

Abstract
Glatiramer acetate is a synthetic, random copolymer widely used as a first-line agent for the treatment of relapsing-remitting multiple sclerosis (MS). While earlier studies primarily attributed its clinical effect to a shift in the cytokine secretion of CD4+ T helper (Th) cells, growing evidence in MS and its animal model, experimental autoimmune encephalomyelitis (EAE), suggests that glatiramer acetate treatment is associated with a broader immunomodulatory effect on cells of both the innate and adaptive immune system. To date, glatiramer acetate-mediated modulation of antigen-presenting cells (APC) such as monocytes and dendritic cells, CD4+ Th cells, CD8+ T cells, Foxp3+ regulatory T cells and antibody production by plasma cells have been reported; in addition, most recent investigations indicate that glatiramer acetate treatment may also promote regulatory B-cell properties. Experimental evidence suggests that, among these diverse effects, a fostering interplay between anti-inflammatory T-cell populations and regulatory type II APC may be the central axis in glatiramer acetate-mediated immune modulation of CNS autoimmune disease. Besides altering inflammatory processes, glatiramer acetate could exert direct neuroprotective and/or neuroregenerative properties, which could be of relevance for the treatment of MS, but even more so for primarily neurodegenerative disorders, such as Alzheimer’s or Parkinson’s disease. In this review, we provide a comprehensive and critical overview of established and recent findings aiming to elucidate the complex mechanism of action of glatiramer acetate.
1. Introduction
Glatiramer acetate (copolymer-1) is a mixture of synthetic peptides of 40–100 residues randomly composed of four amino acids (L-glutamic acid, L-lysine, L-alanine and L-tyrosine) in a pre-defined molar ratio (4.2 : 3.4 : 1.4 : 1.0). Glatiramer acetate was initially developed almost 40 years ago to mimic a major component of the myelin sheath, myelin basic protein, and to induce experimental autoimmune encephalomyelitis (EAE), the animal model of multiple sclerosis (MS). However, unexpectedly, glatiramer acetate inhibited development of EAE in both rodents and monkeys.[1–4] Subsequent early open-label studies suggested that glatiramer acetate treatment may exert a clinical benefit in MS patients with a relapsing-remitting (RR) disease course. Such benefit was ultimately confirmed in 1995 when a phase III, double-blind, placebo-controlled, multicentre trial including 251 RR-MS patients demonstrated a mean reduction in the relapse rate of around 30%.[5] Primarily based on this trial, glatiramer acetate was approved for treating RR-MS patients by the US FDA in 1996 and is now approved in many countries worldwide. Recently, two independent studies including a total of more than 3000 RR-MS patients compared existing immunomodulatory agents for the treatment of MS ‘head to head’. Both studies suggested that the clinical efficacy of glatiramer acetate was comparable to other first-line treatments for MS, interferon (IFN)β-1a (REGARD [REbif vs Glatiramer Acetate in Relapsing MS Disease] study) and IFNβ-1b (BEYOND [Betaferon Efficacy Yielding Outcomes of a New Dose] study) administered at a high frequency and dose.[6,7]
While these studies undoubtedly confirmed the clinical efficacy of glatiramer acetate in the treatment of RR-MS, the mechanism by which glatiramer acetate mediates this benefit remains a matter of ongoing debate. Potential mechanisms of action proposed to date include a preferential differentiation of CD4+ T cells into T helper (Th)-2 cells,[8–10] an increase in frequency and function of CD25+FoxP3+ regulatory T cells,[11,12] modulation of CD8+ T cells,[13] as well as development of regulatory type II antigen presenting cells (APC);[10,12,14–18] most recent studies indicate that glatiramer acetate may also exert an immunomodulatory effect on B cells.[19,20] In addition to its immunomodulatory properties, some evidence suggests that glatiramer acetate may also have direct neuroprotective[21–24] and/or remyelinating properties,[25,26] presumably through an upregulation of neurotrophic factors.
In the following sections, we aim to provide a thorough review of experimental and clinical studies evaluating these diverse effects mediated by glatiramer acetate. These studies were identified by a literature search of PubMed, with no date limitations. Search terms used were ‘glatiramer acetate’, ‘copaxone’, ‘multiple sclerosis’, ‘experimental autoimmune encephalitis’, ‘T cells’, ‘antigen-presenting cells’, ‘neuroprotection’ and ‘animal models’.
2. Mechanism(s) of Action of Glatiramer Acetate: An Update
2.1 Immunomodulatory Effects
Many investigations have attempted to address the immunological basis for the clinical effects of glatiramer acetate in MS and MS models.[ 27,28] Early investigations attributed the immunomodulatory activity of glatiramer acetate to alterations in T-cell antigen reactivity, focusing on its influence on the adaptive immune response, while more recent studies indicate that glatiramer acetate acts on both the innate and the adaptive immune system.
2.1.1 Alteration of Immune Cells of the Adaptive Immune System
Early in vitro studies revealed that glatiramer acetate can bind to MHC class II molecules on the surface of APC.[29,30] In association with MHC class II molecules, glatiramer acetate is recognized by T cells via their T-cell receptor. As glatiramer acetate binding to MHC class II was shown to inhibit the activation of T-cell lines specific for myelin basic protein,[31] it was concluded that glatiramer acetate may compete with myelin antigens for binding to MHC class II. A later study demonstrated, however, that the stereoisomer of glatiramer acetate, D-glatiramer acetate, which contains solely D-amino acids, failed to suppress EAE,[32] while it could effectively bind to MHC class II,[33] indicating that MHC class II antagonism might not be the primary mechanism of glatiramer acetate.
In most MS patients, glatiramer acetate treatment induces a population of CD4+ glatiramer acetate-reactive Th2 cells in the periphery.[8,9,34,35] Several studies in EAE and MS generated the concept that glatiramer acetate-reactive Th2 cells may be reactivated within the CNS through cross-recognition of myelin antigen[9,36] followed by release of anti-inflammatory cytokines in a process termed ‘bystander suppression’. This assumption was primarily supported by the observation that glatiramer acetate-reactive Th2 cells could be identified in the CNS of glatiramer acetate-treated mice,[37] and that in some, but not all studies,[3] glatiramer acetate-specific Th2 cell lines generated from MS patients or mice crossreacted with myelin basic protein at the level of cytokine secretion.[9,36–39] Newer studies indicate, however, that glatiramer acetate suppresses CNS autoimmune disease in an antigen-independent manner[12,40] and thereby question whether recognition of myelin antigen is a requirement for glatiramer acetate-induced anti-inflammatory T-cell populations to exert clinical benefit.
CD4+CD25+Foxp3+ regulatory T cells (Treg) engage in the maintenance of immunological tolerance by actively suppressing self-reactive lymphocytes.[41,42] Similar to other autoimmune conditions,[43] effector function and frequency of Treg were found to be decreased in the peripheral blood of patients with MS.[44] Several studies provided evidence that glatiramer acetate treatment may restore CD4+CD25+ Treg function. In the treatment of MS, glatiramer acetate promoted conversion of CD4+CD25− toCD4+CD25+ Treg and led to a significant increase in the Foxp3 expression of CD4+ T cells.[11] In glatiramer acetate-reactive CD4+CD25+ T-cell lines generated from glatiramer acetate-treated MS patients, high levels of Foxp3 correlated with increased T-cell regulation.[11] Thus, besides the preferential Th2 differentiation of T cells, glatiramer acetate normalizes the frequency and function of Treg in MS, which may represent an additional immunomodulatory effect of glatiramer acetate.
Glatiramer acetate treatment also promotes development of CD8+ glatiramer acetate-reactive T cells. Untreated MS patients, not unlike healthy individuals, often reveal a pre-existing population of glatiramer acetate-reactive CD8+ T cells. Upon glatiramer acetate treatment, the frequency of these cells is significantly increased[13] with an enhanced release of IFNγ by these cells.[45] Although the in vivo function and clinical relevance of these cells is not yet entirely understood, one report indicated that glatiramer acetate-reactive CD8+ T cells may suppress proinflammatory effector T-cell function in a manner similar to CD4+CD25+ Treg.[46,47]
Besides these effects on CD4+ and CD8+ T cells, glatiramer acetate treatment appears to also alter the function of B cells. In earlier reports, glatiramer acetate was shown to induce a humoral response to itself in most patients.[48] Interestingly, glatiramer acetate-treated patients preferentially develop high titres of IgG4 antibodies against glatiramer acetate,[49] which may be a consequence of the induction of glatiramer acetate-reactive Th2 cells, as isotype switching towards IgG4 is promoted by the Th2 cytokine, interleukin (IL)-4. Unlike antibodies against IFNβ,[50] antibodies against glatiramer acetate may not dampen its clinical effect.[51] In this regard, Brenner et al.[52] reported that relapse-free patients displayed higher antibody titres against glatiramer acetate than patients with an active disease course under glatiramer acetate treatment. In an animal model of CNS demyelinating disease, glatiramer acetate-specific antibodies were shown to promote myelin repair,[53] thus indicating a beneficial rather than harmful role of antibodies against glatiramer acetate.
