

Abstract
Purpose
Inflammation and angiogenesis are important contributors to vascular disease. We evaluated imaging both of these biological processes, using Arg–Gly–Asp (RGD)-conjugated human ferritin nanoparticles (HFn), in experimental carotid and abdominal aortic aneurysm (AAA) disease.
Procedures
Macrophage-rich carotid lesions were induced by ligation in hyperlipidemic and diabetic FVB mice (n=16). AAAs were induced by angiotensin II infusion in apoE−/− mice (n=10). HFn, with or without RGD peptide, was labeled with Cy5.5 and injected intravenously for near-infrared fluorescence imaging.
Results
RGD-HFn showed significantly higher signal than HFn in diseased carotids and AAAs relative to non-diseased regions, both in situ (carotid: 1.88±0.30 vs. 1.17±0.10, p=0.04; AAA: 2.59±0.24 vs. 1.82±0.16, p=0.03) and ex vivo. Histology showed RGD-HFn colocalized with macrophages in carotids and both macrophages and neoangiogenesis in AAA lesions.
Conclusions
RGD-HFn enhances vascular molecular imaging by targeting both vascular inflammation and angiogenesis, and allows more comprehensive detection of high-risk atherosclerotic and aneurysmal vascular diseases.
Keywords: Vascular disease, Nanoparticles, Inflammation, Angiogenesis, Atherosclerosis, Aneurysms, RGD, Ferritin
Introduction
Atherosclerosis is now widely accepted as a chronic systemic vascular inflammatory disorder [1]. The monocyte-derived macrophage is one of the key cellular mediators of this inflammatory process. Macrophages produce proteases such as matrix metalloproteinases and cathepsins, which can degrade extracellular matrix, thin the fibrous cap, and destabilize atherosclerotic plaques [2, 3]. Abdominal aortic aneurysm (AAA) disease, which is a common and highly lethal vascular degenerative disease, is also associated with an inflammatory cell infiltrate and enzymatic degradation of the vessel wall [4]. Biological markers of inflammation have been associated with risk of AAA rupture [5].
Angiogenesis has also been noted as a key biological contributor to vascular disease. In atherosclerosis, intraplaque angiogenesis has been implicated in rapid plaque growth and plaque rupture [6]. For AAA, we and others have shown that vascular angiogenesis is present in animal models of AAA [7, 8], and human data demonstrate increased mural neovascularization within aneurysm tissue and at areas of its rupture foci [9, 10]. Therefore, both vascular macrophage infiltration and neoangiogenesis have emerged as key biological processes for targeted imaging and potential therapy for vascular disease.
The αvβ3 integrin, a cell-surface glycoprotein receptor, is a promising target for detecting both inflammation and angiogenesis. Its expression has been shown to be upregulated on angiogenic vascular endothelial cells [11]. In addition, macrophages infiltrating atherosclerotic plaques express high levels of αvβ3 integrin [12]. Arg–Gly–Asp (RGD) is an extensively studied short amino acid sequence binder of the αvβ3 integrin and RGD-based probes have been used for imaging in models of atherosclerosis [13–15] but not aneurysms.
We and others have shown that nanoscale protein cage architectures, such as virus capsids [16–18], ferritins [19–21], and heat shock proteins [22, 23], are useful platforms for loading and/or synthesis of imaging agents within the interior cavity of their protein cages. In particular, we have shown that engineered human ferritin nanoparticles can be used to image plaque macrophages in murine carotid arteries [24]. As protein cages are genetically encoded materials, a targeting peptide such as RGD can be added to these nanoparticles using well-established genetic engineering techniques. We have previously shown the targeting abilities of RGD-conjugated human ferritin nanoparticles toward macrophages in vitro using fluorescence-activated cell sorting, demonstrating higher interaction than non-targeted human ferritin [25]. In this study, we tested the hypothesis that RGD could enhance human ferritin imaging of experimental vascular inflammation and angiogenesis in murine carotid and AAA disease.
Materials and Methods
Preparation of RGD-Conjugated Human Ferritin Nanoparticles
We used the recombinant human heavy-chain ferritin (HFn) protein cage, which was expressed and purified from Escherichia coli as described previously [19, 20]. The HFn protein cage nanoparticle is composed of 24 identical subunits. We genetically introduced the RGD-4C peptide (CDCRGDCFC) to the subunits as an N-terminal fusion, so that 24 copies of the RGD-4C peptide were displayed on the exterior surface of the HFn protein cage [20, 26]. In transmission electron microscopy and dynamic light scattering analyses, both RGD-4C peptide conjugated HFn (RGD-HFn) and HFn are indistinguishable in size and morphology (12–14 nm in diameter) [20].
In Vitro Study of Aortic Endothelial Cells
Bovine aortic endothelial cells were cultured in Bovine Endothelial Cell Growth Medium (Cell Applications Inc, San Diego, CA) at 37°C and 5% CO2. Cells were plated in a 96-well microplate (5 × 104 cells/well), and cultured for 24 h. Cells were stimulated with lipopolysaccharide (LPS, 1 μg/mL) for 4 h [27], and then incubated with RGD-HFn (RGD+ group, six wells) or HFn (RGD− group, six wells) labeled using fluorescein isothiocyanate (FITC, 54 nmol/mL) as in our prior in vitro experiment [25]. After incubation for 1 h, the fluorescence from the cells (arbitrary units, a.u.) was measured with a fluorescence microplate reader (GENios, Tecan Austria GmbH, Grödig, Austria) calibrated for excitation at 490 nm and emission at 535 nm. Cell-only wells (n=6) were compared as a negative control group.
