling: CHEK1, JNK, and MAPKAPK2/MK2 (Bode and Dong, 2004). It is odd that CHEK2 should be activated in many nevi and melanomas yet apparently fails to phosphorylate p53. Perhaps, p53 abundance is so low that phosphorylation still fails to give a detectable level.
A second defect in p53 senescence signaling in pigmented lesions is confirmed here: lack of upregulation of p21 by p53, at the protein level. It is known that melanomas can express p53 yet little or no p21 protein (Gray-Schopfer et al., 2006); here, we show that even ‘activated’ [phospho(Ser20)] p53 is not apparently correlated with p21 expression. Thus, in none of six stages of melanoma progression was there evidence for p53/p21-mediated senescence, except perhaps in parts of some MIV melanomas. Several common alterations in melanoma have been proposed as mediating a failure of p53 to activate p21 transcription, including upregulation of TBX2, mutation of STK11/LKB1 and others (Bennett, 2008). Degradation of p21 may also contribute to its paucity, p21 being a proteasome target (Jung et al., 2010).
Dysfunction in both the p16 and the p53 pathways helps to explain why even the extensive DDS seen here in some melanomas is apparently unable to stop cell division. Loss of both pathways in other cells results in extreme telomere shortening and telomeric crisis with its resultant genomic instability and DDS. Indeed, evidence for telomeric crisis in VGP melanomas was recently described (Soo et al., 2011). Immortalization with telomere maintenance may be a later event, as discussed (Soo et al., 2011).
In summary, we report evidence for DDS in all classes of nevi and melanomas, which could be explained by advancing telomere shortening, although more evidence is needed on that. We describe two apparently inherent defects in p53-mediated senescence in melanocytic lesions. First, CHEK2 activation does not lead to p53 phosphorylation and activation in nevi. Second, p-p53, although abundant in MIV and VGP melanomas, still fails to upregulate p21 protein and arrest the cells. Nucleolin is found in all classes of lesion and may contribute to p53 indolence by limiting ATM phosphorylation of p53. Overall, the findings reveal two separate mechanisms for p53 bypass in melanoma development.
Methods
Immunohistochemistry
Formalin-fixed, paraffin-embedded specimens of melanocytic lesions were obtained and immunostained at the Royal Surrey County Hospital, with ethical approval from the Hospital's Local Research Ethics Committee. Serial adjacent sections (4 μm) were cut from each lesion, of which one was used for hematoxylin and eosin staining for rediagnosis and the rest treated as follows. Sections were mounted on ChemMate Capillary Gap microscope slides (75 μm) and oven-dried below 60°C for 1–24 h. They were dewaxed, rehydrated, and subjected to antigen retrieval in boiling citrate buffer (10 mM, pH 6.0) at 83 kPA for 2 min and then cooled in water. They were placed in Tris-buffered saline (50 mM, pH 7.6) for 5 min, then transferred to a DAKO Techmate 500 Plus Autostainer (DakoCytomation, Glostrup, Denmark). They were stained using primary antibodies as below, followed by the ChemMate biotin–streptavidin–peroxidase Detection System K5001 (DakoCytomation), according to the manufacturer's instructions, followed by Mayer's hematoxylin (1 min).
All slides were assessed by ADMR working with MGC and independently by HC. Proportion of lesional cells stained in each sections was assessed, usually in the categories <5, 5–50, and >50%, giving very good (90%) agreement between assessors (weighted kappa statistic of 0.843). In cases of disagreement, sections were re-examined to enable consensus.
Primary antibodies
All antibodies were well characterized for correct specificity. Anti-p16 monoclonal antibody NCL-p16-432 from Novocastra (Newcastle, UK) was used at 1:20 dilution. Its specificity was shown by Gray-Schopfer et al. (2006). Monoclonal anti-phospho-CHEK2 (Thr68; 2661, Cell Signaling, now Merck Millipore, Watford, UK) was characterized by Lukas et al. (2003) and used at 1:50. Polyclonal anti-phospho-p53 (Ser20; 9287, Cell Signaling) characterized by Chehab et al. (2000) was used at 1:50. Monoclonal anti-p21 (556430; BD Pharmingen, Oxford, UK) was described by Sviderskaya et al. (2003) and used at 1:100. The monoclonal antibody to nucleolin was sold by Novocastra as ‘NCL-hTERT’ but has been shown to react only with nucleolin, not TERT (Wu et al., 2006). It was used at 1:60.