Recent studies indicate that glatiramer acetate may also influence cellular B-cell function. This is of particular interest in light of a recent EAE study[54] and MS clinical trials[55,56] indicating that anti-CD20-mediated depletion of B cells could be of remarkable benefit in the treatment of CNS autoimmune disease. These investigations also suggested that the clinical effect may primarily relate to abrogation of B-cell-mediated activation of encephalitogenic T cells. B cells may, however, have a dual role in CNS autoimmunity, serving as APC promoting development of Th1 and Th17 cells, but also as regulatory cells[57–59] promoting development of Treg [58] and inhibiting maturation and pro-inflammatory differentiation of other APC in vivo.[60] In the treatment ofMS, it may thus be most desirable to shift proinflammatory APC function towards anti-inflammatory B-cell properties. In this regard, two recent studies in mice revealed that glatiramer acetate treatment promoted B-cell production of anti-inflammatory cytokines, such as IL-10,[19] and inhibited release of IL-6, IL-12 and tumour necrosis factor (TNF).[20] Whereas these studies suggest that B-cell immunomodulation contributes to the clinical benefit of glatiramer acetate in CNS autoimmune disease, it remains to be determined whether this effect is primarily achieved by an altered B-cell APC function or provision of anti-inflammatory cytokines for regulation of T cells and other APC.
2.1.2 Effects on the Innate Immune System
Emerging evidence supports the concept that glatiramer acetate also acts on APC and that this property may be a rather early, if not the primary, event in the immunological cascade mediating the clinical benefit in glatiramer acetate treatment. Several groups reported that glatiramer acetate leads to a broad antigen nonspecific alteration of APC function in vitro.[10,14,15,18,61–64] In vitro glatiramer acetate treatment inhibited lipopolysaccharide- mediated expression of several activation markers on freshly isolated human monocytes, such as CD150/signalling lymphocytic activation molecule (SLAM), CD25 and CD69, and significantly lowered monocytic release of proinflammatory TNF and IL-12.[14] Another study demonstrated that glatiramer acetate treatment not only reduced the release of proinflammatory cytokines, but also enhanced production of anti-inflammatory IL-10 by monocytes.[15] Similarly, in vitro-generated human dendritic cells released less TNF[10] and IL-12[62] upon glatiramer acetate exposure.
Based on these in vitro findings, two independent studies investigated whether glatiramer acetate may similarly affect monocytes in treated patients with MS. In these studies, monocytes were isolated from glatiramer acetate-treated patients without any additional in vitro exposure to glatiramer acetate. Paralleling the in vitro findings described above, monocytes from glatiramer acetate-treated MS patients expressed significantly lower levels of the activation marker CD150/SLAM and released less TNF upon lipopolysaccharide stimulation.[14] Kim and colleagues[15] reported that the production of IL-12 was reduced in monocytes from glatiramer acetate-treated patients, whereas the release of IL-10 was significantly enhanced, which they referred to as an anti-inflammatory ‘type II’ monocyte phenotype. A recent study identified an alteration of the monocytic IL-1 system as a mechanism by which glatiramer acetate may promote development of type II monocytes.[18] In mice with EAE, as well as in patients with RR-MS, in vivo glatiramer acetate treatment enhanced blood levels of the secreted IL-1 receptor antagonist, the natural inhibitor of IL-1β. Intracellular signalling pathways induced by glatiramer acetate and controlling secreted IL-1 receptor antagonist (sIL-1Ra) expression in human monocytes were recently identified. These pathways involve the activation of PI3Kδ, Akt, MEK1/2 and ERK1/2, demonstrating that both PI3Kδ/Akt and MEK/ERK pathways govern sIL-1Ra expression in glatiramer acetate-treated human monocytes.[65]
2.1.3 Interplay of Innate and Adaptive Immune Systems in Glatiramer Acetate-Mediated Immune Modulation
In contrast to a former understanding in which antigen presentation simplistically triggered pro-inflammatory activation of the adaptive immune system, distinct APC phenotypes have been demonstrated more recently to exert an anti-inflammatory role in the regulation of T-cell-mediated autoimmunity. Reciprocally, differentiated T cells modify APC function. Experimental evidence suggests that APC may be the primary target of glatiramer acetate and that glatiramer acetate-induced type II APC may direct a secondary T-cell differentiation bias. In a recent publication, Weber et al.[12] dissected the immunological properties of glatiramer acetate on monocytes from its the effect on T cells, applying glatiramer acetate treatment back to EAE. Paralleling the observation inMS patients, clinically beneficial glatiramer acetate treatment of mice with EAE was associated with the development of type II monocytes, Th2 differentiation of T cells and expansion of Treg. Monocytes isolated from glatiramer acetate-treated mice secreted less proinflammatory TNF and IL-12, but more anti-inflammatory IL-10 and transforming growth factor-β (TGFβ), a cytokine with key function for the generation of Foxp3+ Treg.[66] Furthermore, type II monocytes secreted less IL-23 and IL-6, two cytokines that promote development of encephalitogenic Th17 cells.[66–68] Importantly, a comparable cytokine shift occurred in genetically altered mice, which lack mature B and T lymphocytes,[69] excluding that in vivo type II monocyte development was T-cell-dependent. In order to investigate whether type II monocytes may be responsible for the development of Th2 cells and Treg upon glatiramer acetate treatment, in vivo-induced type II monocytes were co-cultured with untreated naïve T cells without further in vitro glatiramer acetate exposure. Strikingly, naïve T cells activated by type II monocytes preferentially differentiated into IL-4-secreting Th2 cells and Foxp3 expressing Treg independent of the respective T-cell antigen specificity.[12] When type II monocytes were adoptively transferred into mice with established EAE, recipients revealed an ameliorated disease course, which was associated with a decreased frequency of Th1 and Th17 cells and expanded populations of Th2 cells and Treg.
These findings indicate that (i) type II monocyte development is of clinical relevance; and (ii) the type II phenotype of monocytes by itself is sufficiently capable of promoting development of regulatory T-cell populations in vivo. Although these findings are suggestive of a scenario in which T cells may not be the primary target of glatiramer acetate, they are most likely the effector cells of glatiramer acetate-mediated immune modulation. In EAE, adoptive transfer of glatiramer acetate-reactive T cells alone can ameliorate EAE in a manner similar to glatiramer acetate treatment itself. Furthermore, glatiramer acetate-reactive T cells accumulate in the CNS of animals benefiting from treatment.[70] Thus, whereas glatiramer acetate may mediate a primary effect on APC independent of T cells, type II APC-induced regulatory T-cell populations could be the effector cells of glatiramer acetate-mediated immune modulation.
Based on the assumption that APC are the primary target of glatiramer acetate treatment, one would anticipate that Th2 deviation and/or induction of Treg should not be restricted to glatiramer acetate-reactive T cells. In this regard, Kantengwa et al.[40] reported that glatiramer acetate inhibited Th1 differentiation of CD4+ T cells at various T-cell maturation stages and in an antigen-independent manner. Another study investigated the phenotype of T-cell lines specific for various antigens generated from MS patients before and after glatiramer acetate treatment onset.[71] T-cell differentiation was evaluated by the ratio between IFNγ and IL-5 release as Th1 and Th2 hallmark cytokines, respectively. In vivo glatiramer acetate treatment biased differentiation of all T-cell lines towards a Th2 phenotype, indicating that Th2 differentiation occurred independent of T-cell antigen specificity.[71] It needs to be noted, however, that another study did not observe an antigen-independent Th2 deviation of established T-cell responses upon glatiramer acetate treatment and reported that Th2 deviation may indeed primarily occur in glatiramer acetate-reactive T cells.[45] Although intuitively conflicting, both findings may be valid. Whereas Th2 deviation and expansion of Treg may in principle not be restricted to T cells specific for glatiramer acetate, it is plausible that an APC-driven Th2 deviation may be pronounced in glatiramer acetate-reactive T cells, as every APC that presents glatiramer acetate should have been in contact with glatiramer acetate and undergone type II differentiation prior to T-cell activation. Most importantly and of clinical relevance, the concept of an antigen nonspecific effect of glatiramer acetate is further supported by the fact that glatiramer acetate treatment has been shown to be clinically beneficial in other models of autoimmune or inflammatory conditions,[72–74] which will be discussed in detail in section 2.3.2.