Animals
All animal procedures were approved by the Administrative Panel on Laboratory Animal Care at Stanford University. All animals were anesthetized with inhaled 2% isoflurane for surgical and imaging procedures and recovered with free access to food and water.
Carotid
Macrophage-rich carotid lesions were induced in FVB mice as described previously [24, 28]. In brief, after 8-week-old male mice were fed a high-fat diet for 4 weeks and rendered diabetic by administration of five daily intraperitoneal injections of streptozotocin, the left common carotid artery was ligated below the bifurcation [29] to develop macrophage-rich neointimal proliferation.
AAA
Murine AAAs were induced in apolipoprotein E-deficient (apoE−/−) mice, as described previously [7]. Male apoE−/− mice on a C57BL/6 background, 11 to 14 weeks of age, were subject to continuous angiotensin II infusion via subcutaneous implanted osmotic mini-pumps [30]. Transabdominal 40 MHz B-mode ultrasound (US) imaging (Vevo 770 Imaging System and RMV 704 microvisualization scan head, Visualsonics Inc, Toronto, ON, Canada) was performed to monitor aortic diameter in vivo (in the longitudinal scan plane) for up to 21 days, as previously described [7], with >25% diameter increase from baseline (0.9–1.0 mm) defined as AAA.
Fluorescence Imaging
We chemically modified the targeted (RGD-HFn) and non-targeted (HFn) nanoparticles to be near-infrared fluorescence (NIRF) imaging agents by conjugating Cy5.5 mono NHS ester (GE Healthcare UK Limited, Buckinghamshire, UK) as described previously [24]. The injected fluorophore molecular weight was adjusted to 8 nmol of Cy5.5 per animal, resulting in 1.0–1.6-mg protein cage per animal.
Carotid
Two weeks after carotid ligation, mice underwent baseline imaging by in vivo fluorescence molecular tomography (FMT) using the FMT™ 2500 system (Visen Inc, Bedford, MA) at 680/700-nm excitation/emission wavelength. Then, they were injected with RGD-HFn-Cy5.5 (RGD+ group, n=8) or HFn-Cy5.5 (RGD− group, n=8) via tail vein and imaged up to 48 h by in vivo FMT. At 48 h, left and right carotid arteries were surgically exposed under inhalational anesthesia (2% isoflurane) and in situ fluorescence imaging was performed using the Maestro™ NIRF imaging system (CRi Inc, Woburn, MA) at 675/690-nm excitation/emission wavelength. Animals were then sacrificed and the carotid arteries were carefully removed en bloc followed by ex vivo fluorescence imaging (Maestro) and immunohistochemistry (see below). Major organs (n=6 in each group for lung, spleen, liver, gall bladder, and kidney; n=4 in each group for colon) were also removed for ex vivo fluorescence imaging to assess the bio-distribution of RGD-HFn and HFn.
AAA
At 14–21 days after mini-pump implantation, AAA mice were injected with RGD-HFn-Cy5.5 (RGD+ group, n=5) or HFn-Cy5.5 (RGD− group, n=5) via tail vein. At 48 h, all mice underwent in situ fluorescence imaging using Maestro after surgical abdominal exposure under inhalational anesthesia (2% isoflurane). Then, the aortae were carefully explanted en bloc followed by ex vivo fluorescence imaging (Maestro) and immunohistochemistry (see below).
Image Analysis
Carotid
For in vivo FMT imaging, fluorescence signal intensity around the carotid region was qualitatively evaluated on two- and three-dimensional images. For in situ and ex vivo imaging, quantitative analysis was performed by placing regions of interest (ROIs) on the ligated left carotid artery and contralateral non-ligated right carotid artery, calculating mean signal intensity within each ROI divided by exposure time (photon counts per second). Similarly, ROIs were placed on ex vivo organs to calculate mean signal intensity divided by exposure time.
AAA
For in situ and ex vivo imaging of AAA mice, ROIs were placed on the aneurysmal segment and a non-aneurysmal remote aortic segment, calculating mean signal intensity within each ROI divided by exposure time.
To assess the enhancement of diseased carotid and AAA relative to non-diseased regions, the mean signal for the left carotid and AAA were normalized to the mean signal of the non-ligated right carotid or non-aneurysmal remote aorta, respectively.
Histology
Carotid
Carotid arteries were cut into two 3-mm sections. These sections were embedded immediately in optimum cutting temperature compound (Sakura Finetek USA, Inc., Torrance, CA) and flash frozen in liquid nitrogen. Frozen sections (5 μm thick) were fixed in acetone for 10 min at 4°C. After sections were washed in PBS, they were incubated overnight at 4°C with anti-mouse Mac-3 antibody to stain for macrophages (BD Biosciences, San Jose, CA) or anti-mouse CD-31 antibody to stain for endothelial cells (BD Biosciences) or anti-mouse smooth muscle actin (SMA) antibody to stain for smooth muscle cells (SMCs) (Abeam Inc., Cambridge, MA). Sections were then incubated for 30 min at room temperature with biotinylated secondary antibodies. Antigen–antibody conjugates were detected with avidinbiotin-horseradish peroxidase complex (Vector Laboratories, Burlingame, CA) according to the manufacturer's instructions. 3-amino-9-ethylcarbazole was used as chromogen. Sections were counterstained with hematoxylin. For immunofluorescence double staining to examine colocalization of RGD-HFn-Cy5.5 or HFn-Cy5.5 and macrophages or SMCs, sections were incubated with anti-mouse Mac-3 antibody or anti-mouse SMA antibody overnight at 4°C and stained with Alexa Fluor 488-conjugated anti-rat IgG (Molecular Probes, Eugene, OR) for 2 h at room temperature. Sections were stained with DAPI (Sigma-Aldrich) to visualize cell nuclei, and then observed under confocal microscopy (Zeiss LSM 510, Carl Zeiss AG, Oberkochen, Germany). ImageJ 1.43 software (NIH Image) was used to quantify the colocalization (from five sections of each group), with the area of double staining expressed as a percentage of the total area of macrophage or SMC staining.