Statistical analysis
Nonparametric statistical tests were used. Intercategory differences were tested by the Kruskal–Wallis one-way analysis of variance by rank (Siegel and Castellan, 1998) using http://department.obg.cuhk.edu.hk/researchsupport/KruskallWallis.ASP.
Trends in prevalence of markers with progression, and correlations between prevalence of two markers, were evaluated with Spearman's rank correlation test, using http://faculty.vassar.edu/lowry/corr_rank.html. Here, the diagnostic categories were given numerical values in order of perceived progression: 1 = BCN, 1.5 = BIN, 2 = DN, 3 = MIS, 4 = MIV, and 5 = VGP. Charts were prepared using Microsoft Excel.
Conflict of interest
The authors state no conflict of interest.
Acknowledgments
We are indebted to Dr Jan Poloniecki (SGUL) for advice on statistics and Dr M Green (RSCH) for expert assistance. ADMR was supported by a fellowship from the St George's NHS Trust Charitable Foundation, and MH by an MRC studentship.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Table S1. Numbers of successfully immunostained lesions by antibody and diagnostic category
Figure S1. Established ATM/CHEK2 pathways: summary
Figure S2. Supporting data for Figure 2
Figure S3. RGB blue channel from all panels of Figure 4
References
- d'Adda di Fagagna F. Living on a break: cellular senescence as a DNA-damage response. Nat. Rev. Cancer. 2008;8:512–522. doi: 10.1038/nrc2440. [DOI] [PubMed] [Google Scholar]
- Alonso SR, Ortiz P, Pollan M, et al. Progression in cutaneous malignant melanoma is associated with distinct expression profiles – a tissue microarray-based study. Am. J. Pathol. 2004;164:193–203. doi: 10.1016/s0002-9440(10)63110-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders L, Ke N, Hydbring P, et al. A systematic screen for CDK4/6 substrates links FOXM1 phosphorylation to senescence suppression in cancer cells. Cancer Cell. 2011;20:620–634. doi: 10.1016/j.ccr.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artandi SE, DePinho RA. Telomeres and telomerase in cancer. Carcinogenesis. 2010;31:9–18. doi: 10.1093/carcin/bgp268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baird DM, Rowson J, Wynford-Thomas D, Kipling D. Extensive allelic variation and ultrashort telomeres in senescent human cells. Nat. Genet. 2003;33:203–207. doi: 10.1038/ng1084. [DOI] [PubMed] [Google Scholar]
- Bandyopadhyay D, Timchenko N, Suwa T, Hornsby PJ, Campisi J, Medrano EE. The human melanocyte: a model system to study the complexity of cellular aging and transformation in non-fibroblastic cells. Exp. Gerontol. 2001;36:1265–1275. doi: 10.1016/s0531-5565(01)00098-5. [DOI] [PubMed] [Google Scholar]
- Bartkova J, Lukas J, Guldberg P, Alsner J, Kirkin AF, Zeuthen J, Bartek J. The p16-cyclin D/Cdk4-pRb pathway as a functional unit frequently altered in melanoma pathogenesis. Cancer Res. 1996;56:5475–5483. [PubMed] [Google Scholar]
- Bataille V, Kato BS, Falchi M, et al. Nevus size and number are associated with telomere length and represent potential markers of a decreased senescence in vivo. Cancer Epidemiol. Biomarkers Prev. 2007;16:1499–1502. doi: 10.1158/1055-9965.EPI-07-0152. [DOI] [PubMed] [Google Scholar]
- Bennett DC. How to make a melanoma: what do we know of the primary clonal events? Pigment Cell Melanoma Res. 2008;21:27–38. doi: 10.1111/j.1755-148X.2007.00433.x. [DOI] [PubMed] [Google Scholar]