Taken together, glatiramer acetate treatment triggers a coordinated concert of immunomodulatory effects on T cells, B cells and APC. Independent of the question of whether one particular cell type may be the primary target of glatiramer acetate, it remains a great challenge to better understand how this drug modulates immune cell function on the molecular level.
2.2 Potential Neuroprotective and Remyelinating Effects
Neuroprotective factors are proteins involved in the regulation of survival, growth and differentiation of neural cells including neurons and oligodendrocytes. The neurotrophic factor that has received most attention in glatiramer acetate studies is brain-derived neurotrophic factor (BDNF), although neurotrophin (NT)-3 and NT-4 may similarly be augmented in the CNS of glatiramer acetate-treated mice. BDNF is involved in the survival and differentiation of neurons and glial cells.[75,76] Interestingly, BDNF is also constitutively expressed in inflammatory cells including T cells, B cells and monocytes.[77] In MS lesions, BDNF is expressed in immune cells and reactive astrocytes in close proximity to BDNF receptor (trkB)-expressing neurons.[78] Furthermore, BDNF was found to be upregulated in glatiramer acetate-specific T-cell lines in vitro.[21–24] BDNF is inducible by glatiramer acetate not only in anti-inflammatory (Th2) but also in pro-inflammatory (Th1) cell lines.
Based on these observations made by independent laboratories, the hypothesis emerged that glatiramer acetate could have not only immunomodulatory properties but also neuroprotective properties, limiting damage and promoting neuronal repair through BDNF release. This possible neuroprotective effect of glatiramer acetate was further assessed on the survival of retinal ganglion cells, neurons that form the axons of the optic nerve.[79] After myelin oligodendrocyte glycoprotein (MOG)-induced EAE in a rat model, optic neuritis was monitored by recording visual evoked potentials and the function of retinal ganglion cells was measured by electroretinogram. Glatiramer acetate treatment exerted protective effects on retinal ganglion cells, as evaluated by measuring neurodegeneration and neuronal function.[79] In another study, EAE was induced by MOG, either in yellow fluorescent protein (YFP) 2.2 transgenic mice, which selectively express YFP on their neuronal population, or in C57BL/6 mice.[80] Neuroprotection and neurogeneration were quantified by measuring the expression of assorted neuronal antigens and markers for in vivo proliferation. Glatiramer acetate treatment in various stages of the disease course led to a sustained reduction in the neuronal/axonal damage. Cell proliferation, migration and differentiation were augmented and extended by glatiramer acetate treatment in EAE mice compared with EAE-untreated mice. These findings may suggest a direct linkage between immunomodulation, neurogenesis and an in situ therapeutic consequence in the CNS.[80]
In general, glatiramer acetate-stimulated T cells are able to enter theCNS.[37] Mice adoptively transferred with glatiramer acetate-specific T cells[70] or treated daily with glatiramer acetate[21] reveal an upregulation of BDNF in the CNS. An EAE study has further shown that glatiramer acetate-treated mice show less axonal damage as measured by a reduction of 85% in SMI-32 staining and a 63%reduction in amyloid precursor protein (APP) staining when compared with untreated mice.[81] In vitro, glatiramer acetate decreases neuronal cell death induced by staurosporine and oxidative stress. This neuroprotective effect was shown to be associated with an upregulation of BDNF.[82] The ability of glatiramer acetate to induce BDNF is not only observed in vitro and in EAE but also in MS patients. Here, initially decreased levels of BDNF in the serum and the CSF of MS patients were shown to be restored by glatiramer acetate treatment.[83] In addition, glatiramer acetate-treated patients were shown to have a decreased number of MRI ‘black holes’, brain lesions in which severe tissue and axon disruption has occurred.[84] A neuroprotective effect of glatiramer acetate in MS patients was further supported by another clinical study using brain proton magnetic resonance spectroscopy (1H-MRS) to perform in vivo examination of axonal integrity. Quantifying the ratio of the neuronal marker N-acetylaspartate (NAA) to creatine over a 2-year period, the authors were able to demonstrate that glatiramer acetate treatment supported axonal integrity.[85]
The beneficial effect of glatiramer acetate is not limited to its effect on neurons but also involves a reduction of demyelination and an increase of myelin repair. In this regard, glatiramer acetate-treated mice reveal a substantial decrease of demyelination, which correlates with an improved clinical score.[26] Recovery from immunemediated demyelination in EAE and MS is initiated by remyelination of axons within CNS lesions. This repair process requires recruitment and migration of oligodendrocyte precursor cells (OPCs) into demyelinated lesions and their differentiation into myelin sheath-forming oligodendrocytes.[ 86,87] In the spinal cord of glatiramer acetate-treated mice, expression of OPC markers indicating their activation and proliferation was found to be elevated. Promoted remyelination by glatiramer acetate was thus attributed to an increase in proliferation, differentiation and survival of OPCs, along with their recruitment into injury sites.[26] The hypothesis of a beneficial effect of glatiramer acetate on oligodendrogenesis and myelin repair is further supported by the recent demonstration that lysolecithin-induced demyelination of the spinal cord in mice is associated with an elevation of not only BDNF, but also insulin-like growth factor-1, a protein that promotes maturation of OPCs. After spinal cord insult, mice treated with glatiramer acetate further revealed less demyelination at the site of injury and an increased occurrence of OPCs.[25]
Whether the reduction in axonal and myelin damage observed in glatiramer acetate-treated mice is truly and exclusively related to a release of neurotrophic factors, or alternatively a consequence of the limitation of CNS inflammation, is still being debated. It is indeed very difficult to dissociate whether a decrease of neuronal and myelin injury occurs secondary to a blockade of inflammatory pathways or by a direct release of neurotrophic factor, especially when both effects occur simultaneously. The difficulty in demonstrating a direct neuroprotective effect of glatiramer acetate is also based on the absence of firmly validated markers that would allow quantification of axonal loss and myelin damage in prospective clinical studies. To date, such evidence relies mainly on in vitro findings and animal studies. This concern is also highlighted by recent clinical data: glatiramer acetate was tested in the primary progressive (PP) form of MS, a subtype of the disease affecting around 15% of patients with MS. PP-MS is considered to be associated with an early and sustained process of neurodegeneration that typically does not respond to immunomodulatory or immunosuppressive drugs.[88,89] The study failed to show a beneficial effect of glatiramer acetate in PP-MS.[90] On the one hand, these results reinforce the importance of a cautious interpretation of data when not based on clinical studies. On the other hand, the absence of efficacy of glatiramer acetate in PP-MS patients does not formally exclude a superimposed neuroprotective effect of glatiramer acetate in the treatment of RR-MS, since the pathophysiology of neurodegeneration seen in RR-MS may substantially differ from the one present in PP-MS.
2.3 Glatiramer Acetate Effects: Lessons from Other Animal Models
2.3.1 Neurodegenerative Diseases of the CNS
Evaluation of neuroprotection and myelin repair mediated by glatiramer acetate may be most promising in animal models that are not primarily associated with a deregulation of the immune system. Accordingly, glatiramer acetate was also studied in three different animal models, mimicking Alzheimer’s disease, Parkinson’s disease and amyotrophic lateral sclerosis (ALS).