AAA
The frozen sections of aneurysmal aortic segments were also prepared as above. Sections were incubated overnight at 4°C with anti-mouse CD-11b antibody to stain for macrophages (BD Biosciences) or anti-mouse CD-31 antibody to stain for endothelial cells (BD Biosciences) or anti-mouse SMA antibody to stain for SMCs (Abeam Inc.), followed by incubation with biotinylated secondary antibodies, color development, and counterstaining as in the carotid model. For immunofluorescence double staining to examine colocalization of RGD-HFn-Cy5.5 or HFn-Cy5.5 and macrophages or endothelial cells or SMCs, after incubation with anti-mouse CD-11b antibody or anti-mouse CD-31 antibody or anti-mouse SMA antibody overnight at 4°C, sections were stained and observed under microscopy as in the carotid model. As with the carotids, ImageJ software was used to quantify the colocalization (from five sections of each group), with the area of double staining expressed as a percentage of the total area of macrophage or endothelial cell or SMC staining.
Statistical Analysis
All data were expressed as mean±SEM (standard error of the mean). A paired Student's t test was used to compare ligated left carotid vs. non-ligated right carotid arteries and to compare AAA vs. non-aneurysmal remote segments. An unpaired t test was used to compare RGD+ vs. RGD− groups. All analyses were performed using JMP 7.0.1 statistical software (SAS Institute Inc, Cary, NC). A two-tailed P value of <0.05 was considered statistically significant.
Results
RGD Enhances Binding to Aortic Endothelial Cells In Vitro
Incubating non-targeted HFn with activated aortic endothelial cells did not show a significant change in fluorescence signal compared to cell-only controls (0.016±0.003 vs. 0.015±0.002 a.u., p = NS). However, when endothelial cells were incubated with RGD-HFn, there was a significant increase in fluorescence signal (0.040±0.002 a.u., p<0.0001 vs. both HFn and controls).
Fluorescence Imaging After In Vivo Administration of RGD-HFn and HFn
Carotid
FMT showed strong in vivo carotid signal in RGD+ mice compared to more limited signal in RGD− mice (Fig. 1). In situ quantitative imaging demonstrated significantly higher signal of ligated left carotid arteries compared to the non-ligated right carotid arteries (Fig. 2a) in the RGD+ group (0.023±0.005 vs. 0.015±0.003 counts/s, p=0.01), while the difference did not reach statistical significance in the RGD− group (0.018±0.003 vs. 0.015±0.002 counts/s, p=0.17). Ex vivo imaging further demonstrated significantly higher signal from the left vs. right carotids (Fig. 2a) in the RGD+ group (0.011±0.002 vs. 0.003±0.001 counts/s, p=0.005), as well as in the RGD− group (0.007±0.003 vs. 0.004±0.001 counts/s, p=0.02). Comparing RGD+ and RGD− groups (Fig. 2b), normalized signal intensity of the ligated left carotid lesions was significantly higher in the RGD+ group, both in situ (p=0.04) and ex vivo (p=0.03). The organ bio-distribution analysis, based on ex vivo fluorescence, showed similar distribution between RGD-HFn and HFn (p = NS), with the dominant accumulation in the liver, gall bladder, and kidney (Supplemental Fig. 1).
Fig. 1.

In vivo fluorescence molecular tomography (FMT) of RGD-HFn-Cy5.5 and HFn-Cy5.5 in murine carotid arteries. Both two-and three-dimensional images showed strong fluorescence signal (arrows) at 48 h around the ligated left carotid artery in RGD+ mice (injected with RGD-HFn-Cy5.5), but more limited carotid signal in RGD− mice (injected with HFn-Cy5.5).
Fig. 2.

In situ and ex vivo fluorescence imaging of RGD-HFn-Cy5.5 and HFn-Cy5.5 in murine carotid arteries. a Both in situ and ex vivo images showed enhanced fluorescence signal from the ligated left carotid artery (LCA) in RGD+ mice (injected with RGD-HFn-Cy5.5), compared to RGD− mice (injected with HFn-Cy5.5). (In situ images: supine position with the head on the left, RCA right carotid artery). b Quantitative analysis showed RGD+ mice with significantly higher LCA signal (normalized to the non-ligated RCA) compared to RGD− mice, both in situ and ex vivo. *p=0.04 vs. RGD− group, † p=0.03 vs. RGD− group.