- Bode AM, Dong ZG. Post-translational modification of p53 in tumorigenesis. Nat. Rev. Cancer. 2004;4:793–805. doi: 10.1038/nrc1455. [DOI] [PubMed] [Google Scholar]
- Bond JA, Haughton MF, Rowson JM, Smith PJ, Gire V, Wynford-Thomas D, Wyllie FS. Control of replicative life span in human cells: barriers to clonal expansion intermediate between M1 senescence and M2 crisis. Mol. Cell. Biol. 1999;19:3103–3114. doi: 10.1128/mcb.19.4.3103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chehab NH, Malikzay A, Appel M, Halazonetis TD. Chk2/hCds1 functions as a DNA damage checkpoint in G1 by stabilizing p53. Genes Dev. 2000;14:278–288. [PMC free article] [PubMed] [Google Scholar]
- Chin K, Solorzano de CO, Knowles D, et al. In situ analyses of genome instability in breast cancer. Nat. Genet. 2004;36:984–988. doi: 10.1038/ng1409. [DOI] [PubMed] [Google Scholar]
- Clark WH, Elder DE, Guerry D, Braitman LE, Trock BJ, Schultz D, Synnestvedt M, Halpern AC. Model predicting survival in stage I melanoma based on tumor progression. J. Natl Cancer Inst. 1989;81:1893–1904. doi: 10.1093/jnci/81.24.1893. [DOI] [PubMed] [Google Scholar]
- Collado M, Serrano M. Senescence in tumours: evidence from mice and humans. Nat. Rev. Cancer. 2010;10:51–57. doi: 10.1038/nrc2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gershenwald JE, Soong SJ, Balch CM AJCC Melanoma Staging Committee. 2010 TNM staging system for cutaneous melanoma… and beyond. Ann. Surg. Oncol. 2010;17:1475–1477. doi: 10.1245/s10434-010-0986-3. [DOI] [PubMed] [Google Scholar]
- Gisselsson D, Höglund M. Connecting mitotic instability and chromosome aberrations in cancer–can telomeres bridge the gap? Semin. Cancer Biol. 2005;15:13–23. doi: 10.1016/j.semcancer.2004.09.002. [DOI] [PubMed] [Google Scholar]
- Gorgoulis VG, Vassiliou LV, Karakaidos P, et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005;434:907–913. doi: 10.1038/nature03485. [DOI] [PubMed] [Google Scholar]
- Gray-Schopfer VC, Bennett DC. The genetics of melanoma. In: Nordlund JJ, Boissy RE, Hearing VJ, King RA, Oetting WS, Ortonne JP, editors. The Pigmentary System. Physiology and Pathophysiology. 2nd edn. New York: Blackwell Publishing; 2006. pp. 472–488. [Google Scholar]
- Gray-Schopfer VC, Cheong SC, Chow J, Moss A, Abdel-Malek ZA, Marais R, Wynford-Thomas D, Bennett DC. Cellular senescence in naevi and immortalisation in melanoma: a role for p16? Br. J. Cancer. 2006;95:496–505. doi: 10.1038/sj.bjc.6603283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha L, Ichikawa T, Anver M, et al. ARF functions as a melanoma tumor suppressor by inducing p53-independent senescence. Proc. Natl Acad. Sci. USA. 2007;104:10968–10973. doi: 10.1073/pnas.0611638104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha L, Merlino G, Sviderskaya EV. Melanomagenesis: overcoming the barrier of melanocyte senescence. Cell Cycle. 2008;7:1944–1948. doi: 10.4161/cc.7.13.6230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haferkamp S, Tran SL, Becker TM, Scurr LL, Kefford RF, Rizos H. The relative contributions of the p53 and pRb pathways in oncogene induced melanocyte senescence. Aging. 2009;1:542–556. doi: 10.18632/aging.100051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of Cancer: the Next Generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Herbig U, Sedivy JM. Regulation of growth arrest in senescence: telomere damage is not the end of the story. Mech. Ageing Dev. 2006;127:16–24. doi: 10.1016/j.mad.2005.09.002. [DOI] [PubMed] [Google Scholar]