Alzheimer’s disease is clinically characterized by a cognitive decline associated with histopathological plaque formation and neuronal loss. Its pathogenesis is supposed to be primarily degenerative, although a role for accompanying inflammation is not ruled out. In Alzheimer’s disease double-transgenic (APP/PS1) mice, glatiramer acetate treatment resulted in a decrease of plaque formation along with neurogenesis.[91] Remarkably, the main effect suggested to explain this favourable outcome was an induction of dendritic cells to produce neuroprotective insulinlike growth factor-1. Another approach to treat this Alzheimer’s disease model was a nasal vaccination composed of a proteosome-based adjuvant plus glatiramer acetate. The results showed a decrease of plaques that correlated with microglial activation.[92]
Parkinson’s disease can be mimicked in 1-methyl- 4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-intoxicated mice. Here, adoptive transfer of glatiramer acetate-treated immune cells to MPTP recipient mice led to a T-cell accumulation within the substantia nigra, which was associated with suppression of microglial activation and an increased expression of astrocyte-associated glial cell line-derived neurotrophic factor. This treatment protocol resulted in a significant protection of nigrostriatal neurons against MPTP-induced neurodegeneration.[93]
ALS is a rapidly progressing neurodegenerative disorder in which mediators of toxicity such as metabolites of glutamate pathways and oxidative stress are thought to be involved. This devastating disease affects both upper and lower motor neurons, and leads to death in a mean of 3–5 years after diagnosis. Mice that express the mutant human gene Cu/Zn superoxide dismutase (SOD1) develop an ALS-like disease. Glatiramer acetate vaccination was evaluated in this animal model (in a protocol different from the ones used in the treatment of CNS autoimmune disease) and was found to significantly prolong life span and motor activity, and to protect motor neurons from acute or chronic degeneration.[94]
2.3.2 Inflammatory Conditions Other than CNS Autoimmune Disease
Accumulating evidence suggests that the mechanism of action of glatiramer acetate is not limited to counteracting myelin-specific T cells but that glatiramer acetate rather induces a broad, antigen-independent immunomodulatory effect on CD4+ T cells, CD8+ T cells, B cells and APC. These findings also imply that immunomodulation by glatiramer acetate should not be restricted to CNS autoimmune disease. In this regard, recent experimental studies reported remarkable results on the effect of glatiramer acetate on immune-mediated disease models other than EAE.
Experimental autoimmune uveoretinitis is an organ-specific, Th1-mediated disease that targets the neural retina. In mice, in which this disease model can be induced by interphotoreceptor retinoid-binding protein, glatiramer acetate was shown to inhibit the development of experimental autoimmune uveoretinitis, with a 53% reduction of the disease severity.[72]
Crohn’s disease is an inflammatory bowel disease (IBD) that shares with EAE and MS a proinflammatory cytokine predominance. The first description of a beneficial effect of glatiramer acetate in IBD was observed with trinitrobenzene sulfonic acid (TNBS)-induced colitis, a murine model that resembles human Crohn’s disease.[95] Experimental colitis was induced by rectal instillation of TNBS in three mice strains: BALB/c, SJL/J and (SJL/JXBALB/c)F1. Glatiramer acetate treatment significantly suppressed TNBS-induced colitis as demonstrated by a substantial reduction in the macroscopic colonic damage, preservation of the microscopic colonic structure, reduced weight loss and improved long-term survival. Glatiramer acetate inhibited proliferation of local mesenteric lymphocytes to syngeneic colon extract and the detrimental TNF and TGFβ.[95] The beneficial effect of glatiramer acetate in IBD was further confirmed with the demonstration that glatiramer acetate suppresses both proinflammatory T-cell responses as well as natural killer cell activity.[73,74] Interestingly, as shown in EAE, the therapeutic effect of glatiramer acetate can be adoptively replaced by glatiramer acetate-specific T cells that are recruited to the injury site, i.e. the intestine.[ 96] In addition, a recent case report describes a patient with active Crohn’s disease in whom glatiramer acetate may have promoted disease remission.[97]
Graft rejection was also suppressed by glatiramer acetate as evaluated in the following two systems: (i) by prolongation of skin graft survival; and (ii) by inhibition of deterioration of thyroid grafts. In these transplantation models, glatiramer acetate treatment inhibited the detrimental secretion of inflammatory cytokines and induced an anti-inflammatory response associated with clinical benefit.[74]
Collagen-induced arthritis (CIA) is an animal model of rheumatoid arthritis, which is also supposed to be primarily Th1 mediated. Surprisingly, glatiramer acetate treatment was shown to exacerbate CIA, leading to a faster onset and a more severe, longer-lasting disease course. The mechanisms underlying the exacerbation of CIA by glatiramer acetate included enhanced activation and cytokine secretion by auto-reactive T cells along with an increased production of auto-reactive antibodies.[98]
Systemic lupus erythematosus (SLE) is an autoimmune disease with a rather unpredictable course that typically affects skin, joint, heart, lungs, liver, kidney, blood vessels and the nervous system. Borel et al.[99] recently evaluated the effect of glatiramer acetate in an animal model of SLE. Male (NZB × BXSB) F1 mice are a lupusprone mouse strain, which is associated with auto-antibodies as well as a monocytosis accelerating disease progression. These mice were treated with glatiramer acetate before disease onset until death, and both mortality rate and immunological parameters were assessed. Glatiramer acetate exerted no significant effect on the median survival. Humoral and cellular parameters used as markers for lupus progression, such as anti-chromatin, anti-double-strandedDNA (dsDNA) and anti-erythrocyte antibodies, haematocrit and monocytosis, were found to be unchanged. Many hypotheses can be raised about the absence of efficacy of glatiramer acetate in these lupus-prone mice, including the fact that the animal model used in this study is one among several spontaneous, transgenic or toxic SLE models. In addition, glatiramer acetate is known to promote the development of Th2 cells that may potentially stimulate B cells to secrete auto-antibodies, which may ultimately favour SLE progression. Finally, glatiramer acetate treatment was administered at an identical concentration to that used in the treatment of EAE models; it cannot be excluded that other doses may provide a different clinical outcome.
Taken together, these studies demonstrate a clinical benefit of glatiramer acetate in several, but not all, animal models of inflammatory diseases other than EAE. Besides their clinical implication, these observations strengthen the hypothesis that glatiramer acetate exerts its immunomodulatory effect in an antigen-independent manner.
3. Conclusion
Glatiramer acetate is a well tolerated first-line treatment for RR-MS. Similar to all currently available agents, glatiramer acetate is only partially effective in the prevention of relapses and accumulation of disability throughout the disease course. Historically, glatiramer acetate was one of the first drugs developed and has proven to be effective using the animal model of MS, EAE. Early investigations attributed the clinical benefit mediated by glatiramer acetate to an interference with the development of self-reactive, proinflammatory T cells through competitive binding ofMHC class II. Studies in MS and EAE provided broad evidence that glatiramer acetate treatment is associated with a preferential Th2 differentiation of glatiramer acetate-reactive T cells. These Th2 cells may enter the CNS, providing anti-inflammatory cytokines at the site of inflammation, possibly upon cross-recognition of CNS auto-antigen. Later investigations widened the scenario, with immunomodulatory effects on various immune cells, such as monocytes and dendritic cells, CD8+ T cells and Foxp3+ regulatory T cells. To date, the majority of existing data point towards a scenario in which an interplay between the innate and adaptive immune systems, namely APC and T cells, may result in a well organized ‘concert’ of effects providing the immunological basis for the clinical benefits of glatiramer acetate (see figure 1). Recent studies indicate that glatiramer acetate can also modulate cellular B-cell function. This is of particular interest as B cells may play an important proinflammatory role in CNS autoimmune disease, which is reflected by the encouraging results testing anti-CD20 B-cell depletion in MS and EAE. B cells may, however, also serve as regulatory cells, promoting development of Treg and inhibiting proinflammatory activity of other APC. Immune modulation of B cells as a general therapeutic concept may thus be a most promising strategy in the treatment of MS.
Fig. 1.
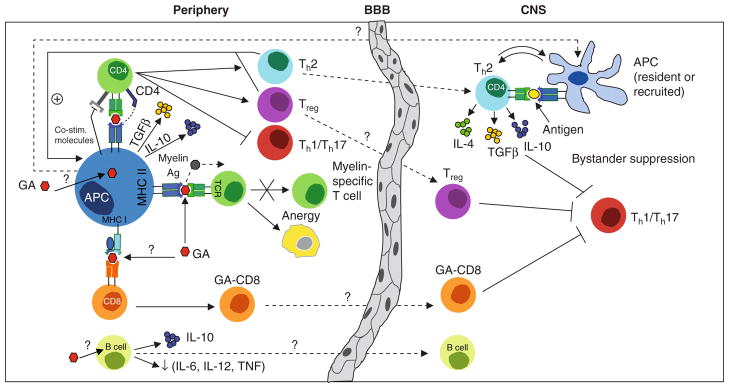
Glatiramer acetate (GA) mediates pleiotropic immunomodulatory effects. GA treatment promotes development of anti-inflammatory type II antigen presenting cells (APC) predominantly producing interleukin (IL)-10 and transforming growth factor-β (TGFβ). Upon encounter of naïve CD4+ T cells, type II APC deviate T-cell differentiation towards preferential development of T helper (Th)-2 cells and Foxp3+ regulatory T cells (Treg). Type II APC and Th2/Treg cells may facilitate each other’s development in a positive feedback mechanism, as T cell-derived anti-inflammatory cytokines may in return foster type II differentiation of APC. GA-promoted anti-inflammatory CD4+ T cells may cross the blood-brain- barrier (BBB) and are thought to be locally reactivated within the CNS. In response, these cells may secrete anti-inflammatory cytokines and neurotrophic factors and dampen neighbouring inflammation (‘bystander suppression’). Another feedback loop between APC and T cells may develop within the CNS itself, as T cell-derived cytokines could also promote type II differentiation of resident or recruited CNS APC. In addition, GA can bind to MHC molecules and may thereby compete with myelin antigens (Ag) for their presentation to T cells. Furthermore, GA treatment is associated with induction of GA-reactive CD8+ T cells and most recent findings indicate that GA may also exert an immunomodulatory effect on B cells promoting secretion of anti-inflammatory cytokines. The in vivo function of GA-affected CD8 cells and B cells, and whether these cells may enter the CNS, remains to be determined. co-stim = costimulatory; TCR= T cell receptor; TNF = tumour necrosis factor; ? indicates assumed mechanism; ↓ indicates decrease.