AAA
In situ and ex vivo imaging (Fig. 3a) both showed that AAAs had significantly higher signal than non-aneurysmal remote segments, both in the RGD+ group (in situ, 0.057±0.006 vs. 0.022±0.001 counts/s, p=0.003; ex vivo, 0.042±0.006 vs. 0.013±0.002 counts/s, p=0.006) and the RGD− group (in situ, 0.052±0.005 vs. 0.029±0.003 counts/s, p=0.006; ex vivo, 0.029±0.002 vs. 0.012±0.002 counts in situ, p=0.004). Comparing RGD+ and RGD− groups (Fig. 3b), normalized signal intensity of AAAs was significantly higher in the RGD+ group, both in situ (p=0.03) and ex vivo (p=0.03). Note the US-determined AAA diameter obtained just prior to imaging was not significantly different between the RGD+ and RGD− groups (1.82±0.10 mm vs. 1.65±0.13 mm, p=0.32).
Fig. 3.

In situ and ex vivo fluorescence imaging of RGD-HFn-Cy5.5 and HFn-Cy5.5 in murine abdominal aortic aneurysms (AAA). a Both in situ and ex vivo images showed enhanced AAA signal in RGD+ mice (injected with RGD-HFn-Cy5.5) compared to RGD− mice (injected with HFn-Cy5.5). (In situ images: supine position with the head on the left, dotted lines outline aortic position). b Quantitative analysis showed RGD+ mice with significantly higher AAA signal (normalized to non-diseased aorta) compared to RGD− mice, both in situ and ex vivo. *p=0.03 vs. RGD− group.
Histology
Carotid
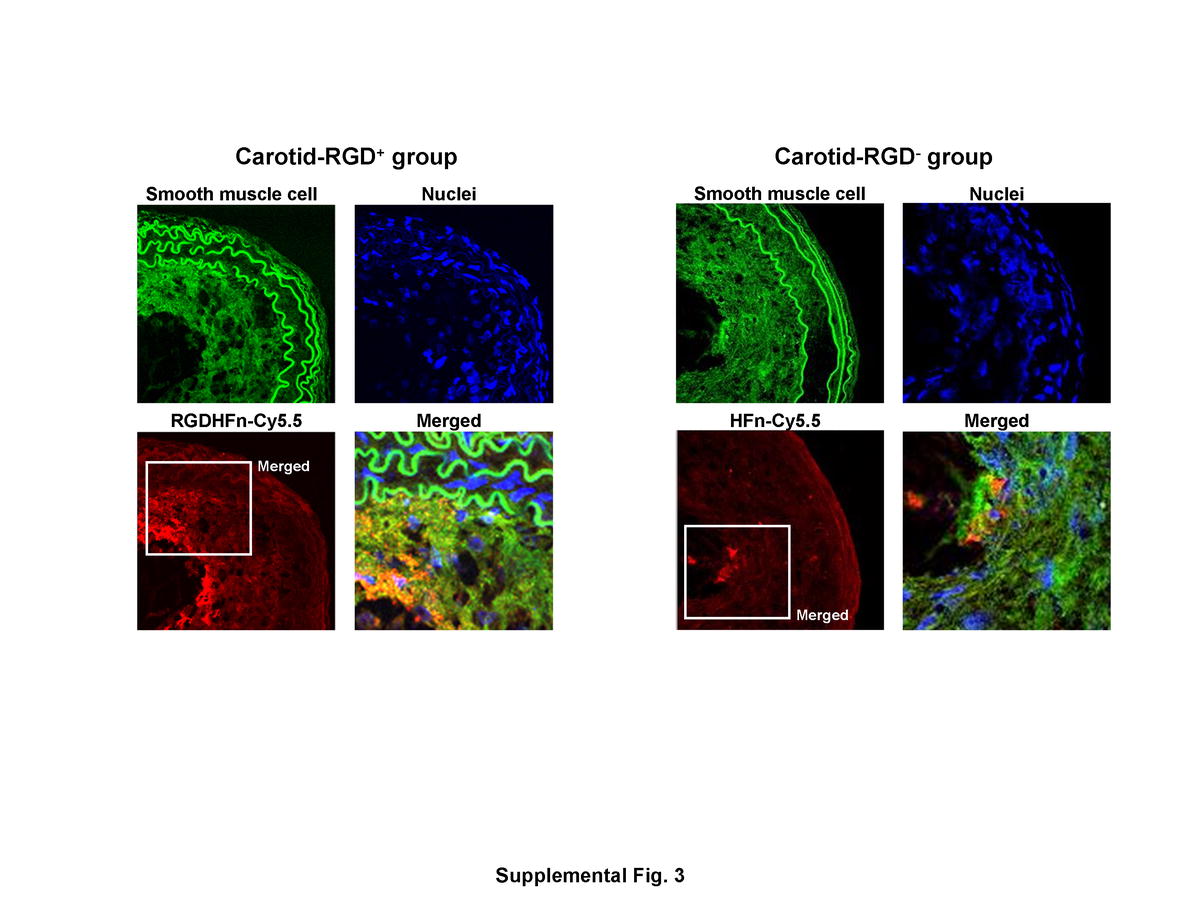
Mac-3 immunohistochemistry demonstrated substantial macrophage infiltration in the neointima and adventitia of the ligated left carotid arteries (Fig. 4). Immunofluorescence staining exhibited stronger Cy5.5 signal of the neointima in the RGD+ group than in the RGD− group (Fig. 4), with greater colocalization of Cy5.5 and macrophages in the RGD+ group (38±3 area% vs. 19±2 area% for RGD−, p=0.0002). Immunofluorescence staining of non-diseased right carotid arteries confirmed minimal macrophage and Cy5.5 signal (Supplemental Fig. 2). CD-31 immunohistochemistry showed positive staining only in the expected endothelial cell lining of the vessel lumen, but was nearly absent in the neointima and adventitia (Fig. 4). Carotid SMCs showed more limited colocalization (Supplemental Fig. 3), which was enhanced in the RGD+ group (15±2 area% vs. 7±2 area% for RGD−, p=0.005).