- Hsu M-Y, Elder DE, Herlyn M. Melanoma: the Wistar melanoma (WM) cell lines. In: Masters JRW, Palsson B, editors. Human Cell Culture. Vol. 3. London: Kluwer Academic Publishers; 2000. pp. 259–274. [Google Scholar]
- Jacobs JJL, Lange de T. p16INK4a as a second effector of the telomere damage pathway. Cell Cycle. 2005;4:1364–1368. doi: 10.4161/cc.4.10.2104. [DOI] [PubMed] [Google Scholar]
- Jung YS, Qian YJ, Chen XB. Examination of the expanding pathways for the regulation of p21 expression and activity. Cell. Signal. 2010;22:1003–1012. doi: 10.1016/j.cellsig.2010.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang TW, Yevsa T, Woller N, et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature. 2011;479:547–551. doi: 10.1038/nature10599. [DOI] [PubMed] [Google Scholar]
- Kaufmann WK, Nevis KR, Qu P, et al. Defective cell cycle checkpoint functions in melanoma are associated with altered patterns of gene expression. J. Invest. Dermatol. 2008;128:175–187. doi: 10.1038/sj.jid.5700935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller-Melchior R, Schmidt R, Piepkorn M. Expression of the tumor suppressor gene product p16INK4 in benign and malignant melanocytic lesions. J. Invest. Dermatol. 1998;110:932–938. doi: 10.1046/j.1523-1747.1998.00211.x. [DOI] [PubMed] [Google Scholar]
- Kiyono T, Foster SA, Koop JI, McDougall JK, Galloway DA, Klingelhutz AJ. Both RB/p16INK4a inactivation and telomerase activity are required to immortalize human epithelial cells. Nature. 1998;396:84–88. doi: 10.1038/23962. [DOI] [PubMed] [Google Scholar]
- Lassam NJ, From L, Kahn HJ. Overexpression of p53 is a late event in the development of malignant melanoma. Cancer Res. 1993;53:2235–2238. [PubMed] [Google Scholar]
- Lukas C, Falck J, Bartkova J, Bartek J, Lukas J. Distinct spatiotemporal dynamics of mammalian checkpoint regulators induced by DNA damage. Nat. Cell Biol. 2003;5:255–260. doi: 10.1038/ncb945. [DOI] [PubMed] [Google Scholar]
- Michaloglou C, Vredeveld LC, Soengas MS, Denoyelle C, Kuilman T, Horst van der CM, Majoor DM, Shay JW, Mooi WJ, Peeper DS. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature. 2005;436:720–724. doi: 10.1038/nature03890. [DOI] [PubMed] [Google Scholar]
- Molven A, Grimstvedt MB, Steine SJ, Harland M, Avril MF, Hayward NK, Akslen LA. A large Norwegian family with inherited malignant melanoma, multiple atypical nevi, and CDK4 mutation. Genes Chromosom. Cancer. 2005;44:10–18. doi: 10.1002/gcc.20202. [DOI] [PubMed] [Google Scholar]
- Mooi WJ, Krausz T. Pathology of Melanocytic Disorders. 2nd edn. London: Hodder Arnold; 2007. [Google Scholar]
- Nalabothula N, Chakravarty D, Pierce A, Carrier F. Over expression of nucleophosmin and nucleolin contributes to the suboptimal activation of a G2/M checkpoint in Ataxia Telangiectasia fibroblasts. Mol. Cell. Pharmacol. 2010;2:179–189. [PMC free article] [PubMed] [Google Scholar]
- Passos JF, Nelson G, Wang C, et al. Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol. Syst. Biol. 2010;6:347. doi: 10.1038/msb.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peeper DS. Oncogene-induced senescence and melanoma: where do we stand? Pigment Cell Melanoma Res. 2011;24:1107–1111. doi: 10.1111/j.1755-148X.2011.00933.x. [DOI] [PubMed] [Google Scholar]
- Peters G. Tumor suppression for ARFicionados: the relative contributions of p16INK4a and p14ARF in melanoma. J. Natl Cancer Inst. 2008;100:757–795. doi: 10.1093/jnci/djn156. [DOI] [PubMed] [Google Scholar]