The immunomodulatory effect of glatiramer acetate may not be restricted to CNS autoimmune disease, as several studies demonstrate clinical benefit in models of other inflammatory conditions, such as uveoretinitis, Crohn’s disease and graft rejection, reinforcing the hypothesis that glatiramer acetate may promote immunomodulation in an antigen-independent manner. Glatiramer acetate has further proven some clinical benefit in models of neurodegenerative diseases with little or no apparent inflammation, including ALS, Alzheimer’s disease and Parkinson’s disease. Whereas these findings are in favour of a direct neuroprotective effect, it is still a matter of debate whether glatiramer acetate exerts such a property independent of its immunomodulatory activity.
Acknowledgments
No sources of funding were used to assist in the preparation of this review. P.H.L. is supported by grants from the Swiss National Foundation (#310000-132705), the Swiss Multiple Sclerosis Society and the Alliance SEP association. D.B. is supported by the Swiss Multiple Sclerosis Society and the Hans Wilsdorf Foundation. R.H. received personal compensation for activities such as advisory boards, consultancy fees from TEVA/Sanofi-Aventis, Bayer/Schering, Merck-Serono, Biogen-Idec and Novartis. R.H. received grant support from Deutsche Forschungsgemeinschaft (DFG), BMBF-KKNMS, TEVA, Bayer/Schering, Merck-Serono, Biogen-Idec and Novartis. S.S.Z. is supported by grants from the National Institute of Health (RO1 AI073737, RO1 AI059709 and RO1 NS063008), the National Multiple Sclerosis Society (NMSS; RG 3622 and 3913), Dana Foundation, Guthy Jackson Charitable Foundation and the Maisin Foundation. M.S.W. received grant support from the NMSS (RG 445A1/T), TEVA, the Else Kröner Fresenius Stiftung (A69/2010), the Kommission für Klinische Forschung (KKF) of the Technische Universität München and the DFG (WE 3547/4-1).
Footnotes
The other authors have no conflicts of interest to declare.
References
- 1.Teitelbaum D, Meshorer A, Hirshfeld T, et al. Suppression of experimental allergic encephalomyelitis by a synthetic polypeptide. Eur J Immunol. 1971 Aug;1( 4):242–8. doi: 10.1002/eji.1830010406. [DOI] [PubMed] [Google Scholar]
- 2.Teitelbaum D, Webb C, Bree M, et al. Suppression of experimental allergic encephalomyelitis in Rhesus monkeys by a synthetic basic copolymer. Clin Immunol Immunopathol. 1974 Nov;3( 2):256–62. doi: 10.1016/0090-1229(74)90012-9. [DOI] [PubMed] [Google Scholar]
- 3.Lisak RP, Zweiman B, Blanchard N, et al. Effect of treatment with Copolymer 1 (Cop-1) on the in vivo and in vitro manifestations of experimental allergic encephalomyelitis (EAE) J Neurol Sci. 1983 Dec;62( 1–3):281–93. doi: 10.1016/0022-510x(83)90205-8. [DOI] [PubMed] [Google Scholar]
- 4.Teitelbaum D, Fridkis-Hareli M, Arnon R, et al. Copolymer 1 inhibits chronic relapsing experimental allergic encephalomyelitis induced by proteolipid protein (PLP) peptides in mice and interferes with PLP-specific T cell responses. J Neuroimmunol. 1996 Feb;64( 2):209–17. doi: 10.1016/0165-5728(95)00180-8. [DOI] [PubMed] [Google Scholar]
- 5.Johnson KP, Brooks BR, Cohen JA, et al. Copolymer 1 reduces relapse rate and improves disability in relapsing-remitting multiple sclerosis: results of a phase III multicenter, double-blind, placebo-controlled trial. Neurology. 1995;45:1268–76. doi: 10.1212/wnl.45.7.1268. [DOI] [PubMed] [Google Scholar]
- 6.Mikol DD, Barkhof F, Chang P, et al. Comparison of subcutaneous interferon beta-1a with glatiramer acetate in patients with relapsing multiple sclerosis (the REbif vs Glatiramer Acetate in Relapsing MS Disease [REGARD] study): a multicentre, randomised, parallel, open-label trial. Lancet Neurol. 2008 Oct;7( 10):903–14. doi: 10.1016/S1474-4422(08)70200-X. [DOI] [PubMed] [Google Scholar]
- 7.O’Connor P, Filippi M, Arnason B, et al. 250 mug or 500 mug interferon beta-1b versus 20 mg glatiramer acetate in relapsing-remitting multiple sclerosis: a prospective, randomised, multicentre study. Lancet Neurol. 2009 Oct;8( 10):889–97. doi: 10.1016/S1474-4422(09)70226-1. [DOI] [PubMed] [Google Scholar]
- 8.Duda PW, Schmied MC, Cook SL, et al. Glatiramer acetate (Copaxone) induces degenerate, Th2-polarized immune responses in patients with multiple sclerosis. J Clin Invest. 2000;105( 7):967–76. doi: 10.1172/JCI8970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Neuhaus O, Farina C, Yassouridis A, et al. Multiple sclerosis: comparison of copolymer-1-reactive T cell lines from treated and untreated subjects reveals cytokine shift from T helper 1 to T helper 2 cells. Proc Natl Acad SciUS A. 2000;97( 13):7452–7. doi: 10.1073/pnas.97.13.7452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vieira PL, Heystek HC, Wormmeester J, et al. Glatiramer acetate (copolymer-1, copaxone) promotes Th2 cell development and increased IL-10 production through modulation of dendritic cells. J Immunol. 2003 May 1;170( 9):4483–8. doi: 10.4049/jimmunol.170.9.4483. [DOI] [PubMed] [Google Scholar]
- 11.Hong J, Li N, Zhang X, et al. Induction of CD4+CD25+ regulatory T cells by copolymer-I through activation of transcription factor Foxp3. Proc Natl Acad Sci U S A. 2005 May 3;102( 18):6449–54. doi: 10.1073/pnas.0502187102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weber MS, Prod’homme T, Youssef S, et al. Type II monocytes modulate T cell-mediated central nervous system autoimmune disease. Nat Med. 2007 Aug;13( 8):935–43. doi: 10.1038/nm1620. [DOI] [PubMed] [Google Scholar]
- 13.Karandikar NJ, Crawford MP, Yan X, et al. Glatiramer acetate (Copaxone) therapy induces CD8(+) T cell responses in patients with multiple sclerosis. J Clin Invest. 2002 Mar;109( 5):641–9. doi: 10.1172/JCI14380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weber MS, Starck M, Wagenpfeil S, et al. Multiple sclerosis: glatiramer acetate inhibits monocyte reactivity in vitro and in vivo. Brain. 2004 Jun;127( Pt 6):1370–8. doi: 10.1093/brain/awh163. [DOI] [PubMed] [Google Scholar]
- 15.Kim HJ, Ifergan I, Antel JP, et al. Type 2 monocyte and microglia differentiation mediated by glatiramer acetate therapy in patients with multiple sclerosis. J Immunol. 2004 Jun 1;172( 11):7144–53. doi: 10.4049/jimmunol.172.11.7144. [DOI] [PubMed] [Google Scholar]
- 16.Stasiolek M, Bayas A, Kruse N, et al. Impaired maturation and altered regulatory function of plasmacytoid dendritic cells in multiple sclerosis. Brain. 2006 May;129( Pt 5):1293–305. doi: 10.1093/brain/awl043. [DOI] [PubMed] [Google Scholar]
- 17.Stuve O, Youssef S, Weber MS, et al. Immunomodulatory synergy by combination of atorvastatin and glatiramer acetate in treatment of CNS autoimmunity. J Clin Invest. 2006 Apr;116( 4):1037–44. doi: 10.1172/JCI25805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burger D, Molnarfi N, Weber MS, et al. Glatiramer acetate increases IL-1 receptor antagonist but decreases T cell-induced IL-1beta in human monocytes and multiple sclerosis. Proc Natl Acad Sci U S A. 2009 Mar 17;106( 11):4355–9. doi: 10.1073/pnas.0812183106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kala M, Rhodes SN, Piao WH, et al. B cells from glatiramer acetate-treated mice suppress experimental autoimmune encephalomyelitis. Exp Neurol. 2010 Jan;221( 1):136–45. doi: 10.1016/j.expneurol.2009.10.015. [DOI] [PubMed] [Google Scholar]