Fig. 4.

Macrophage and endothelial cell immunostaining of representative ligated left carotid arteries. Immunohistochemical staining of the ligated left carotid lesions showed substantial macrophages infiltrating the neointima (two-headed arrows) and peri-adventitia (arrows). Immunofluorescence carotid staining of the outlined region demonstrated more intense RGD-HFn-Cy5.5 signal (red), colocalizing with neointimal macrophages (green; arrowheads), as compared to HFn-Cy5.5 (see text for quantitative colocalization analysis). Immunohistochemical carotid staining also showed positive endothelial cell staining along the luminal border, but few endothelial cells in the neointima and adventitia.
AAA
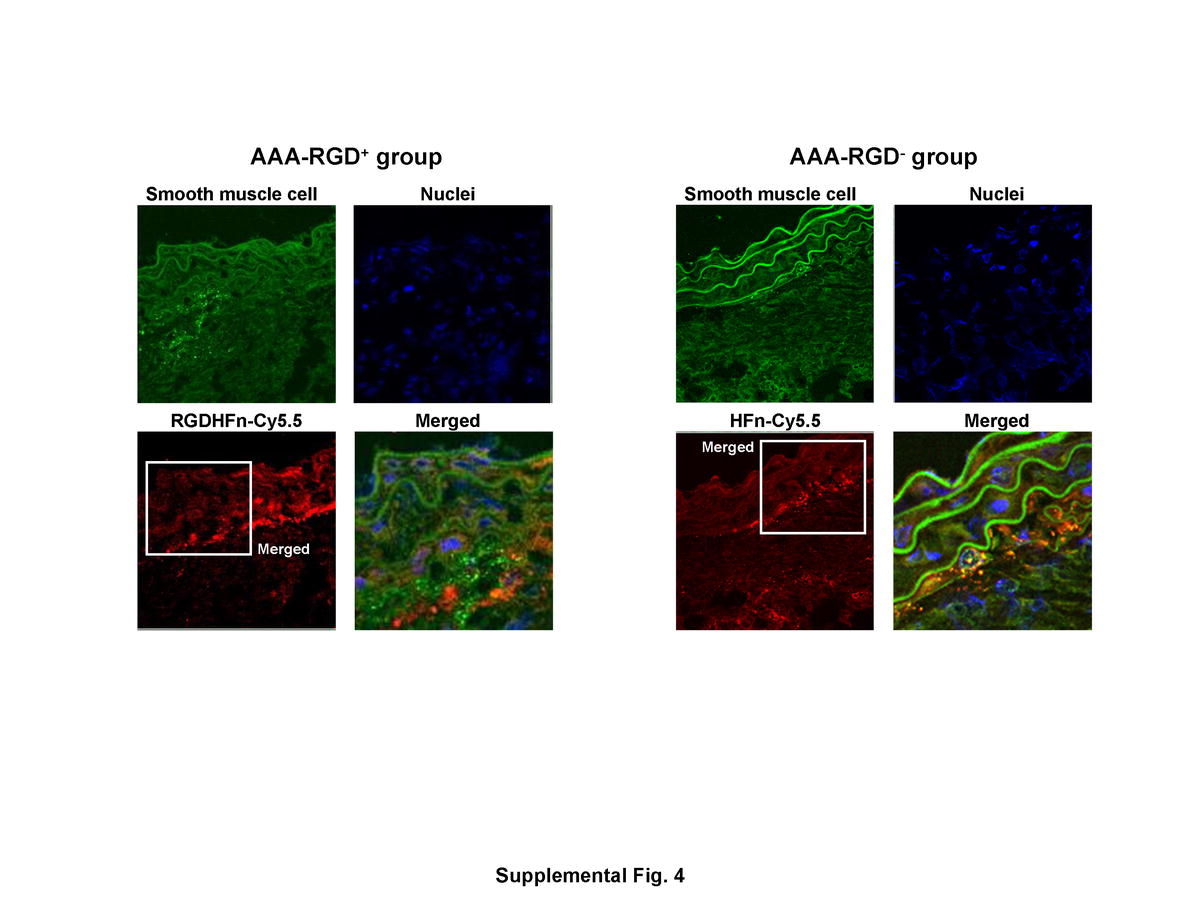
CD-11b immunohistochemistry demonstrated mural macrophage infiltration in the AAA (Fig. 5). Immunofluorescence staining exhibited stronger Cy5.5 signal of the media and adventitia in the RGD+ group than in the RGD− group (Fig. 5), with greater colocalization of Cy5.5 and macrophages in the RGD+ group (55±7 area% vs. 26±2 area% for RGD−, p=0.003). CD-31 immunohistochemistry demonstrated the expression of endothelial cells along the lumen and within the AAA wall (Fig. 6), with immunofluorescence staining exhibiting stronger Cy5.5 signal within the adventitia in the RGD+ group and greater colocalization (52±5 area% vs. 29±4 area% for RGD−, p=0.007). AAA SMCs showed more limited colocalization (Supplemental Fig. 4), which was enhanced in the RGD+ group (17±2 area% vs. 10±2 area% for RGD−, p=0.03).