- Siegel S, Castellan NJ. Nonparametric Statistics for the Behavioural Sciences. 2nd edn. London: McGraw Hill; 1998. [Google Scholar]
- Sirigu P, Piras F, Minerba L, Murtas D, Maxia C, Colombari R, Corbu A, Perra MT, Ugalde J. Prognostic prediction of the immunohistochemical expression of p16 and p53 in cutaneous melanoma: a comparison of two populations from different geographical regions. Eur. J. Histochem. 2006;50:191–198. [PubMed] [Google Scholar]
- Soo JK, MacKenzie Ross AD, Kallenberg DM, et al. Malignancy without immortality? Cellular immortalization as a possible late event in melanoma progression. Pigment Cell Melanoma Res. 2011;24:490–503. doi: 10.1111/j.1755-148X.2011.00850.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanaki C, Stefanaki K, Antoniou C, Argyrakos T, Stratigos A, Patereli A, Katsambas A. G1 cell cycle regulators in congenital melanocytic nevi. Comparison with acquired nevi and melanomas. J. Cutan. Pathol. 2008;35:799–808. doi: 10.1111/j.1600-0560.2007.00912.x. [DOI] [PubMed] [Google Scholar]
- Sviderskaya EV, Hill SP, Evans-Whipp TJ, Chin L, Orlow SJ, Easty DJ, Cheong SC, Beach D, DePinho RA, Bennett DC. p16Ink4a in melanocyte senescence and differentiation. J. Natl Cancer Inst. 2002;94:446–454. doi: 10.1093/jnci/94.6.446. [DOI] [PubMed] [Google Scholar]
- Sviderskaya EV, Gray-Schopfer VC, Hill SP, et al. p16/cyclin-dependent kinase inhibitor 2A deficiency in human melanocyte senescence, apoptosis, and immortalization: possible implications for melanoma progression. J. Natl Cancer Inst. 2003;95:723–732. doi: 10.1093/jnci/95.10.723. [DOI] [PubMed] [Google Scholar]
- Talve L, Sauroja I, Collan Y, Punnonen K, Ekfors T. Loss of expression of the p16INK4/CDKN2 gene in cutaneous malignant melanoma correlates with tumor cell proliferation and invasive stage. Int. J. Cancer. 1997;74:255–259. doi: 10.1002/(sici)1097-0215(19970620)74:3<255::aid-ijc4>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- Vredeveld LC, Possik PA, Meissl K, et al. Abrogation of BRAFV600E-induced senescence by PI3K pathway activation contributes to melanomagenesis. Genes Dev. 2012;26:1055–1069. doi: 10.1101/gad.187252.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vugt van MATM, Medema RH. Getting in and out of mitosis with Polo-like kinase-1. Oncogene. 2005;24:2844–2859. doi: 10.1038/sj.onc.1208617. [DOI] [PubMed] [Google Scholar]
- Wu YL, Dudognon C, Nguyen E, et al. Immunodetection of human telomerase reverse-transcriptase (hTERT) re-appraised: nucleolin and telomerase cross paths. J. Cell Sci. 2006;119:2797–2806. doi: 10.1242/jcs.03001. [DOI] [PubMed] [Google Scholar]
- Yang A, Shi G, Zhou C, Lu R, Li H, Sun L, Jin Y. Nucleolin maintains embryonic stem cell self-renewal by suppression of p53 protein-dependent pathway. J. Biol. Chem. 2011;286:43370–43382. doi: 10.1074/jbc.M111.225185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu G, Montgomery GW, James MR, Trent JM, Hayward NK, Martin NG, Duffy DL. A genome-wide scan for naevus count: linkage to CDKN2A and to other chromosome regions. Eur. J. Hum. Genet. 2007;15:94–102. doi: 10.1038/sj.ejhg.5201729. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Numbers of successfully immunostained lesions by antibody and diagnostic category
Figure S1. Established ATM/CHEK2 pathways: summary
Figure S2. Supporting data for Figure 2
Figure S3. RGB blue channel from all panels of Figure 4