- 20.Begum-Haque S, Sharma A, Christy M, et al. Increased expression of B cell-associated regulatory cytokines by glatiramer acetate in mice with experimental autoimmune encephalomyelitis. J Neuroimmunol. 2010 Feb 26;219( 1–2):47–53. doi: 10.1016/j.jneuroim.2009.11.016. [DOI] [PubMed] [Google Scholar]
- 21.Aharoni R, Eilam R, Domev H, et al. The immunomodulator glatiramer acetate augments the expression of neurotrophic factors in brains of experimental autoimmune encephalomyelitis mice. Proc Natl Acad Sci U S A. 2005 Dec 27;102( 52):19045–50. doi: 10.1073/pnas.0509438102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen M, Valenzuela RM, Dhib-Jalbut S. Glatiramer acetatereactive T cells produce brain-derived neurotrophic factor. J Neurol Sci. 2003 Nov 15;215( 1–2):37–44. doi: 10.1016/s0022-510x(03)00177-1. [DOI] [PubMed] [Google Scholar]
- 23.Kipnis J, Yoles E, Porat Z, et al. T cell immunity to copolymer 1 confers neuroprotection on the damaged optic nerve: possible therapy for optic neuropathies. Proc Natl Acad Sci U S A. 2000 Jun 20;97( 13):7446–51. doi: 10.1073/pnas.97.13.7446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ziemssen T, Kumpfel T, Klinkert WE, et al. Glatiramer acetate-specific T-helper 1- and 2-type cell lines produce BDNF: implications for multiple sclerosis therapy – brain-derived neurotrophic factor. Brain. 2002 Nov;125( Pt 11):2381–91. doi: 10.1093/brain/awf252. [DOI] [PubMed] [Google Scholar]
- 25.Skihar V, Silva C, Chojnacki A, et al. Promoting oligodendrogenesis and myelin repair using the multiple sclerosis medication glatiramer acetate. Proc Natl Acad Sci U S A. 2009 Oct 20;106( 42):17992–7. doi: 10.1073/pnas.0909607106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aharoni R, Herschkovitz A, Eilam R, et al. Demyelination arrest and remyelination induced by glatiramer acetate treatment of experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 2008 Aug 12;105( 32):11358–63. doi: 10.1073/pnas.0804632105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Neuhaus O, Farina C, Wekerle H, et al. Mechanisms of action of glatiramer acetate in multiple sclerosis. Neurology. 2001;56( 6):702–8. doi: 10.1212/wnl.56.6.702. [DOI] [PubMed] [Google Scholar]
- 28.Farina C, Weber MS, Meinl E, et al. Glatiramer acetate in multiple sclerosis: update on potential mechanisms of action. Lancet Neurol. 2005 Sep;4( 9):567–75. doi: 10.1016/S1474-4422(05)70167-8. [DOI] [PubMed] [Google Scholar]
- 29.Fridkis-Hareli M, Strominger JL. Promiscuous binding of synthetic copolymer 1 to purified HLA-DR molecules. J Immunol. 1998 May 1;160( 9):4386–97. [PubMed] [Google Scholar]
- 30.Fridkis-Hareli M, Teitelbaum D, Gurevich E, et al. Direct binding of myelin basic protein and synthetic copolymer 1 to class II major histocompatibility complex molecules on living antigen-presenting cells: specificity and promiscuity. Proc Natl Acad Sci U S A. 1994 May 24;91( 11):4872–6. doi: 10.1073/pnas.91.11.4872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Teitelbaum D, Milo R, Arnon R, et al. Synthetic copolymer 1 inhibits human T-cell lines specific for myelin basic protein. Proc Natl Acad Sci U S A. 1992 Jan 1;89( 1):137–41. doi: 10.1073/pnas.89.1.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Webb C, Teitelbaum D, Herz A, et al. Molecular requirements involved in suppression of EAE by synthetic basic copolymers of amino acids. Immunochemistry. 1976 Apr;13( 4):333–7. doi: 10.1016/0019-2791(76)90344-x. [DOI] [PubMed] [Google Scholar]
- 33.Aharoni R, Schlegel PG, Teitelbaum D, et al. Studies on the mechanism and specificity of the effect of the synthetic random copolymer GLAT on graft-versus-host disease. Immunol Lett. 1997 Jul;58( 2):79–87. doi: 10.1016/s0165-2478(97)00032-1. [DOI] [PubMed] [Google Scholar]
- 34.Wiesemann E, Klatt J, Sonmez D, et al. Glatiramer acetate (GA) induces IL-13/IL-5 secretion in naive T cells. J Neuroimmunol. 2001 Sep 3;119( 1):137–44. doi: 10.1016/s0165-5728(01)00379-4. [DOI] [PubMed] [Google Scholar]
- 35.Dhib-Jalbut S. Mechanisms of action of interferons and glatiramer acetate in multiple sclerosis. Neurology. 2002 Apr 23;58(8 Suppl 4):S3–9. doi: 10.1212/wnl.58.8_suppl_4.s3. [DOI] [PubMed] [Google Scholar]
- 36.Aharoni R, Teitelbaum D, Sela M, et al. Copolymer 1 induces T cells of the T helper type 2 that crossreact with myelin basic protein and suppress experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 1997;94( 20):10821–6. doi: 10.1073/pnas.94.20.10821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aharoni R, Teitelbaum D, Leitner O, et al. Specific Th2 cells accumulate in the central nervous system of mice protected against experimental autoimmune encephalomyelitis by copolymer 1. Proc Natl Acad Sci U S A. 2000;97( 21):11472–7. doi: 10.1073/pnas.97.21.11472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen M, Gran B, Costello K, et al. Glatiramer acetate induces a Th2-biased response and crossreactivity with myelin basic protein in patients with MS. Mult Scler. 2001 Aug;7( 4):209–19. doi: 10.1177/135245850100700401. [DOI] [PubMed] [Google Scholar]
- 39.Aharoni R, Teitelbaum D, Sela M, et al. Bystander suppression of experimental autoimmune encephalomyelitis by T cell lines and clones of the Th2 type induced by co-polymer 1. J Neuroimmunol. 1998;91( 1–2):135–46. doi: 10.1016/s0165-5728(98)00166-0. [DOI] [PubMed] [Google Scholar]
- 40.Kantengwa S, Weber MS, Juillard C, et al. Inhibition of naive Th1 CD4+ T cells by glatiramer acetate in multiple sclerosis. J Neuroimmunol. 2007 Apr;185( 1–2):123–9. doi: 10.1016/j.jneuroim.2006.12.014. [DOI] [PubMed] [Google Scholar]
- 41.Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003 Feb 14;299( 5609):1057–61. doi: 10.1126/science.1079490. [DOI] [PubMed] [Google Scholar]
- 42.Brunkow ME, Jeffery EW, Hjerrild KA, et al. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet. 2001 Jan;27( 1):68–73. doi: 10.1038/83784. [DOI] [PubMed] [Google Scholar]
- 43.Kukreja A, Cost G, Marker J, et al. Multiple immunoregulatory defects in type-1 diabetes. J Clin Invest. 2002 Jan;109( 1):131–40. doi: 10.1172/JCI13605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Viglietta V, Baecher-Allan C, Weiner HL, et al. Loss of functional suppression by CD4+CD25+ regulatory T cells in patients with multiple sclerosis. J Exp Med. 2004 Apr 5;199( 7):971–9. doi: 10.1084/jem.20031579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Farina C, Then Bergh F, Albrecht H, et al. Treatment of multiple sclerosis with Copaxone (COP): Elispot assay detects COP-induced interleukin-4 and interferon-gamma response in blood cells. Brain. 2001 Apr;124( Pt 4):705–19. doi: 10.1093/brain/124.4.705. [DOI] [PubMed] [Google Scholar]