Fig. 5.

Macrophage immunostaining of representative abdominal aortic aneurysms (AAA). Immunohistochemical AAA staining showed mural macrophage infiltration, with immunofluorescence staining of the outlined region demonstrating the more intense RGD-HFn-Cy5.5 signal (red), colocalizing with medial and adventitial macrophages (green; arrowheads) as compared to HFn-Cy5.5 (see text for quantitative colocalization analysis).
Fig. 6.

Endothelial cell immunostaining of representative abdominal aortic aneurysms (AAA). Immunohistochemical AAA staining showed mural endothelial cell expression, with immunofluorescence staining of the outlined region demonstrating the more intense RGD-HFn-Cy5.5 signal (red), colocalizing with the adventitial endothelial cells (asterisk, green; arrowheads), as compared to HFn-Cy5.5 (see text for quantitative colocalization analysis).
Discussion
Using experimental models of carotid and AAA disease, we have demonstrated that HFn molecular imaging of vascular macrophages and angiogenic endothelial cells can be enhanced with RGD targeting. To the best of our knowledge, this is the first study to show a genetically engineered human protein cage for targeted vascular inflammation and angiogenesis imaging. It is also the first use of RGD for targeted molecular imaging of aortic aneurysm disease. Our current results provide further evidence that the protein cage nanoparticles, HFn in particular, are versatile platforms for molecular/cellular imaging of vascular disease. Thus, targeting of HFn with RGD may allow the assessment of both vulnerable atherosclerotic plaques and AAAs prone to rupture.
We and others have reported previously that ferritin itself accumulates in atherosclerotic macrophages [24, 31, 32] and may serve as an intrinsic macrophage imaging agent even without additional macrophage targeting moieties [19, 24]. While we also saw macrophage uptake of non-targeted HFn in the current study, this was significantly enhanced by targeting with RGD, in line with our prior in vitro data, where RGD-HFn was internalized by macrophages [25].
Our results in vitro and in the AAA model also show that RGD-HFn has the added advantage of being able to detect activated or “angiogenic” endothelial cells. The αvβ3 integrin is a biomarker of angiogenic endothelial cells as it is upregulated on activated, proliferating endothelial cells involved in angiogenesis while essentially unexpressed on mature, quiescent cells [33]. This matched our histological findings in the AAA model, where RGD-HFn colocalized with the adventitial CD-31+ cells but not the endothelial cells along the lumen.
As inflammation and angiogenesis can coexist in vascular lesions, it may be difficult to distinguish their relative contribution to HFn uptake. Inflammation and angiogenesis may work synergistically to increase the risk of plaque or aortic aneurysm rupture, so it may be advantageous to detect these two high-risk features of vascular lesions with a single imaging examination using RGD-HFn. The αvβ3 integrin can also be expressed on blood vessel cells other than macrophages and endothelial cells, such as SMCs [11, 12]. We did find SMCs contributed to RGD-HFn binding, but to a limited degree.
In the current study, we examined the imaging capabilities of RGD-HFn and HFn by conjugating to Cy5.5. This was detected noninvasively in the carotid model using FMT, which is more sensitive than conventional NIRF imaging [34], but more challenging to quantify due to limited spatial resolution, tissue penetration, and lack of anatomic reference. The deeper AAA has been difficult to detect by FMT in our experience. Co-registration of FMT with CT or MRI could help colocalize vasculature and fluorescence signals [35]. The ex vivo imaging provided the most conclusive data, as even with in situ imaging the background signal can reduce the differences between diseased and non-diseased vessels.
Fluorescence attenuation with depth is a clear limitation to human translation at present. Beyond fluorescence, there has been a strong interest in more clinically available imaging techniques (e.g., MRI, PET, SPECT) for molecular/cellular vascular imaging. HFn can incorporate other imaging agents, such as iron oxides for MRI, as shown previously [24].
Based our prior work with HFn [24], we focused on the 48-h time point for comparing RGD-HFn to HFn. However, serial studies of multiple time points are needed to identify the optimal vascular uptake of RGD-HFn in carotids and AAA, which would be facilitated by a more quantitative noninvasive imaging method. Also, more detailed analysis is needed to determine if RGD-HFn is externally bound vs. internalized in endothelial cells. While the bio-distribution analysis showed similar organ clearance between RGD-HFn and HFn, more data on in vivo elimination rates are needed.
Short-term animal models are a limitation for studying human diseases. While our murine carotid ligation model provides a macrophage-rich vascular lesion, human atherosclerosis is more chronic and complex. In addition, while this model develops abundant inflammation, it does not appear to develop intraplaque angiogenesis. We have shown previously that the angiotensin II-induced AAA model develops both inflammation and angiogenesis, but it is recognized that aortic dissection plays a role. However, aortic surgery is not required in this model, so nonspecific inflammation and angiogenesis are minimized.
Conclusions
In conclusion, the present study demonstrates that targeting human ferritin nanoparticles with RGD significantly enhances vascular inflammation and angiogenesis imaging in experimental models of carotid and AAA disease. Targeted human ferritin is a promising nanoscale imaging platform that may enable comprehensive assessment of high-risk vascular diseases.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We thank Dr. Matthew Bogyo and his laboratory for their assistance with mouse fluorescence molecular tomography. We thank support from the National Institutes of Health (R21 EB005364, R01 HL078678, P50 HL083800).