- 46.Tennakoon DK, Mehta RS, Ortega SB, et al. Therapeutic induction of regulatory, cytotoxic CD8+ T cells in multiple sclerosis. J Immunol. 2006 Jun 1;176( 11):7119–29. doi: 10.4049/jimmunol.176.11.7119. [DOI] [PubMed] [Google Scholar]
- 47.Biegler BW, Yan SX, Ortega SB, et al. Glatiramer acetate (GA) therapy induces a focused, oligoclonal CD8+ T-cell repertoire in multiple sclerosis. J Neuroimmunol. 2006 Nov;180( 1–2):159–71. doi: 10.1016/j.jneuroim.2006.07.015. [DOI] [PubMed] [Google Scholar]
- 48.Teitelbaum D, Brenner T, Abramsky O, et al. Antibodies to glatiramer acetate do not interfere with its biological functions and therapeutic efficacy. Mult Scler. 2003 Dec;9( 6):592–9. doi: 10.1191/1352458503ms963oa. [DOI] [PubMed] [Google Scholar]
- 49.Farina C, Vargas V, Heydari N, et al. Treatment with glatiramer acetate induces specific IgG4 antibodies in multiple sclerosis patients. J Neuroimmunol. 2002 Feb;123( 1–2):188–92. doi: 10.1016/s0165-5728(01)00490-8. [DOI] [PubMed] [Google Scholar]
- 50.Kappos L, Clanet M, Sandberg-Wollheim M, et al. Neutralizing antibodies and efficacy of interferon beta-1a: a 4-year controlled study. Neurology. 2005 Jul 12;65( 1):40–7. doi: 10.1212/01.wnl.0000171747.59767.5c. [DOI] [PubMed] [Google Scholar]
- 51.Karussis D, Teitelbaum D, Sicsic C, et al. Long-term treatment of multiple sclerosis with glatiramer acetate: natural history of the subtypes of anti-glatiramer acetate antibodies and their correlation with clinical efficacy. J Neuroimmunol. 2010 Mar 30;220( 1–2):125–30. doi: 10.1016/j.jneuroim.2010.01.009. [DOI] [PubMed] [Google Scholar]
- 52.Brenner T, Arnon R, Sela M, et al. Humoral and cellular immune responses to Copolymer 1 in multiple sclerosis patients treated with Copaxone. J Neuroimmunol. 2001 Apr 2;115( 1–2):152–60. doi: 10.1016/s0165-5728(01)00250-8. [DOI] [PubMed] [Google Scholar]
- 53.Ure DR, Rodriguez M. Polyreactive antibodies to glatiramer acetate promote myelin repair in murine model of demyelinating disease. FASEB J. 2002 Aug;16( 10):1260–2. doi: 10.1096/fj.01-1023fje. [DOI] [PubMed] [Google Scholar]
- 54.Weber MS, Prod’homme T, Patarroyo JC, et al. B-cell activation influences T-cell polarization and outcome of anti-CD20 B-cell depletion in central nervous system autoimmunity. Ann Neurol. 2010 Sep;68( 3):369–83. doi: 10.1002/ana.22081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hauser SL, Waubant E, Arnold DL, et al. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N Engl J Med. 2008 Feb 14;358( 7):676–88. doi: 10.1056/NEJMoa0706383. [DOI] [PubMed] [Google Scholar]
- 56.Bar-Or A, Fawaz L, Fan B, et al. Abnormal B-cell cytokine responses a trigger of T-cell-mediated disease in MS? Ann Neurol. 2010 Apr;67( 4):452–61. doi: 10.1002/ana.21939. [DOI] [PubMed] [Google Scholar]
- 57.Fillatreau S, Sweenie CH, McGeachy MJ, et al. B cells regulate autoimmunity by provision of IL-10. Nat Immunol. 2002 Oct;3( 10):944–50. doi: 10.1038/ni833. [DOI] [PubMed] [Google Scholar]
- 58.Mann MK, Maresz K, Shriver LP, et al. B cell regulation of CD4+ CD25+ T regulatory cells and IL-10 via B7 is essential for recovery from experimental autoimmune encephalomyelitis. J Immunol. 2007 Mar 15;178( 6):3447–56. doi: 10.4049/jimmunol.178.6.3447. [DOI] [PubMed] [Google Scholar]
- 59.Bouaziz JD, Yanaba K, Tedder TF. Regulatory B cells as inhibitors of immune responses and inflammation. Immunol Rev. 2008 Aug;224:201–14. doi: 10.1111/j.1600-065X.2008.00661.x. [DOI] [PubMed] [Google Scholar]
- 60.De Smedt T, Van Mechelen M, De Becker G, et al. Effect of interleukin-10 on dendritic cell maturation and function. Eur J Immunol. 1997 May;27( 5):1229–35. doi: 10.1002/eji.1830270526. [DOI] [PubMed] [Google Scholar]
- 61.Li Q, Milo R, Panitch H, et al. Glatiramer acetate blocks the activation of THP-1 cells by interferon-gamma. Eur J Pharmacol. 1998 Jan 26;342( 2–3):303–10. doi: 10.1016/s0014-2999(97)01509-4. [DOI] [PubMed] [Google Scholar]
- 62.Hussien Y, Sanna A, Soderstrom M, et al. Glatiramer acetate and IFN-beta act on dendritic cells in multiple sclerosis. J Neuroimmunol. 2001 Dec 3;121( 1–2):102–10. doi: 10.1016/s0165-5728(01)00432-5. [DOI] [PubMed] [Google Scholar]
- 63.Jung S, Siglienti I, Grauer O, et al. Induction of IL-10 in rat peritoneal macrophages and dendritic cells by glatiramer acetate. J Neuroimmunol. 2004 Mar;148( 1–2):63–73. doi: 10.1016/j.jneuroim.2003.11.014. [DOI] [PubMed] [Google Scholar]
- 64.Sanna A, Fois ML, Arru G, et al. Glatiramer acetate reduces lymphocyte proliferation and enhances IL-5 and IL-13 production through modulation of monocyte-derived dendritic cells in multiple sclerosis. Clin Exp Immunol. 2006 Feb;143( 2):357–62. doi: 10.1111/j.1365-2249.2006.02997.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Carpintero R, Brandt KJ, Gruaz L, et al. Glatiramer acetate triggers PI3Kdelta/Akt and MEK/ERK pathways to induce IL-1 receptor antagonist in human monocytes. Proc Natl Acad Sci U S A. 2010 Oct 12;107( 41):17692–7. doi: 10.1073/pnas.1009443107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bettelli E, Carrier Y, Gao W, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006 May 11;441( 7090):235–8. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 67.Korn T, Mitsdoerffer M, Croxford AL, et al. IL-6 controls Th17 immunity in vivo by inhibiting the conversion of conventional T cells into Foxp3+ regulatory T cells. Proc Natl Acad Sci U S A. 2008 Nov 25;105( 47):18460–5. doi: 10.1073/pnas.0809850105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Korn T, Oukka M, Kuchroo V, et al. Th17 cells: effector T cells with inflammatory properties. Semin Immunol. 2007 Dec;19( 6):362–71. doi: 10.1016/j.smim.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mombaerts P, Iacomini J, Johnson RS, et al. RAG-1-deficient mice have no mature B and T lymphocytes. Cell. 1992 Mar 6;68( 5):869–77. doi: 10.1016/0092-8674(92)90030-g. [DOI] [PubMed] [Google Scholar]
- 70.Aharoni R, Kayhan B, Eilam R, et al. Glatiramer acetate-specific T cells in the brain express T helper 2/3 cytokines and brain-derived neurotrophic factor in situ. Proc Natl Acad Sci U S A. 2003 Nov 25;100( 24):14157–62. doi: 10.1073/pnas.2336171100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Allie R, Hu L, Mullen KM, et al. Bystander modulation of chemokine receptor expression on peripheral blood T lymphocytes mediated by glatiramer therapy. Arch Neurol. 2005 Jun;62( 6):889–94. doi: 10.1001/archneur.62.6.889. [DOI] [PubMed] [Google Scholar]
- 72.Zhang M, Chan CC, Vistica B, et al. Copolymer 1 inhibits experimental autoimmune uveoretinitis. J Neuroimmunol. 2000 Mar 1;103( 2):189–94. doi: 10.1016/s0165-5728(99)00239-8. [DOI] [PubMed] [Google Scholar]