Footnotes
Disclosures. Dr. McConnell's laboratory receives research support from GE Healthcare and he is on a scientific advisory board for Kowa, Inc. Dr. Dalman's laboratory receives AAA research support from Medtronic AVE and Carolus Pharmaceuticals. The other authors have no potential conflicts of interest.
Electronic supplementary material The online version of this article (doi:10.1007/s11307-011-0495-1) contains supplementary material, which is available to authorized users.
References
- 1.Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–874. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- 2.Galis ZS, Sukhova GK, Lark MW, Libby P. Increased expression of matrix metalloproteinases and matrix degrading activity in vulnerable regions of human atherosclerotic plaques. J Clin Invest. 1994;94:2493–2503. doi: 10.1172/JCI117619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sukhova GK, Shi GP, Simon DI, Chapman HA, Libby P. Expression of the elastolytic cathepsins S and K in human atheroma and regulation of their production in smooth muscle cells. J Clin Invest. 1998;102:576–583. doi: 10.1172/JCI181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shimizu K, Mitchell RN, Libby P. Inflammation and cellular immune response in abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol. 2006;26:987–994. doi: 10.1161/01.ATV.0000214999.12921.4f. [DOI] [PubMed] [Google Scholar]
- 5.Lindholt JS, Vammen S, Fasting H, Henneberg EW, Heickendorff L. The plasma level of matrix metalloproteinase 9 may predict the natural history of small abdominal aortic aneurysms. A preliminary study. Eur J Vasc Endovasc Surg. 2000;20:281–285. doi: 10.1053/ejvs.2000.1151. [DOI] [PubMed] [Google Scholar]
- 6.Virmani R, Kolodgie FD, Burke AP, et al. Atherosclerotic plaque progression and vulnerability to rupture: angiogenesis as a source of intraplaque hemorrhage. Arterioscler Thromb Vasc Biol. 2005;25:2054–2061. doi: 10.1161/01.ATV.0000178991.71605.18. [DOI] [PubMed] [Google Scholar]
- 7.Tedesco MM, Terashima M, Blankenberg FG, et al. Analysis of in situ and ex vivo vascular endothelial growth factor receptor expression during experimental aortic aneurysm progression. Arterioscler Thromb Vasc Biol. 2009;29:1452–1457. doi: 10.1161/ATVBAHA.109.187757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Saraff K, Babamusta F, Cassis LA, Daugherty A. Aortic dissection precedes formation of aneurysms and atherosclerosis in angiotensin II-infused. Apolipoprotein E deficient mice. Arterioscler Thromb Vasc Biol. 2003;23:1621–1426. doi: 10.1161/01.ATV.0000085631.76095.64. [DOI] [PubMed] [Google Scholar]
- 9.Paik DC, Fu C, Bhattacharya J, Tilson MD. Ongoing angio-genesis in blood vessels of the abdominal aortic aneurysm. Exper Mol Med. 2004;36:524–533. doi: 10.1038/emm.2004.67. [DOI] [PubMed] [Google Scholar]
- 10.Choke E, Thompso MM, Dawson J, et al. Abdominal aortic aneurysm rupture is associated with increased medial neovascularization and overexpression of proangiogenic cytokines. Arterioscler Thromb Vasc Biol. 2006;26:2077–2082. doi: 10.1161/01.ATV.0000234944.22509.f9. [DOI] [PubMed] [Google Scholar]
- 11.Hoshiga M, Alpers CE, Smith LL, Giachelli CM, Schwartz SM. Alpha-v beta-3 integrin expression in normal and atherosclerotic artery. Circ Res. 1995;77:1129–1135. doi: 10.1161/01.res.77.6.1129. [DOI] [PubMed] [Google Scholar]
- 12.Antonov AS, Kolodgie FD, Munn DH, Gerrity RG. Regulation of macrophage foam cell formation by alphaVbeta3 integrin: potential role in human atherosclerosis. Am J Pathol. 2004;165:247–258. doi: 10.1016/s0002-9440(10)63293-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Waldeck J, Häger F, Höltke C, et al. Fluorescence reflectance imaging of macrophage-rich atherosclerotic plaques using an alphav-beta3 integrin-targeted fluorochrome. J Nucl Med. 2008;49:1845–1851. doi: 10.2967/jnumed.108.052514. [DOI] [PubMed] [Google Scholar]
- 14.Laitinen I, Saraste A, Weidl E, et al. Evaluation of alphavbeta3 integrin-targeted positron emission tomography tracer 18F-galacto-RGD for imaging of vascular inflammation in atherosclerotic mice. Circ Cardiovasc Imaging. 2009;2:331–338. doi: 10.1161/CIRCIMAGING.108.846865. [DOI] [PubMed] [Google Scholar]
- 15.Winter PM, Morawski AM, Caruthers SD, et al. Molecular imaging of angiogenesis in early-stage atherosclerosis with alpha (v) beta3-integrin-targeted nanoparticles. Circulation. 2003;108:2270–2274. doi: 10.1161/01.CIR.0000093185.16083.95. [DOI] [PubMed] [Google Scholar]
- 16.Liepold L, Anderson S, Willits D, et al. Viral capsids as MRI contrast agents. Magn Reson Med. 2007;58:871–879. doi: 10.1002/mrm.21307. [DOI] [PubMed] [Google Scholar]
- 17.Hooker JM, Datta A, Botta M, Raymond KN, Francis MB. Magnetic resonance contrast agents from viral capsid shells: a comparison of exterior and interior cargo strategies. Nano Lett. 2007;7:2207–2210. doi: 10.1021/nl070512c. [DOI] [PubMed] [Google Scholar]
- 18.Anderson EA, Isaacman S, Peabody DS, Wang EY, Canary JW, Kirshenbaum K. Viral nanoparticles donning a paramagnetic coat: conjugation of MRI contrast agents to the MS2 capsid. Nano Lett. 2006;6:1160–1164. doi: 10.1021/nl060378g. [DOI] [PubMed] [Google Scholar]
- 19.Uchida M, Terashima M, Cunningham CH, et al. A human ferritin iron oxide nano-composite magnetic resonance contrast agent. Magn Reson Med. 2008;60:1073–1081. doi: 10.1002/mrm.21761. [DOI] [PubMed] [Google Scholar]
- 20.Uchida M, Flenniken ML, Allen M, et al. Targeting of cancer cells with ferrimagnetic ferritin cage nanoparticles. J Am Chem Soc. 2006;128:16626–16633. doi: 10.1021/ja0655690. [DOI] [PubMed] [Google Scholar]
- 21.Aime S, Frullano L, Geninatti, Crich S. Compartmentalization of a gadolinium complex in the apoferritin cavity: a route to obtain high relaxivity contrast agents for magnetic resonance imaging. Angew Chem Int Ed. 2002;41:1017–1019. doi: 10.1002/1521-3773(20020315)41:6<1017::aid-anie1017>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 22.Flenniken ML, Willits DA, Brumfield S, Young MJ, Douglas T. The small heat shock protein cage from methanococcus jannaschii is a versatile nanoscale platform for genetic and chemical modification. Nano Lett. 2003;3:1573–1576. [Google Scholar]
- 23.Liepold LO, Abedin MJ, Buckhouse ED, Frank JA, Young MJ, Douglas T. Supramolecular protein cage composite MR contrast agents with extremely efficient relaxivity properties. Nano Lett. 2009;9:4520–4526. doi: 10.1021/nl902884p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Terashima M, Uchida M, Kosuge H, et al. Human ferritin cages for imaging vascular macrophages. Biomaterials. 2011;32:1430–1437. doi: 10.1016/j.biomaterials.2010.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Uchida M, Willits DA, Muller K, et al. Intracellular distribution of macrophage targeting ferritin–iron oxide nanocomposite. Adv Mater. 2009;21:458–462. [Google Scholar]
- 26.Arap W, Pasqualini R, Ruoslahti E. Cancer treatment by targeted drug delivery to tumor vasculature in a mouse model. Science. 1998;279:377–380. doi: 10.1126/science.279.5349.377. [DOI] [PubMed] [Google Scholar]
- 27.Kułdo JM, Ogawara KI, Asgeirsdóttir SA, et al. Molecular pathways of endothelial cell activation for (targeted) pharmacological intervention of chronic inflammatory diseases. Curr Vasc Pharmacol. 2005;3:11–39. doi: 10.2174/1570161052773898. [DOI] [PubMed] [Google Scholar]
- 28.Terashima M, Ehara S, Yang E, et al. In vivo bioluminescence imaging of inducible nitric oxide synthase gene expression in vascular inflammation. Mol Imaging Biol. 2010 doi: 10.1007/s11307-010-0451-5. doi:10.1007/s11307-010-0451-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kumar A, Lindner V. Remodeling with neointima formation in the mouse carotid artery after cessation of blood flow. Arterioscler Thromb Vasc Biol. 1997;17:2238–2244. doi: 10.1161/01.atv.17.10.2238. [DOI] [PubMed] [Google Scholar]
- 30.Daugherty A, Manning MW, Cassis LA. Angiotensin II promotes atherosclerotic lesions and aneurysms in apolipoprotein E-deficient mice. J Clin Invest. 2000;105:1605–1612. doi: 10.1172/JCI7818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pang JH, Jiang MJ, Chen YL, et al. Increased ferritin gene expression in atherosclerotic lesions. J Clin Invest. 1996;97:2204–2212. doi: 10.1172/JCI118661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li W, Xu LH, Forssell C, Sullivan JL, Yuan XM. Over-expression of transferrin receptor and ferritin related to clinical symptoms and destabilization of human carotid plaques. Exp Biol Med. 2008;233:818–826. doi: 10.3181/0711-RM-320. [DOI] [PubMed] [Google Scholar]
- 33.Brooks PC, Clark RA, Cheresh DA. Requirement of vascular integrin alpha v beta 3 for angiogenesis. Science. 1994;264:569–571. doi: 10.1126/science.7512751. [DOI] [PubMed] [Google Scholar]
- 34.Ntziachristos V, Ripoll J, Wang LV, Weissleder R. Looking and listening to light: the evolution of whole-body photonic imaging. Nat Biotechnol. 2005;23:313–320. doi: 10.1038/nbt1074. [DOI] [PubMed] [Google Scholar]
- 35.Nahrendorf M, Waterman P, Thurber G, et al. Hybrid in vivo FMT-CT imaging of protease activity in atherosclerosis with customized nanosensors. Arterioscler Thromb Vasc Biol. 2009;29:1444–1451. doi: 10.1161/ATVBAHA.109.193086. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.