- 73.Gur C, Karussis D, Golden E, et al. Amelioration of experimental colitis by Copaxone is associated with class-II-restricted CD4 immune blocking. Clin Immunol. 2006 Feb-Mar;118(2–3):307–16. doi: 10.1016/j.clim.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 74.Arnon R, Aharoni R. Mechanism of action of glatiramer acetate in multiple sclerosis and its potential for the development of new applications. Proc Natl Acad Sci U S A. 2004 Oct 5;101(Suppl 2):14593–8. doi: 10.1073/pnas.0404887101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kerschensteiner M, Stadelmann C, Dechant G, et al. Neurotrophic cross-talk between the nervous and immune systems: implications for neurological diseases. Ann Neurol. 2003 Mar;53( 3):292–304. doi: 10.1002/ana.10446. [DOI] [PubMed] [Google Scholar]
- 76.Riley CP, Cope TC, Buck CR. CNS neurotrophins are biologically active and expressed by multiple cell types. J Mol Histol. 2004 Nov;35( 8–9):771–83. doi: 10.1007/s10735-004-0778-9. [DOI] [PubMed] [Google Scholar]
- 77.Kerschensteiner M, Gallmeier E, Behrens L, et al. Activated human T cells, B cells, and monocytes produce brain-derived neurotrophic factor in vitro and in inflammatory brain lesions: a neuroprotective role of inflammation? J Exp Med. 1999 Mar 1;189( 5):865–70. doi: 10.1084/jem.189.5.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Stadelmann C, Kerschensteiner M, Misgeld T, et al. BDNF and gp145trkB in multiple sclerosis brain lesions: neuroprotective interactions between immune and neuronal cells? Brain. 2002 Jan;125( Pt 1):75–85. doi: 10.1093/brain/awf015. [DOI] [PubMed] [Google Scholar]
- 79.Maier K, Kuhnert AV, Taheri N, et al. Effects of glatiramer acetate and interferon-beta on neurodegeneration in a model of multiple sclerosis: a comparative study. Am J Pathol. 2006 Oct;169( 4):1353–64. doi: 10.2353/ajpath.2006.060159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Aharoni R, Arnon R, Eilam R. Neurogenesis and neuroprotection induced by peripheral immunomodulatory treatment of experimental autoimmune encephalomyelitis. J Neurosci. 2005 Sep 7;25( 36):8217–28. doi: 10.1523/JNEUROSCI.1859-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gilgun-Sherki Y, Panet H, Holdengreber V, et al. Axonal damage is reduced following glatiramer acetate treatment in C57/bl mice with chronic-induced experimental autoimmune encephalomyelitis. Neurosci Res. 2003 Oct;47( 2):201–7. doi: 10.1016/s0168-0102(03)00217-7. [DOI] [PubMed] [Google Scholar]
- 82.Liu J, Johnson TV, Lin J, et al. T cell independent mechanism for copolymer-1-induced neuroprotection. Eur J Immunol. 2007 Nov;37( 11):3143–54. doi: 10.1002/eji.200737398. [DOI] [PubMed] [Google Scholar]
- 83.Azoulay D, Vachapova V, Shihman B, et al. Lower brain-derived neurotrophic factor in serum of relapsing remitting MS: reversal by glatiramer acetate. J Neuroimmunol. 2005 Oct;167( 1–2):215–8. doi: 10.1016/j.jneuroim.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 84.Filippi M, Rovaris M, Rocca MA, et al. Glatiramer acetate reduces the proportion of new MS lesions evolving into ‘black holes’. Neurology. 2001 Aug 28;57( 4):731–3. doi: 10.1212/wnl.57.4.731. [DOI] [PubMed] [Google Scholar]
- 85.Khan O, Shen Y, Bao F, et al. Long-term study of brain 1H-MRS study in multiple sclerosis: effect of glatiramer acetate therapy on axonalmetabolic function and feasibility of long- Term H-MRS monitoring in multiple sclerosis. J Neuroimaging. 2008 Jul;18( 3):314–9. doi: 10.1111/j.1552-6569.2007.00206.x. [DOI] [PubMed] [Google Scholar]
- 86.Franklin RJ. Why does remyelination fail in multiple sclerosis? Nat Rev Neurosci. 2002 Sep;3( 9):705–14. doi: 10.1038/nrn917. [DOI] [PubMed] [Google Scholar]
- 87.Lalive PH, Paglinawan R, Biollaz G, et al. TGF-beta-treated microglia induce oligodendrocyte precursor cell chemotaxis through the HGF-c-Met pathway. Eur J Immunol. 2005 Mar;35( 3):727–37. doi: 10.1002/eji.200425430. [DOI] [PubMed] [Google Scholar]
- 88.Noseworthy JH, Lucchinetti C, Rodriguez M, et al. Multiple sclerosis. N Engl J Med. 2000;343( 13):938–52. doi: 10.1056/NEJM200009283431307. [DOI] [PubMed] [Google Scholar]
- 89.Wolinsky JS. The diagnosis of primary progressive multiple sclerosis. J Neurol Sci. 2003 Feb 15;206( 2):145–52. doi: 10.1016/s0022-510x(02)00346-5. [DOI] [PubMed] [Google Scholar]
- 90.Wolinsky JS, Narayana PA, O’Connor P, et al. Glatiramer acetate in primary progressive multiple sclerosis: results of a multinational, multicenter, double-blind, placebocontrolled trial. Ann Neurol. 2007 Jan;61( 1):14–24. doi: 10.1002/ana.21079. [DOI] [PubMed] [Google Scholar]
- 91.Butovsky O, Koronyo-Hamaoui M, Kunis G, et al. Glatiramer acetate fights against Alzheimer’s disease by inducing dendritic-like microglia expressing insulin-like growth factor 1. Proc Natl Acad Sci U S A. 2006 Aug 1;103( 31):11784–9. doi: 10.1073/pnas.0604681103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Frenkel D, Maron R, Burt DS, et al. Nasal vaccination with a proteosome-based adjuvant and glatiramer acetate clears beta-amyloid in a mouse model of Alzheimer disease. J Clin Invest. 2005 Sep;115( 9):2423–33. doi: 10.1172/JCI23241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Benner EJ, Mosley RL, Destache CJ, et al. Therapeutic immunization protects dopaminergic neurons in a mouse model of Parkinson’s disease. Proc Natl Acad Sci U S A. 2004 Jun 22;101( 25):9435–40. doi: 10.1073/pnas.0400569101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Angelov DN, Waibel S, Guntinas-Lichius O, et al. Therapeutic vaccine for acute and chronic motor neuron diseases: implications for amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2003 Apr 15;100( 8):4790–5. doi: 10.1073/pnas.0530191100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Aharoni R, Kayhan B, Arnon R. Therapeutic effect of the immunomodulator glatiramer acetate on trinitrobenzene sulfonic acid-induced experimental colitis. Inflamm Bowel Dis. 2005 Feb;11( 2):106–15. doi: 10.1097/00054725-200502000-00003. [DOI] [PubMed] [Google Scholar]
- 96.Aharoni R, Sonego H, Brenner O, et al. The therapeutic effect of glatiramer acetate in a murine model of inflammatory bowel disease is mediated by anti-inflammatory T-cells. Immunol Lett. 2007 Oct 15;112( 2):110–9. doi: 10.1016/j.imlet.2007.07.009. [DOI] [PubMed] [Google Scholar]
- 97.Neesse A, Michl P, Kunsch S, et al. Glatiramer acetate: a novel therapeutic approach in Crohn’s disease? Inflamm Bowel Dis. 2009 Jan;15( 1):156–7. doi: 10.1002/ibd.20537. [DOI] [PubMed] [Google Scholar]
- 98.Zheng B, Switzer K, Marinova E, et al. Exacerbation of autoimmune arthritis by copolymer-I through promoting type 1 immune response and autoantibody production. Autoimmunity. 2008 Aug;41( 5):363–71. doi: 10.1080/08916930801931001. [DOI] [PubMed] [Google Scholar]
- 99.Borel P, Benkhoucha M, Weber MS, et al. Glatiramer acetate treatment does not modify the clinical course of (NZB x BXSB)F1 lupus murine model. Int Immunol. 2008 Oct;20( 10):1313–9. doi: 10.1093/intimm/dxn086. [DOI] [PubMed] [Google Scholar]